
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantifying transient interactions between amide groups and the guanidinium cation†
V.
Balos
,
M.
Bonn
and
J.
Hunger
*
Max Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: hunger@mpip-mainz.mpg.de
First published on 9th October 2015
Abstract
We study the interaction of the guanidinium cation, a widely used protein denaturant, with amide groups, the common structural motif of proteins. Our results provide evidence for direct contact between guanidinium and ∼2 amide groups, but the interaction is transient and weaker than for other cations with high charge-density.
The mechanism and molecular interactions by which ions affect the tertiary structure of proteins is a longstanding, yet poorly understood topic in biochemical science. The first systematic study dates back to the work of Hofmeister in 1888.1 In his work Hofmeister qualitatively studied the effect of salts on proteins and ordered them according to their efficiency to promote protein stability or denaturation. For cations, an increase in charge density results in an increasing denaturation tendency, while cations with low charge density promote protein folding.
Even though this trend holds for most cations, the Gdm+ cation (C(NH2)3+) is, despite its bulky chemical structure and correspondingly large degree of charge delocalization, one of the most efficient protein denaturation agents, having an even higher denaturation tendency than bivalent cations like Mg2+ and Ca2+. This is even more surprising, given that transport properties and association with anions for Gdm+ cations are similar to the Na+ cation.2,3 These remarkable and peculiar properties, make Gdm+ salts the most commonly used denaturants in biotechnological processes to reversibly unfold and re-fold proteins.4,5
The anomalous behaviour of Gdm+, together with its technological relevance has stimulated much research aimed at elucidating the specific molecular interactions that make Gdm+ such a powerful protein denaturant. Although still somewhat controversially discussed, there is growing evidence that denaturants like Gdm+ and urea directly bind to proteins.6 Based on molecular dynamics simulations, several mechanisms and protein interaction sites for Gdm+ have been suggested. These mechanisms include binding of Gdm+ to the carbonyl oxygen of the protein's amide backbone and the carboxylate groups of the peptide side chains,7 the pairing of Gdm+ with the positively charged arginine side chains8 and the coating of the hydrophobic fragments of the protein.9 However, denaturation experiments show no dependence on the amino-acid sequence but the interaction of Gdm+ with proteins scales with the protein's available surface area.10 Consequently, it was suggested that there is no specific interaction with the protein's side chains, but denaturation occurs through interaction with the general structural motif of proteins, i.e. the amide backbone.10
Here we study the cation–amide interaction, using the rotational dynamics of N-methylacetamide (NMA), in aqueous solution as a probe. We use NMA, a small amide-rich molecule, as a model compound mimicking the protein's backbone, as it is sufficiently small to readily detect its rotation in salt solutions. We investigate these dynamics in the presence of the chloride salts of K+, Na+, Li+, Mg2+ and Gdm+ as representative cations of the Hofmeister series, while keeping the concentration of NMA constant at 2 mol L−1 (i.e. varying the molar concentration of water). The dynamics are studied using dielectric spectroscopy,11–13 which probes the rotation of permanent electric dipoles of water and NMA as a response to an externally applied alternating electric field. The total polarization of the sample is probed as a function of the field frequency, ν, and expressed in terms of complex permittivity, ![[small epsilon, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e107.gif) (ν) = ε′(ν) − iε′′(ν). The dispersive (in-phase) polarization of the sample is described by ε′, where the low frequency plateau in ε′ corresponds to the static permittivity. The imaginary part, ε′′, represents absorptive (out-of-phase) polarization components. Here, we cover frequencies 0.8 ≤ ν/GHz ≤ 36 using a frequency domain reflectometer based on a vector network analyzer and frequencies at 0.3 ≤ ν/THz ≤ 1.6 using a transmission THz time domain spectrometer.14
(ν) = ε′(ν) − iε′′(ν). The dispersive (in-phase) polarization of the sample is described by ε′, where the low frequency plateau in ε′ corresponds to the static permittivity. The imaginary part, ε′′, represents absorptive (out-of-phase) polarization components. Here, we cover frequencies 0.8 ≤ ν/GHz ≤ 36 using a frequency domain reflectometer based on a vector network analyzer and frequencies at 0.3 ≤ ν/THz ≤ 1.6 using a transmission THz time domain spectrometer.14
For mixtures of dipolar molecules the rotational relaxation of each dipolar species gives rise to a characteristic dispersion in ε′ and a peak in ε′′.15,16 The peak position corresponds to the characteristic rotation frequency and the peak amplitude – the dielectric strength – scales with the molar concentration of the dipoles and the squared dipole moment.16 In Fig. 1a we show the dielectric spectrum for an aqueous solution of NMA (2 mol L−1) together with the individual contributions of H2O and NMA (for details see below).
 | ||
| Fig. 1 (a) Complex permittivity spectrum of an aqueous solution (2 mol L−1) of NMA. The shaded areas indicate the three contributions to the dielectric loss: bulk water, NMA and fast water. (b) Spectra for binary mixtures of water and NMA with increasing concentration of GdmCl. Symbols correspond to experimental data and the solid lines show fits with the dielectric relaxation model (eqn (1)). | ||
As can be seen in Fig. 1b (for other samples see ESI,† Fig. S1 and S2), increasing salt concentration leads to a decrease of the static permittivity of the sample, i.e. less dipoles align to the external field. This phenomenon is generally referred to as depolarization and has both macroscopic and microscopic origins. Macroscopically, depolarization arises from dilution (i.e. non-dipolar salt ions displace dipolar solvent molecules, decreasing the concentration of dipoles). Microscopically, depolarization may originate from strong binding of dipoles in the solvation shell of the ion and is typically observed for cations with surface high charge density.17 Finally, depolarization originates from kinetic depolarization (KD),17,18 which results from the coupling of dipolar rotation to the translation of the ions in the sample (i.e. dipolar molecules align according to the local field of a translating ion). The decrease of the amplitude due to KD is directly proportional to the conductivity of the solution, κ (mobility of the ions) and the relaxation time of the solvent (see ESI†). Both, the formation of rigid hydration shells and KD are a measure of the interaction of the salt with the solvent. We note, in accordance with previous studies, besides dilution KD dominates for aqueous solutions of KCl, NaCl and GdmCl.2,19,20
We first consider the magnitude of the depolarization for the studied samples, as it is directly accessible from the raw data and does not depend on the model used to describe the spectra. Due to the linear scaling of the dominating KD with conductivity,18 we plot the measured depolarization vs. the sample conductivities (Fig. 2) for the ternary samples (salt + NMA + H2O; black symbols) together with the depolarization for the corresponding aqueous electrolyte solution (salt + H2O; red symbols). For aqueous salt solutions the slopes for K+(aq) (Fig. 2a), and Gdm+(aq) (Fig. 2e) are similar and correspond to what would be expected for KD, in agreement with literature reports.2,19,20 For Na+(aq), Li+(aq), Mg2+(aq) the slopes are steeper, in line with rigid binding of ∼4/∼4/∼12 water molecules in the hydration shell of Na+/Li+/Mg2+,17,20–22 respectively (in addition to KD).
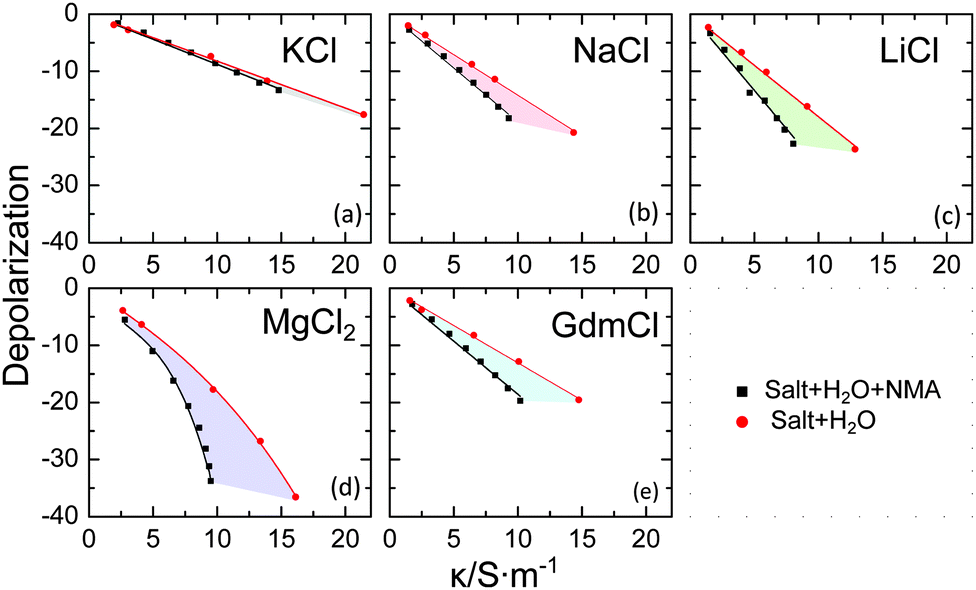 | ||
| Fig. 2 Total depolarization for (a) KCl, (b) NaCl, (c) LiCl, (d) MgCl2 and (e) GdmCl in aqueous (red) and aqueous NMA (2 mol L−1) solutions (black), as a function of the conductivity of each sample. The shaded areas are visual aids to highlight the difference between solutions in water and NMA(aq). | ||
It is important to note, that both the formation of rigid hydration shells and KD require the close proximity of the ion to the dipolar solvent, as the electric field of the ion rapidly decays with distance. For a binary solvent (i.e. aqueous solution of NMA) the magnitude of the depolarization provides information on any preferential interaction of the ions with different solvent components. For our present study it is important to note that the effective dipole moment of NMA is 2.2 times higher than the one of water.15,23 As the dielectric amplitude scales with the squared effective dipole moment, the depolarization is expected to be ∼5 times higher if an ion interacts with a NMA molecule compared to the interaction with a water molecule. It is thus informative to compare the depolarization for aqueous solutions of NMA to the depolarization of aqueous salt solution. For this comparison only hydration and KD are relevant as the dilution effect is very similar in both solvents.
Remarkably, the depolarization for solutions of NMA varies substantially with varying nature of the cation: while for K+ the depolarization in water and in NMA(aq) are very similar, substantially higher depolarization is observed for Gdm+ in NMA(aq) compared to water. A similar difference (indicated by the shaded areas in Fig. 2) is observed for Na+, Li+ and Mg2+. These observations indicate that for KCl the mechanism of depolarization is similar in water and in NMA(aq), and suggests that KCl interacts in NMA(aq) predominantly with water. On the contrary, GdmCl, NaCl, LiCl, and MgCl2 exhibit significant interaction with NMA, which results in a more pronounced depolarization due to the larger dipole moment of NMA. While the marked depolarization qualitatively demonstrates the interaction of Gdm+, Na+, Li+ and Mg2+ with NMA, the strong binding of solvent molecules in the hydration shell of Na+, Li+ and Mg2+ does not allow for a quantitative comparison of the interaction with NMA.
To obtain quantitative information, we decompose the individual contributions of all dipolar molecules to the experimental spectra. The rotational relaxation of NMA in water has been reported15 to obey a Debye type relaxation at ∼4 GHz (red shaded area in Fig. 1a). Pure water exhibits two relaxations in the frequency range relevant to the present study: the intense orientational relaxation mode of water (centred at 20 GHz in neat water at ambient temperature) and a weak fast relaxation at ∼200 GHz (blue shaded areas in Fig. 1a).13 Accordingly, we fit a combination of three relaxation modes to the spectra:
 | (1) |
The above extracted amplitudes scale with the concentration of molecular dipoles that are free to rotate, thus the reduction of SNMA provides information on how many NMA molecules are rotationally hindered by the added salt. However, as can be seen from the contributions of the three relaxation processes indicated in Fig. 1a, the orientational relaxation of water and NMA closely overlap. Hence, to reduce the number of adjustable parameters, we fix the amplitude of the water orientational relaxation to that expected for an ideal solution (i.e. a random distribution of NMA and salt and full hydration of the salt; see ESI†). This can be justified by noting that the concentration of bulk water is at least 20 times higher than the concentration of NMA. The parameters extracted from such fits are shown in the ESI† (Fig. S3 and S4). While the relaxation time of the fast water mode (τfast) is somewhat decreasing upon addition of salt, its amplitude remains fairly constant. We note that the exact molecular origin of this relaxation is poorly understood,2 thus we refrain from further discussion. The orientational relaxation time of NMA (τNMA) increases with increasing concentration (Fig. S4, ESI†). This is in line with a diffusive rotation of NMA as the increase is correlated with an increase of the viscosity of the solutions (Fig. S4, ESI†). Assuming the effective dipole moment‡ of NMA to be constant upon addition of salt, we obtain the concentration of free NMA molecules, cNMA,free, from SNMA (eqn (S4), ESI†).16 In Fig. 3 we show the thus obtained values of cNMA,free. In agreement with the qualitative conclusions from Fig. 2, these results indicate that K+ does not affect NMA. Contrarily, increasing concentration of Gdm+, Na+, Li+ and Mg2+ leads to a reduction of the concentration of rotationally-free NMA molecules. Interestingly, Li+ and Mg2+ strongly affect NMA, while for Na+ and Gdm+ the reduction of cNMA,free is less pronounced. To further quantify the interaction of the different cations with NMA we assume an association equilibrium of nNMA molecules with a cation M+:
| M+ + nNMA ⇆ nNMA·M+ | (2) |
 | ||
| Fig. 3 Concentration of free NMA molecules as a function of salt concentration. The solid lines correspond to fits using eqn (3) with n = 2, while the dotted lines show the corresponding fits assuming n = 1. The dashed lines show literature results obtained using vibrational spectroscopy.25 Error bars correspond to the standard deviation within at least 6 independent measurements. | ||
The corresponding equilibrium constant, K, is accordingly defined as:
 | (3) |
Interestingly, leaving the number of NMA molecules interacting with a cation as a free parameter, we obtain nGdmCl = 2.4 ± 0.4, nNaCl = 2.5 ± 1.2, nLiCl = 2.1 ± 0.2, and nMgCl2 = 1.8 ± 0.1.§ On the other hand a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 association (n = 1) gives a significantly worse description of the data (dotted lines in Fig. 3). Thus, our results indicate that Gdm+, Na+, Li+ and Mg2+ interact with up to ∼2 amide groups. The corresponding association constants K amount to 0.65 L2 mol−2 (Mg2+), 0.33 L2 mol−2 (Li+), 0.09 L2 mol−2 (Na+) and 0.06 L2 mol−2 (Gdm+). Note that for better comparability the values of K correspond to a 1:2 association (n = 2).
:1 association (n = 1) gives a significantly worse description of the data (dotted lines in Fig. 3). Thus, our results indicate that Gdm+, Na+, Li+ and Mg2+ interact with up to ∼2 amide groups. The corresponding association constants K amount to 0.65 L2 mol−2 (Mg2+), 0.33 L2 mol−2 (Li+), 0.09 L2 mol−2 (Na+) and 0.06 L2 mol−2 (Gdm+). Note that for better comparability the values of K correspond to a 1:2 association (n = 2).
Hence, our results show that protein denaturing cations exhibit significant interaction with the amide group, consistent with the notion that their denaturation activity is strongly related to direct interactions with the amide backbone of proteins. This conclusion is in qualitative agreement with vibrational spectroscopy experiments, which indicate strong binding of Li+ and Mg2+ to the amide group, while no interaction is observed for K+ (and Na+).25 Compared to the present results, the interaction of Na+, Li+ and Mg2+ was concluded to be significantly weaker using vibrational spectroscopy,25 (dashed lines in Fig. 3). This discrepancy can be rationalized by considering that, for the interaction to be detectable in the vibrational spectra, the interaction between a cation and the amide group has to be sufficiently strong to result in a shift of the amide vibration, which requires the ion to exert high local fields on the amide group.26,27 As the electric field of the ion rapidly decays with distance such spectral shifts are very sensitive to the valence of the ion, the distance to the amide group and therefore also to the ion's hydration shell. Dielectric spectroscopy can probe much weaker interactions, as the reduction of the mobility of a NMA molecule can be solely caused by the proximity of a translating ion to an NMA molecule (KD). As mentioned above, KD dominates for solutions of KCl, NaCl and GdmCl. Thus, our results show that, even though a negligible fraction of cations strongly bind to the amide group,25 there is substantial weak interaction with transiently formed contacts between Gdm+, Na+, Li+, Mg2+ and NMA.
The association of Gdm+ with NMA of the present study is also in broad concordance with protein denaturation studies using triplet–triplet energy transfer experiments, where a value of K = 0.6 L mol−1 was found for the association of Gdm+ with a single peptide binding site.10 The corresponding 1:1 association constant of the present work amounts to K = 0.22 L mol−1 (n = 1 in eqn (3), blue dotted line in Fig. 3). This comparison suggests that the Gdm+–amide interaction largely explains the denaturation behaviour of the Gdm+ cation. Our results however also indicate that the interaction of Na+, Li+ and Mg2+ with NMA is even stronger, despite their lower denaturation tendency (compared to Gdm+). Thus the interaction of Gdm+ with other protein sites may result in even higher denaturation activity, with interaction of the positively charged Gdm+ with negative proteins residues being the most likely candidate. However, the interaction of Gdm+ with carboxylates is expected to be weak as even association of Gdm+ with the bivalent carbonate anion is weak and comparable to Na+.2,3 Thus, specific interaction with carboxylates seem unlikely, in line with the binding of Gdm+ to proteins scaling with available protein surface.10
Despite the compelling evidence for denaturation originating from cation–amide binging, our results indicate that the strength of the interaction is apparently not the sole criterion that determines the denaturation efficiency, but denaturation is also a result of the competition between the binding strength, configurational freedom of the interaction, and binding kinetics.28 The entropically driven protein denaturation due to Gdm+29 is thus consistent with the intermediate binding and thus conformationally flexible interaction of Gdm+. Such transient interactions are also supported by recent NMR experiments, which indicate that Gdm+–amide H-bonds are too short to affect proton exchange at the amide.30 Interestingly, we find that Gdm+, Na+, Li+ and Mg2+ interact with up to ∼2 amide groups. This finding may be the key to understand the molecular mechanism of the protein denaturation by cations, as thus the cations can efficiently penetrate into the backbone with simultaneous binding to two amide moieties and rupture the protein structure. The intermediate binding strength and thus flexible interaction of Gdm+ with amides thereby potentially facilitates efficient disruption of protein structures.
Conclusions
In summary, we report on rotational mobility of N-methylacetamide in aqueous solution in the presence of the Gdm+ cation and compare the mobility of NMA co-solvated with other representative cations of the Hofmeister series. We find that K+ has virtually no effect on the rotational mobility of NMA, while addition of Gdm+, Na+, Li+ and Mg2+ results in a significant reduction of the rotational freedom of NMA. Our results indicate that each strongly interacting cation can interact with up to ∼2 amides, which is the likely origin of the effective disruption of protein structures. Quantitative analysis reveals high affinities of Li+ and Mg2+ to the amide group, while the association of Gdm+ with NMA is ∼8 times lower and comparable to Na+. While our results indicate that Gdm+ interacts with the amide, consistent with denaturation via interaction with the protein backbone, the Gdm+–amide interaction is relatively weak (compared to Li+ and Mg2+) and of transient nature, in line with the entropically driven denaturation.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG) HU1860/4.Notes and references
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 24, 247 Search PubMed
.
- J. Hunger, S. Niedermayer, R. Buchner and G. Hefter, J. Phys. Chem. B, 2010, 114, 13617–13627 CrossRef CAS PubMed
- J. Hunger, R. Neueder, R. Buchner and A. Apelblat, J. Phys. Chem. B, 2013, 117, 615–622 CrossRef CAS PubMed
- T. Svedberg, Nature, 1937, 139, 1051–1062 CrossRef CAS PubMed
- H. Neurath, G. R. Cooper and O. John, J. Biol. Chem., 1942, 142, 249–263 CAS
- D. R. Canchi and A. E. García, Annu. Rev. Phys. Chem., 2013, 64, 273–293 CrossRef CAS PubMed
- E. P. O'Brien, R. I. Dima, B. Brooks and D. Thirumalai, J. Am. Chem. Soc., 2007, 129, 7346–7353 CrossRef PubMed
- A. Kubíčková, T. Křížek, P. Coufal, E. Wernersson, J. Heyda and P. Jungwirth, J. Phys. Chem. Lett., 2011, 2, 1387–1389 CrossRef
- R. Godawat, S. N. Jamadagni and S. Garde, J. Phys. Chem. B, 2010, 114, 2246–2254 CrossRef CAS PubMed
- A. Möglich, F. Krieger and T. Kiefhaber, J. Mol. Biol., 2005, 345, 153–162 CrossRef PubMed
-
F. Kremer and A. Schönhals, Broadband Dielectric Spectroscopy, Springer, Berlin, 2003 Search PubMed
- U. Kaatze and Y. Feldman, Meas. Sci. Technol., 2006, 17, R17–R35 CrossRef CAS
- T. Fukasawa, T. Sato, J. Watanabe, Y. Hama, W. Kunz and R. Buchner, Phys. Rev. Lett., 2005, 95, 197802 CrossRef
- W. Ensing, J. Hunger, N. Ottosson and H. J. Bakker, J. Phys. Chem. C, 2013, 117, 12930–12935 CAS
- U. Kaatze, H. Gerke and R. Pottel, J. Phys. Chem., 1986, 90, 5464–5469 CrossRef CAS
- E. A. S. Cavell, P. C. Knight and M. A. Sheikh, Trans. Faraday Soc., 1971, 67, 2225–2233 RSC
- N. Ottosson, J. Hunger and H. J. Bakker, J. Am. Chem. Soc., 2014, 136, 12808–12811 CrossRef CAS PubMed
- J. B. Hubbard, L. Onsager, W. M. van Beek and M. Mandel, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 401–404 CrossRef CAS
- T. Chen, G. Hefter and R. Buchner, J. Phys. Chem. A, 2003, 107, 4025–4031 CrossRef CAS
- R. Buchner, G. T. Hefter and P. M. May, J. Phys. Chem. A, 1999, 103, 1–9 CrossRef CAS
- Y.-Z. Wei and S. Sridhar, J. Chem. Phys., 1990, 92, 923–928 CrossRef CAS PubMed
- R. Buchner, T. Chen and G. Hefter, J. Phys. Chem. B, 2004, 108, 2365–2375 CrossRef CAS
- J. Hunger, A. Stoppa, R. Buchner and G. Hefter, J. Phys. Chem. B, 2009, 113, 9527–9537 CrossRef CAS PubMed
- U. Kaatze, J. Phys. Chem., 1987, 91, 3111–3113 CrossRef CAS
- H. I. Okur, J. Kherb and P. S. Cremer, J. Am. Chem. Soc., 2013, 135, 5062–5067 CrossRef CAS PubMed
- H. Kim and M. Cho, Chem. Rev., 2013, 113, 5817–5847 CrossRef CAS PubMed
- E. Pluhařová, M. D. Baer, C. J. Mundy, B. Schmidt and P. Jungwirth, J. Phys. Chem. Lett., 2014, 5, 2235–2240 CrossRef PubMed
- S. K. Jha and S. Marqusee, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4856–4861 CrossRef CAS PubMed
- G. England and J. L. Haran, Annu. Rev. Phys. Chem., 2011, 62, 257–277 CrossRef PubMed
- W. K. Lim, J. Rösgen and S. W. Englander, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2595–2600 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp04619j |
| ‡ Note that the effective dipole moment values may contain dipolar correlations. Given the similar static permittivities and ranges of ionic strengths for each studied salt (except MgCl2), screening effects and thus dipolar correlations are expected to be similar as only the nature of the cation is changed. |
| § Errors correspond to a 50% increase in the sum of the squared deviations. |
| This journal is © the Owner Societies 2015 |