
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA microwave molecular solution based approach towards high-κ-tantalum(V)oxide nanoparticles: synthesis, dielectric properties and electron paramagnetic resonance spectroscopic studies of their defect chemistry†
R. C.
Hoffmann
a,
M.
Kaloumenos
b,
D.
Spiehl
c,
E.
Erdem
d,
S.
Repp
d,
S.
Weber
d and
J. J.
Schneider
*a
aFachbereich Chemie, Eduard-Zintl-Institut für Anorganische und Physikalische Chemie, Technische Universität Darmstadt, Alarich-Weiss-Straße 12, 64287 Darmstadt, Germany. E-mail: joerg.schneider@ac.chemie.tu-darmstadt.de
bIntegrated Electronic Systems Lab, Technische Universität Darmstadt, Merckstraße 25, 64283 Darmstadt, Germany
cDepartment of Printing Technology, Technische Universität Darmstadt, Magdalenenstr. 2, 64283 Darmstadt, Germany
dAlbert-Ludwigs Ludwigs-Universität Freiburg, Institut für Physikalische Chemie, Alberstraße 21, 79104 Freiburg, Germany
First published on 3rd November 2015
Abstract
Stable dispersions of tantalum oxide nanoparticles are accessible from solutions of tantalum(V) complexes with a mixed malonato and alkanolato ligand sphere in ethoxyethanol by microwave processing. The malonato ligand is cleaved during decomposition and acetic acid or acetic acid esters are formed as derived from in situ spectroscopic studies. The solubility of the tantalum precursor and the obtained particle size therefrom depend strongly on the type of alkanolato ligand moiety. Dispersions of the molecular complexes possess good film forming properties. Films with low surface roughness can be obtained by spincoating. These exhibited a dielectric constant of about 15 and disruptive strengths above 1.5 MV cm−1. The electrical measurements indicate that the presence of moisture is detrimental with respect to the dielectric performance of the films. After removal of the solvent from the suspensions of the nanoparticles, the residue can be redispersed in aprotic solvents. The particles can be isolated therefrom by precipitation with pentane. XRD and HRTEM indicate that the material remains amorphous up to temperatures of 750 °C. XPS proved that only Ta2O5 is formed as lower oxidation states of Ta cannot be detected. A detailed EPR study allows us to gain insight into the surface defect chemistry. Multiple types of oxygen vacancies exist at the surface of the Ta2O5 particles which are influenced by additional calcination and annealing in a vacuum.
Introduction
The fabrication of dielectric thin films for capacitors, metal oxide semiconductor (MOS) capacitors or field effect transistors (FETs) by solution deposition remains an immanent challenge due to problems with leakage current and early breakdown.1 However, solution deposition seems to be nevertheless attractive as it offers a significantly reduced instrumental effort compared to physical deposition techniques as well as the employment of cost efficient pattering techniques such as printing or stamping.2 In the manufacturing process, surface adsorbates, defect chemistry and interface effects, however, play a more significant role than those in physical deposition processes employing mostly high vacuum gas phase techniques.3,4For the time being, a more comprehensive knowledge of the functional stability and reliability of electronically important thin film materials processed from solution remains still challenging but needful. Hereby residues from the processing atmosphere, water content and left overs of organic compounds from the solution processes play an important role. When using molecular precursors often a cascade of events is set in motion when a molecule is adsorbed onto a surface. This includes a perturbation of the local charge distribution rendering a rearrangement of the electronic band alignment, altering the permittivity of the interface as well as the electrostatic gating and the gate coupling/capacitance.4,5 Apart from a mere electrical characterisation, other direct methods are needed which allow us to characterize the electronic microstructure especially for surface states and defects.6,7
Thin film deposition can be carried out either by direct deposition of a solution of a suitable metal oxide precursor or by using dispersions of nanoparticles. In both cases a post-treatment is required to achieve final ceramisation of the green body by complete removal of solvents and organic residues. The results are comparable with respect to the dielectric performance.8,9 High-κ dielectric materials such as ZrO2 or HfO2 are preferred in that regard as they lead to higher capacitance at a smaller film thickness compared to more conventional inorganic dielectrics e.g. SiO2 or Al2O3.10–12 Interestingly, much less work has been done so far on the solution deposition of tantalum pentoxide (Ta2O5), which offers a comparable high dielectric constant (κ ∼ 22) as zirconia and hafnia as well as a sufficiently large band gap.13 Sol–gel techniques are well established, but require annealing temperatures above 450 °C, which make them unsuitable for substrates such as conventional glass or polymer materials.14,15 Recently, we reported a microwave-assisted route towards the synthesis of oxide nanoparticles by the decomposition of tailored molecular precursors. Rapid heating by microwaves ensures low particle size in the order of several nanometers. Stabilisation of the nanoparticle dispersions can be achieved by residual molecular fragments remaining on the particle surface.8 This method is more direct and should be advantageous in comparison to earlier reports, which apply especially designed larger and thus sterically demanding capping agents, and necessarily introduce higher amounts of organic residues. Further metathesis reactions of the stabilising small residues are also avoided. The latter would lead to contaminations by alkali metals or halide ions.16,17 Therefore our proposed approach offers a valuable synthetic approach to the hitherto less researched tantalum oxide nanoparticles as solution processed dielectrics.
In the work reported herein, we present a synthetic approach to synthesize stable dispersions of tantalum pentoxide (Ta2O5) nanoparticles. Electrical capacitors were fabricated by employing thin films of this material allowing us to determine the electrical performance. The influence of the environmental conditions on the dielectric properties was monitored in detail. For the first time a dedicated EPR investigation was carried out in order to identity bulk defects and surface adsorbates of the tantalum oxide nanoparticles.
Results and discussion
Precursor synthesis and characterisation
The reaction (Fig. 1) of tantalum ethoxide with two equivalents of malonic acid in the presence of an excess (i.e. either as solvent or as a mixture with tetrahydrofuran) of the alcohol ROH (R = –CH3, –CH(CH3)2 or –C(CH)3) leads to complexes with the nominal composition Ta[(CH2(COO)2)]2(RO). The compounds were crystalline, white powders, which hydrolyse slowly when exposed to air. The solubility in organic solvents increased with the extent of the alkyl moiety R: –C(CH)3 > –CH(CH3)2 ≫ –CH3. To the best of our knowledge, no complexes of tantalum with malonic acid were structurally characterised so far, however, reactions of tantalum alkoxides with various carboxylic acids were reported, which lead to similar stoichiometries.18–20 Also, it is a common approach in sol gel chemistry to decrease the susceptibility to moisture by adding multidentate ligands to metal alkoxides.21 For the microwave reaction described in the following, it was important that no side products from hydrolysis or even nanoscale particles were present, which could serve as nucleation centres. | ||
| Fig. 1 Schematic presentation of synthesis and thermal decomposition of the (alkanolato)bis(malonato)tantalum(V) complexes (1)–(3). | ||
The thermal decomposition of the precursors (1)–(3) was investigated by means of TG coupled with MS and IR (Fig. 2).
 | ||
| Fig. 2 Thermogravimetric (TG) mass loss curve obtained by heating (1)–(3) in oxygen. | ||
The onset of the decomposition increased in the order (3) > (2) > (1). The decay was finished at about 300 °C and only a minor mass loss was observed thereafter. The course of the decomposition occurred in several steps, which was more distinct for (2) and (3), whereas (1) exhibited a gradual decomposition. This finding was a first hint that the decomposition of the three complexes proceeded by different reaction mechanisms (Fig. 1). The corresponding Gram-Schmidt curves showed two maxima in all cases, though (Fig. S1, ESI†). The analysis of the gaseous decomposition products was possible online by means of IR coupling, whereby complex mixtures were observed (Fig. S2, ESI†). According to earlier investigations by other groups, the decomposition of malonic acid proceeds by cleavage, yielding carbon dioxide and acetic acid,22,23 whereas salts of malonic acid with alkali, alkaline earth or transition metals also lead to the formation of acetone and esters as by-products.23,24
In the present investigation the tantalum complexes (1)–(3) lead to a plethora of gaseous products, those were obviously dependent on the nature of the alkoxylato moiety. This suggests that a metal mediated reaction of the malonato and the alkoxylato ligands occurred. The identification was possible on the basis of signals from functional groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC) and the most intense peaks in the fingerprint region. The formation of further products in significantly smaller amounts could not be excluded, though. For (1) the formation of methylacetate (2965, 1783, 1765 and 1252 cm−1) was observed in the first decomposition step, whereas in the second also acetic acid (3589, 1798, 1776, and 1179 cm−1) and methanol (3681, 2950, and 1038 cm−1) could be found. In the case of (2) 2-propylacetate (2986, 1785, 1379, 1245 and 1116 cm−1) was observed, whereby acetic acid and 2-propene (2979, 1665, 1638, 1473, 1455 and 913 cm−1) formed at higher temperatures. The differences of the position of the ν(CO) vibrations between acetic acid and esters are sufficient to allow a clear distinction. Also the fingerprint regimes in the decomposition products vary significantly. In contrast, the decomposition of (3) did not lead to the formation of an ester. Apart from acetic acid the presence of 2-methylpropene (3085, 2942, 1659, 1446, 1388 and 891 cm−1) could be identified. A complete summary of the decomposition sequence is given in Fig. 1.
O and CC) and the most intense peaks in the fingerprint region. The formation of further products in significantly smaller amounts could not be excluded, though. For (1) the formation of methylacetate (2965, 1783, 1765 and 1252 cm−1) was observed in the first decomposition step, whereas in the second also acetic acid (3589, 1798, 1776, and 1179 cm−1) and methanol (3681, 2950, and 1038 cm−1) could be found. In the case of (2) 2-propylacetate (2986, 1785, 1379, 1245 and 1116 cm−1) was observed, whereby acetic acid and 2-propene (2979, 1665, 1638, 1473, 1455 and 913 cm−1) formed at higher temperatures. The differences of the position of the ν(CO) vibrations between acetic acid and esters are sufficient to allow a clear distinction. Also the fingerprint regimes in the decomposition products vary significantly. In contrast, the decomposition of (3) did not lead to the formation of an ester. Apart from acetic acid the presence of 2-methylpropene (3085, 2942, 1659, 1446, 1388 and 891 cm−1) could be identified. A complete summary of the decomposition sequence is given in Fig. 1.
The formation of the gaseous decomposition products, which were identified already by means of IR spectroscopy, could be further confirmed by additional experiments using TG-MS (Fig. S3, ESI†). Thus, besides the masses of less specific species such as carbon dioxide (m/z+ = 44), water (m/z+ = 18) and also acetic acid (m/z+ = 60), the characteristic fragmentation patterns of methylacetate (m/z+ = 74, 59, 43, 29) as well as 2-propylacetate (m/z+ = 61, 59, 43), 2-propene (m/z+ = 41, 39) and 2-methylpropene (m/z+ = 56, 41, 39) could be clearly identified. In the latter cases signals corresponding to the molecule ion itself could not be found due to insufficient intensity.
Synthesis of stable tantalum oxide nanoparticle dispersions
In the next step nanoparticles were synthesized by thermolysis of solutions of the tantalum complexes in ethoxyethanol by heating in a laboratory microwave to 300 °C. The solubility of (1) was not sufficient to obtain high particle concentrations, so this precursor was not further investigated. In the other cases, particle sizes were determined by means of DLS (Fig. 3, Table S1, ESI†). Thereby the influence of the starting concentration of the tantalum complex on the particle size was more pronounced for (3) than for (2). Higher precursor concentrations lead to the formation of gels. Dispersions of particles with a diameter of below 10 nm were transparent and storable over months without flocculation. Higher particle concentrations could be obtained by the evaporation of the solvent under vacuum. Complete removal of the solvent however leads to sticky residues, which could still be redispersed in acetone or THF. The dispersions, though, were only stable for a few hours after which the onset of turbidity was observed. The precipitation could be accelerated by the addition of pentane. The particles were then isolated by centrifugation in the form of off-white powders.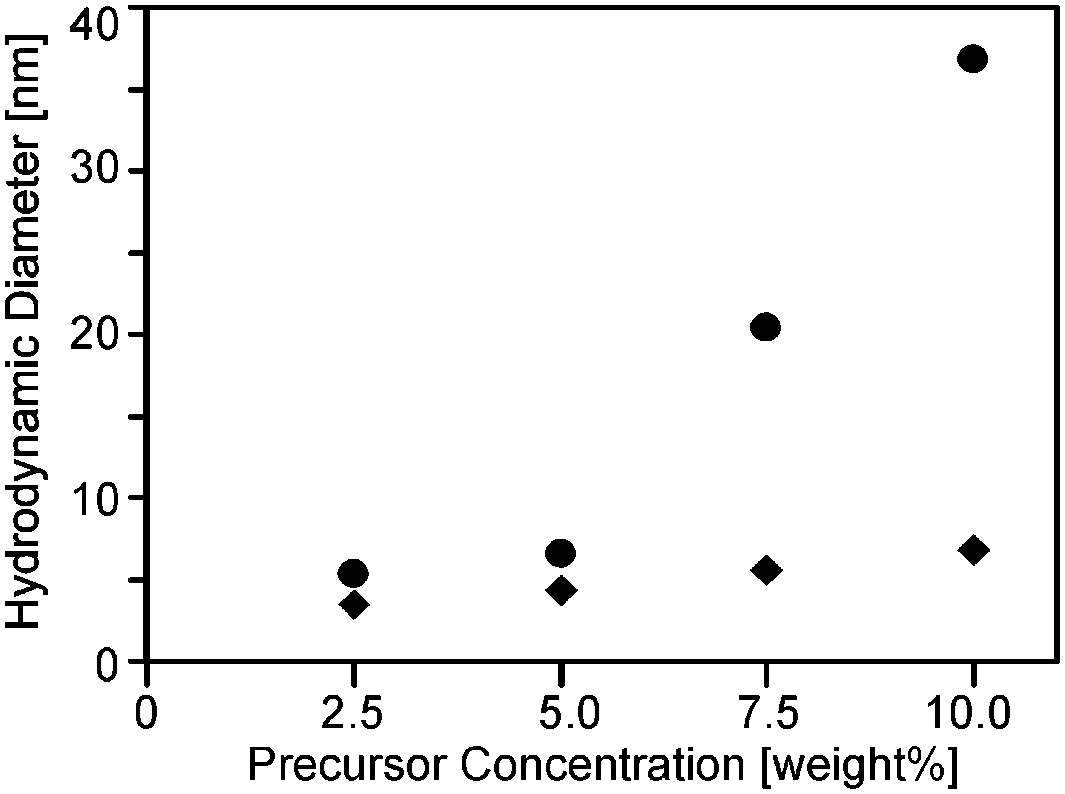 | ||
| Fig. 3 Hydrodynamic diameter from DLS measurements of particles obtained from the microwave reaction of precursors (2) (rhombs) and (3) (circles) in ethoxyethanol. | ||
HRTEM investigations (Fig. 4) showed that the so-obtained material was amorphous, as confirmed by means of XRD (Fig. S4, ESI†). After heating to significantly higher temperatures, i.e. 750–950 °C, however, crystalline products were obtained. The crystallisation behaviour depended on the individual precursor employed in the microwave reaction. Particles obtained from (2) and (3) crystallised after heating to 750 °C in the hexagonal phase of Ta2O5. After heating to 950 °C additional peaks appeared, which indicated a partial transformation to the orthogonal phase of Ta2O5.
 | ||
| Fig. 4 Characteristic HRTEM micrograph of agglomerates of amorphous particles obtained by the microwave reaction of 10 weight% of precursor (2) in ethoxyethanol. | ||
Tantalum oxide exhibits two thermodynamically stable phases. The low-temperature form is orthorhombic, β-Ta2O5. A reversible transition occurs at about 1360 °C yielding the tetragonal α-phase.25 Heating of amorphous Ta2O5 in air leads to a hexagonal modification (δ-Ta2O5), whereby the crystallisation temperature depends on the employed procedure and is in the range of 700 to 800 °C. The hexagonal phase is only metastable at low temperatures and irreversibly transforms to the orthorhombic form at higher temperatures.14,25,26 The XRD diffraction patterns for β-Ta2O5 (JCPDS: 25-0922) and δ-Ta2O5 (JCPDS: 19-1299) are, however, nearly overlapping and difficult to distinguish in the case of an observed line broadening.
The analysis of the tantalum oxide powders obtained by microwave reaction by TG coupled with IR spectroscopy (i.e. for the gases evolving from the powder upon heating, Fig. S5 and S6, ESI†) as well as by IR spectroscopy (i.e. of the powders themselves, Fig. S7, ESI†) showed that detectable amounts of organic residues were present in the particles. Consequently, evidence for functional groups was found which are related to molecular moieties of the solvent and to decomposition products of the ligand (i.e. acetate). It was previously suggested that this surface functionalisation is the reason for the stability of the particle dispersions.8
Manufacturing and properties of tanatalum oxide thin films
After evaporation of the solvent the tantalum oxide nanoparticles agglomerate strongly. The dispersions are therefore highly suitable for the coating of many materials including glass or indium tin oxide. Uniform films with low surface roughness and high transparency were subsequently obtained by spincoating and subsequent annealing (Fig. 5). The absorption edge of films obtained after annealing at 100 °C was situated at about 255 nm (Fig. 6). Processing at higher temperatures, i.e. 350 or 450 °C, leads to a shift of the edge to about 272 nm, which is expected for Ta2O5.27,28 This effect is due to the removal of hydroxyl groups at the surface of the oxide particles, which were also visible in the before mentioned IR spectra of powders, which were heated at different temperatures. | ||
| Fig. 5 AFM image of Ta2O5 nanoparticles on ITO-coated glass. (Spincoating of three consecutive layers from dispersions of microwave processed precursor (2) and annealing at 350 °C. ITO 140 nm, Ta2O5 208 nm, and RMS 0.5 nm.) | ||
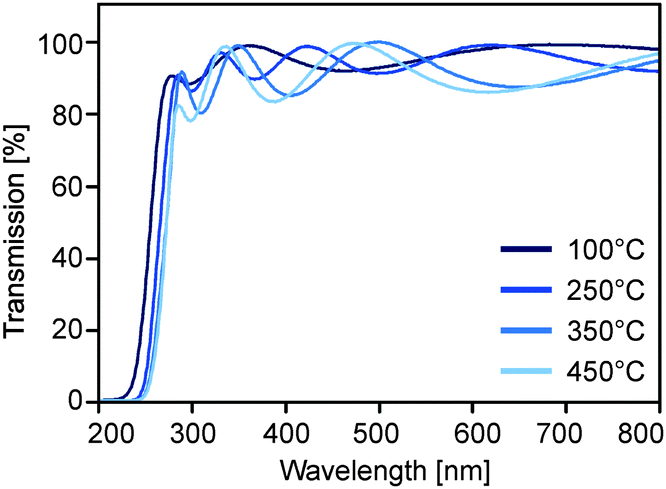 | ||
| Fig. 6 UV/VIS spectra of films obtained from spincoating of particle dispersions obtained from (2) on quartz and additional annealing at various temperatures in air. | ||
XPS spectra (Fig. S8, ESI†) of films calcined at different temperatures in air confirmed that a part of the surface hydroxylation is lost at temperatures above 250 °C. In accordance with the results from the TG measurements only minor carbon residues can be found. The signals for Ta 4f7/2 and 4f5/2 are located at 25.8 and 28.7 eV, respectively. No apparent change was observed depending on the heat treatment temperature. The values are in accordance with previous investigations.27,29 The area below the curves could be fitted with one component. The addition of a second contribution did not lead to a significant amount (Table S2, ESI†). XPS therefore proved that Ta(V) is most likely the sole component present at all temperatures.
In order to investigate point defects in the tantalum oxide films PL measurements were carried out (Fig. 7). The PL spectra of the films consisted of at least three contributions at about 400, 460 and 535 nm. The positions shifted slightly when the films were processed at different temperatures. The results are in accordance with the known emission for Ta2O5, which is a broad signal in the blue range of the visible spectrum. It is generally attributed to oxygen vacancies in the crystal lattice.30,31
 | ||
| Fig. 7 PL spectra of films obtained from spincoating of particle dispersions obtained from (2) on quartz and additional annealing at various temperatures in air. | ||
In addition sintering experiments were carried out at higher temperatures of films on quartz substrates. Thereby particles obtained from the microwave reaction of (3) in ethoxyethanol were spincoated and annealed at 350 °C first. This procedure was repeated to obtain thicker amorphous layers. Sintering in air at 700 to 800 °C, respectively, yielded films (Fig. S9, ESI†) of hexagonal Ta2O5 which exhibited a distinct texture (Fig. S10, ESI†). The (001) peak was most pronounced and had a smaller line width in comparison to the other reflections, which indicated a preferred growth along the z-axis. The transformation from the amorphous to the crystalline phase is presumably initiated at the substrate surface. In the following, randomly oriented nuclei grow and undergo a competitive selection process, in which alignment parallel to the surface normal is preferred.32 Other examples for the synthesis of tantalum(V) oxide coatings with high anisotropy by means of solution deposition were reported earlier33,34 and are relevant for applications in piezoelectric devices.35
Dielectric properties
For the characterisation of the dielectric properties of the Ta2O5 thin films a capacitor was fabricated (Fig. 8a). However, general structural designs of microscale devices could not be readily transferred for these needs. The use of silicon as a substrate and an electrode likewise is not reasonable, as the native oxide layer acts an additional insulating layer, which must be taken into account in the evaluation procedure.36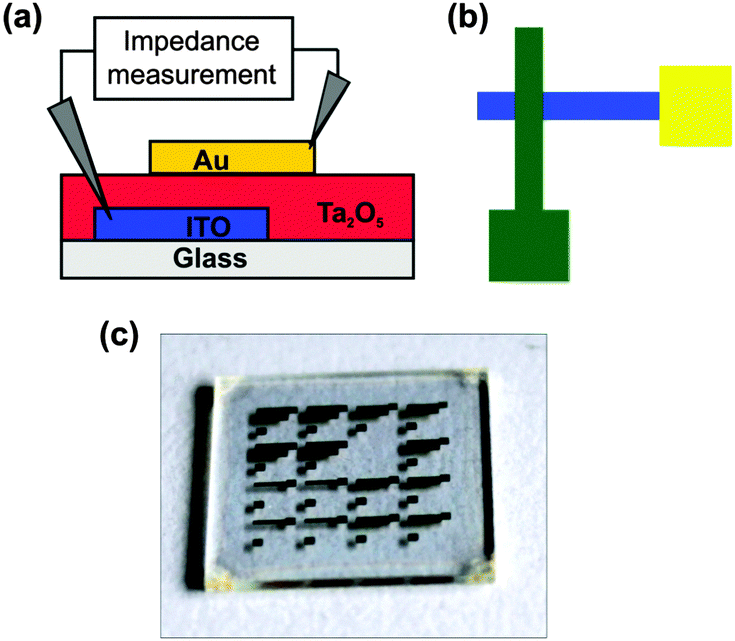 | ||
| Fig. 8 (a) Schematic presentation of the cross-section of the layers in the capacitor (ITO 140 nm, Au 50 nm, and Ta2O5 208 nm for particles from (2) and 162.5 nm for particles from (3)). (b) Schematic presentation of the electrode arrangement. (c) Photograph of a substrate (1.5 × 1.5 cm2) with the top gold electrodes. ITO and Ta2O5 layers are transparent and not discernable. | ||
Metallic contacts, which were deposited by means of sputtering or evaporation in a high vacuum, could not be used as bottom electrodes (e.g. on a glass substrate). Furthermore annealing of the nanoparticle dispersion which was deposited by spincoating caused rupture and delamination of the metal electrodes below. Thus, the employment of structured ITO on glass was established as the material for the bottom electrode.37 Metallic top electrodes were deposited by sputtering. The final set up is shown in Fig. 8b and c.
Measurements of samples in air showed a drastic deviation from the expected behaviour, Fig. 9. A measurement in a glove box after short annealing at 140 °C leads to an improvement and a well-defined characteristic could be observed (Fig. 10). The decrease in the capacity after heating hinted at the removal of adsorbed water, which is a well-known effect,38,39 and could also be responsible for the bias instability.40 From the Bode plot the material properties were extracted. For films obtained from particle dispersion of precursor (2) the dielectric constant was 15.2 ± 0.8 and the dielectric loss (value at 10 Hz) was tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ ∼ 0.073, whereas film from (3) yielded κ ∼ 15.7 ± 1.1 and tanδ ∼ 0.126. Within in the range of the experimental error the performance of the thin films obtained from both precursors is identical. In breakdown measurements, failure of the devices typically occurred between 15 and 20 V (Fig. S11, ESI†), which gives an estimate of the disruptive strength of about 1.5 MV cm−1. Thereby the leakage current density increases gradually (“soft breakdown”) and the failure of the device (“hard breakdown”) occurs much later at higher voltages. This effect can be described by the formation of percolation paths, whereby defects in the oxide connect the electrodes and the defect density is still below a critical threshold.7,41
δ ∼ 0.073, whereas film from (3) yielded κ ∼ 15.7 ± 1.1 and tanδ ∼ 0.126. Within in the range of the experimental error the performance of the thin films obtained from both precursors is identical. In breakdown measurements, failure of the devices typically occurred between 15 and 20 V (Fig. S11, ESI†), which gives an estimate of the disruptive strength of about 1.5 MV cm−1. Thereby the leakage current density increases gradually (“soft breakdown”) and the failure of the device (“hard breakdown”) occurs much later at higher voltages. This effect can be described by the formation of percolation paths, whereby defects in the oxide connect the electrodes and the defect density is still below a critical threshold.7,41
 | ||
| Fig. 9 Bode-plots for tantalum oxide films obtained after annealing at 350 °C in air only. (a) Particles synthesized from (2) and (b) particles synthesized from (3). Blue curves refer to top electrodes with 250 μm width, whereas green electrodes to 1000 μm. ITO bottom electrodes had 250 μm width in all cases. | ||
 | ||
| Fig. 10 Bode-plots for tantalum oxide films obtained after annealing at 350 °C in air and additional treatment in argon at 140 °C. (a) Particles synthesized from (2) and (b) particles synthesized from (3). Blue curves refer to top electrodes with 250 μm width, whereas green electrodes to 1000 μm. ITO bottom electrodes had 250 μm width in all cases. | ||
EPR studies
In addition to the XPS and PL investigations, also EPR spectroscopy (Fig. 11) was applied to study defects in Ta2O5 particles. Specifically, powders precipitated from the dispersions after the microwave reaction as well as samples, which were additionally calcined at 450 or 600 °C, respectively (Fig. 11a), were examined. Further experiments were carried out with such calcined powders after annealing in a vacuum at 140 °C (Fig. 11b). The aim of the vacuum treatment was to reveal the influence of the ambient atmosphere on the defect chemistry of the metal oxide. Possible defect centres in Ta2O5 are (i) vacancy trapped one electron defects (F-centre, VO−), (ii) vacancy trapped two electron defects (VO2−), (iii) defect complexes like Ta4+-VO or Ta4+-VO-Me (Me = impurity atom), and (iv) coordination complexes with hydroxyl groups like Ta4+-OH− (OH− as a molecular ion species located in the site of VO). Similar paramagnetic defect centres in oxide dielectrics or semiconductors are well studied by EPR and described in the literature.42–45 | ||
| Fig. 11 Cw-EPR of Ta2O5 powders measured (a) under air and (b) vacuum. (c) Plot of the EPR peak-to-peak intensity of defect signals with respect to P1/2 for 450 °C and 600 °C annealed Ta2O5 samples measured under vacuum. | ||
All samples showed one signal with a g-factor very close to that of free electrons g = 2.0029. A compilation of g-factors and EPR signal linewidths can be found in Table S3 (ESI†). The as-received sample showed an EPR signal of only very weak intensity, but calcination at elevated temperature leads to a drastic increase of the signal. Evidently, an intensity increase of the EPR signals after annealing of Ta2O5 is related to the formation of structural defects that possess unpaired electron spins. Thereby oxygen vacancies are expected to be the most frequent types of defects.46 According to first-principles calculations, various types of oxygen vacancies were predicted, which correspond to sites with different coordination spheres in the Ta2O5 lattice.47,48 The differences in the g-factors and linewidths from as-received to annealed samples indicate different site symmetries and locations of defect centres, which can be attributed naturally to oxygen vacancies.
The EPR signal did not significantly change further after annealing in a vacuum; the g-factor remained almost constant and the linewidth decreased only slightly. To understand the dependencies of the EPR signals of defect centres on the applied microwave power, we have investigated the EPR saturation behaviour of samples calcined at 450 °C and 600 °C under vacuum conditions (Fig. 11c). The EPR intensities for both samples rise with increasing microwave power. However, the signal of the sample annealed at 450 °C deviates strongly from linearity in the plot at microwave powers above 2.25 mW, thus indicating a characteristic saturation behaviour. In EPR spectroscopy it is well known that systems, which saturate easily, generally have long spin–lattice relaxation times, whereas systems which are hard to saturate relax quite efficiently. This means that the environment of the spins (surface defects) changes by annealing. From microwave-power-dependent measurements it can be concluded that the sample annealed at 450 °C has much longer spin–lattice relaxation times in comparison to the sample annealed at 600 °C.
The temperature dependence of the reciprocal intensity which is related to the magnetic susceptibility of the tantalum oxide material was also investigated. It was found that an increase of the temperature leads to a decrease of the susceptibility of the sample obeying the Curie–Weiss paramagnetism reflected in two different Curie constants for the two different samples studied (Fig. S12, ESI†).
Very recently, first-principles calculations were carried out indicating that oxygen-deficient Ta2O5 has exceptionally long-ranged lattice relaxation.49,50 Our results are now able to confirm this prediction experimentally. By the application of spin counting (for details see ESI†), one can quantitatively determine the defect concentration. It was found that the sample annealed at 450 °C has the highest defect concentration, regardless whether annealed under air or vacuum (Table S3, ESI†). It is worth noting that after vacuuming, annealed samples revealed a higher defect concentration by a factor of roughly three.
According to the deconvolution of the integrated EPR signals we observed the following transformation pattern regarding the EPR lineshapes before and after treatment in a vacuum: an asymmetric line was obtained for the sample calcined at 450 °C which turned into a symmetric Lorentzian shape after annealing in a vacuum. The asymmetric line was composed of two Gaussian lines. On the other hand, a symmetric Lorentzian line was obtained when the sample was calcined at 600 °C which turned into an asymmetric shape composed of two Gaussian lines after vacuum annealing (Fig. S13, ESI†).
Finally, also a commercially available reference sample was investigated. The micrometre-sized crystallites of orthorhombic β-Ta2O5 did not exhibit any EPR signals (Fig. S14, ESI†), even after calcination in air at 450 °C or 600 °C. Thus, the before-mentioned EPR investigations of the powders synthesized by microwave-assisted precursor decomposition proved the existence of different types of oxygen vacancies on the surface of Ta2O5, which have hitherto only been reported in a few theoretical studies.47–50 This is in accordance with photoluminescence studies reported here (Fig. 7) and by other groups revealing multiple defect energy states in the band gap.31,51
Conclusions
The reaction of tantalum complexes with malonic acid in ethoxyethanol at high temperatures under microwave-heating leads to the formation of stable dispersions of nanoparticles of tantalum(V) oxide. The dispersions possess good coating properties and can be employed for the manufacturing of thin films, e.g. in electrical capacitors. Electrical measurements for the characterisation of the dielectric properties showed a dependence of the performance on the device set-up and the environmental conditions. The presence of water has a drastic influence on the electrical device characteristic. An uncritical disregard of this impact would lead to a systematic overvaluation of the capacitance and thus also of the dielectric constant. Further restrictions for long term stability and operational behaviour can be expected.52In addition to the electrical characterisation EPR was employed as a tool for the determination of point defects. Although this method is restricted to paramagnetic species, high sensitivity and the possibility to distinguish between various influences are clear advantages. In this way the interpretation of the electrical measurements can be complemented by a powerful spectroscopic technique.
Experimental
Synthesis of tantalum precursor compounds
All manipulations were carried out under argon and with dried solvents. Complexes were stored and handled under argon after synthesis.Microwave synthesis
Microwave reactions were carried out in a discover (CEM) microwave reactor with original CEM reaction tubes and silicon stops supplied by the manufacturer. The precursor was dissolved in 2-ethoxyethanol. The resulting solution was filtered through a syringe filter and filled into a glass tube (5 mL of solution in 10 mL tubes). The solution was heated to 300 °C with a power of 300 W. A temperature of 250 °C was reached in about 5 minutes, whereas the maximum temperature was obtained after further 30 minutes. The vessel was then kept at this temperature for 5 minutes under constant microwave heating and was afterwards cooled down to room temperature with compressive air. (An overview of the reaction conditions in the microwave is shown in Fig. S15, ESI.†) After finishing the microwave reaction the solvent could be removed by condensation into a cold trap. The complete removal of the solvent resulted in a waxy residue, which was redispersed in acetone or tetrahydrofuran. The addition of pentane to the dispersion caused flocculation of the white precipitate, which could be separated by centrifugation and subsequently dried at 100 °C for about 3 hours.Cleaning and coating of substrates
Dispersions for coating were obtained from solutions of 10 weight% of precursor (2) and 5 weight% of precursor (3), respectively, in ethoxyethanol and reaction in the microwave at 300 °C (conditions as mentioned above). Afterwards the dispersions were concentrated to about 50% of their original volume. Films were produced by spincoating (20 s at 1500 rpm) of the particle dispersions on quartz (2 × 2 cm2), silicon or indium-tin-oxide (ITO) coated glass slides (1.5 × 1.5 cm2). Thicker films could be obtained by iteration of the described coating procedure.The capacitor was fabricated by using patterned indium tin oxide (ITO) on glass (150 nm, OLED grade) as a bottom electrode. A sacrificial gold layer (50 nm) of square geometry was deposited at an edge of the ITO film by sputtering using a shadow mask. Thereafter the tantalum oxide dispersion was spincoated and annealed at 350 °C. This coating procedure was repeated three times for precursor (2) and six times for precursor (3). The film thickness was determined by ellipsometry and verified by SEM cross-sectioning yielding 208.0 nm for (2) and 162.5 nm for (3), respectively. After the deposition of the dielectric material, platinum electrodes (50 nm) were deposited on these dielectric films using a shadow mask by sputter evaporation using a platinum metal source. ITO and platinum electrodes formed perpendicularly overlapping strips with different intersections of varying area sizes. The contact probe was attached to the ITO electrode through the sacrificial gold layer. Due to this procedure the tantalum oxide film is not punctured or damaged by mechanical cracking.
Characterisation
Atomic Force Microscopy (AFM): CP-II (Bruker-Veeco), tapping mode, 1 Hz, silicon cantilevers (T 3.75 μm, L 125 μm, W 35 μm, f 0.300 kHz, k 40 N m−1). Thermogravimetry (TG): TG209F1-Iris (Netzsch). Samples were measured in oxygen at a heating rate of 10 °C min−1 in the range of 30–600 °C in aluminium crucibles. Dynamic Light Scattering (DLS): Zetasizer Nano (Malvern). Samples were measured in 10 mm quartz cuvettes. IR Spectroscopy: Nicolet 6700 (Thermo Scientific). Powders were measured using an attenuated total reflection (ATR) unit. Transmission Electron Microscopy (TEM): CM20 (Philips, FEI) operated at 200 kV. Samples were supported on a lacey-carbon copper grid (300 mesh). UV/VIS Spectroscopy: Lambda 900 (Perkin Elmer). X-Ray Photoelectron Spectroscopy (XPS): ESCALab 250 (Thermo VG Scientific) using monochromated Al Kα radiation (1486.6 eV) in constant analyser energy (CAE) mode with a pass energy of 50 eV for all spectra. X-Ray diffraction (XRD): Miniflex 600 (Rigaku), Cu-Kα radiation, 600 W in Bragg–Brentano geometry.Continuous wave (cw) X-band (9.86 GHz) EPR measurements were performed at room temperature using a Bruker EMX spectrometer using a cylindrical super-high-quality (SHQ) resonator (Bruker). The offset in the magnetic field and the exact g-factors in X-band measurements were determined with a polycrystalline DPPH (2-diphenyl-1-picrylhydrazyl) reference sample with a well-known g-factor (g = 2.0036). The EPR spectral analysis has been performed with the aid of the WINEPR program from Bruker. A turbomolecular pump was used to evacuate the EPR quartz tubes. EPR investigations included a commercial sample as reference (Alfa Aesar Order No. 10881; puratronic 99.993% metal basis excluding niobium, Nb 50 ppm max).
Acknowledgements
EE thanks the DFG for financial support (Grant No: Er 662/1-2). Experimental research of RH, MK and DS was performed in collaboration with the Merck-Lab, a joint academia/industry lab of TU Darmstadt and Merck KGaA.References
- S. J. Kim, S. Yoon and H. J. Kim, Jpn. J. Appl. Phys., 2014, 53, 02BA02 CrossRef.
- S. R. Thomas, P. Pattanasttayavong and T. D. Anthopoulos, Chem. Soc. Rev., 2013, 42, 6910 RSC.
- P. Raghu, N. Rana, C. Yim, E. Shero and F. Shadman, J. Electrochem. Soc., 2003, 150, F186 CrossRef CAS.
- S. J. Kim, D. H. Yoon, Y. S. Rim and H. J. Kim, Electrochem. Solid-State Lett., 2011, 14, E35 CrossRef CAS.
- J. H. Park, Y. B. Yoo, K. H. Lee, W. S. Jang, J. Y. Oh, S. S. Chae, H. W. Lee, S. W. Han and H. K. Baik, ACS Appl. Mater. Interfaces, 2013, 5, 8067 CAS.
- N. Raghavan, K. L. Pey and L. Shubhakar, Microelectron. Reliab., 2014, 54, 847 CrossRef CAS.
- C. H. Ho, S. Y. Kim and K. Roy, Microelectron. Reliab., 2015, 55, 308 CrossRef CAS.
- R. C. Hoffmann, M. Kaloumenos, E. Erdem, S. Weber and J. J. Schneider, Eur. J. Inorg. Chem., 2014, 5554 CrossRef CAS.
- T. A. Cheema and G. Garntweiler, CrystEngComm, 2014, 16, 3366 RSC.
- J. Robertson and R. M. Wallace, Mater. Sci. Eng., R, 2015, 88, 1 CrossRef.
- P. Ortiz, A. Facchetti and T. J. Marks, Chem. Rev., 2010, 110, 205 CrossRef PubMed.
- G. He, Z. Sun and L. Zhang, Crit. Rev. Solid State Mater. Sci., 2012, 37, 131 CrossRef CAS.
- C. Chaneliere, J. L. Autran, R. A. B. Devine and B. Balland, Mater. Sci. Eng., R, 1998, 22, 269 CrossRef.
- M. Epifani, R. Zamani, J. Arbiol, C. Fabrega, T. Andreu, G. B. Pace, P. Siciliano and J. R. Morante, Thin Solid Films, 2014, 555, 39 CrossRef CAS.
- J. C. Sarker, R. Vasan, Y. F. Makableh, S. Lee, A. I. Nusir and M. O. Manasreh, Sol. Energy Mater. Sol. Cells, 2014, 127, 58 CrossRef CAS.
- J. Buha, D. Arcon, M. Niederberger and I. Djerdi, Phys. Chem. Chem. Phys., 2010, 12, 15537 RSC.
- P. J. Bonitatibus, A. S. Torres, B. Kandapallil, B. D. Lee, G. D. Goddard, R. E. Colborn and M. E. Marino, ACS Nano, 2012, 6, 6650 CrossRef CAS PubMed.
- M. Hayatifar, F. Marchetti, G. Pamploni and S. Zacchini, Inorg. Chem., 2013, 52, 4017 CrossRef CAS PubMed.
- T. V. Rogava, M. I. Yanovskaya, D. U. Grudtsyna, D. Yu and E. P. Kovsman, Russ. J. Gen. Chem., 1997, 67, 1078 Search PubMed.
- S. Prakash and P. N. Kapoor, Inorg. Chim. Acta, 1971, 5, 372 CrossRef CAS.
- V. Krishnan, S. Gross, S. Müller, L. Armelao, E. Tondello and H. Bertagnolli, J. Phys. Chem. B, 2007, 111, 7501 CrossRef CAS PubMed.
- A. M. El-Awad and R. M. Mahfouz, J. Therm. Anal., 1989, 35, 1413 CrossRef CAS.
- F. J. Caires, L. S. Lima, C. T. Carvalho, R. J. Giagio and M. Ionashiro, Thermochim. Acta, 2010, 497, 35 CrossRef CAS.
- M. A. Mohamed, A. K. Galwey and S. A. Halawy, Thermochim. Acta, 1998, 323, 27 CrossRef CAS.
- J. Gonzalez, M. C. Del Ruiz, J. B. Rivarola and D. Paquevich, J. Mater. Sci., 1998, 33, 4173 CrossRef CAS.
- T. J. Bright, J. I. Watjen, Z. M. Zhang, C. Muratone, A. A. Voevodin, D. I. Koukis, D. B. Tanner and D. J. Arenas, J. Appl. Phys., 2013, 114, 083515 CrossRef.
- P. Shang, S. Xiong, L. Li, D. Tian and W. Ai, Appl. Surf. Sci., 2013, 285P, 713 CrossRef.
- A. X. Wei, Z. X. Ge, X. H. Zaho, J. Liu and Y. Zhao, J. Alloys Compd., 2011, 509, 9758 CrossRef CAS.
- E. Çetinörgü-Goldenberg, J. E. Klemberg-Sapieha and L. Martinu, Appl. Opt., 2012, 51, 6498 CrossRef PubMed.
- R. S. Devan, C. L. Lin, S. Y. Gao, C. L. Cheng, Y. Liou and Y. R. Ma, Phys. Chem. Chem. Phys., 2011, 13, 13441 RSC.
- M. Zhu, Z. Zhang and W. Miao, Appl. Phys. Lett., 2006, 89, 021915 CrossRef.
- B. Leung, J. Song, Y. Zhang and J. Han, Adv. Mater., 2013, 25, 1285 CrossRef CAS PubMed.
- C. Chaneliere, S. Four, J. L. Auran, R. A. B. Devine and N. P. Sandler, J. Appl. Phys., 1998, 83, 4823 CrossRef CAS.
- E. Atanassova, M. Kalitzova, G. Zollo, A. Paskaleva, A. Peeva, M. Georgieva and G. Vitali, Thin Solid Films, 2003, 426, 191 CrossRef CAS.
- R. Parmentier, F. Lernarchand, M. Cathelinaud, M. Lequime, C. Amra, S. Labat, S. Bozzo, F. Bocquet, A. Charai, O. Thomas and C. Dominici, Appl. Opt., 2002, 41, 3270 CrossRef CAS PubMed.
- C. Chaneliere, S. Four, J. L. Autran and R. A. B. Devine, Microelectron. Reliab., 1999, 39, 261 CrossRef.
- J. J. Schneider, R. C. Hoffmann, A. Issanin and S. Dilfer, Mater. Sci. Eng., C, 2011, 176, 965 CrossRef CAS.
- T. Ramdeen, L. A. Dissado and R. M. Hill, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 352 RSC.
- M. Su, J. Wang and Y. Hao, Mater. Chem. Phys., 2011, 126, 31 CrossRef CAS.
- S. Yang, C. S. Hwang, J. I. Lee, S. M. Yoon, M. K. Ryu, K. I. Cho, S. K. Park, S. H. Kim, C. E. Park and J. Jang, Appl. Phys. Lett., 2011, 98, 103515 CrossRef.
- N. Alimardani, J. M. McGlone, J. F. Wager and J. F. Conley, J. Vac. Sci. Technol., A, 2014, 32, 01A122 Search PubMed.
- H. Kaftelen, K. Ocakoglu, S. Tu, R. Thomann, S. Weber and E. Erdem, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 014113 CrossRef.
- V. V. Laguta, M. I. Zaritskii, M. D. Glinchuk, I. P. Bykov, J. Rosa and L. Jastrabak, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 156 CrossRef CAS.
- J. Strunk, W. C. Vining and A. T. Bell, J. Phys. Chem. C, 2010, 114, 16937 CAS.
- E. Erdem, J. Alloys Compd., 2014, 605, 34 CrossRef CAS.
- R. Ramprasad, J. Appl. Phys., 2003, 94, 5609 CrossRef CAS.
- R. Ramprasad, M. Sadd, D. Roberts, T. Remmel, M. Raymond, E. Luckowski, S. Kalpat, C. Barron and M. Miller, Microelectron. Eng., 2003, 69, 190 CrossRef CAS.
- R. Ramprasad, J. Appl. Phys., 2004, 95, 954 CrossRef CAS.
- Y. Yang, O. Sugino and Y. Kawazoe, Solid State Commun., 2014, 195, 16 CrossRef CAS.
- Y. Guo and J. Robertson, Appl. Phys. Lett., 2014, 104, 112906 CrossRef.
- H. Shin, S. Y. Park, S.-T. Bae, S. Lee, K. S. Hong and H. S. Jung, J. Appl. Phys., 2008, 104, 116108 CrossRef.
- IEEE standard 62860-2013: test methods for the characterization of organic transistors and materials.
Footnote |
| † Electronic supplementary information (ESI) available: MS spectra, XRD data, details of EPR investigations and SEM micrographs. See DOI: 10.1039/c5cp05166e |
| This journal is © the Owner Societies 2015 |