
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of the fragmentation of core-ionised deoxyribose: a study as a function of the tautomeric form
Marie-Anne
Hervé du Penhoat
*a,
Krishna
Kamol Ghose
b,
Marie-Pierre
Gaigeot
bc,
Rodolphe
Vuilleumier
d,
Kentaro
Fujii
e,
Akinari
Yokoya
e and
Marie-Françoise
Politis
b
aIMPMC, Sorbonne Universités - UPMC Univ Paris 06, UMR CNRS 7590, Muséum National d'Histoire Naturelle, IRD UMR 206, 4 Place Jussieu, 75005 Paris, France. E-mail: penhoat@impmc.upmc.fr; Fax: +33 1 44 27 37 85; Tel: +33 1 44 27 75 02
bLAMBE, UMR CNRS 8587, Université d'Evry val d'Essonne, 91025 Evry, France
cInstitut Universitaire de France IUF, 103 Blvd St Michel, 75005 Paris, France
dEcole Normale Supérieure, Département de Chimie, UMR 8640 CNRS-ENS-UPMC, 75005 Paris, France
eAdvance Science Research Center, Japan Atomic Energy Agency, Tokai, Japan
First published on 9th November 2015
Abstract
We have investigated the gas phase fragmentation dynamics following the core ionisation of 2-deoxy-D-ribose (dR), a major component in the DNA chain. To that aim, we use state-of-the-art ab initio Density Functional Theory-based Molecular Dynamics simulations. The ultrafast dissociation dynamics of the core-ionised biomolecule, prior Auger decay, is first modelled for 10 fs to generate initial configurations (atomic positions and velocities) for the subsequent dynamics of the doubly ionised biomolecule in the ground state. The furanose, linear and pyranose conformations of dR were investigated. We show that fragmentation is relatively independent of the atom struck or of the duration of the core vacancy, but depends rather critically on the molecular orbital removed following Auger decay.
1 Introduction
Ionising radiations (electromagnetic radiation, beams of electrons, protons, or heavy ions) are commonly used in cancer therapy to irradiate “risk zones” or tumours. It is well understood that energy deposition by ionising radiation at various DNA sites, such as sugars and bases, can lead to permanent molecular changes, and some of those are known to be the initiating events for the cells inactivation or the formation of chromosomal aberrations. Establishing the origins of radiation induced DNA damage thus seems an important step for the application of these therapies and for the investigation of their possible undesirable side effects.The purpose of this paper is to understand, at the atomic level, the direct effects of ionising radiations on DNA building blocks. Here, we will focus on describing, by means of quantum-classical theoretical methods, the fragmentation processes of core ionised deoxyribose (dR), at the molecular scale, since the induction of core holes in the DNA molecule was shown to lead to major lethal effects in cells.1–6 The overall proposed mechanism is the following: core hole states have a strongly dissociative character. They may induce ultra-fast dissociation reactions in which the nuclear dynamics takes place on time scales comparable to the lifetime of the core vacancies, which is about 8 and 5 fs in the case of carbon and oxygen atoms, respectively.7 Moreover, in the relaxation process following the inner-shell ionisation events, Auger transition probabilities are nearly 100% in DNA constituent atoms, leading to the multi-ionisation of the atom struck and to the emission of two low energy electrons, the photo- and Auger electrons, which can in turn, locally damage DNA.
Many experiments have been performed in the gas phase in order to understand the mechanisms of fragmentation of sugars after K-shell ionisation and/or multiple ionisation events (by ionisation or charge transfer).8–10 Several authors have pointed out that the dissociation process of a molecule after K-shell excitation or ionisation in light atoms does not seem to depend on the position nor on the nature of the core-ionised atom but rather on the molecular orbitals (MO) ionised after Auger decay.10–13 In order to understand this experimental fact, we have simulated core holes dynamics followed, after Auger decay, by double hole dynamics.
Different theoretical approaches have been developed in order to describe the fragmentation processes of doubly ionised biomolecules. In previous studies, we have used Time-Dependent Density Functional Theory (TD-DFT) molecular dynamics (MD) to remove electrons from selected MO and follow in time the nuclear and electrons dynamics.14–16 This method is computationally demanding but provides the physical origin of bond breakage. Born Oppenheimer and Car Parrinello MD of ground state doubly-ionised isolated uracil were also performed at high temperature in order to take into account the energy deposit by the incoming high velocity ion.15 Other methods were recently developed. One of them17 is a simple statistical approach based on the enumeration of assumed equiprobable pathways of breakage of doubly ionised molecules. The other one18 is based, like ours, on molecular dynamics simulations. Initial states are generated with internal energy, i.e. temperature, corresponding to a sampling of energies within a range of estimated energy deposits in the molecule after core ionisation and different Auger decay channels. Starting from these different initial configurations (atomic positions and velocities), ground state double hole MD are performed. These methods provide the different fragmentation pathways but do not describe the driving mechanisms (electron dynamics).
As pointed out in experimental and theoretical works,10 the 2-deoxy-D-ribose molecule may be found under different conformations with regards to the environment (Fig. 1). In the DNA backbone, this monosaccharide is mainly in the furanose form constructed around a 5-membered ring with CH2OH and OH substituents. Such a conformation has been considered in our previous work19 and is worth investigating with regards to the behaviour of DNA building blocks. However, in gas-phase experiments after being heated in an oven, the pyranose form appears dominant20 and comparison with experimental data should take this conformation into account. The 2-deoxy-D-ribose may also exist under a linear form. To understand the impact of the sugars conformation on their dissociation, we have investigated in parallel the fragmentation induced by K-shell ionisation followed by Auger processes for the furanose, pyranose and linear conformations of 2-deoxy-D-ribose.
 | ||
| Fig. 1 Structures of the pyranose, linear and furanose forms of 2-deoxy-D-ribose. | ||
2 Material and methods
We explicitly model core-ionisation of the carbon and oxygen atoms of the sugar as initial electronic conditions, and follow the subsequent dynamics of the ionised sugar by Born–Oppenheimer Molecular Dynamics simulations (BOMD). These simulations were performed with the plane wave Kohn Sham based DFT code CPMD.21 Core electrons were replaced by pseudopotentials of the Trouiller-Martins form.22 The Kleinman–Bylander integration scheme was used for all atom type, and the plane-wave basis set was truncated at an energy cutoff of 90 Rydberg (Ry: 1 Ry = 2.17989 × 1018 J). The exchange and correlation energy was calculated using the generalized gradient approximation (GGA) functional BLYP.23,24 The long-range electrostatic potential was computed using the standard Ewald summation scheme and a compensating background was thus used for the charged simulations. The analysis of the fragments charges were performed in the Bader scheme.25The three different conformations of deoxyribose investigated in this study are presented in Fig. 2. All simulations presented here model the three isomeric forms of 2-deoxy-D-ribose in a cubic box of size L = 20 Å. The initial atomic configurations (positions and velocities) were generated at the end of a 10 ps equilibration run with the Car–Parrinello MD algorithm thermostated at 320 K by a Nosé–Hoover chain. For the pyranose and furanose forms, ten different initial configurations were selected at 484 fs intervals along a neutral BOMD, taking time steps of 0.12 fs. For the linear form, two different initial configurations were selected at 1 ps intervals along a neutral BOMD, taking time steps of 0.12 fs.
 | ||
| Fig. 2 The three deoxyribose conformations investigated: A and B, pyranose; C and D, furanose; and E and F, linear. A, C and E, atom labelling; and B, D and F, HOMO negative (yellow) and positive (orange) contours. C blue, O red, H white. | ||
The dissociation induced by the ionisation of core electrons of dR atoms was modelled in two steps: (1) the ultrafast dissociation reactions induced by the core vacancy, before Auger decay, was simulated first. This step was described by replacing the 1s2 pseudopotential of one atom of the target molecule with a pseudopotential for a 1s1 core-hole state, as detailed in ref. 26. A global charge equal to unity was added to the system to take into account the missing electron. BOMD simulations of the core-ionised system were then performed for 9.6 fs, taking time steps of 0.024 fs. One initial configuration was selected for each dR conformation. Core-holes were placed successively on each of the five carbon and four oxygen atoms of the dR molecule. Consequently, nine core-hole trajectories were generated for each dR conformation. (2) The dissociation dynamics of the doubly-ionized dR molecule, after Auger decay, was then modelled. In the second step, we thus started from atomic configurations generated during the first step (at t = 4.8 fs and t = 9.6 fs) but changing the charge of the system from +1 (step 1) to +2 (step 2, this step). BOMD of ground state dR2+ were performed for several hundreds of femtoseconds, taking time steps of 0.024 fs. Two different dR2+ trajectories were thus generated for each core-hole trajectory, replacing the core hole by a double vacancy at either t = 4.8 fs or t = 9.6 fs.
In parallel, we have investigated the fragmentation of doubly-ionized dR (without prior core ionisation) starting from several different initial configurations and removing the two electrons from the highest occupied MO (HOMO, see Fig. 2). BOMD were performed for several hundreds of femtoseconds, taking time steps of 0.024 fs. These dynamics will provide a benchmark to compare to the fragmentation induced by double ionisation events resulting from core ionisation and subsequent Auger decay.
3 Results and discussion
3.1 Pyranose conformation
The most probable pathway, which corresponds to eight out of ten cases, is illustrated in Fig. 3. In this particular dynamics, the C1C2 and C3O16 bonds first dissociate at 15.3 and 52.5 fs, respectively. In the following, CO and CC bonds will be considered broken when the bond distance exceeds 2 Å. A criteria of 1.5 Å is used for OH bonds.27 A CH2O fragment (30 amu) is thus formed, which is almost neutral (charge about +0.3e). The remaining C4H8O31.7+ fragment is shown to dissociate after 408 fs to form C2H4O20.9+ (60 amu) and C2H4O0.8+ (44 amu) fragments. In all eight cases, the fragmentation of the C1C2 bond is the fastest, between 15.3 to 17.9 fs, while the C3O16 bond breaks after 46 to 168 fs. The second fragmentation step takes place at much longer times and was only observed in two cases, at 408 and 474 fs. The CH2O0.3+ fragment sometimes exited the simulation box. When it was more than 12 Å away from C4H8O31.7+, typically after about 350–410 fs, the C4H8O3 fragment was isolated and BOMD were performed for the doubly ionized system.
 | ||
| Fig. 3 BOMD trajectory initiated from an electronic configuration where two electrons were removed from the HOMO of the deoxyribopyranose molecule (left, t = 0). The C4O3H82+ fragment was isolated during the last 290 fs of the dynamics. Bond breakage positions are marked by arrows. C blue, O red, H white. | ||
The second fragmentation pathway is shown in Fig. 4. In this particular dynamics, the C1C5 bond dissociates at 19.2 fs. This bond was found to dissociate at 18.2 fs, in the other case leading to the same fragmentation pattern. As a consequence, the dR2+ molecule becomes linear and this configuration remains stable within the few picoseconds investigated in this paper.
 | ||
| Fig. 4 BOMD trajectory initiated from an electronic configuration where two electrons were removed from the HOMO of the deoxyribopyranose molecule. C blue, O red, H white. | ||
In conclusion, two different fragmentation pathways are found, initiated by the rupture of either the C1C2 or the C1C5 bond. The HOMO orbital is localized on these two bonds at the beginning of the dynamics (Fig. 2B) and there appears to be a competition in the fragmentation induced in these two bonds by the removal of the HOMO.
In the case of oxygen-K vacancies (labelled O*), O*H bonds remain fairly constant. In contrast, CO* bonds exhibit relatively large increase by 0.08 to 0.33 Å as shown in Table 1. The smallest variations correspond to the localization of the core hole on O16, which is located within the ring, while the largest corresponds to a core hole on O17.
| Atom | Bond | Bond distance Å | ||
|---|---|---|---|---|
| t = 0 | t = 4.8 fs | t = 9.6 fs | ||
| O16 | C2–O16* | 1.47 | 1.51 | 1.59 |
| C3–O16* | 1.44 | 1.48 | 1.61 | |
| C3–O17 | 1.46 | 1.42 | 1.35 | |
| O17 | C3–O17* | 1.46 | 1.55 | 1.79 |
| C2–O16 | 1.47 | 1.49 | 1.54 | |
| C3–O16 | 1.44 | 1.38 | 1.26 | |
| O18 | C5–O18* | 1.46 | 1.52 | 1.68 |
| O19 | C1–O19* | 1.44 | 1.50 | 1.66 |
When the core hole is located on C1, C4 or C5 during either 4.8 fs or 9.6 fs, or on C3 and O17 during 4.8 fs before double ionisation, the dR2+ molecule breaks at the C1C5 bond and remains linear. The dissociation of dR2+ is thus not modified by the slight changes in the molecules geometry induced by the presence of the core vacancy.
When the core hole is located on C2, O16, O18 or O19 during either 4.8 or 9.6 fs, or on C3 during 9.6 fs before double ionisation, the dR2+ molecule breaks at the C1C2 and C3O16 bonds, leading to the formation of CH2O0.3+ (30 amu) and C4H8O31.7+ (104 amu) fragments. In the case of core holes on O16 (4.8 fs), O18 (9.6 fs) and O19 (4.8 and 9.6 fs), the remaining linear fragment further dissociates after about 400 fs to form C2H4O0.8+ (44 amu) and C2H4O20.9+ (60 amu) fragments. These fragmentation patterns were also observed in dR2+, without prior core ionisation, but starting from other initial configurations. It thus appears, that the molecules geometry modifications induced by the presence of these core vacancies favour the second identified dissociation pathway for dR2+ in its ground state.
Finally, when a core hole is placed on O17 during 9.6 fs, the fragmentation pattern is completely different, as shown in Fig. 5. First, the C3O17 bond dissociates at t = 13.7 fs to yield an OH fragment, then the C1C5 bond dissociates (t = 29.1 fs). This is followed by the dissociation of the O18H13 bond at t = 57.4 fs. The H13 atom reacts with the OH fragment to form an H2O molecule. Finally the C5C4 bond breaks at t = 241 fs, releasing a CHO fragment. This whole set of events leads to the formation of 3 fragments: H2O0.2+ (18 amu), CHO0.8+ (29 amu) and C4H7O2+ (87 amu). The presence of a core vacancy on O17 during 9.6 fs induced an increase of the C3O17* bond by about 0.3 Å (see Table 1), which could be at the origin of this new fragmentation pathway for the subsequent dR2+ dynamics. This case corresponds to the largest CO* bond increase observed during the core hole state for deoxyribopyranose.
 | ||
| Fig. 5 Fragmentation dynamics of deoxyribopyranose, after a core hole was placed for 9.6 fs on O17. C blue, O red, H white. | ||
As a conclusion, in all cases but one, the fragmentation pathways are the same as those found for ground state dR2+, without prior core ionisation.
3.2 Furanose conformation
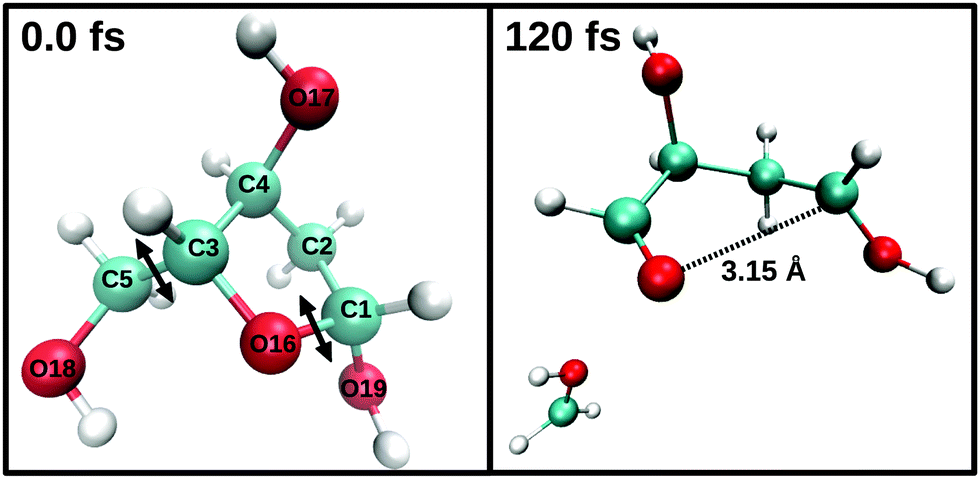 | ||
| Fig. 6 BOMD trajectory initiated from an electronic configuration where two electrons were removed from the HOMO of the deoxyribofuranose molecule. C blue, O red, H white. | ||
In contrast, oxygen core-holes (O*) induce significant CO* bond elongation as summarized in Table 2. The smallest bond elongation is observed in the case of the O16 K-hole, which is located in the furanose ring. This is accompanied by a reduction in the C1O19 bond by 0.14 Å at t = 9.6 fs. The largest increase is observed when a core hole is placed on O19, corresponding to an elongation of the C1O19 bond by about 0.30 Å at 9.6 fs. It also induces variations in the bonds involving O16.
| Atom | Bond | Bond distance Å | ||
|---|---|---|---|---|
| t = 0 | t = 4.8 fs | t = 9.6 fs | ||
| O16 | C1–O16* | 1.47 | 1.54 | 1.63 |
| C3–O16* | 1.49 | 1.51 | 1.65 | |
| C1–O19 | 1.45 | 1.42 | 1.31 | |
| O17 | C4–O17* | 1.48 | 1.54 | 1.68 |
| O18 | C5–O18* | 1.44 | 1.50 | 1.64 |
| O19 | C1–O19* | 1.45 | 1.56 | 1.76 |
| C1–O16 | 1.47 | 1.41 | 1.30 | |
| C3–O16 | 1.49 | 1.52 | 1.57 | |
Most interestingly, we observed a new fragmentation pattern when the K-shell vacancy was placed on O18 during 9.6 fs before the dR2+ dynamics, as shown in Fig. 7. The trajectory reveals that the C3C4 and C1O16 bonds first break at 23.6 and 41.9 fs, respectively. Two fragments are thus formed, C2H4O2 and C3H6O2. The smallest fragment breaks at the C3C5 bond at 102 fs. As a results, this dynamics leads to the formation of CHO0.25+ with mass 29 amu, CH3O0.25+ with mass 31 amu and C3H6O21.5+ with mass 74 amu.
 | ||
| Fig. 7 Fragmentation dynamics of the furanose conformation of dR2+, after a core hole was placed for 9.6 fs on O18. C blue, O red, H white. | ||
3.3 Linear conformation
 | ||
| Fig. 8 BOMD trajectory initiated from an electronic configuration where two electrons were removed from the HOMO of the linear dR conformation. C blue, O red, H white. | ||
In the second pathway, the C1C5 bond breaks leading to the formation of C2O2H5+ and C2O3H5+ fragments.
 | ||
| Fig. 9 Fragmentation of the linear conformation of dR2+, after a core hole was placed on O16 during 9.6 fs. C blue, O red, H white. | ||
Here again, the bonds which first break are the CC bonds on which the HOMO presented σ character (see Fig. 2), and from which the two electrons were removed.
The presence of a core vacancy on O17, O18 or O19 during 9.6 fs, before the double hole dynamics, led to the same fragmentation pattern as that of dR2+ without the presence of a core hole, that is the one depicted on Fig. 8.
When a core hole is placed on O16 during 9.6 fs, the fragmentation pattern was different. As shown on Fig. 9, the C1C5 bond breaks, which is also one of the dissociation patterns found for dR2+ without prior core ionisation, but starting from another initial configuration.
3.4 Excited states and discussion of results
Our results can be compared to the experiment by Ha et al.,10 who irradiated dR with soft X-rays at energies near the C–K and O–K edges and measured charged fragments in coincidence. In these gas phase experiments, the most probable conformation is the pyranose one. For this conformation, we found that the main dissociation pathways (see Table 3) are either a linearisation of the molecule or the production of CH2O and C4H8O32+ fragments. These fragments will not appear on coincidence spectra, as only one of the two fragments is charged. Ha et al. in fact point out that the absence (or weak traces) of fragment CH2O+ (30 amu) is noteworthy considering that these are most likely the moieties that are the precursors of the CHO+ fragments. Here, this fragment is found mostly neutral. It should also be mentioned that CHxO+ (x = 1,…) fragments were shown to desorb from dR films upon soft X-ray impact.28,29 We also find that the C4H8O32+ fragment sometimes dissociates into C2H4O+ (44 amu) and C2H4O2+ (60 amu). Ha et al. have only observed fragments with mass less than 43 amu. It is thus likely that C2H4O+ and C2H4O2+ further fragment at longer times, not accessible in terms of the present work trajectory length times. In one case, we show the production of H2O0.2+ and CHO0.8+. Although the charge of the water molecule is only +0.2e, indicating that it is most often neutral, we find possible (18 amu, 29 amu) coincidence, as measured by Ha et al. Nonetheless, the most probable pathway is the formation of CHO+ (29 amu) together with C4H7O2+ (87 amu). CHO+ has been measured in coincidence with different fragments with masses smaller than 43 amu, which suggests that the 87 amu fragment could undergo fragmentation at longer times.| Conformation | Fragments formed | Ionisation process | ||
|---|---|---|---|---|
| a In some cases, dissociation of the large fragment occurs leading to the formation of C2H4O0.8+ and C2H4O20.9+. | ||||
| Pyranose | CH2O0.3+ (30 amu) | C4H8O31.7+ (104 amu)a | Double ionisation (8/10 cases) | |
| Core holes on O16, O18, O19, and C2 during 4.8 and 9.6 fs | ||||
| Core holes on C3 during 9.6 fs | ||||
| Linear stable (134 amu) | Double ionisation (2/10 cases) | |||
| Core holes on C1, C4 and C5 during 4.8 and 9.6 fs | ||||
| Core holes on O17 and C3 during 4.8 fs | ||||
| CHO0.8+ (29 amu) | H2O0.2+ (18 amu) | C4H7O2+ (87 amu) | Core hole on O17 during 9.6 fs | |
| Furanose | CH3O+ (31 amu) | C4H7O3+ (103 amu) | Double ionisation (10/10 cases) | |
| Core holes, except O18 during 9.6 fs | ||||
| CH3O0.25+ (31 amu) | CHO0.25+ (29 amu) | C3H6O21.5+ (74 amu) | Core hole on O18 during 9.6 fs | |
| Linear | CH3O+ (31 amu) | C4H7O3+ (87 amu) | Double ionisation (1/2 cases) | |
| Core holes on O17, O18 and O19 during 9.6 fs | ||||
| C2H5O2+ (61 amu) | C3H5O2+ (73 amu) | Double ionisation (1/2 cases) | ||
| Core hole on O16 during 9.6 fs | ||||
For the three deoxyribose conformations investigated, the fragmentation induced by the removal of two electrons from the HOMO (without prior core ionisation) appears to be guided by the initial localization of the holes. Fig. 10 shows that the first bond to break is always a CC bond on which the HOMO has a σ-type: C1C2 and C1C5 in the linear case, C1C2 and C1C5 in the pyranose case and C5C3 in the furanose case. In pyranose singly ionised dR+, similar conclusions have been drawn.30 Indeed Ab Initio MD (AIMD) simulation reveals that most of the fragmentation events start with C1C2 bond breaking, which is also attributed to the MO shape of removed electron (i.e. the HOMO). Moreover, when the K-hole dynamics before Auger decay is taken into account, we find that the fragmentation pathway after Auger decay is almost always one of the dissociation schemes found for dR2+ without prior core ionisation. Only in two cases, when the core hole was placed on one of the oxygen atom (O*) for 9.6 fs, was the CO* bond elongation sufficient to open a new dissociation pathway. Due to the very short lifetime of core vacancies (about 5 fs in the case of oxygen atoms), this will only occur for a small fraction of the core-ionised molecules. As a conclusion, the fragmentation process seems mainly driven by the double-hole state following Auger decay.
 | ||
| Fig. 10 Fragmentation of the different conformations of dR2+, after double ionisation in the HOMO. The position of the first bond to break is marked by an arrow. In parenthesis, the fraction of the trajectories starting with this bond break divided by the number of initial configurations investigated. C blue, O red, H white. | ||
To have a complete picture of decay, it would thus be interesting to use a complementary method which allows to follow the dynamics of doubly ionised states in which electrons are ejected from inner valence molecular orbitals. The calculation of the branching ratios of the Auger deexcitation channels would be prohibitive taking into account that the number of 2-combinations from a set of 54 orbitals (in a local spin density approximation picture) is almost 700. Nevertheless, we tested the effect of removing electrons from inner valence molecular orbitals. To that aim, we used Time Dependent-DFT Ehrenferst Molecular Dynamics which allows the description of the nuclear dynamics guided by the evolution of the electronic density, as done in some of our previous works.14–16 Since this method is very computationally consuming, we restrict our tests to the furanose conformation and a few location of double holes at the beginning of the dynamics.
The four cases exhibit a common feature (see Fig. 11): the fragmentation is guided by the location of the holes at the beginning of the trajectory, as when the two electrons are removed from the HOMO (see Fig. 10). The fragmentation is more harmful than in the case of double holes in the HOMO, which is not very astonishing in view of the energy deposit in the system (about 20 eV). This kind of ultrafast multi-fragmentation could explain the fact that no large fragments were measured by Ha et al.,10 even if the conformation in this experiment is the pyranose one. As a conclusion, the fragmentation appears to be mainly guided by the rearrangement of the electronic density during the deexcitation of the doubly excited ionised state leading to double holes in valence bonds.
 | ||
| Fig. 11 Fragmentation of furanose dR2+ after removal of deep molecular orbitals with energy (A) 25.9 eV, (B) 24.9 eV, (C) 24.8 eV and (D) 18.8 eV. The localization of the MO electronic density are schematized as ellipses. Bond breakage positions are marked by arrows. C blue, O red, H white. | ||
This could explain why experimental findings exhibit no difference in fragmentation whether core holes are induced in carbon or oxygen atoms since molecular orbitals are affected in the whole core ionisation and subsequent Auger decay processes. Several authors10,13 have indeed pointed out that the dissociation of a molecule after K-shell ionisation in light atoms and Auger decay does not seem to depend on the position nor on the nature of the K ionised atom. Statistically one cannot directly deduce from the two charged fragments emitted in coincidence where the K holes were induced by the photoionisation process in this kind of polyatomic molecules. On the contrary, in the case of small symmetrical molecules, where measurements and completely quantum theoretical calculations are possible, dissociation pathways could be deduced as an evidence of preferential bond breaking following Auger decays, with photoelectron–photoion–photoion coincidence technique measurements performed for C2H2 molecule irradiated at the C–K threshold.31 In this case, the photoelectron angular distribution (PAD) for non-symmetric fragmentation provided direct evidence of a localized core-hole and preferential bond breaking following Auger decays. In the case of dR, although core holes are not degenerated in energy (so distinguishable), differences coming from the localization of the K hole could not be observed in experimental spectra.10
The furanose conformation study will provide a reference for future works in which we will (step by step) increase the complexity of the molecule investigated and include a liquid water environment. Future work will consider the effect of adding the base at C1′ and the phosphate groups at C3′ and C5′ to the furanose dR conformation to provide a more realistic model of dissociation of core ionized dR in the DNA molecule. Also, as pointed out in previous studies on the uracil molecule, the water environment plays a critical role on the fragmentation induced both by core holes26 and double ionization events.16 We are currently investigating the effect of the liquid water environment on the fragmentation of core-ionized dR.
4 Conclusions
For the three conformations investigated, we find the same fragmentation patterns for ground-state doubly-ionised deoxyribose whether or not the core hole dynamics is taken into account before Auger decay. In most cases, no clear memory of the K-hole location neither of the nature of the atoms struck, apart from the energy induced in the molecule, could be found. For the sake of comparison, we have performed TD-DFT Ehrenferst MD which is the only possible method for representing the very excited doubly-ionised states resulting from K-shell ionisation and subsequent Auger effect. Such a method cannot be used for statistics because of the too large computational cost necessary. But these preliminary dynamics reinforce the conclusion that the main ingredient for the dissociation is the excited state, i.e. the localization of the holes in the molecule, after Auger decay. We have indeed previously shown that doubly-ionised states, created by removing electrons in MO with very near energy but localized in different bonds, lead to very different bond breakage in the case of isolated uracil.15 This fact shows that the energy deposited in the molecule is not the unique criteria to take into account for the fate of a doubly ionised molecule. Moreover, in the gas phase, such polyatomic molecules do not exhibit very large bond elongations during the core hole dynamics, before Auger decay. Here, specific fragmentation was only found in two cases, when the core vacancy was located on an oxygen atom external to the cycle for 9.6 fs. Due to the very short lifetime of O–K vacancies (about 5 fs), this will only occur for a small fraction of the core-ionised molecules. As a conclusion, the location of the K hole does not play a major role in elongating bonds within the cycle in such cyclic polyatomic molecules. The location of the core hole will play a major role to induce Auger effect thus 2 holes in different orbitals of the molecule. The fragmentation will not only depend on the energy but also strongly on the location of the empty orbitals thus created.Acknowledgements
We thank Y. Jeanvoine and J. Y. Salpin for helpful discussions on sugar conformations. This work was performed using HPC resources from GENCI-CINES/IDRIS (Grant 2014-085014) and CCRE UPMC. All calculations have been performed with the CPMD package, Copyright IBM Corp 1990–2011, Copyright MPI fuer Festkoer-perforschung Stuttgart 1997–2001. The present study is partially supported by the Reimei Research Program (Japan Atomic Energy Agency).References
- S. Saigusa, Y. Ejima, K. Kobayashi and M. S. Sasaki, Int. J. Radiat. Biol., 1992, 61, 785–790 CrossRef CAS PubMed.
- M. Watanabe, M. Suzuki, K. Watanabe, K. Suzuki, N. Usami, A. Yokoya and K. Kobayashi, Int. J. Radiat. Biol., 1992, 61, 161–168 CrossRef CAS PubMed.
- M.-A. Hervé du Penhoat, B. Fayard, F. Abel, A. Touati, F. Gobert, I. Despiney-Bailly, M. Ricoul, L. Sabatier, D. L. Stevens, M. A. Hill, D. T. Goodhead and A. Chetioui, Radiat. Res., 1999, 151, 649–658 CrossRef.
- B. Fayard, A. Touati, F. Abel, M.-A. Hervé du Penhoat, I. Despiney-Bailly, F. Gobert, M. Ricoul, A. L'Hoir, M.-F. Politis, M. A. Hill, D. L. Stevens, L. Sabatier, E. Sage, D. T. Goodhead and A. Chetioui, Radiat. Res., 2002, 157, 128–140 CrossRef CAS PubMed.
- F.-N. Gobert, M. Lamoureux, M.-A. Hervé du Penhoat, M. Ricoul, A. Boissière, A. Touati, F. Abel, M.-F. Politis, B. Fayard, J. Guigner, L. Martins, I. Testard, L. Sabatier and A. Chetioui, Int. J. Radiat. Biol., 2004, 80, 135–145 CrossRef CAS PubMed.
- M.-A. Hervé du Penhoat, A. Eschenbrenner, F. Abel, A. Boissière, J.-M. Guigner, A. Chetioui, M.-F. Politis, A. Touati, E. Sage, T. J. Jenner, D. L. Stevens and M. A. Hill, Int. J. Radiat. Biol., 2010, 86, 205–219 CrossRef PubMed.
- J. Campbell and T. PAPP, At. Data Nucl. Data Tables, 2001, 77, 1–56 CrossRef CAS.
- F. Alvarado, S. Bari, R. Hoekstra and T. Schlatholter, Phys. Chem. Chem. Phys., 2006, 8, 1922–1928 RSC.
- F. Alvarado, J. Bernard, B. Li, R. Brédy, L. Chen, R. Hoekstra, S. Martin and T. Schlathölter, ChemPhysChem, 2008, 9, 1254–1258 CrossRef CAS PubMed.
- D. Ha, M. Huels, M. Huttula, S. Urpelainen and E. Kukk, Phys. Rev. A: At., Mol., Opt. Phys., 2011, 84, 033419 CrossRef.
- T. Sekitani, E. Ikenaga, K. Tanaka, K. Mase, M. Nagasono, S. Tanaka and T. Urisu, Surf. Sci., 1997, 390, 107–111 CrossRef CAS.
- E. Sako, Y. Kanameda, E. Ikenaga, M. Mitani, O. Takahashi, K. Saito, S. Iwata, S. Wada, T. Sekitani and K. Tanaka, J. Electron Spectrosc. Relat. Phenom., 2001, 114–116, 591–596 CrossRef CAS.
- P. Bolognesi, M. Castrovilli, P. O'Keeffe, A. Casavola, D. Catone, S. Turchini and L. Avaldi, Nucl. Instrum. Methods Phys. Res., Sect. B, 2012, 279, 118–123 CrossRef CAS.
- I. Tavernelli, M.-P. Gaigeot, R. Vuilleumier, C. Stia, M.-A. Hervé du Penhoat and M.-F. Politis, ChemPhysChem, 2008, 9, 2099–2103 CrossRef CAS PubMed.
- P. López-Tarifa, M.-A. Hervé du Penhoat, R. Vuilleumier, M.-P. Gaigeot, I. Tavernelli, A. L. Padellec, J.-P. Champeaux, M. Alcamí, P. Moretto-Capelle, F. Martín and M.-F. Politis, Phys. Rev. Lett., 2011, 107, 023202 CrossRef PubMed.
- P. López-Tarifa, M.-P. Gaigeot, R. Vuilleumier, I. Tavernelli, M. Alcamí, F. Martín, M.-A. Hervé du Penhoat and M.-F. Politis, Angew. Chem., Int. Ed., 2013, 52, 3160–3163 CrossRef PubMed.
- J.-P. Champeaux, P. Çarçabal, M. Sence, P. Moretto-Capelle and P. Cafarelli, J. Phys. B: At., Mol. Opt. Phys., 2011, 44, 045205 CrossRef.
- E. Kukk, D. T. Ha, Y. Wang, D. G. Piekarski, S. Diaz-Tendero, K. Kooser, E. Itälä, H. Levola, M. Alcamí, E. Rachlew and F. Martín, Phys. Rev. A: At., Mol., Opt. Phys., 2015, 91, 043417 CrossRef.
- M.-A. Hervé du Penhoat, P. López-Tarifa, K. K. Ghose, Y. Jeanvoine, M.-P. Gaigeot, R. Vuilleumier, M.-F. Politis and M.-C. Bacchus-Montabonel, J. Mol. Model., 2014, 20, 2221 CrossRef PubMed.
- L. Guler, Y.-Q. Yu and H. Kenttämaa, J. Phys. Chem. A, 2002, 106, 6754–6764 CrossRef CAS.
- R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471–2474 CrossRef CAS PubMed . CPMD, http://www.cpmd.org/, Copyright IBM Corp 1990–2015, Copyright MPI für Festkörperforschung Stuttgart 1997–2001.
- N. Troullier and J. Martins, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 1993–2006 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules – A Quantum Theory, Oxford University Press, Oxford, UK, 1990 Search PubMed.
- C. R. Stia, M.-P. Gaigeot, R. Vuilleumier, A. O. Fojón, M.-A. Hervé du Penhoat and M.-F. Politis, Eur. Phys. J. D, 2010, 60, 77–83 CrossRef CAS.
- O. Takahashi, M. Odelius, D. Nordlund, A. Nilsson, H. Bluhm and L. G. M. Pettersson, J. Chem. Phys., 2006, 124, 064307 CrossRef PubMed.
- K. Fujii, K. Akamatsu and A. Yokoya, Surf. Sci., 2003, 528, 249–254 CrossRef CAS.
- K. Fujii, K. Akamatsu and A. Yokoya, Radiat. Res., 2004, 161, 435–441 CrossRef CAS PubMed.
- D. Ghosh, A. Golan, L. Takahashi, A. Krylov and M. Ahmed, J. Phys. Chem. Lett., 2012, 3, 97–101 CrossRef CAS.
- J. Adachi, K. Hosaka, T. Teramoto, M. Yamazaki, N. Watanabe, M. Takahashi and A. Yagishita, J. Phys. B: At., Mol. Opt. Phys., 2007, 40, F285 CrossRef CAS.
| This journal is © the Owner Societies 2015 |