
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ spectroelectrochemical and theoretical study on the oxidation of a 4H-imidazole-ruthenium dye adsorbed on nanocrystalline TiO2 thin film electrodes†
Ying
Zhang‡
ab,
Stephan
Kupfer‡
a,
Linda
Zedler
ab,
Julian
Schindler
ab,
Thomas
Bocklitz
a,
Julien
Guthmuller
c,
Sven
Rau
d and
Benjamin
Dietzek
*ab
aInstitute of Physical Chemistry, Friedrich Schiller University Jena, Helmholtzweg 4, 07743 Jena, Germany. E-mail: benjamin.dietzek@uni-jena.de; Fax: +49-3641-948302
bLeibniz Institute of Photonic Technology Jena (IPHT), Albert-Einstein-Stra�e 9, 07745 Jena, Germany
cFaculty of Applied Physics and Mathematics, Gdansk University of Technology, Narutowicza 11/12, 80233 Gdansk, Poland
dInstitute of Inorganic Chemistry I, University Ulm, Albert-Einstein-Allee 11, 89081 Ulm, Germany
First published on 19th October 2015
Abstract
Terpyridine 4H-imidazole-ruthenium(II) complexes are considered promising candidates for use as sensitizers in dye sensitized solar cells (DSSCs) by displaying broad absorption in the visible range, where the dominant absorption features are due to metal-to-ligand charge transfer (MLCT) transitions. The ruthenium(III) intermediates resulting from photoinduced MLCT transitions are essential intermediates in the photoredox-cycle of the DSSC. However, their photophysics is much less studied compared to the ruthenium(II) parent systems. To this end, the structural alterations accompanying one-electron oxidation of the RuIm dye series (including a non-carboxylic RuIm precursor, and, carboxylic RuImCOO in solution and anchored to a nanocrystalline TiO2 film) are investigated via in situ experimental and theoretical UV-Vis absorption and resonance Raman (RR) spectroelectrochemistry. The excellent agreement between the experimental and the TDDFT spectra derived in this work allows for an in-depth assignment of UV-Vis and RR spectral features of the dyes. A concordant pronounced wavelength dependence with respect to the charge transfer character has been observed for the model system RuIm, and both RuImCOO in solution and attached on the TiO2 surface. Excitation at long wavelengths leads to the population of ligand-to-metal charge transfer states, i.e. photoreduction of the central ruthenium(III) ion, while high-energy excitation features an intra-ligand charge transfer state localized on the 4H-imidazole moiety. Therefore, these 4H-imidazole ruthenium complexes investigated here are potential multi-photoelectron donors. One electron is donated from MLCT states, and additionally, the 4H-imidazole ligand reveals electron-donating character with a significant contribution to the excited states of the ruthenium(III) complexes upon blue-light irradiation.
Introduction
Ruthenium polypyridyl dyes have attracted considerable interest in various applications due to their chemical stability, strong absorption of visible light, and unique redox and catalytic properties.1�8 For example, ruthenium polypyridyl complexes are used as photosensitizers in dye-sensitized solar cells (DSSCs). Besides the nanocrystalline semiconductor, the redox electrolyte and the platinum counter electrode the ruthenium polypyridyl complexes are one of the main components of these devices.9,10 In DSSCs, the direct conversion of light into electrical energy occurs via visible light absorption by the dye molecules, followed by a generally very fast electron transfer into the wide-bandgap semiconductor.11,12 Subsequently, the oxidized form of the adsorbed dye is re-reduced by a redox couple in the electrolyte � a process which is dominated by molecular collisions of the redox couple with the adsorbed dye.13�15 The optimization of the performance of DSSCs is based on a detailed understanding of the system and its light-induced dynamics at a molecular level, which allows deeper insight into the structure-dynamics-function interplay in complex systems for the conversion of sunlight into electricity16 and other storable forms of energy.17�19 A significant amount of work focused on improving the light-absorbing properties of the sensitizer,6,20�27 which should cover the visible range of the solar spectrum and stretch far into the IR. In this context, herein the investigated ruthenium complexes coordinating 2-phenyl-4,5-p-tolylimino-4H-imidazole (4H-imidazole) as an organic chromophore, have been introduced as panchromatic dyes.28�30 Aside from the 4H-imidazole a polypyridyl fragment, which consists of 4,4',4''-tri-tert-butyl-2,2':6',2''-terpyridine and an additional monodentate isothiocyanate ligand are coordinated to the ruthenium(II) metal center (RuIm) (Fig. 1A). Due to the direct and strong interaction between each chromophore with the metal center the complexes exhibit the photoelectrochemical and photophysical properties related to the ruthenium(II)-polypyridine fragment while displaying broad and strong absorption in the visible range due to the presence of strong 1MLCT and ligand centered (1LC) transitions.29�34 Furthermore, the redox and photophysical properties of the 4H-imidazole coordinating ruthenium(II) complexes can be easily tuned by varying the substituent pattern and the protonation state of the ligand, as has been recently shown.30,31,33,34 These properties render complexes like RuIm with promising structures for use as molecular sensitizers in DSSCs, in which the electron injection from the photoexcited light harvesting unit into the wide bandgap semiconductor occurs via an anchoring group, typically a carboxylate group (COO-).9,35,36 High electron injection yields are obtained by placing the anchoring group close to the ligand that carries the excess charge density in the photoexcited state,37,38i.e., for RuIm at the 4H-imidazole ligand and the directly connected phenyl ring, since the lowest initially excited 1MLCT state and the thermally relaxed 3MLCT state, from which electron injection in the acceptor states of TiO2 appears, are both dominantly localized on the imidazole fragment.29�31,33,39 Hence, the carboxyl anchoring group has been introduced at the phenyl moiety of the 4H-imidazole ligand in order to facilitate the electronic coupling between both, the 1MLCT and the 3MLCT state and the acceptor states of TiO2 (RuImCOO, Fig. 3C). Furthermore, recently it was reported that injection for closely related complexes occurs partially from hot vibrational states, since the thermalized long-lived relaxed 3MLCT states fall within the bandgap of TiO2.32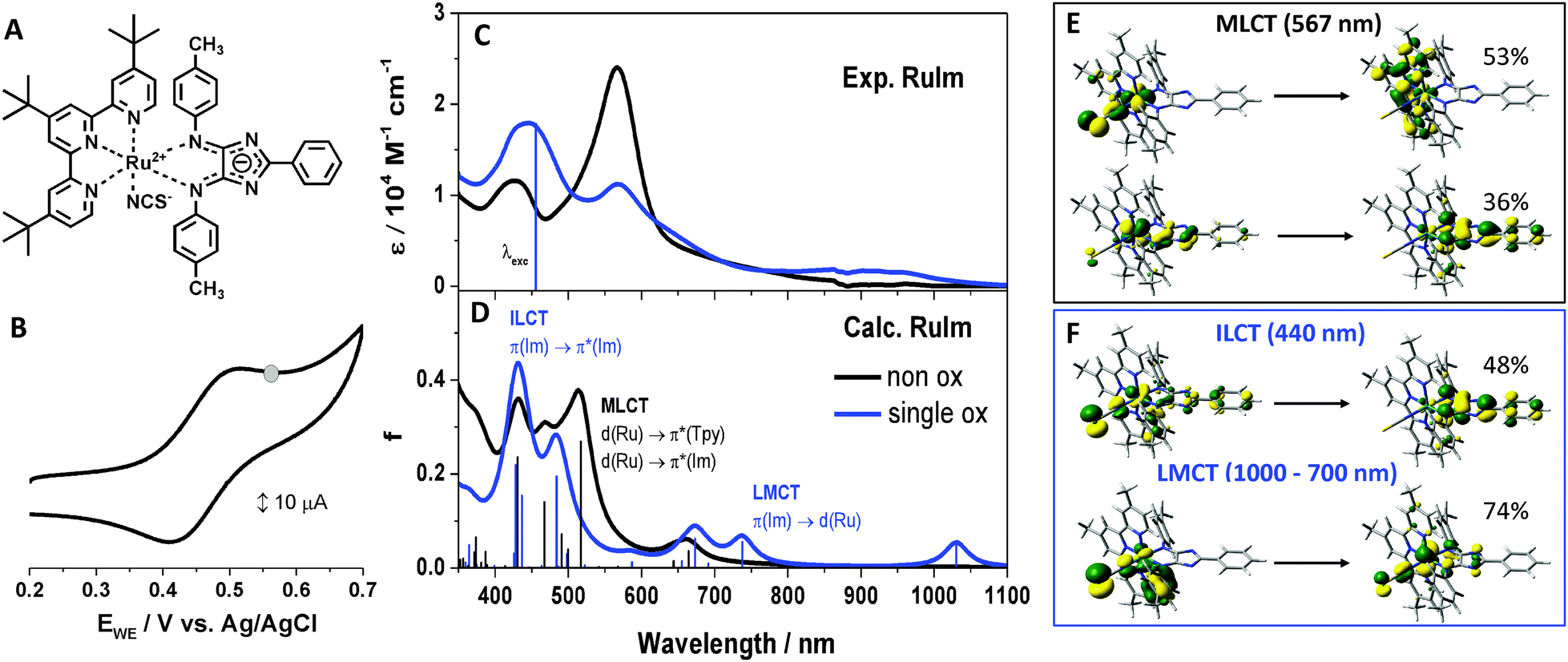 | ||
| Fig. 1 (A) Molecular structure of RuIm. (B) CV obtained for the oxidation of RuIm in ACN with 0.1 M tetrabutylammonium tetrafluoroborate (TBABF4) as supporting electrolyte with a scan rate of 50 mV s-1, the applied oxidation potential (0.55 V vs. Ag/AgCl) during UV-Vis and RR spectroscopy is marked as a grey circle in the curve. (C) UV-Vis spectra of RuIm (black curve) and electrochemically oxidized RuIm (blue curve) in 0.1 M TBABF4/ACN, with an applied potential of 0.55 V vs. Ag/AgCl. The RR excitation wavelength (458 nm) is displayed as a vertical line in the spectrum. (D) Calculated spectra of non-oxidized (black curve) and single oxidized (blue curve) RuIm. (E) Molecular orbitals involved in the S6 MLCT transition of RuIm. (F) Molecular orbitals involved in the ILCT and LMCT transitions of oxidized RuIm. | ||
In this contribution we focus on the spectroscopic properties of the photo-oxidized ruthenium(III) species of RuImCOO, which appear as essential mechanistic intermediates in the photoelectrochemical cycle underlying the function of the DSSC. The electronic transitions in these species, which are exposed to visible light, are studied by a combined experimental-theoretical approach utilizing UV-Vis absorption and resonance Raman (RR) spectroelectrochemistry (SEC) to study structural changes accompanying one-electron oxidation and to characterize photoexcited intermediates of the oxidized terpyridine 4H-imidazole-ruthenium(II) complexes RuIm and RuImCOO in solution. Special emphasis will be put on the identification of the predominant protonation state of the 4H-imidazole ligand sphere in RuImCOO since protonation strongly impacts the redox and photophysical properties of the investigated complexes. Finally, RuImCOO was attached to mesoporous nanocrystalline TiO2 thin films and investigated by means of SEC to derive influences of the interface between the sensitizer and the semiconductor on the oxidation induced spectral shifts.
Results and discussion
Spectroelectrochemistry on the RuIm precursor
To aid the study and rationalization of the electronic properties of RuImCOO on TiO2 surfaces, the complex without an anchoring group, i.e.RuIm, is initially studied as a benchmark system in solution. The cyclic voltammogram (CV) of RuIm (Fig. 1B) exhibits a quasi-reversible oxidation peak at 0.46 V vs. Ag/AgCl. To gain insight into the (electronic) structures of non-oxidized and oxidized species, a combination of spectroelectrochemical UV-Vis and RR techniques is utilized.The absorption spectrum of RuIm recorded in acetonitrile (ACN) exhibits two broad absorption bands in the visible region centered at 567 nm (e = 24?100 M-1 cm-1) and 427 nm (e = 11?600 M-1 cm-1) as well as a shoulder at approximately 700 nm (Fig. 1C). In order to unravel the nature of the electronic transitions underlying these absorption features quantum chemical simulations at the time-dependent density functional theory (TDDFT) level of theory have been performed (Fig. 1D and Table S1, ESI†). Based on the calculations, the first bright absorption band can be assigned to the bright S6 MLCT state (517 nm) with slight contributions from the MLCT states S7 (500 nm) and S8 (491 nm). S6 and S8 feature transitions to the terpyridine as well as to the 4H-imidazole ligand sphere, while S7 merely exhibits transitions to the terpyridine ligand. The long-wavelength shoulder at 700 nm is due to the weakly absorbing S1 (664 nm, localized on the 4H-imizazole) and S2 (644 nm, localized on the terpyridine) MLCT states. The second bright absorption band is assigned to the bright MLCT state S9 (467 nm), featuring transitions towards the 4H-imidazole and terpyridine ligand spheres, as well as to the 4H-imidazole centered intra-ligand charge transfer (ILCT) state S10. Detailed information with respect to the computational results is illustrated in Table S2 (ESI†). As predicted by TDDFT the first bright MLCT band (567 nm) is dominated by transitions to the terpyridine ligand, while transitions towards the 4H-imidazole feature less weight. This finding is contrary to the expectations based on the structurally closely related chloro-complex, where merely the isothiocyanate ligand is replaced by a chloro ligand.29,34 For the previously studied chloro-complex, the first bright MLCT band is dominated by 4H-imidazole transitions; very similar results have been obtained for complexes with variations of the substitution pattern in the periphery of the 4H-imidazole.28,33 In addition, changing the polypyridyl sphere from terpyridine to bipyridine31,32 induces no pronounced alterations to the nature of the excited states underlying the MLCT band, hence, the majority of the MLCT transitions in the visible range involve charge density shifts towards the 4H-imidazole ligand. Herein the reported phenomenon of enhanced terpyridine character of the MLCT band in RuIm can be rationalized by means of the molecular orbital (MO) dxz(187), which is involved in the terpyridine centered MLCT transition of the bright S6 state, see Tables S1 and S2 (ESI†). In contrast to the respective MO of the chloro-complex (Fig. S1, ESI†), dxz(187) features an increased mixing with p-orbitals of the isothiocyanate ligand. This leads in consequence to an increased weight of the terpyridine transition.
Upon electrochemical oxidation of RuIm, the intensity of the MLCT absorption band at 567 nm is quenched significantly, while several new absorption features arise (Fig. 1C): in the low-energy region between 1000 and 700 nm a broad yet rather weak absorption band is formed. In addition, the absorbance of the ILCT is increased substantially. This increase in absorption is accompanied by a red-shift of the band from 427 to 440 nm (~700 cm-1) upon oxidation. Reversibility of electrochemical reduction was confirmed by measuring UV-Vis spectra during the acquisition of a CV of oxidation (Fig. S2, ESI†). The origin of these spectral alterations is elucidated by quantum chemical calculations for the singly oxidized species of RuIm (Fig. 1D), where oxidation was found to be almost entirely localized on the ruthenium center (see Table S3, ESI†). The optimized equilibrium structures of both oxidation states are very similar. However, contrary to the non-oxidized singlet, the singly oxidized doublet exhibits no intense MLCT states in the visible range, which is directly correlated with the formation of the ruthenium(III) species, i.e., to the decreased electron density at the ruthenium ion. Consequently, the intensity of the MLCT band is reduced in the UV-Vis-SEC measurements. Likewise, the formation of the absorption bands between 1000 and 700 nm and 440 nm can be rationalized. TDDFT assigns the low-energy band to three weakly absorbing ligand-to-metal charge transfer (LMCT) states, namely, D3, D4 and D6 localized at 1030, 738 and 673 nm, respectively. The rising absorption band at 440 nm is due to several intense 4H-imidazole centered ILCT states, D16, D23 and D31 at 484, 428 and 409 nm, as well as to a ligand-to-ligand charge transfer (LLCT) state from isothiocyanate to terpyridine (D22 at 437 nm). The ILCT occurs from the terminal tolyl moieties to the central 4H-imidazole fragment. Thus, a pronounced wavelength dependency with respect to the charge transfer (CT) character of the electronic transitions is observed for the absorption spectrum of the oxidized RuIm: low-energy excitation leads to the population of LMCT states and, thus, to the formation of ruthenium(II), while high-energy excitation leads to an ILCT of the 4H-imidazole ligand.
In order to investigate the nature of the excited states of the non-oxidized as well as of the single oxidized RuIm within the respective Franck�Condon points RR-SEC has been applied.39 For these experiments, excitation at 458 nm is employed, which is in resonance with the ILCT band centered at 427 nm (and partially with the MLCT band at 567 nm) of the non-oxidized RuIm.
Based on the agreement of the measured RR-SEC spectra for both redox states (Fig. 2) with the simulated RR intensity pattern (non-oxidized singlet and single oxidized doublet depicted in Fig. S3, ESI† and respective vibrational modes in Table S4, ESI†) almost all intense Raman bands could be assigned to vibrational normal modes. For the calculation of the RR spectrum of the non-oxidized form contributions from the MLCT states S6, S7, S8 and S9 and of the IL state S10 have been taken into account, while the excitation energy for all states has been red-shifted by 1000 cm-1. Following this procedure, the Raman active vibrational bands at 1226, 1240, 1354, 1388, 1492, 1505 and 1564 cm-1 have been assigned to normal modes of the 4H-imidazole ligand at 1221.6, 1241.0, 1342.6, 1380.8, 1493.2, 1576.7 and 1581.7 cm-1 (modes 163, 164, 178, 180, 205 and 207). In addition, the weak shoulders at 1340 and 1537 cm-1 and the feature at 1609 cm-1 have been associated with the terpyridine modes 177, 209 and 217 at 1324.0, 1550.7 and 1609.1 cm-1, respectively. Hence, a maximum absolute deviation (MAD) of 6.9 cm-1 is obtained for the assigned normal modes. In general contributions of the terpyridine ligand to the RR intensity pattern seem to be slightly overestimated, which correlates with the increased terpyridine character to the MLCT transition in the absorption spectrum of RuIm.
 | ||
| Fig. 2 Experimental (A) and calculated (B) RR spectra for non-oxidized (black) and single oxidized (blue) RuIm in 0.1 M TBABF4/ACN, excited at 458 nm. | ||
The excitation wavelength of 458 nm is in resonance with the rising ILCT band centered at 440 nm and the increase in intensity upon single oxidation. Consequently, alterations of the RR spectra are observed upon oxidation, e.g., the most intense band (1354 cm-1) is hypsochromically shifted by 9 cm-1. Furthermore, the intensity of the band at 1388 cm-1 decreases substantially, while the bands at 1492 and 1572 cm-1 become more intense. The origin of the alteration of the RR intensity pattern is unraveled based on the simulations. Taking into account the contributions from D16, D22 and D23 states and shifting the excitation energies of these states by �1000 cm-1, an excellent agreement was achieved with respect to the frequencies as well as to the relative RR intensities (MAD of 6.9 cm-1). Hence, the shift of the most intense resonance Raman band from 1354 to 1363 cm-1 is reproduced by the simulations (from 1342.6 to 1353.6 cm-1), while the underlying vibrational modes of the non-oxidized (178) and single oxidized complex (179) are assigned to 4H-imidazole centered modes (see Fig. S3 and Table S4, ESI†). Likewise the increase in intensity at 1492 and 1572 cm-1 stems from contributions of the intense 4H-imidazole normal modes (205, 212 and 213) at 1490.8, 1566.1 and 1570.9 cm-1. Merely the small shoulder at 1330 cm-1 (mode 177 at 1330.7 cm-1) is associated with the terpyridine ligand. Hence, the contribution of the terpyridine ligand sphere is considerably reduced upon single oxidation, which roots in the electronic nature of the electronic states, which are in resonance with the excitation light at 458 nm, i.e. intense ILCT states D16 and D23.
RuImCOOH
The absorption spectrum of RuImCOO, depicted in Fig. S4 (ESI†), features intense bands at 570 and 442 nm and shoulders at approximately 700 and 420 nm. Thus, its absorption properties are very similar to that of the RuIm precursor. Upon single protonation both absorption features are bathochromically shifted to 582 (?? = -362 cm-1) and 451 nm (?? = -451 cm-1), while the absorbance of red (700 nm) and blue-sided (425 nm) shoulders decreases. Further protonation by TFA yields the double protonated species RuImHCOOH, whose formation is accompanied by a further red-shift of the first absorption band to 603 nm (?? = -960 cm-1 with respect to RuImCOO) and the disappearance of the red-sided shoulder. In contrast, the blue absorption band is shifted to shorter wavelengths (412 nm, ?? = +1648 cm-1 with respect to RuImCOO). In order to obtain insight into the excited states underlying the absorption pattern of the protonated species TDDFT calculations have been performed within the equilibrium structures of RuImCOO, RuImCOOH, RuImHCOO, and RuImHCOOH.
The experimental results obtained for RuImCOO are almost identical to the data of the RuIm precursor, and, hence similar excited states properties are: the first absorption feature (570 nm), shown in Fig. S5 (ESI†), is assigned to the MLCT states S6 (518 nm), S7 (503 nm) and S8 (498 nm) featuring transitions to the 4H-imidazole as well as to the terpyridine ligand. The second absorption band at 442 nm as well as the blue-sided shoulder (420 nm) stem from the bright S10 (469 nm), which is a MLCT state localized on both ligand spheres, and the bright 4H-imidazole centered ILCT state S13 (426 nm). Thus, the carboxylic acid anchoring group introduces only minor alterations to the excited states properties as compared to RuIm, which are based on its electron-withdrawing character.5,41,42 Protonation of RuImCOO can result in the formation of the single protonated forms RuImHCOO and RuImCOOH, the latter of which is more stable by 4000 cm-1. Hence, only the excited states of RuImCOOH are used to interpret the experimental UV-Vis data of the single protonated complex: the slight red-shift of the first absorption band upon protonation is mainly represented by the bright MLCT state S6 (532 nm) and is accompanied by a substantial increase of the 4H-imidazole character. Further contributions to this absorption feature originate from the weakly absorbing MLCT states S7 (497 nm) and S8 (490 nm). The second band (at 451 nm) and the blue-sided shoulder are assigned to the bright 4H-imidazole ILCT state S10 (452 nm) and the weakly absorbing S9 (468 nm) MLCT state, which is localized on the terpyridine ligand.
Double protonation leads to RuImHCOOH; in this protonation state the MLCT band is further shifted to higher wavelengths (i.e., to 603 nm). TDDFT assigns this band to the S4 MLCT state (556 nm), while the bathochromic shift is associated with a further increase of weight of the transition towards the 4H-imidazole. However, the blue-shift of the ILCT band upon double protonation to 412 nm is not correctly reproduced by considering the S6 state (484 nm) in the quantum chemical simulation, a phenomenon which was previously reported for other 4H-imidazole ruthenium(II) complexes.28,29,31�33 This problem is related to the shortcoming of TDDFT to give a balanced description for electronic states of different electronic natures, such as MLCT, IL, ILCT, and LLCT states. As shown in ref. 29 the B3LYP exchange correlation functional gives the most balanced description for the involved excited states. More detailed information concerning the computational results is presented in Tables S5�S8 of the ESI.†
In general, successive protonation from RuImCOOviaRuImCOOH to RuImHCOOH leads to a characteristic red-shift of the MLCT band, which roots in the increasing 4H-imidazole (and decreasing terpyridine) character of the underlying excited states. In addition, wavelength shifts have been observed for the ILCT band with respect to RuImCOO. Hence, the protonation state of the investigated ruthenium complex can be monitored by means of UV-Vis spectroscopy.
In the following, the protonation state of the complex is investigated upon anchoring onto the nanocrystalline TiO2 film. To this end, the UV-Vis absorption of the dye anchored onto TiO2 through the carboxyl group was measured in ACN (depicted in Fig. 3E and Fig. S6, ESI†). The spectrum exhibits strong absorption due to TiO2 nanoparticles below 400 nm,43 an intense MLCT absorbing band at 576 nm and a less intense 4H-imidazole ILCT band at 445 nm. The position of the MLCT band of the dye-sensitized TiO2 film in ACN demonstrates that the 4H-imidazole ring is deprotonated. The comparison with the pH dependent UV-Vis spectra confirms the presence of RuImCOO on the dye-sensitized TiO2 film. Nevertheless, both the MLCT and the ILCT band of the dye-sensitized TiO2 film in ACN (at 576 and 445 nm respectively) are localized at longer wavelengths compared to the free RuImCOO in solution (at 570 and 442 nm, respectively). To illustrate the shifts, the UV-Vis spectra of the dye-sensitized TiO2 film were also recorded under atmospheric conditions (Fig. S6, ESI†), where the spectral shape is broader than that in ACN and the intense MLCT band is bathochromically shifted to 582 nm (?? = -179 cm-1). These alterations indicate that the dye molecules anchored onto the TiO2 film aggregate slightly and exhibit weak intermolecular interactions.44,45 Intermolecular interactions among the dyes can be (partially) quenched by the solvent molecules (ACN) acting as spacers; however, most likely aggregation of the dyes on the surface cannot be suppressed completely. Thus, the bathochromic shifts in the UV-Vis spectra upon anchoring of RuImCOO onto the TiO2 film are ascribed to the aggregation of the dye on the surface.
 | ||
| Fig. 3 (A) and (B) CVs for the oxidation of RuImCOO in solution and on TiO2 surface measured in 0.1 M TBABF4/ACN with a scan rate of 50 mV s-1, the applied oxidation potentials (0.70 and 0.75 V vs. Ag/AgCl) during UV-Vis and RR spectroscopy are marked as grey circles in the curves. (C) and (E) In situ UV-Vis spectra of non-oxidized RuImCOO (black curve) and electrochemically oxidized RuImCOO (blue curve) in solution and on the TiO2 surface measured in 0.1 M TBABF4/ACN. The RR excitation wavelength (458 nm) is displayed as a vertical line in the spectra. (D) Calculated spectra of non-oxidized (black) and single oxidized (blue) RuImCOO in solution. (F) Molecular orbitals involved in the S6 MLCT transition of RuImCOO in solution. (G) Molecular orbitals involved in the ILCT and LMCT transitions of oxidized RuImCOO in solution. | ||
Besides UV-Vis also RR spectroscopy has been proven to be a powerful tool to discriminate different protonated states of ruthenium-polypyridyl-4H-imidazole complexes.29,31�33 As shown in Fig. S7 (ESI†), only the double protonated species RuImHCOOH displays characteristic bands of the protonated 4H-imidazole ring at 1480 and 1516 cm-1; thus the absence of these signatures further corroborates the conclusion that the dye molecule bound to TiO2 is the deprotonated RuImCOO.
UV-Vis-SEC has been applied to study RuImCOO in solution as well as on the TiO2 surface. Firstly, the spectral alterations of the single oxidized RuImCOO complex in solution are rationalized: analogous to the single oxidation of the RuIm precursor, oxidation of RuImCOO is almost entirely localized on the ruthenium atom (see Table S3, ESI†); the absorption spectrum of the oxidized species of RuImCOO features a significantly decreased MLCT band (570 nm) upon oxidation, while a broad absorption band arises at 459 nm; in addition, the absorbance between 1000 and 700 nm increases slightly (Fig. 3C). The slight red-shift of the absorption band with respect to RuIm is associated with the introduction of the electron-withdrawing carboxylic acid group. The evaluation of the excited states of the oxidized complex performed at the TDDFT level of theory enabled the assignment of these spectral alterations to specific electronic transitions (Fig. 3D). The increasing absorption between 1000 and 700 nm stems from the underlying medium bright LMCT states D4, D7 and D9 (at 1037, 754 and 665 nm), which are associated with charge density shifts from the 4H-imidazole ligand to the central ruthenium ion. The increasing absorption band at 459 nm contains dominant contributions from the D25, D27, D37, D38 and D39 states at 485, 481, 441, 435 and 432 nm, respectively. The ILCT states D25 and D27 are mainly localized on the bridging ligand, while the highly mixed D37, D38 and D39 states feature LLCT and ILCT character with contributions of all three ligands. Hence, low-energy excitation leads to the direct population of LMCT states and, thus, to a photoreduction of ruthenium(III). On the other hand, excitation under blue light leads to an ILCT towards the central moiety of the 4H-imidazole.
The UV-Vis-SEC of RuImCOO on the TiO2 surface resembles the respective data in solution with increasing LMCT and ILCT bands and a decreasing MLCT band (Fig. 3E). Therefore, oxidized RuImCOO on the TiO2 surface also shows the same wavelength dependence of the CT features; with low-energy excitation yielding ruthenium(II) and high-energy excitation leading to an ILCT located on the 4H-imidazole ligand. Hence, anchoring onto nanocrystalline TiO2 does not affect the photophysical properties of the dye significantly.
In order to investigate the potential applications of RuImCOO as sensitizers in DSSCs, RR-SEC has been applied to study the photophysical properties of the excited states. These measurements have been performed for the complex bound to the TiO2 surface as well as in solution with an excitation wavelength of 458 nm, while the quantum chemical simulations � due to the complexity associated with the inclusion of the semiconductor surface � have been carried out exclusively for RuImCOO in solution. Hence, at first the RR-SEC data for the complex on TiO2 will be discussed. The RR spectrum of the non-oxidized RuImCOO on the TiO2 film features intense Raman bands at 1356, 1392, 1502, 1568 and 1607 cm-1 as well as weak bands and shoulders at 1227, 1477, 1517 and 1537 cm-1 (Fig. 4A). Analogous to RuIm, the bands at 1227, 1356, 1392, 1502 and 1568 cm-1 are assigned to vibrational normal modes of the 4H-imidazole ligand (normal mode displacements of the respective modes are summarized in the ESI†), and the other bands at 1477 and 1607 cm-1 are associated with the terpyridine ligand (see Tables S4 and S9, ESI†). Upon electrochemical oxidation of the dye-sensitized TiO2 film, the intense bands at 1356 and 1568 cm-1 are slightly shifted to higher frequencies, i.e. to 1359 and 1576 cm-1. Furthermore, the band at 1392 cm-1 decreases upon oxidation, while the weak RR signals and shoulders arise at 1336, 1517 and 1478 cm-1. Likewise the most intense bands (i.e., at 1359, 1502 and 1576 cm-1) are correlated with normal modes associated with the 4H-imidazole ligand. Hence, the contribution of the 4H-imidazole ligand is considerable for both the non-oxidized and oxidized anchored RuImCOO.
 | ||
| Fig. 4 (A) RR spectra of non-oxidized (black) and oxidized (blue) RuImCOO on TiO2 film, immersed in 0.1 M TBABF4/ACN; (B) and (C) Experimental and calculated RR spectra of non-oxidized (black) and oxidized (blue) RuImCOO in solution, measured in 0.1 M TBABF4/ACN; all the excitation wavelengths were 458 nm. | ||
The RR spectrum of the non-oxidized RuImCOO in solution is illustrated in Fig. 4B and features intense Raman bands at 1356, 1389, 1496 and 1567 cm-1 as well as less intense bands and shoulders at 1227, 1328, 1504, 1537 and 1609 cm-1. Based on the computed RR intensity pattern (Fig. 4C) which takes into account contributions from the excited states S6, S10, and S13, the respective excitations energies have been slightly red-shifted by 1000 cm-1 (Fig. S8 and Table S9, ESI†). Almost all bands could be assigned to vibrational normal modes with a MAD of 8.4 cm-1. All RR signals besides the weak bands at 1537 and 1609 cm-1 and the shoulders at 1328 and 1504 cm-1 are assigned to vibrational normal modes of the 4H-imidazole ligand. Upon single oxidation the intense 4H-imidazole bands at 1356, 1496 and 1567 cm-1 are slightly shifted to higher frequencies at 1359, 1502 and 1571 cm-1, while the formation of the weak RR signals (and shoulders) at 1336, 1402, 1478 and 1540 cm-1 is assigned to vibrational modes localized on the terpyridine ligand. Analogous to the procedure for the oxidized RuIm, the energies of the states contributing to the RR intensity pattern (D25, D27, D37, D38 and D39) have been blue-shifted by 1000 cm-1. A comparison of the RR-SEC spectra of the anchored complex RuImCOO and RuImCOO in solution yields merely minor alterations of the RR intensity pattern. Thus, the excited states in both RuImCOO systems (on TiO2 and in solution) are centered on the 4H-imidazole ligand.
Hence, as shown by UV-Vis-SEC, RR-SEC in combination with quantum chemical simulations the oxidized RuImCOO both on TiO2 and in solution features an ILCT state localized on the 4H-imidazole upon blue-light irradiation, while photoexcitation with red light leads to the regeneration of the ruthenium(II) species.
Experimental section
Sample preparation
The complexes RuIm and RuImCOOH were provided by Prof. Rau.40 All samples were dissolved in anhydrous ACN (Sigma-Aldrich, spectroscopic grade), which was dried using calcium hydride (Sigma-Aldrich, 98%) and distilled twice. Dye solutions were prepared with a concentration of 0.25 mM in ACN. To obtain the non-protonated RuIm complex, additional traces of DBU (Sigma-Aldrich, 98%) were added into the solution to deprotonate the sample which is partly protonated due to the preparation procedure.40 Protonation and deprotonation states of RuImCOOH were adjusted by addition of aliquots of 0.02 M TFA (Sigma-Aldrich, >99.5%) and DBU. 0.1 mM TBABF4 in ACN was used as an electrolyte for the electrochemical and spectroelectrochemical experiments. Nanocrystalline and transparent TiO2 films were prepared according to Kallioinen et al.50 from diluted anatase paste (Ti-Nanoxide HT) consisting of 8�10 nm diameter TiO2 particles (Solaronix SA, Switzerland). The paste was spread on a 1 mm thick, 7 mm width ITO glass slide. After drying at room temperature, the slide was calcined at 450 �C for 15 min and then cooled off slowly. Afterwards the film was stained in a dye bath containing 0.5 mM RuImCOOH in ACN for about 16 hours. After sensitization of the TiO2, physisorbed dye was wiped off by purging the film with ACN. All measurements were carried out just directly after the preparation of the films to minimize the effect of aging of the samples.Spectroscopic measurements
UV-Vis spectra were recorded on a double-beam Cary 5000 UV-Vis spectrometer (Varian, USA) at room temperature. RR experiments were performed through excitation by the visible lines of an Innova 300C Argon ion laser (Coherent, USA) and detected using an Acton SpectraPro 2758i spectrometer (Princeton Instruments, USA) with an entrance slit width of 100 �m, focal length 750 mm, and grating 1800 Blz/500 nm. The Raman signals were recorded using a liquid-nitrogen-cooled SPEC-10 CCD detector (Princeton Instruments, USA). The Raman spectra were subjected to a SNIP baseline correction.51 The iteration parameter was set to 37 and pre-smoothing was applied to allow for an adaptive baseline estimate, which is not prone to noise artefacts. The ACN solvent spectrum was subtracted from all RR spectra.UV-Vis-SEC, RR-SEC and CV measurements were performed either in a three-electrode thin-layer spectroelectrochemical cell with a pathlength of 1 mm (Bioanalytical Systems, USA), for the measurement of free dye in solution, or in a standard 5 mm pathlength cuvette, for the measurement on the TiO2 film. The three-electrode system contains a Pt counter electrode, an Ag/AgCl pseudo-reference electrode and either a Pt-gauze working electrode or a dye-sensitized TiO2 film on ITO glass. The potential was adjusted using a PC-controlled potentiostat. The potentials were measured with respect to the Ag/AgCl reference electrode, against which the oxidation of ferrocene was measured to be +0.42 V under the same experimental conditions. CVs were obtained at a scan rate of 50 mV s-1. The insulating properties of nanocrystalline TiO2 films in the probe potential range provide a background free platform for monitoring the oxidation reactions of adsorbed dye molecules. The spectra of the oxidized dye were taken after a certain time until the spectra remain constant, allowing for saturation of the dye oxidation processes. UV-Vis and RR spectra were recorded before and after each spectroelectrochemical measurement to check the quality and stability of the samples.
Computational details
In order to reduce the computational cost of the simulations without affecting the spectroscopic properties of the complexes, the three tert-butyl groups of the terpyridine ligand have been approximated in the calculations by methyl groups. The structural and electronic data for the RuIm precursor as well as for the respective complex featuring a carboxylic acid anchoring group in the deprotonated (RuImCOO), single protonated (RuImCOOH and RuImHCOO) and double protonated form (RuImHCOOH) were obtained from quantum chemical simulations performed using the Gaussian 09 program.52 All complexes have been investigated in their non-oxidized singlet form, while RuIm and RuImCOO have been studied additionally as single oxidized doublets. The geometries, vibrational frequencies and normal coordinates of the electronic ground state (non-oxidized: singlet, single oxidized: doublet) were calculated at the density functional theory level of theory using the B3LYP53,54 exchange�correlation functional and employing the 6-31G(d) double-? basis set55 for all main group elements. For the ruthenium atom the 28-electron relativistic core potential MWB56 was applied with its basis set describing the valence electrons (4s, 4p, 4d and 5s) explicitly and the inner shells by means of a core potential. In order to correct for the lack of anharmonicity and the approximate treatment of electron correlation the harmonic vibrational frequencies were scaled by a factor of 0.97.57 The vertical excitation energies, oscillator strengths and analytic Cartesian energy derivatives of the excited states were obtained by TDDFT within the adiabatic approximation and by utilizing the same exchange�correlation functional, basis set and pseudopotential. As shown in ref. 28 and 29, B3LYP provides the most balanced description of the absorption features for this class of complexes.The simulated absorption spectra have been determined based on the excitation energies and oscillator strengths of the 120 lowest excited states of the respective ground state multiplicity. The integral equation formalism of the polarizable continuum model58 was applied to take interactions with a solvent (ACN: e = 35.688, n = 1.344) into account with respect to the equilibrium geometry, vibrational frequencies, excitation energies, transition dipole moments and excited state gradients. For the calculations of the excitation energies and excited state gradients, where only the fast reorganization of the solvent is important, the non-equilibrium procedure of solvation was used.
The RR spectra have been calculated within the independent mode displace harmonic oscillator model (IMDHOM) assuming that the electronic ground and excited states potentials are harmonic oscillators and merely displaced in the equilibrium position and, hence, share the same set of vibrational modes.59,60 Detailed information with respect to the computational method can be found in ref. 39 and 61 and the references therein.
In order to calculate the RR intensities of the present ruthenium complexes upon excitation at 458 nm a dumping factor of G = 1500 cm-1 describing homogeneous broadening was assumed in the simulations to reproduce the experimental broadening.
The RR intensity pattern for the non-oxidized form of RuIm has been simulated by taking into account contributions from the S6, S7, S8, S9 and S10, while all excitation energies have been shifted by -1000 cm-1. The respective RR spectrum of the oxidized form was obtained based on the excited doublet states D16, D22 and D23, which have been hypsochromically shifted by 1000 cm-1. Likewise a shift of 1000 cm-1 has been applied for the S6, S10 and S13 states of the non-oxidized RuImCOO and of -1000 cm-1 for the D25, D37, D38 and D39 of the single oxidized doublet. To reproduce the RR intensity pattern for the non-oxidized single and double protonated forms the excitation energies of the states S6, S7, S8, S9 and S10 of RuImCOOH and S4, S6, S11 and S12 of RuImHCOOH have been bathochromically shifted by 1000 cm-1.
The baseline estimation of the experimental Raman spectra was carried out applying the statistical language R62 using the �peaks� package.63
Conclusion
A joint theoretical-experimental SEC study on the oxidation of black absorbing dyes was reported. The photophysical and photochemical properties of the precursors RuIm and RuImCOO as well as their photoexcited intermediates have been studied by using UV-Vis-SEC and RR-SEC methods. In addition, alterations of the spectroscopic properties of RuImCOO upon anchoring onto nanocrystalline TiO2 have been investigated. Consequently, a concordant pronounced wavelength dependency with respect to the CT character has been determined for RuIm, the soluted RuImCOO and RuImCOO on the TiO2 surface, that is, low-energy excitation leads to the population of LMCT states, thus yields the formation of ruthenium(II), while high-energy excitation localizes the excited state in the 4H-imidazole ligand. Therefore, these 4H-imidazole ruthenium complexes are potential multi-photoelectron donors: one electron is donated from MLCT states, and additionally, the 4H-imidazole ligand reveals electron-donating character with a significant contribution to the excited states of the ruthenium(III) complexes upon blue-light irradiation. Future measurements will aim at the determination of the photon to electron conversion efficiency (IPCE) since those experiments would provide information about the potential of 4H-imidazole complexes as multi-photoelectron donors in dye-sensitized solar cells.Acknowledgements
Financial support from the CSCC, Fonds der Chemischen Industrie (B.D., J.S.), DAAD (Y.Z.), 7th Framework Programme of the European Union (J.G., grant No. 321971) and the COST Action PERSPECT-H2O, CM1202, is gratefully acknowledged. All calculations have been performed at the Universit�tsrechenzentrum of the Friedrich Schiller University Jena and at the HP computers of the Theoretical Chemistry group in Jena.Notes and references
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, in Photochemistry and Photophysics of Coordination Compounds I SE - 133, ed. V. Balzani and S. Campagna, Springer, Berlin Heidelberg, 2007, vol. 280, pp. 117�214 Search PubMed.
- V. Balzani and A. Juris, Coord. Chem. Rev., 2001, 211, 97–115 CrossRef CAS.
- H.-W. Tseng, R. Zong, J. T. Muckerman and R. Thummel, Inorg. Chem., 2008, 47, 11763–11773 CrossRef CAS PubMed.
- C. Busche, P. Comba, A. Mayboroda and H. Wadepohl, Eur. J. Inorg. Chem., 2010, 1295–1302 CrossRef CAS PubMed.
- A. Islam, H. Sugihara and H. Arakawa, J. Photochem. Photobiol., A, 2003, 158, 131–138 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef PubMed.
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS PubMed.
- J.-K. Lee and M. Yang, Mater. Sci. Eng., B, 2011, 176, 1142–1160 CrossRef CAS PubMed.
- L. J. Antila, P. Myllyperki�, S. Mustalahti, H. Lehtivuori and J. Korppi-Tommola, J. Phys. Chem. C, 2014, 118, 7772–7780 CAS.
- G. Boschloo and A. Hagfeldt, Acc. Chem. Res., 2009, 42, 1819–1826 CrossRef CAS PubMed.
- E. A. Gibson, L. Le Pleux, J. Fortage, Y. Pellegrin, E. Blart, F. Odobel, A. Hagfeldt and G. Boschloo, Langmuir, 2012, 28, 6485–6493 CrossRef CAS PubMed.
- T. Daeneke, A. J. Mozer, T.-H. Kwon, N. W. Duffy, A. B. Holmes, U. Bach and L. Spiccia, Energy Environ. Sci., 2012, 5, 7090–7099 CAS.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115–164 RSC.
- L. Sun, L. Hammarstr�m, B. �kermark and S. Styring, Chem. Soc. Rev., 2001, 30, 36–49 RSC.
- A. Magnuson, M. Anderlund, O. Johansson, P. Lindblad, R. Lomoth, T. Polivka, S. Ott, K. Stensj�, S. Styring, V. Sundstr�m and L. Hammarstr�m, Acc. Chem. Res., 2009, 42, 1899–1909 CrossRef CAS PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- C.-Y. Chen, M. Wang, J.-Y. Li, N. Pootrakulchote, L. Alibabaei, C. Ngoc-le, J.-D. Decoppet, J.-H. Tsai, C. Gr�tzel, C.-G. Wu, S. M. Zakeeruddin and M. Gr�tzel, ACS Nano, 2009, 3, 3103–3109 CrossRef CAS PubMed.
- P. Wang, S. M. Zakeeruddin, J. E. Moser, R. Humphry-Baker, P. Comte, V. Aranyos, A. Hagfeldt, M. K. Nazeeruddin and M. Gr�tzel, Adv. Mater., 2004, 16, 1806–1811 CrossRef CAS PubMed.
- D. Kuang, S. Ito, B. Wenger, C. Klein, J.-E. Moser, R. Humphry-Baker, S. M. Zakeeruddin and M. Gr�tzel, J. Am. Chem. Soc., 2006, 128, 4146–4154 CrossRef CAS PubMed.
- N. Robertson, Angew. Chem., Int. Ed., 2006, 45, 2338–2345 CrossRef CAS PubMed.
- M. K. Nazeeruddin, P. Pechy and M. Gr�tzel, Chem. Commun., 1997, 1705–1706 RSC.
- Y. Ooyama and Y. Harima, Eur. J. Org. Chem., 2009, 2903–2934 CrossRef CAS PubMed.
- A. Mishra, M. K. R. Fischer and P. B�uerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed.
- S. Altobello, R. Argazzi, S. Caramori, C. Contado, S. Da Fr�, P. Rubino, C. Chon�, G. Larramona and C. A. Bignozzi, J. Am. Chem. Soc., 2005, 127, 15342–15343 CrossRef CAS PubMed.
- M. W�chtler, S. Kupfer, J. Guthmuller, S. Rau, L. Gonz�lez and B. Dietzek, J. Phys. Chem. C, 2012, 116, 25664–25676 Search PubMed.
- S. Kupfer, J. Guthmuller, M. W�chtler, S. Losse, S. Rau, B. Dietzek, J. Popp and L. Gonz�lez, Phys. Chem. Chem. Phys., 2011, 13, 15580–15588 RSC.
- M. W�chtler, M. Maiuri, D. Brida, J. Popp, S. Rau, G. Cerullo and B. Dietzek, ChemPhysChem, 2013, 14, 2973–2983 CrossRef PubMed.
- S. Kupfer, M. W�chtler, J. Guthmuller, J. Popp, B. Dietzek and L. Gonz�lez, J. Phys. Chem. C, 2012, 116, 19968–19977 CAS.
- J. Schindler, S. Kupfer, M. W�chtler, J. Guthmuller, S. Rau and B. Dietzek, ChemPhysChem, 2015, 16, 1061–1070 CrossRef CAS PubMed.
- M. W�chtler, S. Kupfer, J. Guthmuller, J. Popp, L. Gonz�lez and B. Dietzek, J. Phys. Chem. C, 2011, 115, 24004–24012 Search PubMed.
- L. Zedler, S. Kupfer, I. R. de Moraes, M. W�chtler, R. Beckert, M. Schmitt, J. Popp, S. Rau and B. Dietzek, Chem. � Eur. J., 2014, 20, 3793–3799 CrossRef CAS PubMed.
- G. J. Meyer, Inorg. Chem., 2005, 44, 6852–6864 CrossRef CAS PubMed.
- C. P�rez Le�n, L. Kador, B. Peng and M. Thelakkat, J. Phys. Chem. B, 2006, 110, 8723–8730 CrossRef PubMed.
- K. J. Young, L. A. Martini, R. L. Milot, R. C. Snoeberger III, V. S. Batista, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2012, 256, 2503–2520 CrossRef CAS PubMed.
- K. Sayama, S. Tsukagoshi, T. Mori, K. Hara, Y. Ohga, A. Shinpou, Y. Abe, S. Suga and H. Arakawa, Sol. Energy Mater. Sol. Cells, 2003, 80, 47–71 CrossRef CAS.
- M. W�chtler, J. Guthmuller, L. Gonz�lez and B. Dietzek, Coord. Chem. Rev., 2012, 256, 1479–1508 CrossRef PubMed.
- S. Losse, Redoxaktive metallorganische Farbstoffkomplexe zur Verwendung in Photovoltaik und Photokatalyse, Diss. Friedrich-Schiller-Universit�t Jena, 2010 Search PubMed.
- M. K. Nazeeruddin, S. M. Zakeeruddin, R. Humphry-Baker, M. Jirousek, P. Liska, N. Vlachopoulos, V. Shklover, C.-H. Fischer and M. Gr�tzel, Inorg. Chem., 1999, 38, 6298–6305 CrossRef CAS PubMed.
- E. Badaeva, V. V. Albert, S. Kilina, A. Koposov, M. Sykora and S. Tretiak, Phys. Chem. Chem. Phys., 2010, 12, 8902–8913 RSC.
- M. M. Ba-Abbad, A. A. H. Kadhum, A. B. Mohamad, M. S. Takriff and K. Sopian, Int. J. Electrochem. Sci., 2012, 7, 4871–4888 CAS.
- R. Katoh, A. Furube, M. Kasuya, N. Fuke, N. Koide and L. Han, J. Mater. Chem., 2007, 17, 3190–3196 RSC.
- M. Murai, A. Furube, M. Yanagida, K. Hara and R. Katoh, Chem. Phys. Lett., 2006, 423, 417–421 CrossRef CAS PubMed.
- A. Ehret, L. Stuhl and M. T. Spitler, J. Phys. Chem. B, 2001, 105, 9960–9965 CrossRef CAS.
- M. Choi, K. Jo and H. Yang, Bull. Korean Chem. Soc., 2013, 34, 421–425 CrossRef CAS.
- J. R. Lenhard and B. R. Hein, J. Phys. Chem., 1996, 100, 17287–17296 CrossRef CAS.
- E. J. F. Dickinson, J. G. Limon-Petersen, N. V Rees and R. G. Compton, J. Phys. Chem. C, 2009, 113, 11157–11171 CAS.
- J. Kallioinen, G. Benk�, P. Myllyperki�, L. Khriachtchev, B. Sk�rman, R. Wallenberg, M. Tuomikoski, J. Korppi-Tommola, V. Sundstr�m and A. P. Yartsev, J. Phys. Chem. B, 2004, 108, 6365–6373 CrossRef CAS PubMed.
- C. G. Ryan, E. Clayton, W. L. Griffin, S. H. Sie and D. R. Cousens, Nucl. Instrum. Methods Phys. Res., Sect. B, 1988, 34, 396–402 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- J. P. Merrick, D. Moran and L. Radom, J. Phys. Chem. A, 2007, 111, 11683–11700 CrossRef CAS PubMed.
- B. Mennucci, C. Cappelli, C. A. Guido, R. Cammi and J. Tomasi, J. Phys. Chem. A, 2009, 113, 3009–3020 CrossRef CAS PubMed.
- J. B. Page and D. L. Tonks, J. Chem. Phys., 1981, 75, 5694–5708 CrossRef CAS PubMed.
- W. L. Peticolas and T. Rush, J. Comput. Chem., 1995, 16, 1261–1270 CrossRef CAS PubMed.
- J. Guthmuller and L. Gonz�lez, Phys. Chem. Chem. Phys., 2010, 12, 14812–14821 RSC.
- R Core Team, R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria, 2014 Search PubMed.
- M. Morhac, Peaks: Peaks, R package version 0.2, 2012 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp04484g |
| ‡ Both authors contributed equally to this work. |
| This journal is © the Owner Societies 2015 |