
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral modification of platinum: ab initio study of the effect of hydrogen coadsorption on stability and geometry of adsorbed cinchona alkaloids†
Konstanze R.
Hahn
a,
Ari P.
Seitsonen
b and
Alfons
Baiker
*c
aDepartment of Physics, University of Cagliari, Cittadella Universitaria, I-09048 Monserrato, Italy
bDepartment of Chemistry, Ecole Normale Superieur, F-75005 Paris, France
cDepartment of Chemistry and Applied Biosciences, ETH Zurich, Hönggerberg, HCI, CH-8093 Zurich, Switzerland. E-mail: alfons.baiker@chem.ethz.ch
First published on 22nd September 2015
Abstract
The cinchona alkaloids cinchonidine and cinchonine belong to the most efficient chiral modifiers for the noble metal-catalyzed enantioselective hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC bonds. Under reaction conditions these modifiers are coadsorbed on the noble metal surface with hydrogen. Using density functional theory, we studied the effect of coadsorbed hydrogen on the adsorption mode of cinchonidine and cinchonine on a Pt(111) surface at different hydrogen coverages. The theoretical study indicates that the presence of coadsorbed hydrogen affects both the adsorption geometry as well as the stability of the adsorbed cinchona alkaloids. At all hydrogen coverages the cinchona alkaloids are found to be adsorbed via anchoring of the quinoline moiety. In the absence of hydrogen as well as at low hydrogen coverage the quinoline moiety adsorbs nearly parallel to the surface, whereas at higher hydrogen coverage it becomes tilted. Higher hydrogen coverage as well as partial hydrogenation of the quinoline part of the cinchona alkaloid and hydrogen transfer to the CC double bond at 10, 11 position of the quinuclidine moiety destabilize the adsorbed cinchona alkaloid, whereas hydrogen transfer to the nitrogen atom of the quinoline and the quinuclidine moiety stabilizes the adsorbed molecule. The stability as well as the adsorption geometry of the cinchona alkaloids are affected by the coadsorbed hydrogen and are proposed to influence the efficiency of the enantiodifferentiating ability of the chirally modified platinum surface.
O and CC bonds. Under reaction conditions these modifiers are coadsorbed on the noble metal surface with hydrogen. Using density functional theory, we studied the effect of coadsorbed hydrogen on the adsorption mode of cinchonidine and cinchonine on a Pt(111) surface at different hydrogen coverages. The theoretical study indicates that the presence of coadsorbed hydrogen affects both the adsorption geometry as well as the stability of the adsorbed cinchona alkaloids. At all hydrogen coverages the cinchona alkaloids are found to be adsorbed via anchoring of the quinoline moiety. In the absence of hydrogen as well as at low hydrogen coverage the quinoline moiety adsorbs nearly parallel to the surface, whereas at higher hydrogen coverage it becomes tilted. Higher hydrogen coverage as well as partial hydrogenation of the quinoline part of the cinchona alkaloid and hydrogen transfer to the CC double bond at 10, 11 position of the quinuclidine moiety destabilize the adsorbed cinchona alkaloid, whereas hydrogen transfer to the nitrogen atom of the quinoline and the quinuclidine moiety stabilizes the adsorbed molecule. The stability as well as the adsorption geometry of the cinchona alkaloids are affected by the coadsorbed hydrogen and are proposed to influence the efficiency of the enantiodifferentiating ability of the chirally modified platinum surface.
1 Introduction
Cinchona alkaloids (cinchonidine, cinchonine, quinine and quinidine) play an important role as organocatalysts in many reactions.1,2 They are also the most efficient organic molecules for bestowing chirality to noble metal catalysts used for asymmetric hydrogenation.3–8 Key for the enantiodifferentiating power of these chirally modified catalysts is the adsorptive anchoring and conformational structure of the cinchona alkaloid on the noble metal surface. Subtle changes in the adsorptive anchoring and conformation can induce dramatic changes in the enantioselection.9Cinchona alkaloids show a rich conformational behavior and the population of the different conformers depends on the polarity of the reaction medium10 and temperature. Attenuated total reflection (ATR-IR)11 and reflection–adsorption (IRAS)12 infrared spectroscopy studies revealed that at least three different adsorption modes of cinchonidine can be distinguished on a polycrystalline Pt surface. Depending on surface coverage the following adsorption modes were uncovered: (i) anchoring by nearly parallel adsorption via aromatic π-bonding of the quinoline moiety, (ii) tilted adsorption of the quinoline moiety via the N-lone pair where the α-H is abstracted, and (iii) tilted adsorption via N-lone pair bound quinoline.11
Besides their experimental investigation, the conformations of cinchonidine adsorbed on a clean Pt(111) surface have also been studied theoretically using first principles studies.13 The adsorption mode of the modifier is considered to be crucial for the enantiodifferentiating interaction between the adsorbed chiral modifier and the substrate and thus for achieving high enantioselectivity in the asymmetric hydrogenation of activated ketones over cinchona-modified noble metals.
Time lapsed scanning tunneling microscopy (STM) studies of cinchonidine and cinchonine adsorption on Pt(111) surfaces indicated considerable mobility of the adsorbed cinchona alkaloids, which increased when the hydrogen pressure was increased.14 There exists considerable evidence that the adsorption geometry of the cinchona alkaloid may change when other species involved in the reaction are coadsorbed, such as substrates (activated ketones)15 and hydrogen.14 In the latter case, destabilization of the adsorptive binding due to partial hydrogenation of the aromatic quinoline ring has been observed.16
Recently, Motobayashi et al. investigated the effect of hydrogen on the orientation of cinchonidine (CD) adsorbed on platinum using attenuated total reflection-surface enhanced infrared reflection adsorption spectroscopy (ATR-SEIRAS).17 This study showed that the adsorbed quinoline moiety of CD changed from a π-bonded nearly parallel orientation to an N-bonded upright one in the presence of hydrogen. The origin of the reorientation was ascribed by the authors to the repulsive interaction of the quinoline π-orbital and the negative surface charge induced by dissociative adsorption of H2 and hydrogenation of the CC double bond at C10–C11 (Scheme 1). Thus there exists considerable experimental evidence that hydrogen coadsorption affects the adsorption of the cinchona alkaloids, which in turn can greatly affect the enantiodifferentiating ability and stability of these chiral modifiers. To our knowledge there exists no systematic theoretical study focusing on the effect of hydrogen coadsorption on the adsorption geometry and stability of cinchona alkaloids.
 | ||
| Scheme 1 Chemical structure of the diastereomers cinchonidine and cinchonine. | ||
With this in mind we have studied the coadsorption of the most frequently used chiral modifiers, cinchonidine and cinchonine (CN), with hydrogen on Pt(111) using density functional theory (DFT). These two chiral modifiers are diastereomers and only differ in the chirality at C8 and C9 (see Scheme 1). Pt(111) was chosen as the model surface because structure sensitivity studies of the Pt-catalyzed asymmetric hydrogenation have indicated that the Pt(111) face is one of the most active in these reactions.18
2 Methods
DFT calculations of this study have been performed using the Quickstep module19 of the CP2K program package,20 a suite of programs developed to efficiently perform electronic structure and molecular dynamics calculations based on the Gaussian and plane waves formalism.21Interactions with the frozen atomic cores are described with norm-conserving pseudopotentials while valence electrons are treated explicitly.22 For O, N and C atoms, respectively six, five and four electrons have been considered in the valence shell and a triple-ζ valence plus double polarization basis set has been adopted for their valence electrons. The same kind of basis set has been used for H. Pt atoms have been simulated with 18 valence electrons and a triple-ζ basis set. The energy cutoff of the electron density has been set to 600 Ry and the relative cutoff to 50 Ry. Preliminary calculations showed the default value of 40 Ry for the relative cutoff to be insufficient for reliable results. Exchange–correlation potentials are modelled within the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) functional.23 Spin-polarization has been considered in all calculations. With these configurations, the lattice parameter of Pt has been optimized to be 3.965 Å.
The Pt surface, exposing the (111) crystallographic plane, has been simulated with a slab of four atomic layers and a total of 36 atoms in the surface plane resulting in a total number of 144 Pt atoms in the slab. The top two layers of the slab were allowed to relax. A vacuum of 25·ds, where ds is the slab thickness, has been added to avoid interactions with the periodic images of the slabs.
The binding energy EB of CD and CN has been calculated according to eqn (1), where EPt+mol is the total energy of the (hydrogenated) Pt(111) slab with the CD or CN molecule adsorbed, EPt refers to the total energy of the (hydrogenated) Pt(111) surface without adsorbate and Emol,gas is the energy of the corresponding molecule in the gas phase.
| EB = EPt+mol − (EPt + Emol,gas) | (1) |
The binding energy of hydrogen has been calculated accordingly, however, here, the reference state in gas phase is the H2 molecule and EB,H2 is normalized to the number n of H atoms (eqn (2)).
 | (2) |
With this definition negative values of the binding energy indicate an exothermic adsorption process. The more negative EB, the stronger and thus more stable is the adsorbed state of the molecule.
The relative energy shown in the figures represents the relative binding energy with respect to the reference state according to
| ΔEB,rel = EB − EB,ref. | (3) |
3 Results and discussion
3.1 Adsorption of hydrogen
Adsorption of H on Pt(111) has been simulated for isolated H atoms (θH = 1/36 ML) and at higher coverages of 0.5 and 1 ML. The binding energy EB,H2 has been calculated with respect to molecular H2 in the gas phase as described in detail in the Methods section. The preferred adsorption site for isolated H is found to be on top of a surface Pt atom with a binding energy of −0.52 eV (Table 1). On the fcc site it is marginally less stable with EB,H2 = −0.51 eV. The hcp site is least stable with EB,H2 = −0.47 eV. These trends are in excellent agreement with previous DFT calculations where H was reported to be most stable on a top site followed by the fcc site where it was marginally less stable by 0.01 eV. On hcp it was least stable with an energy difference of 0.04 eV with respect to the top site.24| θ H | E B,H2 [eV] | Distribution | ||
|---|---|---|---|---|
| top | fcc | hcp | ||
| 1/36 | −0.52 | −0.51 | −0.47 | — |
| 0.5 | −0.45 | — | — | Homogeneous [01![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ] ] |
| −0.45 | — | — | Homogeneous [12] |
|
| −0.43 | — | — | Segregated | |
| 1 | −0.37 | −0.39 | −0.33 | — |
The binding energies on top, fcc and hcp sites result in −2.84, −2.83 and −2.79 eV, respectively, when evaluated with respect to atomic H in the gas phase. This is in very good agreement with the binding energy derived experimentally using helium beam diffraction (EB,H = −2.78 eV).25
Increasing coverage leads to destabilization of adsorbed H atoms. At 0.5 ML and 1 ML, the binding energy of H adsorbed on a top site increases to −0.45 and −0.37 eV, respectively, indicating repulsive interactions between adsorbed H atoms. Lateral repulsion between adsorbed species has been reported also experimentally.26
In the case of partially hydrogenated Pt (θH = 0.5 ML), different distributions of H atom on the surface have been considered. When H atoms are segregated on one part of the surface, the binding energy increases (destabilization) slightly to −0.43 eV. The alignment of homogeneously distributed H atoms does not affect the binding energy. Both alignment along the [01] and the [12] direction resulted in EB,H2 = −0.45 eV.
At 1 ML, the stability of H has been additionally investigated on fcc and hcp sites. The most stable configuration of H at 1 ML is found on an fcc site. This is only slightly more stable (ΔEB,H2 = 0.02 eV) than on a top site (Table 1). In the following calculations, H adsorption has been considered on top Pt sites.
3.2 Adsorption of quinoline
Previous experimental investigations showed that the quinoline part of the cinchona alkaloids plays an important role in the adsorption interaction between the cinchona alkaloids and the Pt surface.11,12,17 Therefore, we first investigated the stability of quinoline on clean Pt(111) for several configurations adopted from previous calculations.27 The most stable configuration with a binding energy of −1.88 eV is shown in Fig. 1(a1). Adsorption of quinoline has additionally been calculated on partially and fully hydrogenated Pt(111). In general, hydrogenation of the surface leads to a destabilization of the quinoline molecule. However, the H distribution on the surface has to be considered as well since it affects the availability of active sites. | ||
| Fig. 1 Relative binding energy and orientation of quinoline adsorbed on (hydrogen covered) Pt(111). The relative energy is calculated with respect to the binding energy of the reference state a1 where quinoline is adsorbed on the clean Pt(111) surface. Results of configurations where no H transfer to the molecule takes place are shown in green. Results where H transfer to N takes place are shown in blue. The lower panel shows sample configurations of quinoline adsorption on Pt without H transfer at θH = 0 (a1), 1/36 (a2) and 0.5 ML (a3) and configurations where H transfer to N takes place at θH = 1/36 (b2), 0.5 (b3) and 1 ML (b4). H atoms that are transferred to the molecule are shown in black. | ||
At 0.5 ML coverage, several distributions of H atoms have been investigated. The quinoline molecule is found to be most stable (EB = −1.14 eV) when H atoms are segregated leaving enough active sites for quinoline to adsorb (Fig. S1, ESI†). When H atoms are distributed homogeneously on the surface, several factors have to be considered that lead to a change in stability. These include, for example, repulsive interactions between the surface H atoms and the quinoline molecule, diffusion of H atoms to different surface sites and H transfer to the molecule. An elaborate study of such, however, is not viable in the context of this work. Nevertheless, here, it is noted that the quinoline molecule tends to incline when surface Pt sites underneath the molecule are occupied by H atoms. For a homogeneous distribution of H atoms at 0.5 ML coverage, a binding energy of −0.62 eV is calculated for a configuration where the quinoline molecule forms an angle αQ–Pt of 46° with the surface plane of Pt(111) (Table 2, Fig. 1(a3)). The elevated H coverage can additionally lead to hydrogenation of the quinoline molecule. In the sample configurations calculated here hydrogen transfer to the N atom results in a binding energy of −0.93 eV indicating a stabilization by 0.31 eV. In this configuration (Fig. 1(b3)), the quinoline molecule is with 22° less inclined compared to the configuration without H transfer.
| θ H | E B [eV] | α Q–Pt [°] | H uptake | E B,H2 [eV] | ||
|---|---|---|---|---|---|---|
| On C | On N | |||||
| 0 ML | −1.88 | 0 | — | — | — | Fig. 1(a1) |
| 0.5 ML | −1.14 | 0 | — | — | −0.41 | Fig. S1 (ESI) |
| −0.93 | 22 | — | 1 | −0.40 | Fig. 1(b3) | |
| −0.62 | 46 | — | — | −0.38 | Fig. 1(a3) | |
| 1 ML | −0.91 | 78 | 2 | 1 | −0.35 | Fig. 1(b4) |
On a fully hydrogenated Pt surface, a stable configuration has been found with a binding energy of −0.91 eV where the quinoline molecule is inclined by 78° (Fig. 1(b4)). In this configuration, one H atom is transferred to the N atom. Compared to the corresponding configuration at half H-coverage where H is transferred to N (EB = −0.93 eV), the configuration at full coverage is marginally destabilized by 0.02 eV. With respect to the configuration at 0.5 ML where no H transfer takes place, the quinoline molecule on a fully hydrogenated Pt surface is stabilized by 0.29 eV (see Fig. 1). In this configuration, additional H transfer to several C atoms of the quinoline molecule takes place. Comparing the total energies of these configurations thus has to be done with caution since different factors are present that affect the stability of the molecule.
3.3 Adsorption of cinchonidine
The effect of H transfer from the surface to the CD molecule has been investigated with one H atom on surface, corresponding to a hydrogen coverage of 1/36 ML. In the reference configuration, the H atom has been placed furthest away possible from the CD molecule to minimize interactions with the adsorbate. The binding energy of CD in this reference state has been calculated to be −2.27 eV (Table 3, “on Pt”). Even though the H atom is relatively far away from the CD molecule, the CD molecule is slightly destabilized (ΔEB = 0.04 eV) compared to adsorption on clean Pt(111) (−2.31 eV).
| H position | CD | CN | ||
|---|---|---|---|---|
| E B [eV] | ΔEB [eV] | E B [eV] | ΔEB [eV] | |
| On Pt | −2.27 | — | −2.32 | — |
| On N′ | −2.56 | −0.29 | −2.57 | −0.25 |
| On N | −2.45 | −0.18 | −2.75 | −0.43 |
| On C9′ | −1.83 | 0.44 | −1.90 | 0.42 |
| On C11 | −1.94 | 0.33 | −1.54 | 0.78 |
To study the effect of H transfer, the H atom has been placed explicitly next to specific N and C atoms of the CD molecule (Fig. 2, blue lines). In particular, the H atom has been attached to the N atom of the quinuclidine group, the N′ atom of the quinoline moiety, the C11 atom of the terminal vinyl group and to the C9′ atom of the quinoline group (cf.Scheme 1).
 | ||
| Fig. 2 Relative binding energy and orientation of CD and CN adsorbed on Pt(111) in the presence of one H atom (θH = 1/36 ML). The relative energy is calculated with respect to binding energy of CD adsorbed on Pt(111) with one H atom adsorbed on a top Pt site. The results for CD adsorption are represented in blue and for CN adsorption in red. The shown configurations correspond to adsorption of CD [CN] on Pt(111) when the H atom is bound to N (a1 [a2]), to N′ (b1 [b2]) and to C11 (c1 [c2]). | ||
Hydrogen transfer to nitrogen leads to a stabilization of the adsorbed molecule. It is most pronounced when the H atom binds to the N′ atom of the quinoline group (ΔEB = −0.29 eV) where the binding energy is −2.56 eV (Table 3). When the H atom is transferred to the N atom of the quinuclidine group, the molecule is stabilized by 0.18 eV (Fig. 2(a1)). H transfer to the C atoms of the quinoline group, on the other hand, is endothermic, thus unfavorable. Transfer to the C9′ atom results in a destabilization by 0.44 eV (EB = −1.83 eV) and H transfer to the CC double bond (C11) leads to a destabilization by 0.33 eV. In all configurations, the quinoline group is adsorbed parallel to the Pt(111) surface (αQ–Pt = 0°).
| θ H | E B [eV] | H to | α Q–Pt [°] | E B,H2 [eV] | |
|---|---|---|---|---|---|
| as: indicates that H atoms are located on active Pt sites needed for the CD molecule to adsorb. | |||||
| 0 ML | −2.31 | — | 0 | — | Fig. S2(a) (ESI) |
| 0.5 ML | −1.62 | — | 0 | −0.41 | Fig. 3(b) |
| −1.97 | N′ | 0 | −0.43 | Fig. 3(d) | |
| −1.34 | C9′ | 0 | −0.40 | Fig. S2(d) (ESI) | |
| −1.29 | C6′ | 0 | −0.39 | Fig. S2(c) (ESI) | |
| −1.03 | C11 | 0 | −0.38 | Fig. 3(c) | |
| −0.57 | as | 29 | −0.35 | Fig. 3(a) | |
| −1.18 | N′, as | 26 | −0.39 | Fig. 4(d) | |
| −1.34 | N′, C11 | 0 | −0.39 | Fig. 4(a) | |
| −0.47 | C11, as | 31 | −0.35 | Fig. 4(b) | |
| −0.95 | N′, C11, as | 29 | −0.37 | Fig. 4(c) | |
| 1 ML | −0.61 | N′, C4′ | 20 | −0.33 | Fig. 3(f) |
| −0.16 | N′, C4′, C11 | 22 | −0.31 | Fig. 3(e) | |
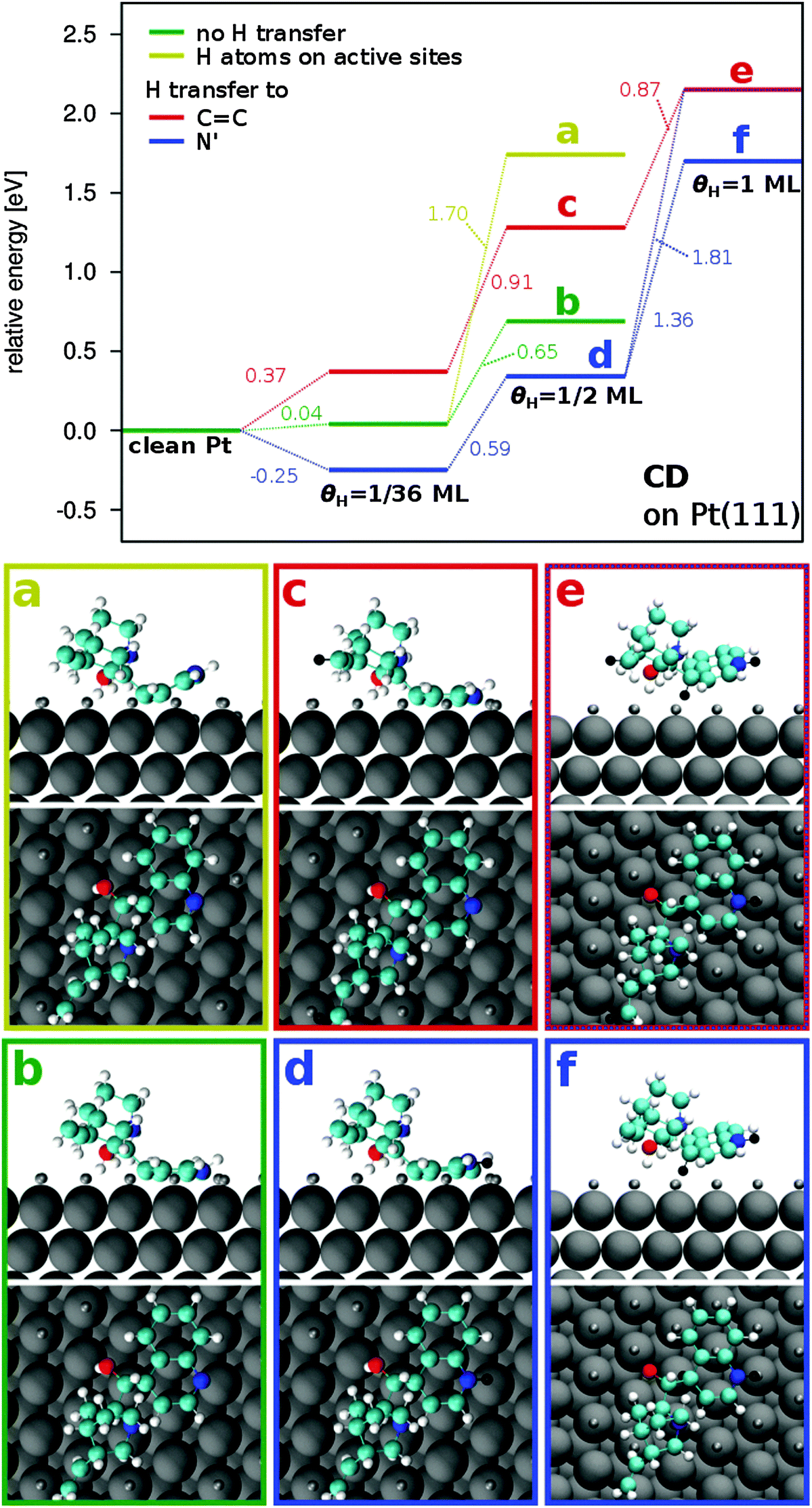 | ||
| Fig. 3 Relative binding energy and orientation of CD adsorbed on (hydrogen covered) Pt. The relative energy is calculated with respect to the binding energy of CD on clean Pt(111). Green lines show the results where no H transfer to the CD molecule takes place. Blue lines show the results for H transfer to the quinuclidine N′ atom and red lines for H transfer to the C11 atom. Configurations of CD are shown at θH = 0.5 ML with H atoms preadsorbed on active sites (a (light green)), at θH = 0.5 ML without interaction between CD and H (b), at θH = 0.5 ML with H transfer to C11 (c), at θH = 0.5 ML with H transfer to the quinoline N′ atom (d), at θH = 1 ML with H transfer to N′, C11 and C4′ (e) and at θH = 1 ML with H transfer to N′ and C4′ (f). | ||
Simulation of different configurations confirmed the stabilization of CD adsorption by hydrogen transfer to the N′ atom (Fig. 3, blue line). At θH = 0.5 ML, the molecule is stabilized by 0.35 eV, resulting in a binding energy of −1.97 eV (Fig. 4, Table 4). This is similar to the results found if only one H atom is present on the surface (ΔEB = −0.29 eV, Fig. 2(b1)). Instead, when the H atom is attached to one of the C atoms of the quinoline group (C9′ and C6′), the molecule is destabilized by ΔEB = 0.28 and 0.33 eV, with binding energies −1.34 and −1.29 eV, respectively (Table 4).
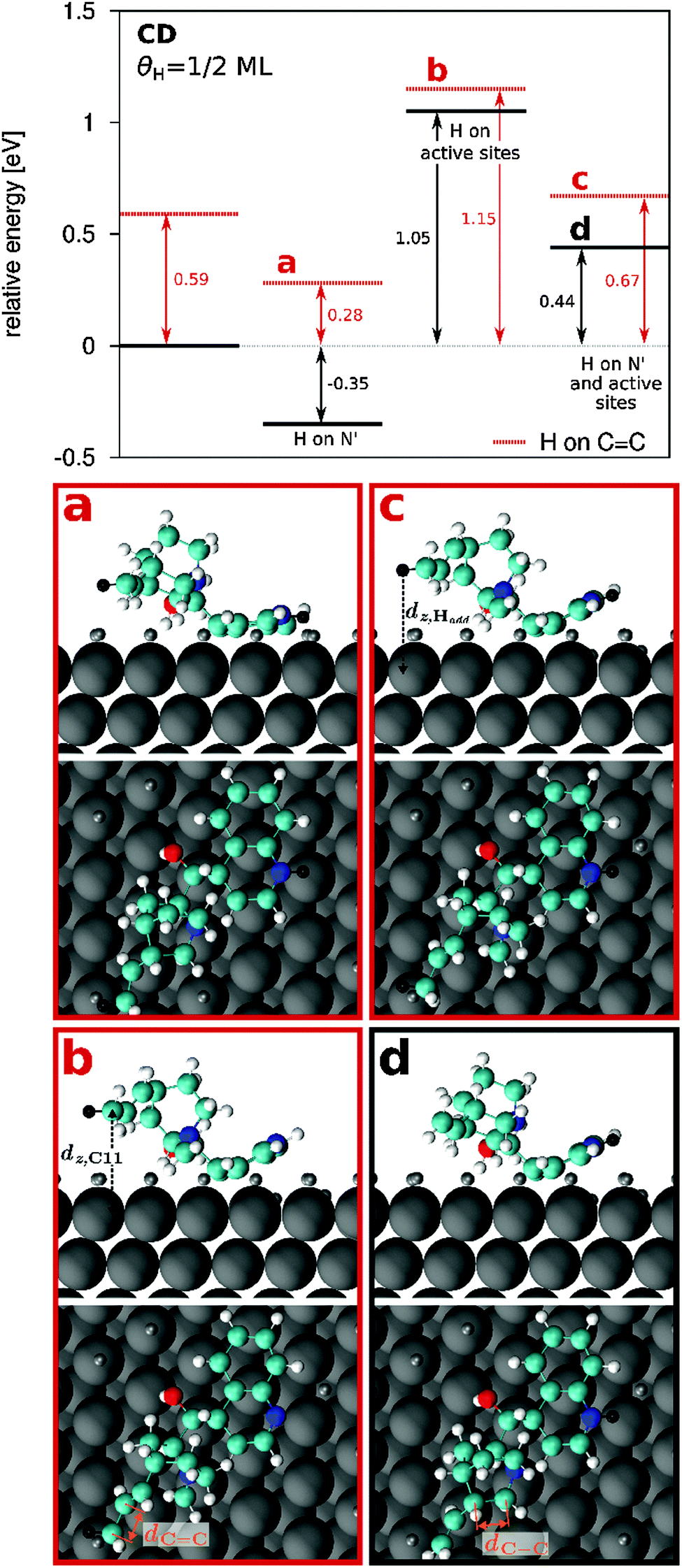 | ||
| Fig. 4 Relative binding energy and orientation of CD adsorbed on Pt at θH = 0.5 ML. The relative energy is calculated with respect to the binding energy of adsorbed CD where no H transfer to the CD molecule takes place and H atoms are distributed on Pt sites that are not relevant for CD adsorption (segregated H distribution). Black lines show the results where no H transfer to the C11 atom takes place and red dashed lines represent results where H transfer to the C11 atom takes place. Additional H transfer is indicated in the graph. Configurations are shown for H transfer to C11 and N′ (a), H transfer to C11 and preadsorbed H on active sites (b), H transfer to C11, N′ and preadsorbed H on active sites (c) and for H transfer to N′ and preadsorbed H on active sites (d). | ||
On partially hydrogenated Pt(111) surface with a hydrogen coverage of 0.5 ML, it is likely that H atoms cover active sites necessary for the CD molecule to adsorb. To account for these effects, H atoms have been specifically placed underneath the molecule. When two or more H atoms are transferred from unaffected top sites to active sites underneath the molecule, the quinoline group of CD bends away from the Pt surface. A binding energy of −0.57 eV is calculated when two H atoms cover the active sites underneath the CD molecule and no H transfer to the molecule takes place (Table 4, H to “as”). In this configuration the molecule is bent by 29° and it is destabilized with respect to the reference configuration at 0.5 ML by 1.05 eV (Fig. 3(a), Table 4).
The relatively low stability of this configuration suggests that, during the adsorption process, the CD displaces the H atoms from active sites. Considering their high mobility, H atoms are likely to diffuse to unaffected sites resulting in more stable configurations such as the reference state shown in Fig. 3(b). Furthermore, it could also be possible that the H atoms desorb as molecular H2. To examine this scenario a geometry optimization has been carried out using the configuration of Fig. 3(a) as initial state and manually moving the two H atoms from active sites to the gas phase. During the simulation the CD molecule bends back with the quinoline group parallel to the surface. The energy of the optimized configuration with one H2 molecule in the gas phase is by 0.34 eV more stable than the one with the two H atoms underneath the molecule. This indicates that at high H coverage (θH ≥ 0.5 ML) adsorption of the cinchona alkaloid is likely to displace H atoms from active sites which would then either diffuse to unaffected sites or desorb from the surface as molecular H2.
Hydrogen transfer to the quinoline N′ atom in such a configuration leads to a stabilization of the molecule by 0.61 eV with a binding energy of −1.18 eV forming an inclination angle of 26° (Fig. 4(d), Table 4). In a similar configuration, which is slightly less stable (EB = −1.05 eV) as a result of a stronger inclination of the quinoline group towards the surface (αQ–Pt = 33°), a comparable degree of stabilization upon H transfer to N′ (ΔEB = −0.59 eV) is observed (not shown). The effect of H transfer to N′ in these cases is stronger than for a configuration where the quinoline group is adsorbed planar on the surface (ΔEB = 0.35 eV). This can possibly result from the vicinity of the N′ atom to the surface which leads to repulsive interactions between the transferred H atom and the Pt surface when the molecule is adsorbed parallel to the surface (Fig. 3(d)). These interactions are less pronounced when the molecule is inclined, thus, leading to a higher degree of stabilization upon H transfer to N′ (Fig. 4).
A significant destabilization by 1.36 eV of the CD molecule is observed when the hydrogen coverage increases from θH = 0.5 to 1 ML (Fig. 3). The occupation of active Pt sites by H atoms repels the CD molecule from the surface. Furthermore, the probability of H transfer is increased, leading to hydrogenation of C atoms of the CD molecule. This is the case in the most stable configuration (Fig. 3(f)) where, in addition to H transfer to N′, one H atom is transferred to the C4′ atom. Hydrogen transfer to the C11 atom, leading to elimination of the CC double bond, additionally destabilizes the molecule by 0.45 eV (Fig. 3(e), EB = −0.16 eV). It should be noted that the energy levels represented in Fig. 3 do not refer to the actual binding energy of the corresponding configuration but are calculated with respect to the reference state of CD adsorbed on clean Pt(111).
In summary it is found that preadsorbed H on Pt(111) leads to a destabilization of the CD molecule. With increasing H coverage, the binding energy of CD on Pt(111) changes from −2.31 eV (θH = 0 ML) to −0.61 eV (θH = 1 ML) indicating a destabilization of the system. If H atoms are adsorbed on active Pt sites required for the CD molecule to adsorb, the quinoline group is inclined forming an angle αQ–Pt > 0° with the Pt(111) surface. The inclination angle increases with the number of H atoms underneath the molecule. H transfer to C atoms leads to an additional destabilization of the system in agreement with the results discussed above for isolated H atoms (θH = 1/36 ML). At all coverages, H transfer to the quinoline N′ atom leads to a stabilization of the CD molecule. The effect of the stabilization of CD upon H transfer to N′ is more pronounced when active sites are occupied by H atoms leading to an inclination of the CD molecule with respect to the Pt surface.
C double bond) has been evaluated based on the energy differences of several configurations at θH = 0.5 ML as shown in Fig. 4. Changes in structural properties in these configurations have been analyzed by the distance of the C11 atom (dz,C11) and the transferred H atom (dz,Hadd) from the surface and by the C–C bond length of C10 and C11 (dCC) and of C2 and C3 (dC–C) (cf.Scheme 1, Fig. 4).
When the CD molecule is adsorbed with the quinoline group parallel to the Pt(111) surface (Fig. 3(b), CDref), H transfer to the CC double bond leads to a notable destabilization by 0.59 eV (CDref-hyd, Table 5). In this configuration, the terminal CH2 moves closer to the Pt surface, reflected in a smaller distance of C11 (dz,C11 = 4.01 Å) to the Pt(111) surface plane with respect to the reference configuration (CDref, dz,C11 = 4.30 Å). Elimination of the CC double bond leads to an elongation of the C11–C10 bond length by 0.12 Å. The CC bond destabilization also affects the neighboring C3–C2 bond which is elongated by 0.17 Å (Table 5). Similar effects both in terms of stability and structure are observed when H is transferred to quinoline N′ prior to the H transfer to the C11 atom (CDN′, Fig. 4(a)). Details are represented in Table 5.
C double bond. The hydrogenated configurations have been created by transferring one surface H atom to the terminal vinyl group. Geometric properties include the height of the C11 atom (dz,C11) and the added H atom (dz,Hadd) above the Pt surface, the bond length between the C10 and the C11 atom (dCC) and the bond length between the C2 and the C3 atom (dC–C) (cf.Fig. 4)
| Label | E B [eV] | ΔE [eV] | Distances [Å] | α Q–Pt [°] | |||
|---|---|---|---|---|---|---|---|
| d z,C11 | d z,Hadd |
d
CC
|
d C–C | ||||
| CDref | −1.62 | — | 4.30 | — | 1.34 | 1.57 | 0 |
| CDref-hyd | −1.03 | 0.59 | 4.01 | 4.00 | 1.46 | 1.74 | 0 |
| CDN′ | −1.97 | — | 4.32 | — | 1.34 | 1.57 | 0 |
| CDN′-hyd | −1.34 | 0.63 | 3.97 | 3.90 | 1.47 | 1.72 | 0 |
| CDas | −0.57 | — | 4.25 | — | 1.34 | 1.57 | 29 |
| CDas-hyd | −0.47 | 0.10 | 4.77 | 4.73 | 1.50 | 3.42 | 31 |
| CDN′,as | −1.18 | — | 4.23 | — | 1.34 | 1.57 | 27 |
| CDN′,as-hyd | −0.95 | 0.23 | 4.70 | 4.59 | 1.50 | 3.45 | 29 |
Significant differences in the effect of H transfer to the CC double bond are observed when the quinoline group is inclined with respect the Pt(111) surface (CDas and CDN′,as). In these cases, the C2–C3 bond is broken and the destabilization effect is less pronounced. When H is added to the quinoline N′ atom, additional H transfer to C11 leads to a destabilization of the system by 0.23 eV (Fig. 4(c)) and even less (0.10 eV, Fig. 4(b)) when no H atom has been transferred to the CD molecule prior to the H transfer to the CC group. The smaller destabilization effect possibly results from a higher flexibility and mobility of the quinuclidine moiety. In particular, the quinuclidine group moves away from the surface indicated by a larger distance of the C11 atom and of the added H atom from the surface (see Table 5).
3.4 Adsorption of cinchonine
Hydrogen transfer to N and N′ results in a stabilization of the CN molecule by 0.43 and 0.25 eV, respectively (Fig. 2(a2 and b2), Table 3). The different absolute configuration at the C8 center in the CN molecule, (R), results in a change in the orientation of the quinuclidine group compared to the CD isomer. In the CD molecule, where C8 has (S) character, the OH group points towards the surface (Fig. 2(a1)). In contrast, in CN, the OH group is oriented away from the surface and the quinuclidine N atom points downward. When H is transferred to the N atom, the H atom points towards the Pt surface (Fig. 2(a2)) indicating attractive interactions reflected in stronger stabilization (ΔEB = −0.43 eV) compared to the situation in CD (ΔEB = −0.18 eV). The orientation of the quinoline group, on the other hand, is identical with respect to the surface in both CN and CD. Thus, H transfer to the quinoline N′ atom has a similar effect in CN (Fig. 2(b2)) and CD (Fig. 2(b1)) with a stabilization by 0.25 and 0.29 eV, respectively (Table 3).
Hydrogen transfer to a C atom instead destabilizes the CN molecule equivalent to what is found for the CD molecule. If attached to the C9′ atom of the quinoline group, CN is destabilized by 0.42 eV. Even more pronounced is the effect of H transfer to the CC double bond (Fig. 2(c2)). It results in a destabilization by 0.78 eV. In the case of CD, CC bond elimination has less effect. There, the adsorbed molecule is destabilized by only 0.33 eV.
 | ||
| Fig. 5 Relative binding energy and orientation of CN adsorbed on (hydrogen covered) Pt. The relative energy is calculated with respect to the binding energy of CN on clean Pt(111). Green lines show the results where no H transfer to the CN molecule takes place. Blue lines show the results for H transfer to the quinuclidine N′ atom, light blue for H transfer to the quinoline N atom and red for H transfer to the C11 atom. Configurations of CN are shown at θH = 0.5 ML with H transfer to C11 (a), at θH = 0.5 ML without interaction between CN and H (b), at θH = 1 ML with H transfer to C11 (c), at θH = 1 ML without H transfer to the CN molecule (d), at θH = 1 ML with H transfer to N′ (e) and at θH = 1 ML with H transfer to N (f). | ||
 | ||
| Fig. 6 Relative binding energy and orientation of CN adsorbed on Pt at θH = 0.5 ML. The relative energy is calculated with respect to the binding energy of adsorbed CN where no H transfer to the CN molecule takes place and H atoms are distributed on Pt sites that are not relevant for CD adsorption (segregated H distribution). Black lines show the results where no H transfer to the C11 atom takes place and red dashed lines represent results where H transfer to the C11 atom takes place. Additional H transfer is indicated in the graph. Configurations are shown for H transfer to C11 (a), H transfer to N′ (b), H transfer to C11 and with preadsorbed H on active sites (c), no H transfer but with preadsorbed H on active sites (d), H transfer to C11, N′ and with preadsorbed H on active sites (e) and for H transfer to N′ and with preadsorbed H on active sites (f). | ||
When H atoms are adsorbed on active Pt sites underneath the CN molecule bending of the quinoline group is observed similar to the case of CD. For such a configuration (without any H transfer) a binding energy of −0.66 eV has been calculated (Fig. 6(d)), corresponding to a destabilization by 0.90 eV with respect to the reference state. In this configuration the quinoline group is bent by 28° (Table 6).
| θ H | E B [eV] | H to | α Q–Pt [°] | E B,H2 [eV] | |
|---|---|---|---|---|---|
| as indicates that H atoms are located on active Pt sites needed for the CD molecule to adsorb.a Negative values of αCD–Pt indicate an inclination of the quinoline group in the other direction (formation of a roof-like structure). | |||||
| 0 ML | −2.37 | — | 0 | — | Fig. S3(a) (ESI) |
| 0.5 ML | −1.56 | — | 0 | −0.40 | Fig. 5(b) |
| −1.84 | N′ | 0 | −0.42 | Fig. 6(b) | |
| −0.69 | C11 | 0 | −0.36 | Fig. 5(a) | |
| −0.66 | as | 28 | −0.35 | Fig. S3(b) (ESI) | |
| −1.12 | N′, as | 26 | −0.38 | Fig. 6(f) | |
| −0.99 | N′, C11 | 0 | −0.37 | Fig. 6(a) | |
| 0.22 | C11, as | 27 | −0.31 | Fig. 6(c) | |
| −0.21 | N′, C11, as | 26 | −0.33 | Fig. 6(e) | |
| 1 ML | 0.22 | C11 | −22 | −0.30 | Fig. 5(c) |
| −0.24 | as | −26 | −0.32 | Fig. 5(d) | |
| −0.96 | N′ | −51 | −0.34 | Fig. 5(e) | |
| −1.09 | N | 14 | −0.34 | Fig. 5(f) | |
For CD the hydrogen transfer to quinoline N′ of the inclined molecule stabilizes the molecule more (ΔEB = −0.61 eV) than in the case of planar adsorption (ΔEB = −0.35 eV). This trend is also observed for CN, however, less prominent. Upon H transfer to N′, the inclined molecule is stabilized by 0.46 eV (EB = −1.12 eV, Fig. 6(f)) while in the planar configuration the energy gain has been calculated to be 0.28 eV (Fig. 6(b)).
Increasing the hydrogen coverage leads to additional destabilization of the adsorbed CN molecule. In the case where hydrogen is transferred to quinoline N′, the CN molecule is destabilized by 0.88 eV (Fig. 5, blue lines) for the H coverage increasing from θH = 0.5 ML to 1 ML. Significant differences are observed in the adsorption modes of CD and CN at 1 ML. In CD, no configuration is found without H transfer to one of the C atoms of the quinoline moiety. Furthermore, all stable configurations have similar binding mechanisms with respect to the ones at 0.5 ML, which is with the C4′ atom closest to the surface and the quinoline group bent away (see Fig. 3). The most stable configuration is obtained when H is transferred to quinoline N′ and C4′. For CN, on the other hand, in the most stable configuration H is transferred to the quinuclidine N atom (Fig. 5(f)), resulting in a binding energy of −1.09 eV (Table 6). The bending angle in this configuration is 14°. The CN molecule becomes slightly less stable when H is transferred to the quinoline N′ instead resulting in a binding energy of −0.96 eV. In this configuration, the CN molecule is bent in the other direction, forming a “roof-like” shape with an inclination angle of 51° between the quinoline group and the Pt surface. Negative values of αQ–Pt given in Table 6 refer to configurations with a roof-like shape where the quinoline group is inclined in the opposite direction as the quinoline moiety in CD. Both configurations are more stable compared to the CD molecule (EB = −0.61 eV).
When no H atom is transferred to the CN molecule, a binding energy of −0.24 eV is calculated (Fig. 5(d)) with the molecule again in a roof-like configuration forming an angle of 26° with the Pt surface. Accordingly, hydrogen transfer to quinuclidine N and quinoline N′ leads to a stabilization with respect to the latter configuration by 0.85 and 0.72 eV, respectively, which is more pronounced than at 1/36 ML (0.43 and 0.25 eV). Hydrogen transfer to the C11 atom (Fig. 5(c)), on the other hand, results in a destabilization by 0.46 eV and the adsorption process becomes endothermic with a positive binding energy of 0.22 eV.
In summary, general stabilization trends are similar for the two adsorbed diastereomers CN and CD. Both molecules are destabilized with increasing H coverage. Hydrogen transfer to the N′ (and N) atoms, instead, leads to a stabilization of the adsorbed cinchona alkaloids. In CN, the range of binding energies for the calculated sample configurations is larger than for CD. This indicates a higher flexibility of the molecule and a smoother potential energy surface in the case of CN. Furthermore, the CN molecule tends to bind in a roof-like configuration at high H coverage.
C double bond at C10, C11 position has been studied for CN at θH = 0.5 ML in analogy to CD. The investigated configurations are CN adsorbed with the quinoline group parallel to the surface and no H transfer (CNref), with H transfer to quinoline N′ (CNN′), with H atoms adsorbed on active sites leading to a bending of the quinoline group (CNas) and with both H transfer to N′ and H atoms on active sites (CNN′,as). The required energy for CC bond elimination and subsequent C–H bond formation is similar in all configurations (0.88 ± 0.03 eV, Table 7, Fig. 6) and it is notably higher than that for the CD molecule (ΔEB,max = 0.61 eV).
C double bond. The hydrogenated configurations have been created by transferring one surface H atom to the C11 atom of the terminal vinyl group. The geometric properties are compared to the corresponding configuration without H transfer to the vinyl group. See Table 5 and Fig. 4 for specification of geometrical parameters
| Label | E B [eV] | ΔE [eV] | Distances [Å] | α Q–Pt [°] | |||
|---|---|---|---|---|---|---|---|
| d z,C11 | d z,Hadd |
d
CC
|
d C–C | ||||
| CNref | −1.56 | — | 5.62 | — | 1.34 | 1.56 | 0 |
| CNref-hyd | −0.69 | 0.87 | 4.93 | 4.22 | 1.47 | 1.67 | 0 |
| CNN′ | −1.84 | — | 5.62 | — | 1.34 | 1.56 | 0 |
| CNN′-hyd | −0.99 | 0.85 | 4.78 | 4.00 | 1.48 | 1.67 | 0 |
| CNas | −0.66 | — | 5.54 | — | 1.34 | 1.56 | 28 |
| CNas-hyd | 0.22 | 0.88 | 4.76 | 4.02 | 1.47 | 1.67 | 27 |
| CNN′,as | −1.12 | — | 5.61 | — | 1.34 | 1.56 | 26 |
| CNN′,as-hyd | −0.21 | 0.91 | 4.68 | 3.90 | 1.48 | 1.66 | 26 |
In contrast to CD, in CN, the C2–C3 bond of the quinuclidine remains stable in all calculated configurations. This signifies a reduced flexibility of the quinuclidine moiety with respect to the cases where the C2–C3 bond is broken. Hydrogen transfer to C11 in CN simply leads to a destabilization of the C2–C3 bond indicated by an elongation of the bond 0.11 ± 0.006 Å.
At the same time, the C10–C11 bond is elongated by 0.14 Å to 1.47 Å (in CNref-hyd and CNas-hyd) and 1.48 Å (in CNN′-hyd and CNN′,as-hyd, see Table 7). Hydrogen transfer to the CC bond furthermore leads to a bending of the CH3 group towards the surface as demonstrated by the decrease in dz,C11. However, the distance between the CH3 and the Pt surface remains longer than that for CD. More structural and energetic properties of all calculated configurations are represented in Table 7.
3.5 Comparison of adsorption behavior of the cinchona alkaloids
Density functional theory has been used to study the coadsorption of the cinchona alkaloids (CD and CN) with hydrogen on the Pt(111) surface. Adsorption has been simulated focusing on the effect of preadsorbed hydrogen on Pt(111) and possible H transfer to the adsorbed cinchona alkaloids.On the clean Pt(111) surface, both diastereomers adsorb with the quinoline group parallel to the Pt(111) surface plane. In this configuration, CN is slightly more stable with a binding energy of −2.37 eV compared to the pseudo-enantiomer CD which is adsorbed with a binding energy of −2.31 eV.
The effect of H transfer to the C9′ atom of the quinoline moiety is again similar for both molecules. It leads to a destabilization by 0.44 and 0.42 eV in adsorbed CD and CN, respectively. Elimination of the CC double bond of the terminal vinyl group, on the other hand, destabilizes the adsorbed CD molecule by only 0.33 eV while the CN molecule is destabilized by 0.78 eV. As a matter of fact, in this configuration, the CD molecule is more stable (EB = −1.94 eV) than the CN molecule (EB = −1.54 eV). Apparently, the different chirality of the C8 center and the resulting different orientation of the quinuclidine moiety significantly affect hydrogenation processes and the resulting stability of the two diastereomers on the Pt(111) surface.
At all coverages, the most stable configurations are found when H transfer to N takes place. In CD, the transfer to N′ of the quinoline moiety is more stable, in CN the transfer to N of the quinuclidine group. In fact, at low H coverage (θH = 1/36 ML) this is even more stable than adsorption on a clean Pt(111) surface (Fig. 3 and 5 and Tables 4 and 6). In the interest of a better comparison to CD, Fig. 5 shows the energy levels of CN when H transfer to N′ takes place (blue line) even though transfer to N is expected to result in the highest stability at all coverages.
With increasing H coverage, more Pt sites become occupied by H reducing the number of available sites for the cinchona alkaloids to adsorb. As a results, at high H coverage (θH ≥ 0.5 ML), the molecules tend to anchor on less sites and become lifted away from the surface. In these cases, a remarkable destabilization of the molecules is observed and the quinoline group bends away from the surface. At 0.5 ML, the adsorbed CD and CN become destabilized by 1.05 and 0.90 eV, respectively, when H atoms cover active sites underneath the molecule. In these configurations the quinoline group forms an angle of 29° (CD) and 28° (CN) with the Pt surface.
As a result of the high mobility of H atoms on the Pt surface, it is likely that H atoms diffuse to unaffected sites upon adsorption of the molecules. Furthermore, the H atoms could desorb as molecular H2 as has been shown in the case of CD.
At full coverage (1 ML), all active sites are covered by H atoms and the molecules are destabilized even more. Despite the simulation of numerous initial configurations, only a limited number of stable configurations has been found for CD at 1 ML coverage (Fig. 3). In these configurations, H atoms are transferred to N′ and one C atom of the quinoline group (C4′). Additional H transport to the C11 atom, resulting in the elimination of the CC double bond, leads to a destabilization by 0.45 eV. A similar inclination angle of the quinoline group is observed for these configurations. It results in 20° for H transport to N′ and C4′ and in 22° when H is additionally transferred to the C11 atom. For CN, on the other hand, several stable configurations with H transfer to different atomic sites of the molecule have been found. These include the H transfer to N, N′ and C11. CN is most stable, when one H atom is transferred to N (EB = −1.09 eV, Fig. 5 and Table 6). In this configuration the quinoline group forms an angle of 14°. In all other stable configurations at 1 ML, the quinoline group is inclined in the other direction forming a roof-like structure (Fig. 5(c–e)). Even without H transfer to the molecule, CN is stable at θH = 1 ML, however, with a positive binding energy (0.22 eV) indicating endothermic adsorption behavior. The notable range of different configurations and their binding energies found for CN at 1 ML points to a smooth potential energy surface and a higher flexibility of the molecule compared to CD.
C double bond is eliminated (Fig. 4 and 6), which has been studied at a partially hydrogen covered Pt surface (θH = 0.5 ML). For a parallel configuration of CD, hydrogen transfer to C11 leads to a destabilization of the system by 0.59 eV. Elimination of the CC double bond results in an elongation of the C–C bond length from 1.34 to 1.46 Å (Table 5). In addition, the C2–C3 bond length is elongated by 0.14 Å and the CH3 group moves towards the surface as indicated by a reduced distance of the C11 atom from the surface (Δdz,C11 = −0.29 Å). A similar behavior is observed when H transfer to N′ occurs (see Table 5 for details).
The situation changes drastically when the quinuclidine group is inclined towards the surface. In this case, hydrogen transfer to the C11 group leads to a breaking of the C2–C3 bond in the quinuclidine moiety and the system is destabilized only by 0.10 eV. A similar elongation of the C10–C11 bond length is observed (ΔdC10–C11 = 0.16 Å) compared to the situation described above where the C2–C3 bond remains intact. However, as a result of the higher flexibility of the quinuclidine part after breaking of the C2–C3 bond, the CH3 is further away from the Pt surface as before (Δdz,C11 = 0.52 Å).
When hydrogen is transferred to quinoline N′ in the inclined molecule (CDN′,as, Fig. 4(d)), additional H transfer to C11 has similar effects. The C2–C3 bond of the quinuclidine group is broken and the terminal CH3 group moves away from the surface by 0.47 Å. The destabilization of the system upon H transfer to C11 is slightly stronger (ΔEB = 0.23 eV) than that when no H is transferred to N′ (ΔEB = 0.10 eV).
Hydrogen transfer to C11 in the CN molecule is in contrast to the situation of CD. Destabilization is stronger and almost equivalent in all calculated configurations (0.88 ± 0.03 eV, Fig. 6). As a result of the stronger destabilization, the CN molecule becomes less stable than the CD isomer. Upon H transfer to C11, the terminal CH3 moves towards the surface by 0.81 ± 0.1 Å. However, it is still further away from the surface than in the CD molecule. For CN, no C2–C3 bond breaking has been observed. The quinuclidine ring remains stable in all calculated configurations.
3.6 Comparison of theoretical results with experimental observations
The results of the DFT calculations of the coadsorption of cinchona alkaloids and hydrogen substantiate the experimental observations made by in situ IR spectroscopy11,12,17 and Raman spectroscopy28 as well as catalytic tests,3,4,15,16,18 but also provide new insight into the adsorption behavior of cinchona alkaloids. The IR studies as well as the DFT results indicate that adsorption occurs via interaction of the quinoline ring of the cinchona alkaloids with the Pt surface. The IR studies further showed that at low cinchona alkaloid concentration (low coverage) adsorption occurs via π-bonding resulting in nearly parallel adsorption and at higher coverage via quinoline N′-lone-pair bonding yielding a tilted adsorption mode. Interestingly, the DFT results reveal that tilting of the quinoline anchoring unit can also be induced by coadsorption of hydrogen as recently evidenced by ATR-SEIRAS studies.17 This change of the adsorption mode alters the position of the stereogenic centers around C8 and C9 with respect to the Pt surface and thus the chiral space. Whether this tilting has a bearing on the enantiodifferentiating ability of the cinchona alkaloid is not clear yet and likely will depend on the structure of the substrate. However, the DFT results show that it has a strong influence on the stability of the adsorbed cinchona alkaloid.Experimental investigations showed considerable propensity of the adsorbed cinchona alkaloids to undergo partial hydrogenation in the presence of coadsorbed hydrogen.15,16 This propensity affects the stability of the adsorbed cinchona alkaloid under reaction conditions of the asymmetric hydrogenation. The DFT calculation indicate that in the presence of coadsorbed hydrogen the cinchona alkaloids are affected by possible partial hydrogenation, in agreement with experimental observations.3,4,15,16 While partial hydrogenation of the quinoline ring (C6′ and C9′) as well as H transfer to the CC double bond at C10, C11 position of the quinuclidine part destabilize the adsorbed cinchona alkaloid, H transfer to the quinoline N′ atom and quinuclidine N atom stabilize the adsorbed cinchona alkaloid. The DFT calculation confirm that the hydrogen transfer from the surface to the quinuclidine N is energetically feasible. This hydrogen transfer is crucial for the formation of the N–H⋯O hydrogen bond between cinchona alkaloid and substrate as experimentally evidenced in recent operando ATR-IR studies.29 Under reaction conditions of the enantioselective hydrogenation of ketones there are two possible scenarios for the hydrogen addition to the quinuclidine N:H transfer from the Pt surface (governing mechanism in apolar and aprotic solvents) or protonation in protic solvents (e.g. acetic acid).3
For the two diastereomers (pseudoenantiomers) CD and CN, which only differ in the absolute configuration at C8 and C9 (Scheme 1) similar adsorption energies are calculated. The two diastereomers show comparable behavior concerning stabilization and destabilization effects caused by hydrogen coadsorption and transfer. However, there are some striking differences concerning the effect of hydrogen transfer to the N′ atom of the quinoline moiety and the N atom of the quinuclidine part as well as the stability of the C2–C3 bond of the quinuclidine moiety. Whether these differences are responsible for the slightly different enantiodifferentiating ability of CD and CN observed in catalytic tests cannot be answered based on the present data, but the theoretical calculations reveal significant differences of the adsorption geometry and stability for the two diastereomers, which likely have a bearing on the catalytic behavior (enantiodifferentiating ability, activity and stability under reaction conditions).
4 Conclusions
The theoretical study of the coadsorption of cinchona alkaloids (CD and CN) and hydrogen on Pt(111) indicates that the presence of hydrogen significantly changes the adsorption geometry and stability of the cinchona alkaloids. While on a clean Pt surface the quinoline moiety of the cinchona alkaloids is adsorbed nearly parallel to the surface, at higher hydrogen coverage (0.5 ML) the quinoline part becomes tilted leading to different spatial arrangement of the stereogenic centers around C8 and C9. Hydrogen transfer from the surface to the N′ atom of the quinoline part as well as to the N atom of the quinuclidine moiety stabilizes the adsorbed cinchona alkaloids, whereas partial hydrogenation of the quinoline ring as well as hydrogen transfer to the CC double bond at C10, C11 position of the quinuclidine destabilizes it.
The results of the DFT calculations are in good agreement with experimental observations. In addition, they provide information about the stability of the different adsorption geometries of the cinchona alkaloids induced by coadsorption with hydrogen and corroborate that the coadsorption with hydrogen significantly changes the adsorption mode of the cinchona alkaloids, which should be taken into account in future mechanistic investigations of the asymmetric hydrogenation of cinchona-modified platinum.
Acknowledgements
AB thanks the foundation Claude & Giuliana for supporting this work.References
- K. Kacprzak and J. Gawroński, Synthesis, 2001, 0961–0998 CrossRef CAS.
- T. Marcelli and H. Hiemstra, Synthesis, 2010, 1229–1279 CrossRef CAS.
- T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS PubMed.
- H.-U. Blaser and M. Studer, Acc. Chem. Res., 2007, 40, 1348–1356 CrossRef CAS PubMed.
- M. Bartok, Curr. Org. Chem., 2006, 10, 1533–1567 CrossRef CAS.
- D. Y. Murzin, P. Mäki-Arvela, E. Toukoniitty and T. Salmi, Catal. Rev.: Sci. Eng., 2005, 47, 175–256 CAS.
- F. Zaera, J. Phys. Chem. C, 2008, 112, 16196–16203 CAS.
- G. Kyriakou, S. K. Beaumont and R. M. Lambert, Langmuir, 2011, 27, 9687–9695 CrossRef CAS PubMed.
- A. Vargas, D. Ferri, N. Bonalumi, T. Mallat and A. Baiker, Angew. Chem., Int. Ed., 2007, 46, 3905–3908 CrossRef CAS PubMed.
- T. Bürgi and A. Baiker, J. Am. Chem. Soc., 1998, 120, 12920–12926 CrossRef.
- D. Ferri and T. Bürgi, J. Am. Chem. Soc., 2001, 123, 12074–12084 CrossRef CAS PubMed.
- J. Kubota and F. Zaera, J. Am. Chem. Soc., 2001, 123, 11115–11116 CrossRef CAS.
- A. Vargas and A. Baiker, J. Catal., 2006, 239, 220–226 CrossRef CAS PubMed.
- M. von Arx, M. Wahl, T. A. Jung and A. Baiker, Phys. Chem. Chem. Phys., 2005, 7, 273 RSC.
- E. Schmidt, T. Mallat and A. Baiker, J. Catal., 2010, 272, 140–150 CrossRef CAS PubMed.
- E. Schmidt, W. Kleist, F. Krumeich, T. Mallat and A. Baiker, Chem. – Eur. J., 2010, 16, 2181–2192 CrossRef CAS PubMed.
- K. Motobayashi, R. Tomioka, T. Uchida and M. Osawa, Chem. Lett., 2015, 44, 770–772 CrossRef CAS.
- E. Schmidt, A. Vargas, T. Mallat and A. Baiker, J. Am. Chem. Soc., 2009, 131, 12358–12367 CrossRef CAS PubMed.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS PubMed.
- CP2K Developers, version 2.4, 2012, http://www.cp2k.org Search PubMed.
- G. Lippert, J. Hutter and M. Parrinello, Mol. Phys., 1997, 92, 477–487 CrossRef CAS PubMed.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- D. C. Ford, Y. Xu and M. Mavrikakis, Surf. Sci., 2005, 587, 159–174 CrossRef CAS PubMed.
- B. Poelsema, G. Mechtersheimer and G. Comsa, Surf. Sci., 1981, 111, 519–544 CrossRef CAS.
- N. M. Marković, B. N. Grgur and P. N. Ross, J. Phys. Chem. B, 1997, 101, 5405–5413 CrossRef.
- G. Santarossa, M. Iannuzzi, A. Vargas and A. Baiker, ChemPhysChem, 2008, 9, 401–413 CrossRef CAS PubMed.
- R. J. LeBlanc, W. Chu and C. T. Williams, J. Mol. Catal. A: Chem., 2004, 212, 277–289 CrossRef CAS PubMed.
- F. Meemken, K. Hungerbühler and A. Baiker, Angew. Chem., Int. Ed., 2014, 53, 8640–8644 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting information provides additional configurations of the molecules adsorbed on Pt(111) that are not shown in the main manuscript. See DOI: 10.1039/c5cp04792g |
| This journal is © the Owner Societies 2015 |