
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanometer-scale hydrogen ‘portals’ for the control of magnesium hydride formation†
Chia-Jung
Chung‡
,
Chinmay
Nivargi‡
and
Bruce
Clemens
*
Department of Materials Science and Engineering, Stanford University, Stanford, CA - 94305, USA. E-mail: bmc@stanford.edu
First published on 29th September 2015
Abstract
Magnesium and Mg-based material systems are attractive candidates for hydrogen storage but limited by unsuitable thermodynamic and kinetic properties. In particular, the kinetics are too slow at room temperature and atmospheric pressure. To study the hydride formation kinetics in a controlled way, we have designed a unique ‘nanoportal’ structure of Pd nanoparticles deposited on epitaxial Mg thin films, through which the hydride will nucleate only under Pd nanoparticles. We propose a growth mechanism for the hydrogenation reaction in the nanoportal structure, which is supported by scanning electron microscopy (SEM) images of hydrogenated samples exhibiting consistent results. Interestingly, the grain boundaries of Mg films play an important role in hydride nucleation and growth processes. Kinetic modeling based on the Johnson–Mehl–Avrami–Kolmogorov (JMAK) formalism seems to agree with the two-dimensional nucleation and growth mechanism hypothesized and the overall reaction rate is limited by hydrogen flux through the interface between the Pd nanoparticle and the underlying Mg film. The fact that in our structure Mg can be transformed completely into MgH2 with only a small percentage of Pd nanoparticles offers possibilities for future on-board storage applications.
1 Introduction
Magnesium hydride is an attractive material for hydrogen storage due to its high reversible hydrogen mass capacity of 7.6 wt%. However, a high desorption temperature (∼265–320 °C for 1 bar H2 pressure depending on ref. 1 and 2) and slow room-temperature hydrogen sorption kinetics pose difficulties for on-board applications.3 Thermodynamic destabilization of the system can be achieved through alloying with other elements,4–6 exploiting the excess interfacial energy in multilayers7 or intermixing energy with a catalyst,8 leading to an increase in the equilibrium hydrogenation pressure at room temperature for practical use.The Mg hydrogenation reaction is also sluggish on its own and is further restricted due to the presence of a native oxide on the Mg surface. High temperatures of at least 400 °C are needed to crack the MgO layer and dissociate the hydrogen, and even then the absorption/desorption of hydrogen takes place over a period of hours.9,10 The hydrogenation is often catalyzed at lower temperatures by Pd deposited on its surface, in contact with the pristine Mg, enabling dissociation of the H2 molecules into H atoms which then diffuse through and react with Mg. In fact, in our experiments, we did not see a significant rate of hydrogen sorption at room temperature without the presence of the Pd catalyst. The nucleation occurs at Pd–Mg (or the catalyst/additive-Mg interface) followed by growth due to the diffusion of H atoms to the growth front. After nucleation, the kinetics are limited by the slow diffusion of H atoms to the reaction front, since the diffusion of H atoms through the already-grown magnesium hydride is eight to ten orders of magnitude slower than diffusion through a metal.11,12 The formation of a ‘blocking’ layer of MgH2 has been described previously in the literature and hence the diffusion length scale for complete reaction needs to be kept below 30–50 μm.1,13–15
The hydrogenation kinetics can be substantially improved by reducing the diffusion length scale using nanosized materials.10,16–18 The kinetics are also enhanced upon addition of the catalyst or alloying additives. Transition metals or their oxides are the most investigated. A good overview of the research on Mg based materials for hydrogen storage was provided in previous reviews.19,20 However, the connection between the nanostructure and its effect on the kinetic properties of these materials is still unclear. Different studies use a variety of materials, catalysts and geometries, making identification of the underlying mechanisms very difficult.
In order to critically examine the effects of the nanostructure on the kinetics of nucleation and growth, we have designed a Pd nanoparticle ‘nanoportal’ structure, illustrated in Fig. 1(a). This unique structure enables a physical and temporal decoupling of nucleation and growth processes, by allowing the nucleation to occur only under the Pd nanoparticles, followed by growth due to hydrogen flux through the Pd catalyst nanoparticle ‘portals’. As a result, it can be employed to study hydrogenation reactions under a wide variety of carefully controllable experimental conditions. This has potential to provide great insight into the underlying reaction mechanisms.
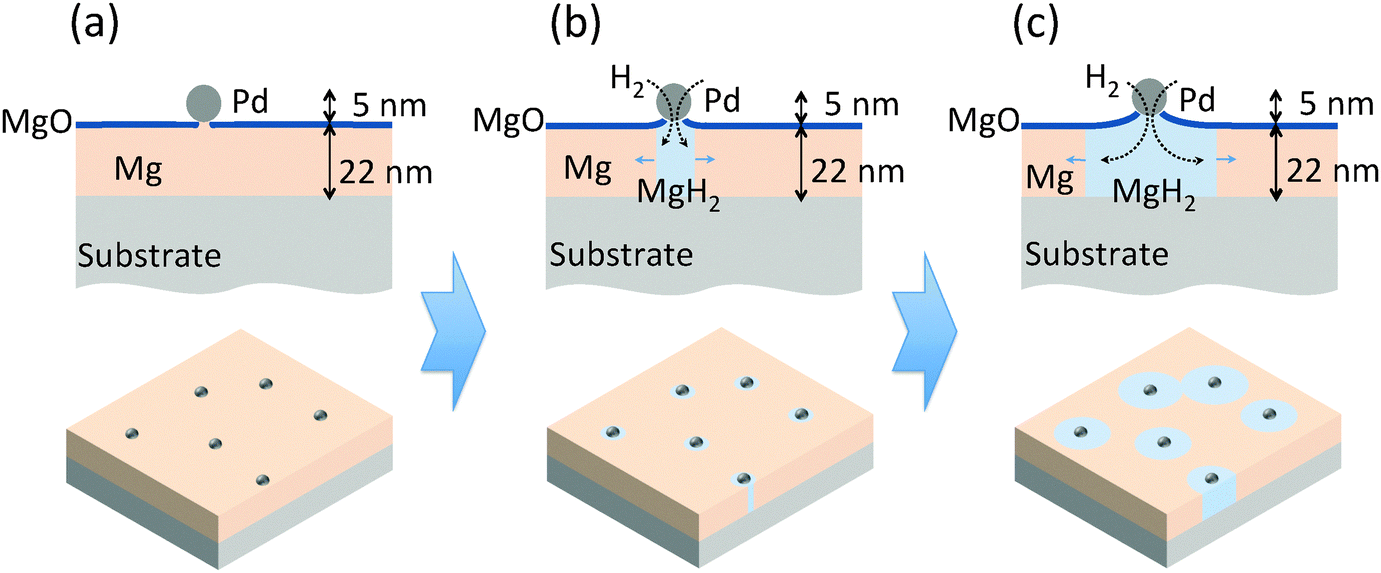 | ||
| Fig. 1 A schematic diagram of the novel Pd nanoparticle nano-portal structure and the predicted hydrogenation mechanism. The upper part of the figure is the structure in cross-section and the lower part is the structure visualized in three dimensions. (a) The structure of the as-grown sample. (b) The nucleation of MgH2. (c) The growth process of the nucleated MgH2. | ||
The nanoportal structure also allows the examination of the reaction using Scanning Electron Microscopy (SEM) and an optical transmission technique based on hydrogenography,21 since the catalyst nanoparticles do not obscure the observation of the underlying phase transformation. We propose a reaction mechanism for this structure and then correlate a kinetic model that we have developed based on the Johnson–Mehl–Avrami–Kolmogorov (JMAK) formalism22 with the experimental observations. The results suggest that the overall reaction rate depends on the flux of hydrogen atoms through the interface between the Pd catalyst nanoparticle and the underlying Mg film.
We found that, even for the thin Mg films studied here, Pd nanoparticles representing a mass fraction of only a few percent can be effective catalysts, increasing reaction rates by orders of magnitude over bare Mg. The investigation of reaction mechanisms with this novel nanostructure can pave the way to rational designs of hydrogen storage systems with better storage properties for future on-board applications.
2 Experimental
2.1 Sputtering of Pd nanoparticles and sample preparation
Samples were prepared in a UHV sputtering chamber consisting of two sputtering zones; one was for conventional thin film growth and the second was a high-pressure condensation zone for producing nanoparticles.23 Epitaxial Mg films of 22 nm thickness in the (001) orientation were grown on (001) Al2O3 at room temperature (for more details see ref. 24) using DC magnetron sputtering in a chamber zone with a base pressure of 5 × 10−9 torr and an Ar deposition pressure of 1.3 millitorr. In order to investigate the effect of the grain structure on hydrogenation kinetics, non-epitaxial Mg films of the same thickness were grown on oxidized (100) Si wafers using the same growth conditions. Pd nanoparticles were generated by a nanoparticle source from Mantis Deposition Ltd, consisting of a sputter source operating in a condensation zone held at high (hundreds of millitorr) Ar pressure, and a quadrupole mass filter. By controlling the condensation zone length and pressure, and by using the mass filter, particle sizes in the range of 1–20 nm can be produced. The generated Pd nanoclusters were then impinged upon the as-deposited Mg film. A detailed description of the sputtering conditions is provided in the ESI.†2.2 X-Ray diffraction
Symmetric θ − 2θ scans and rocking curve scans were performed to examine the overall structure and preference of out-of-plane texture, while ϕ scans after aligning on known peaks away from the sample normal were used to determine the presence of in-plane orientation induced by epitaxial alignment with the substrate. More information is provided in the ESI.†2.3 Optical transmission measurement
The samples were charged with hydrogen (99.999%) in an optical measurement chamber. A high-power LED with a wavelength of 625 nm was used as the light source in the measurements. The LED output was modulated at 316 Hz to allow for lock-in detection in order to increase the signal-to-noise ratio. Since the 635 nm light was strongly absorbed by Mg but not MgH2, monitoring the optical transmission allowed real-time, direct measurement of the fraction of the Mg converted to hydride.21 The schematic of the setup and the pictures of the sample before/after hydrogenation can be found in the ESI.†3 Results and discussion
3.1 Construction of the nanoportal structure
The nanoportal structure consists of Pd nanoparticle catalysts forming portals for the dissociated H atoms to diffuse through and then nucleate and grow the hydride in the underlying Mg film. To form this structure we first deposit a Mg film using conventional UHV sputter deposition onto either sapphire or oxidized Si substrates (the Mg growth process is described in more detail in ref. 24). This is followed by the deposition of Pd nanoparticles, formed by inert gas condensation23 of a sputtered Pd atom flux, combined with in situ mass filtering to produce uniformly distributed, size-selected Pd nanoparticles. The Pd nanoparticle deposition immediately follows the Mg film growth in the same deposition environment without breaking vacuum, leading to an intimate contact (and presumably some reaction8) between the Pd nanoparticles and the Mg film. The resulting structure is then exposed to air, whereupon a ∼2 nm thick native MgO layer forms on the remaining Mg film that is not covered by the Pd nanoparticles. This oxide layer effectively blocks hydrogen from reacting with the underlying Mg. The final structure is shown schematically in Fig. 1(a).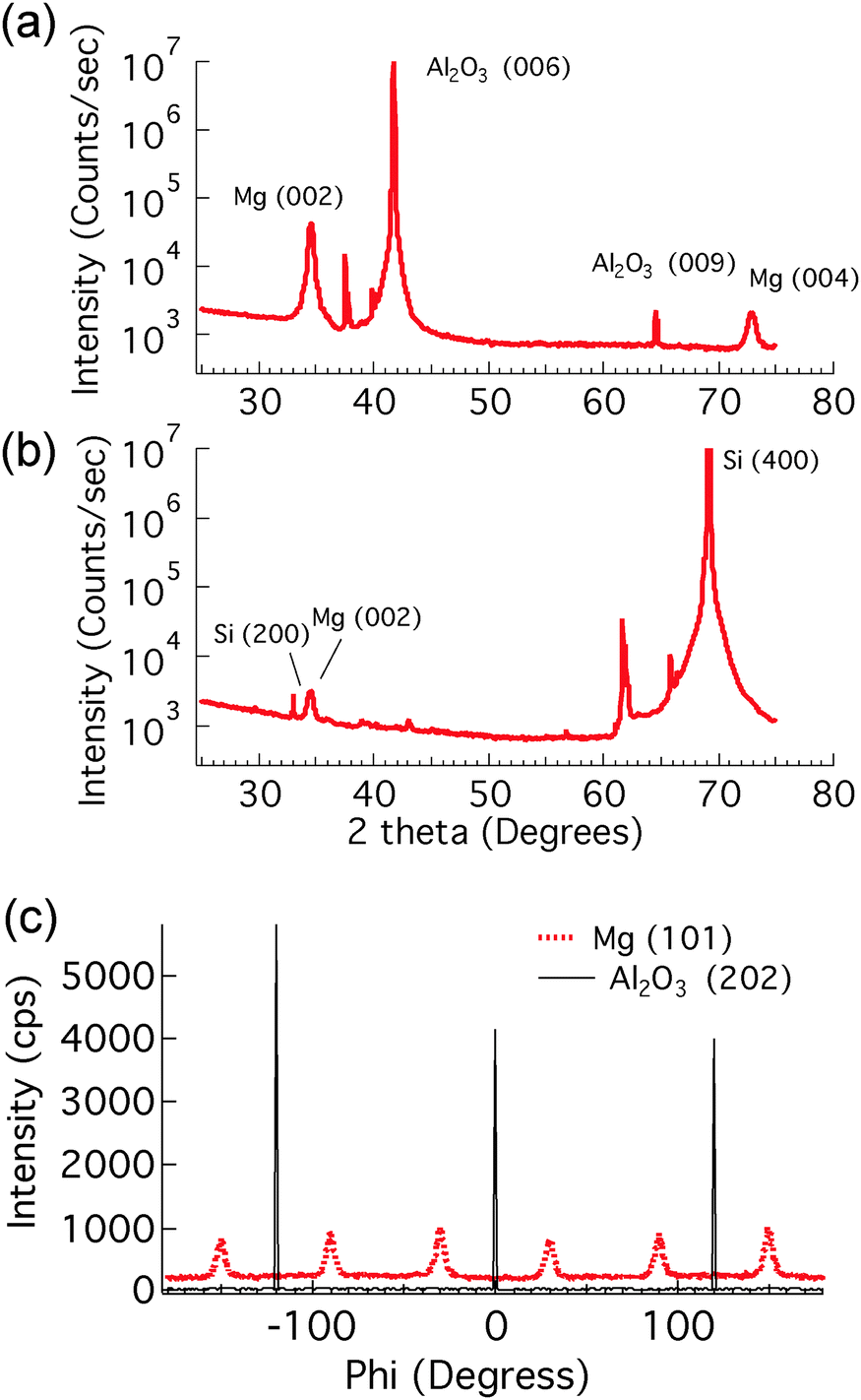 | ||
| Fig. 2 X-ray diffraction θ − 2θ scans of as-deposited 22 nm Mg with Pd nanoparticles. (a) Samples that were grown on a sapphire substrate. (b) Samples that were grown on a silicon substrate. The unlabeled peaks are all from substrates. (c) X-ray diffraction phi scans of an epitaxial Mg film. The sample was grown on a (0001) Al2O3 substrate with Pd nanoparticles deposited on the surface. | ||
Localizing the nucleation sites results in an ability to study reaction kinetics with unprecedented control. By controlling the density of nanoportals we can vary the nucleation density and examine new growth behaviors. For example, when the distance between nucleation sites is large compared to the film thickness (true for all the samples discussed here) the hydride growth will occur as two-dimensional lateral growth for most of the reaction processes, since vertical growth will rapidly be limited by the film thickness (Fig. 1(b) and (c)). By following this growth process we can examine the limiting mechanisms as well as explore nucleation behavior.
3.2 Visualization of the reaction progress
In order to visualize the nucleation and growth processes of MgH2, we used field emission scanning electron microscopy (FEI Magellan XHR SEM) to examine the morphology and the size of the hydride at different stages of the hydrogenation reaction. To minimize beam damage to the hydride, we imaged the samples at a relatively low voltage of 2 kV with a probe current of 13 pA. Owing to the electronic property difference between Mg and MgH2, the hydride phase, which had a lower secondary electron yield, appeared darker in the SEM than the untransformed metallic Mg. Therefore, the SEM allowed direct observation of the shape and the size of the hydride regions.Fig. 3 shows SEM images of the hydrogenated Mg films grown on sapphire and oxidized Si substrates, respectively, with an areal Pd nanoparticle coverage of 1.4%. Clearly the structural difference in the parent Mg phase, as shown by X-ray diffraction, altered both the extent of the hydrogenation reaction and the morphology of the hydride. For the epitaxial Mg films, the hydride regions were nearly circular, while showing some overlap of neighboring regions. These phenomena indicate that the hydride reaction rate was roughly independent of the in-plane crystallographic direction within a grain. However, the hydride regions on the non-epitaxial Mg films were more irregular in shape and much smaller. It appears that the shape of the hydride region in the non-epitaxial Mg films was influenced by the underlying Mg grain structure, coupled with a variation in the difficulty with which the growing hydride was able to cross the different grain boundaries between adjacent grains. The shape of the hydride regions appears to mimic the grain structure of the parent Mg phase, indicating that the hydride growth is impaired during transit across grain boundaries. Further examination can explore further the connection between this growth impediment and grain boundary orientation, but it is clear that the small angle grain boundaries present in the sample grown on sapphire offer much less interference to hydride growth.
 | ||
| Fig. 3 SEM micrographs of hydride growth in Mg films. (a) and (c) SEM of hydride growth in an epitaxial Mg film grown on a sapphire substrate. (b) and (d) SEM of hydride growth in a non-epitaxial Mg film grown on a silicon substrate. Both samples were hydrogenated under 700 torr H2 at room temperature for 8.5 minutes. Hydride regions are dark due to the lower electron yield. Pd nanoparticles are small bright spots in the pictures. ∼59% of the area is reacted for the epitaxial film, compared to ∼52% for the non-epitaxial film. | ||
We note that for both epitaxial and non-epitaxial samples, the spacing between the hydride regions is much larger than the spacing between the Pd nanoparticles, and that there are Pd nanoparticles over unreacted Mg regions of the film, or more than one Pd nanoparticle on each nucleated hydride patch. This implies that only a fraction of the Pd nanoparticles are effective in nucleating a hydride region. We suspect that the failure of many nanoparticles to result in hydride formation is due to the lack of direct contact between the Pd nanoparticle and the Mg film, perhaps due to oxidation in the imperfect vacuum of the deposition environment, or oxidation of the interface subsequent to nanoparticle deposition and exposure to air.
It is also notable that the hydride regions in a given film at a given stage of reaction are approximately the same size, which indicates that each hydride region begins to grow at approximately the same time. This result indicates that nucleation takes place early in the reaction process, and that once the effective nucleation sites are used up, no further nucleation occurs. Finally, we note that the spacing between hydride regions is much larger than the film thickness, so that much of the transformation occurs via two-dimensional growth in the plane of the film after the hydride spans the thickness of the film, which is consistent with our predicted growth mechanism.
To extract the time-dependence of the hydride growth behavior we used SEM to observe the size of the hydride patches in samples with the same nominal nanoportal areal density (∼1.7%) but exposed to hydrogen for varying reaction times. The average radius (extracted by image analysis using ImageJ25) as a function of reaction time is shown in Fig. 4. We see that the growth rate dr/dt decreases greatly with time. Fitting to a growth law r ∼ tm leads to unsatisfactory fits and to a value of m ∼ 0.3, which is incompatible with growth limited by either diffusion through the growing hydride or by flux through the nanoparticle–film interface,15 both of which would produce a r ∼ t1/2 behavior for two-dimensional growth (see ESI†). At longer times, the observed growth rate decreases more rapidly than the r ∼ t0.3 of the fitted function, leading to the conclusion that the effectiveness of the nanoportals might be decreasing with time. We speculate that this decrease may be due to further oxidation of the nanoparticle–film interface, resulting in a decrease in the effective nanoportal area. This possibility is discussed more fully below. Note that these observations of nanoparticle diameter are taken in the time and nucleation density regime before coalescence occurs, so the decreased growth rate is not due to the impingement of growing hydride regions.
 | ||
| Fig. 4 Model fits to explain the time dependence of the measured average radii of the hydride patches grown in epitaxial Mg samples. The agreement is seen to be much better for the models incorporating the oxidation under the portals. | ||
To explore the effect of oxidation of a nanoportal on the hydride growth rate, we considered both the interfacial and diffusive flux limited encroachment of the oxide under the nanoportal. This is illustrated schematically in Fig. 5. This leads to a simple expression for the nanoportal area and hence the flux of the H atoms through the nanoportals as a function of time, which in turn can be used to obtain an expression of the dependence of the radius of an individual hydride patch as a function of time. Balancing the nanoportal flux and the hydride growth in a time increment dt for mass conservation, we find:
| jHAnpdt = cH(2πrh)dr |
so r ∼ t1/2 as mentioned above. Allowing the area of the nanoportal to decrease with time with either interface or diffusion limited oxide encroachment as shown in Fig. 5 gives relatively simple analytical expressions with different time dependencies (see ESI†) and a much better fit to the hydride radius behavior as shown in Fig. 4. The dependence is very similar to a power law at small times, before slowing down as the portal shrinks, and eventually pinches off. As can be seen, for the interface limited oxide encroachment, assuming the average initial radius of the nanoportal to be 5 nm, the nanoportal pinches off at ∼516 s.

 | ||
| Fig. 5 The portal pinches off under the Pd nanoparticle. On the right is an exaggerated top view showing a decreased area of the portal and hence a lowered H flux. | ||
3.3 Optical transmission measurement of reaction progress
In order to further examine hydride growth behavior using the nanoportal mechanism, an optical transmission technique was used to monitor the hydrogenation kinetics of Mg films with varying Pd nanoparticle surface coverage. This monitoring is possible due to the difference in optical transmission of MgH2, which is essentially transparent for light at λ = 625 nm as compared to the Mg metal which is reflective. The reaction fraction was calculated by taking the light transmission of the sample at a given reaction time and then normalized by the total change of transmission when the sample was fully reacted, when it became fully transparent. The reacted fraction as a function of time is shown in Fig. 6. Samples with a higher coverage of Pd nanoparticles had faster hydrogenation kinetics, as seen in Fig. 6, due to the increased Pd nanoparticle density and hence a higher density of nucleation sites. Thus a higher coverage of Pd nanoparticles results in a higher density of effective nanoportals, even though, as mentioned above, only a fraction of Pd nanoparticles are effective as nucleation sites. We also observe that samples that are exposed to air for a longer period of time do not react as quickly as those that have shorter air exposure. This is consistent with the growth behavior discussed above, which we explain by a decrease in the effective area of a nanoportal over time, even in the hydrogenation environment. These observations show that the nature of the interface between the nanoparticle and the underlying film is critical for an effective nanoportal function. Future work will explore ways to improve nanoparticle–film interaction.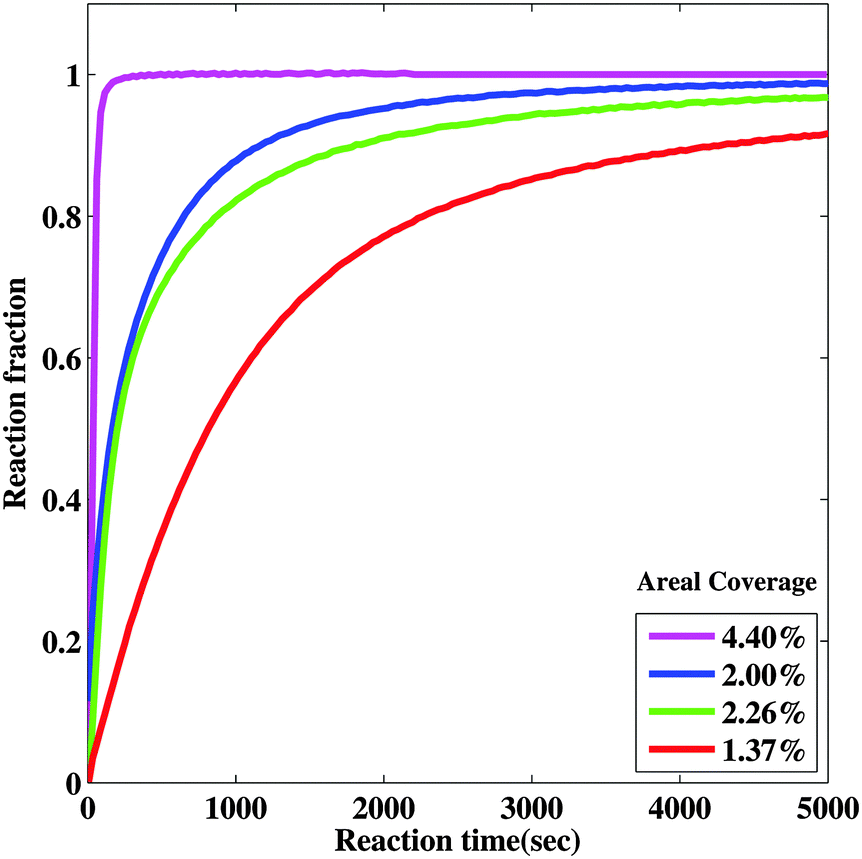 | ||
| Fig. 6 Effect of Pd nanoparticle coverage on the hydrogenation reaction rate. Hydrogenation reaction curves of samples with different Pd nanoparticle coverages (∼1.7 to ∼6.3%) grown on epitaxial Mg films. | ||
3.4 Kinetic modeling of the reaction progress
In order to further understand the limiting mechanisms of the growth behavior, we constructed a kinetic model based on the well-known JMAK formalism to understand the limiting steps for the reaction. Here we follow Christian's treatment26 to apply this approach to our nanoportal mechanism. Considering first the nucleation behavior, we observe that since nucleation occurs under active nanoportals, we initially have a fixed number (per area) of possible sites where nucleation can occur. If the sites nucleate with average time constant τN, then we find for the nucleation rate per area:where N0 is the initial number density of available nucleation sites and N is its (decreasing) value as a function of time.
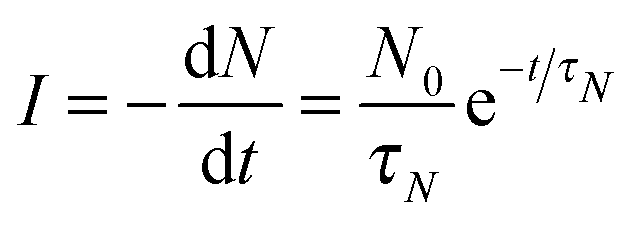
Next we consider growth of the nuclei as a function of time. A nucleus that begins growth at time τ and grows as a two-dimensional disk will have area at time t given by:

| Aτ(t) = c[(t − τ)1/2]2 = c(t − τ) |
If we add up all the area of growing hydride regions, ignoring impingement and counting phantom nuclei that are present in already transformed regions we find the extended area:
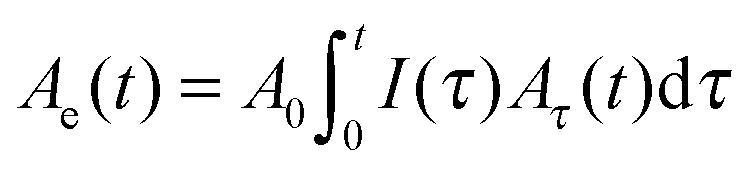
| A(t) = A0(1 − e−Ae(t)/A0) |
Comparing this model with the observed transformed fraction f = A(t)/A0 behavior we can investigate the nucleation behavior and growth limiting mechanisms. To explore nucleation behavior we first fit the short-time (t ≤ 700 s) experimental data using a power law growth behavior (r ∼ tm). We find relatively good agreement with the optical behavior as shown in Fig. 7, with an exponent of 0.2 ≤ m ≤ 0.5 which is roughly consistent with the fits we obtained for the radius behavior discussed above. We find that we are unable to extract meaningful nucleation times τN for samples with low nanoparticle density, and that the nucleation time we extract for the sample with high density is short (∼54 s), but comparable to the reaction time of around 100 seconds. This indicates that the nucleation is complete in the low nanoparticle density samples before significant reaction takes place, while for the high nanoparticle density samples there is nucleation and growth occurring during the reaction. The quality of the fit decreases if we include longer reaction times, indicating that the power law growth behavior is not good for a long time. This is consistent with the hydride radius behavior discussed above and with the idea that the nanoportals become less effective with time, perhaps due to pinching off by MgO growth under the nanoportal.
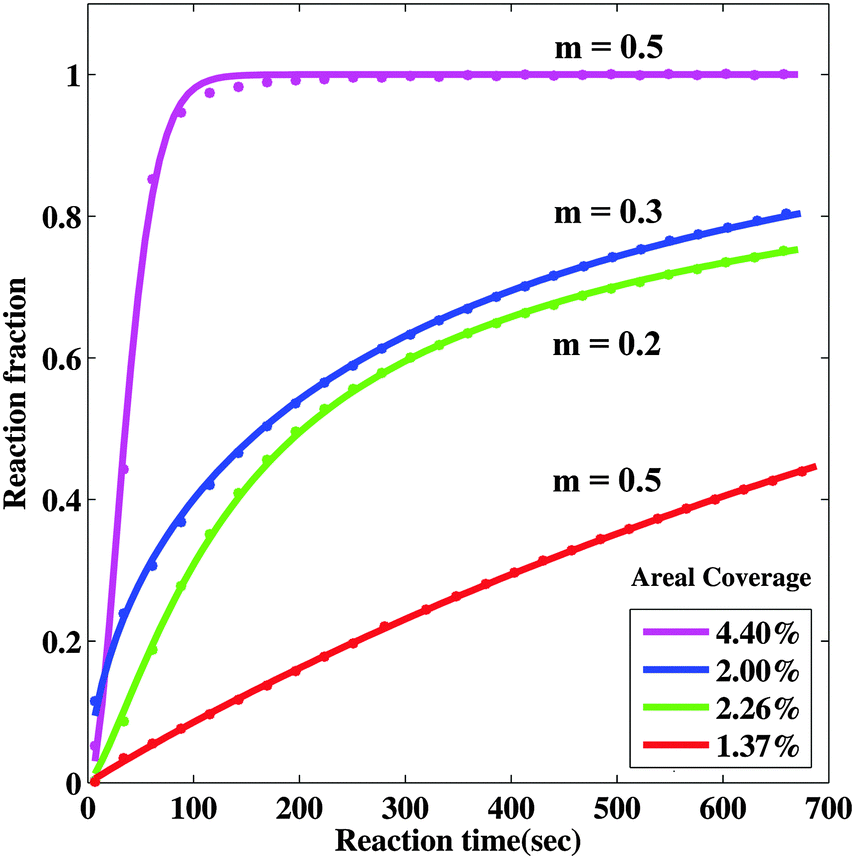 | ||
| Fig. 7 Model fits to observed short-time behavior using power-law growth behavior. The solid lines are fits to the data and the best fit to the parameter m is shown. | ||
To explore this we incorporated the hydride growth behavior associated with the decreasing nanoportal area due to the oxide encroachment discussed above (see ESI,† for the model). The oxide encroachment was modeled as both interfacial as well as diffusive flux limited growth, leading to a decreasing area of the nanoportal, and hence a slowdown of the growth of the hydride patch from the expected r ∼ t1/2 behavior. The resulting fits for the interfacial flux limited model are shown in Fig. 8. As can be seen, the model is able to describe the behavior fairly adequately over the entire reaction time. The diffusive flux limited model shows somewhat similar behavior. This model was considered only for the low coverage samples for which the oxide encroachment before reaction completion was significant. The fast reacting sample essentially reacted fully before encroachment became appreciable.
 | ||
| Fig. 8 Model fits to observed behavior incorporating portal pinch-off and an additional leak flux through MgO, for the low coverage samples. The solid lines are fits to the data. | ||
Using this approach, it was again found that the nucleation time is short compared to the reaction times, and hence nucleation is effectively instantaneous. This results in hydride patches of about the same size, as observed in the SEM. The time of nanoportal pinch-off varies across samples with different nanoportal densities. It is also presumably affected by the amount of time it was exposed to air before hydrogenation and the specific variation of nanoparticle sizes across samples, which is hard to completely account for in the modeling. However, for a similar nanoparticle density, it was found that the time for the nanoportals to completely pinch-off was comparable for the fits to the transmission measurements (∼350−1340 s depending on interfacial or diffusive flux limitation) and the radius measurements (∼516−950 s). These times are very sensitive to the initial radius of the nanoportals, which, of course, have a size distribution that is not included in the modeling.
Although the portals eventually pinch off, the reaction is still seen to proceed slowly, indicating that the MgO layer on top of Mg and under the nanoparticle might be slightly permeable to H atoms dissociated by Pd, through cracks or defects. This is likelier after appreciable nucleation and growth, as opposed to at the beginning of the reaction where there is no significant nucleation yet. This is because of the growing hydride having a 32% larger volume,8 compared to Mg, that might contribute to a decreased integrity of the oxide layer on top. A constant flux term was incorporated into the fits to account for this behavior, leading to much better agreement at long and short times, as shown. A better engineering of the Mg–Pd nanoparticle interface could help avoid this slowdown and pinch-off. A potential way would be to accelerate the nanoparticles towards the sample by means of a voltage bias during deposition so as to have a more intimate contact with the underlying Mg film – to prevent oxidation or cause more Mg–Pd intermixing. Investigations to verify this hypothesis are ongoing in our lab.
4 Conclusions
In summary, we have developed a novel Pd nanoportal structure that enables us to study the hydride nucleation and growth mechanism by physically controlling the nucleation sites of the hydride. X-ray diffraction analysis and SEM observations show the effect of the Mg crystallographic structure on the hydride growth – high-angle grain boundaries seemed to impede hydride growth. The SEM micrographs exhibited consistent hydride growth behavior as predicted. We developed a simple kinetic model based on the JMAK formalism, which was able to provide significant insight into the nucleation and growth behaviors. It seemed to suggest that the reaction is limited by flux of the H atoms across the interface between the Pd nanoportals and the MgH2 nuclei. The nanoportals get constricted and eventually might pinch off over time due to oxidation underneath the portals during sample transfer or the hydrogenation reaction. A modification to the model incorporating effect of this oxidation gave better agreement with the determined radius of the hydride patches as a function of time as well as the optical transmission through the sample that indicates the reacted fraction.The amount of Pd used in this study is much less than the conventional Pd/Mg thin film structures. With only a small percentage of Pd nanoparticles, the Mg film can be fully transformed into hydride. The separation between the nanoparticles is less than 30–50 μm of a blocking layer, thus enabling full conversion. Therefore, the storage capacity is significantly increased with this nanoportal structure compared to Pd thin film catalyst structures. This makes it promising for future on-board storage applications. We hope to do further work to apply this novel structure to understand and design other hydrogen storage systems.
Acknowledgements
The authors would like to thank Stephen T. Kelly for useful discussions. This work was supported as part of the Center on Nanostructuring for Efficient Energy Conversion (CNEEC) at Stanford University, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Basic Energy Sciences under Award Number DE-SC0001060. Part of this work was performed at the Stanford Nano Shared Facilities (SNSF) at Stanford University.References
- B. Vigeholm, J. Kjøller, B. Larsen and A. Pedersen, J. Less-Common Met., 1983, 89, 135–144 CrossRef CAS.
- B. Tanguy, J. Soubeyroux, M. Pezat, J. Portier and P. Hagenmuller, Mater. Res. Bull., 1976, 11, 1441–1448 CrossRef CAS.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- J. J. Vajo, F. Mertens, C. C. Ahn, R. C. Bowman and B. Fultz, J. Phys. Chem. B, 2004, 108, 13977–13983 CrossRef CAS.
- X. Tan, L. Wang, C. M. B. Holt, B. Zahiri, M. H. Eikerling and D. Mitlin, Phys. Chem. Chem. Phys., 2012, 14, 10904–10909 RSC.
- C. Zhou, Z. Z. Fang, J. Lu, X. Luo, C. Ren, P. Fan, Y. Ren and X. Zhang, J. Phys. Chem. C, 2014, 118, 11526–11535 CAS.
- P. Kalisvaart, B. Shalchi-Amirkhiz, R. Zahiri, B. Zahiri, X. Tan, M. Danaie, G. Botton and D. Mitlin, Phys. Chem. Chem. Phys., 2013, 15, 16432–16436 RSC.
- C. J. Chung, S.-C. Lee, J. Groves, E. Brower, R. Sinclair and B. M. Clemens, Phys. Rev. Lett., 2012, 108, 106102 CrossRef.
- A. Zaluska, L. Zaluski and J. Ström Olsen, J. Alloys Compd., 1999, 288, 217–225 CrossRef CAS.
- W. Oelerich, T. Klassen and R. Bormann, J. Alloys Compd., 2001, 322, L5–L9 CrossRef CAS.
- J. Čermák and L. Král, Acta Mater., 2008, 56, 2677–2686 CrossRef PubMed.
- J. Renner and H. Grabke, Z. Metalkd., 1978, 69, 639–642 CAS.
- C. M. Stander, Z. Phys. Chem., 1977, 104, 229–238 CrossRef CAS.
- P. Kalisvaart, E. Luber, H. Fritzsche and D. Mitlin, Chem. Commun., 2011, 47, 4294–4296 RSC.
- S. T. Kelly and B. M. Clemens, J. Appl. Phys., 2010, 108, 013521 CrossRef PubMed.
- W. Oelerich, T. Klassen and R. Bormann, J. Alloys Compd., 2001, 315, 237–242 CrossRef CAS.
- M. Fichtner, J. Engel, O. Fuhr, O. Kircher and O. Rubner, J. Mater. Sci. Eng. B, 2004, 108, 42–47 CrossRef PubMed.
- A. Borgschulte, R. J. Westerwaal, J. H. Rector, B. Dam and R. Griessen, Appl. Phys. Lett., 2004, 85, 4884 CrossRef CAS PubMed.
- K.-F. Aguey-Zinsou and J.-R. Ares-Fernández, Energy Environ. Sci., 2010, 3, 526 CAS.
- C. Webb, J. Phys. Chem. Solids, 2015, 84, 96–106 CrossRef CAS PubMed.
- R. Gremaud, C. Broedersz, D. Borsa, A. Borgschulte, P. Mauron, H. Schreuders, J. Rector, B. Dam and R. Griessen, Adv. Mater., 2007, 19, 2813–2817 CrossRef CAS PubMed.
- M. Avrami, J. Chem. Phys., 1939, 7, 1103 CrossRef CAS PubMed.
- M. Gracia-Pinilla, E. Martínez, G. S. Vidaurri and E. Pérez-Tijerina, Nanoscale Res. Lett., 2010, 5, 180–188 CrossRef CAS PubMed.
- R. Kelekar, H. Giffard, S. T. Kelly and B. M. Clemens, J. Appl. Phys., 2007, 101, 114311 CrossRef PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- J. Christian, The Theory Of Transformations In Metals And Alloys, Oxford University Press, Oxford, 3rd edn, 1975 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp04515k |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2015 |