
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phonon anomalies predict superconducting Tc for AlB2-type structures
Jose A.
Alarco
ab,
Peter C.
Talbot
ab and
Ian D. R.
Mackinnon
 *a
*a
aInstitute for Future Environments, Queensland University of Technology (QUT), Brisbane, QLD 4001, Australia. E-mail: ian.mackinnon@qut.edu.au
bScience and Engineering Faculty, School of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology (QUT), Brisbane, QLD 4001, Australia
First published on 28th August 2015
Abstract
We show that the well-known Kohn anomaly predicts Tc for ordered AlB2-type structures. We use ab initio density functional theory to calculate phonon dispersions for Mg1−xAlxB2 compositions and identify a phonon anomaly with magnitude that predicts experimental values of Tc for all x. Key features of these anomalies correlate with the electronic structure of Mg1−xAlxB2. This approach predicts Tc for other known AlB2-type structures as well as new compositions. We predict that Mg0.5Ba0.5B2 will show Tc = 63.6 ± 6.6 K. Other forms of the Mg1−xBaxB2 series will also be superconductors when successfully synthesised. Our calculations predict that the end-member composition, BaB2, is likely to show a Tc significantly higher than currently achieved by other diborides although an applied pressure ∼16 GPa may be required to stabilise the structure.
Introduction
Magnesium diboride, with alternating layers of Mg and B atoms of hexagonal symmetry, is a superconductor below the transition temperature,1Tc ∼ 39 K. Within the MgB2 structure, shown in Fig. 1a and c, the boron layer is critical to the superconducting properties of MgB2. Theory2–4 and experiment5,6 demonstrate close links, or coupling, between phonons and electrons.7 Phonons are collective excitations of atoms or molecules in a solid that oscillate at a single frequency and, by their nature, are temperature dependent. Boron vibration frequencies in MgB2 vary with direction in real space. A dominant collective excitation of these atoms – a phonon mode – is designated the E2g mode and is readily detected by Raman spectroscopy.8,9 The wave vectors and the quantized energies of all atom vibrations are depicted in a phonon dispersion (PD) plot as shown in Fig. 1d. This plot identifies primary reciprocal space directions along the abscissa and, along the ordinate axis, the phonon frequency (or energy). The two in-plane orthogonal E2g modes near the reciprocal space point G are also shown schematically in Fig. 1. These phonon modes are a key to understanding the superconducting properties of MgB2 and metal-substituted forms10–12 and, as we will show, provide a simple unambiguous method to calculate Tc.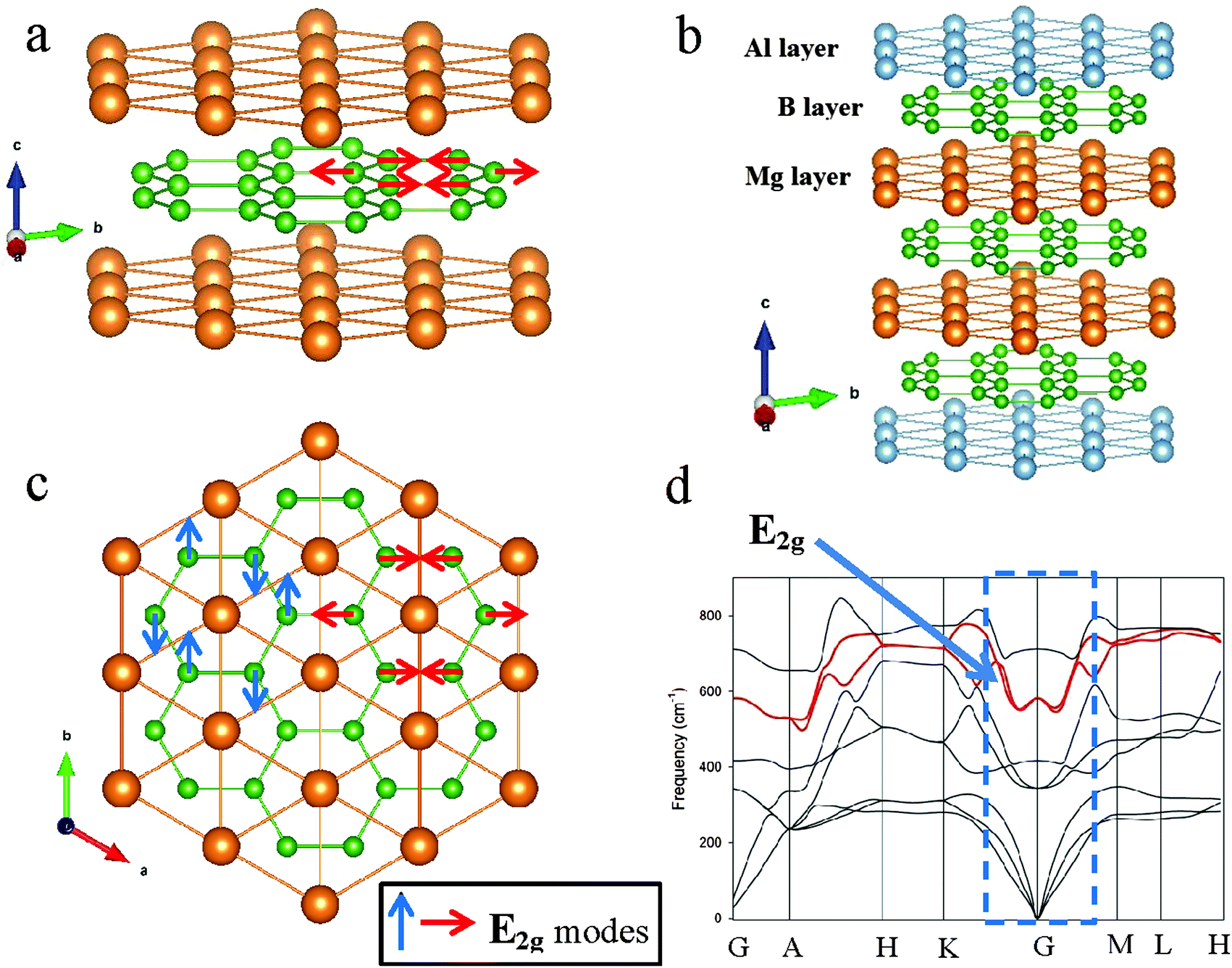 | ||
| Fig. 1 Schematic of the AlB2-type structure and relationship of key atom vibration modes to real and reciprocal space directions. (a) Alternating layers of Mg (gold spheres) and B (green spheres) for MgB2 viewed at an angle to the a-axis direction. (b) Schematic of the Mg2AlB6 superlattice structure showing alternating layers of Mg, B and Al. For both (a) and (b) the c-axis is elongated by ∼30% and the a-axis tilted toward the viewer to highlight the hexagonal arrangements of atoms. (c) Projection of MgB2 down the c* direction showing the two orthogonal E2g phonon modes. (d) PD plot in reciprocal space of phonon modes in the MgB2 structure showing the frequencies (or energies) of vibration with principal direction. The principal direction denoted G is at the origin of the unit cell (i.e. [0, 0, 0]). Phonon branches that contain the E2g phonon modes are highlighted in red. The dotted rectangle along the G direction is the location of the phonon (or Kohn) anomaly and shown in detail in Fig. 2 for MgB2, Mg2AlB6 and Mg1−xBaxB2 compositions. | ||
The density functional theory (DFT), which describes the electronic structure of solids in terms of an electron density distribution rather than a many-electron wave function13,14 underpins our current understanding of many compounds and, in principle, allows prediction of bulk physical properties.14 Analysis and prediction is facilitated by major improvements in computational capacity and use of approximations to address the physics of electronic structure in order to describe electron density for specific crystal structures.14,15 For superconductors, many computational methods have been used2,3,16,17 to estimate physical properties such as Tc. However, predictions of Tc based on DFT band structure calculations alone typically involve estimates of adjustable parameters to ensure theory conforms with experiment.2,7 Floris et al.16 have shown that superconducting DFT (SCDFT)13 can be employed, post facto, to estimate from the electronic band structure a calculated Tc that closely matches experimental data for MgB2. However, this notionally ab initio method employs modifications to standard DFT models13,16,18 including new approximations and additional parameterization to close the gap between theory and experiment.
We outline in this paper a method to determine phonon-mediated physical properties using ab initio DFT calculations and interpretation of PDs for AlB2-type structures. We describe an approach to predict Tc that adds no new functionals to standard DFT models,19 does not invoke free or adjustable parameters4,6,7 and accommodates metal atom substitutions. We use the Mg1−xAlxB2 system to demonstrate that ab initio DFT calculation19,20 predicts Tc across the compositional range 0.0 < x < 1.0 based on the magnitude of a phonon anomaly defined by PDs associated with the E2g mode around the G-point in reciprocal space. This phonon anomaly is described as a Kohn anomaly along G–M based on inelastic X-ray scattering (IXS) experiments5 on MgB2, which show a softening and broadening of the E2g mode close to G.
Computational methods
DFT calculations were undertaken using the CASTEP19,20 module of Materials Studio 7.0 and a more recent version, Materials Studio 8.0. This module provides the functionality to calculate vibrational properties for a wide range of materials. The linear response within the local density approximation (LDA) and generalized gradient approximations (GGA) with a dense k-grid mesh is used as detailed in our earlier work.9,21Calculations are undertaken with an ultra-fine cut-off typically >990 eV. Convergence criteria for most calculations are as follows: energy at 5 × 10−6 eV per atom; maximum force at 0.01 eV Å−1; maximum stress at 0.02 GPa and maximum atom displacement at 5 × 10−4 Å. For BaB2 calculations, the maximum stress value is varied.
Optimal calculation conditions using CASTEP for the AlB2-type structure are given in earlier work.9 Ordered superlattice structures for non-end-member compositions within the Mg1−xAlxB2 and Mg1−xBaxB2 series are based on geometry optimization of unit cell parameters with P6/mmm symmetry. These optimized parameters correlate with experimentally determined values for Al substitutions.22 Experimentally determined values for Ba substitutions in the MgB2 structure are not available. For end-member compositions, optimized unit cell parameters are based on literature values for MgB2 and AlB2 as shown in earlier work.9,21 Schematic models of crystal structures shown in Fig. 1 are built using optimized cell parameters from CASTEP as input to the program VESTA.23 Each schematic shows a unit cell in the c-axis direction only.
Calculations are undertaken with the High Performance Computing facility at QUT using <200 cores that are multiples of the k-grid mesh in a and b reciprocal space directions. Within these conditions, PD calculations for the more complex structures may require 7–10 days to achieve dispersion bands with positive values and convergence of the calculation. This requirement may force a compromise in the choice of k-grid mesh density, the accuracy of results and identification of the anomaly in PD plots.
Electronic band structure calculations for all compositions are consistent with earlier work, in particular, those that invoke a dense k-grid mesh.2,4,16 Electronic structure calculations of substituted compositions also requires consideration of the k-grid mesh density2 and, for MgB2, influences resolution of the tubular sections of Fermi surfaces.
For each composition, calculations on a range of k-grid mesh densities are undertaken to determine the lowest k-value to achieve convergence. For extended superlattice models such as Mg4AlB10 or Mg5AlB12, computations for k = 0.02, 0.025, 0.027 and 0.03, respectively, are evaluated. The lowest k-value to achieve convergence is listed in Table 1 and, for all compositions, k ≤ 0.03 Å−1 although the computational cost increases by a cube power as grid size is reduced.
| Al | Grid | Optimised unit cell parameters | DFT type | Fermi energy | DFT modelled values | Average valuesa | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E2g at G | δ | T δ | δ av | T δ (av.) | σ T(error) | |||||||
| x | k | a (Å) | c (Å) | (eV) | (cm−1) | (cm−1) | (meV) | (K) | (meV) | (K) | (K) | |
| a Average for both DFT models: LDA and GGA. | ||||||||||||
| 0.00 | 0.020 | 3.0391 | 3.4866 | LDA | 8.3976 | 582.3 | 127.0 | 15.7 | 40.6 | 16.3 | 42.0 | 3.3 |
| 0.00 | 0.020 | 3.0796 | 3.5538 | GGA | 8.1076 | 554.9 | 135.8 | 16.8 | 43.4 | |||
| 0.125 | 0.029 | 3.0297 | 27.6213 | LDA | 8.4968 | 579.4, 619.5, 652.4, 817.0 | 120.4 | 14.9 | 38.5 | 15.3 | 39.4 | 3.2 |
| 0.125 | 0.030 | 3.0682 | 28.1065 | GGA | 8.2262 | 543.0, 569.8, 594.6, 786.3 | 125.9 | 15.6 | 40.3 | |||
| 0.167 | 0.025 | 3.0287 | 20.6121 | LDA | 8.5908 | 563.1, 654.5, 853.7 | 104.1 | 12.9 | 33.3 | 14.3 | 35.5 | 3.9 |
| 0.167 | 0.024 | 3.0639 | 21.0239 | GGA | 8.3043 | 541.4, 581.9, 821.3 | 116.4 | 14.4 | 37.2 | |||
| 0.20 | 0.027 | 3.0253 | 17.1504 | LDA | 8.5786 | 549.2, 710.0, 810.7 | 91.7 | 11.4 | 29.3 | 12.2 | 31.3 | 3.4 |
| 0.20 | 0.027 | 3.0623 | 17.4416 | GGA | 8.3135 | 514.4, 646.0, 782.8 | 104.3 | 12.9 | 33.3 | |||
| 0.25 | 0.025 | 3.0233 | 13.6459 | LDA | 8.6535 | 607.7, 850.8 | 78.2 | 9.7 | 25.0 | 10.4 | 26.7 | 2.1 |
| 0.25 | 0.025 | 3.0593 | 13.8730 | GGA | 8.3911 | 569.5, 822.5 | 89.1 | 11.0 | 28.5 | |||
| 0.333 | 0.020 | 3.0169 | 10.1709 | LDA | 8.7013 | 574.0, 850.8 | 57.9 | 7.2 | 18.5 | 6.3 | 16.2 | 3.3 |
| 0.333 | 0.026 | 3.0514 | 10.3285 | GGA | 8.4612 | 570.1, 828.9 | 43.4 | 5.4 | 13.9 | |||
| 0.50 | 0.020 | 3.0241 | 6.5758 | LDA | 8.8714 | 893.6 | 12.1 | 1.5 | 3.9 | 1.8 | 4.6 | 2.5 |
| 0.50 | 0.030 | 3.0381 | 6.7710 | GGA | 8.6299 | 851.3 | 16.8 | 2.1 | 5.4 | |||
| 0.667 | 0.030 | 2.9978 | 9.9116 | LDA | 9.0831 | 935.4, 947.5 | 2.2 | 0.3 | 0.7 | 0.2 | 0.5 | 0.3 |
| 0.667 | 0.030 | 3.0273 | 10.0250 | GGA | 8.8601 | 914.3, 925.7 | 0.7 | 0.1 | 0.2 | |||
| 1.00 | 0.020 | 2.9795 | 3.2539 | LDA | 9.3125 | 963.9 | na | na | 0.0 | 0.0 | 0.0 | 0.0 |
| 1.00 | 0.020 | 3.0039 | 3.2806 | GGA | 9.0828 | 947.6 | na | na | 0.0 | |||
The values for the k-grid mesh density used in this work are comparable to, or higher than, that used for many PD calculations due to earlier studies4,8,9,24 that identified key changes in PD characteristics with this parameter. These changes include shifts in E2g frequency values at specific reciprocal lattice points8,9,24 and the appearance of vibration mode branches in PD plots.9
Computational DFT methods are limited by fundamental assumptions on delocalisation and static correlation embedded in functional approximations25 as well as the degree of complexity of material composition and structure,17 particularly structures containing transition metals.15 In this work, practical limits include the extent of superlattice models, extension of DFT calculations to include transition metal diborides with substituted compositions and, as noted above, the trade-off between k-grid mesh density, PD detail and convergence. Indications of computational limit for a particular composition in the Mg1−xAlxB2 series are shown by (a) failure to converge, (b) inconsistent or irregular format of the anomaly in PD plots (e.g. multiple lows and highs of an E2g mode within one branch) and (c) negative phonon frequency values.
Stoichiometry and superlattices
We use superlattices as a computational strategy to model compositions for which x is not an integer. CASTEP19 allows fractional occupancy of specific atoms in a structure for electronic band structure calculations. This structural description is an alternative method to represent intermediate compositions in the Mg1−xAlxB2 series, but is not possible for PD calculations using CASTEP. Random fractional occupancy of atoms in a solid solution is an alternative approach for CASTEP calculations of band structure. However, this approach induces significant changes in electronic band structures that do not match experimental data. As noted by Kortus,2 the approach is suited to low levels of metal doping (e.g. x < 0.1) but is expected to fail at higher doping concentrations.Use of fractional occupancies may invoke inconsistent or uninterpretable shifts in the calculated electronic band structure, particularly in the proximity of the Fermi level. Hence, our PD calculations on ordered compositions do not utilise fractional site occupancies but invoke a superlattice along the c-axis. For superlattice unit cell calculations, appropriate multiples of the end member parameters are used and weighted combinations of these parameters are used as input for mixed compositions containing both Al and Mg or Ba and Mg. A schematic of the ordered composition for x = 0.33, in which an Al layer is sandwiched between two Mg layers, resulting in a 3x superlattice along the c axis, is shown in Fig. 1b. For x = 0.125, the cell size for a DFT calculation can increase to a ∼ 3.07 Å and c ∼ 28.0 Å. This size cell is the maximum we are able to optimise to then calculate a PD with convergence within a reasonable computation time when limited to <200 cores.
For each superlattice, the degrees of freedom per atom within the sub-lattice unit cell is nine although the number of phonon branches in calculated PDs will depend on the multiples of the sub-lattice used to form the P6/mmm unit cell for a specific composition. For structures used in these calculations, Z = 2, 3, 4 or 5 for the superlattice constructs while for the basic P6/mmm structure, such as MgB2, Z = 1.26 The Fermi energy corresponds to the average electron density of a structure and measures the highest energy (in the ground state) of valence electrons in the conduction band as free or nearly free electrons. Therefore, to compare phonon anomalies of end member and intermediate compositions, each superlattice is normalized to a single unit cell.
Error estimates
We have described intrinsic sources of error for PD calculations in earlier work.21 Estimates of error in the calculation of Tδ are obtained by measurement of δ for both branches of the E2g mode in the G–M and G–K reciprocal directions for each calculation (i.e. for LDA and GGA). Our analysis and estimate of δ is dependent on measurement of calculated vibrational frequencies that show a mean relative error27 of about ±5% for crystal structures of similar size and complexity to MgB2. These values of δ are converted to Tδ using eqn (1) below. The error estimate is one standard deviation for the values of Tδ determined from all the measured values of δ. For some compositions, the value of Tδ is an average of up to eight separate estimates of δ obtained by measurement of the frequency difference on two branches of E2g either side of G for both LDA and GGA models. The individual values for frequency measurements are not shown in Table 1.Results
Phonon anomaly for Mg1−xAlxB2
An example of the phonon anomaly for MgB2 calculated using the LDA model with P6/mmm symmetry is given in Fig. 1d and, in closer detail, in Fig. 2a. Fig. 2a shows a portion of the total PD across the G–M (right-hand side of the diagram) and G–K (left-hand side of the diagram) reciprocal lattice directions. The phonon bands or branches that contain the degenerate E2g modes at the G point and define the double parabolic phonon anomaly are shown in red. The upper limit of the anomaly is defined in this case by the B2g mode at higher energy. For substituted compositions, the E2u or the B2g mode defines the upper extent of the phonon anomaly for E2g bands in the G–M and G–K directions, as shown in Fig. 2b. For MgB2, we show that these upper modes are important for energy conservation through conversion of phonon energies by coherent relaxation.9,21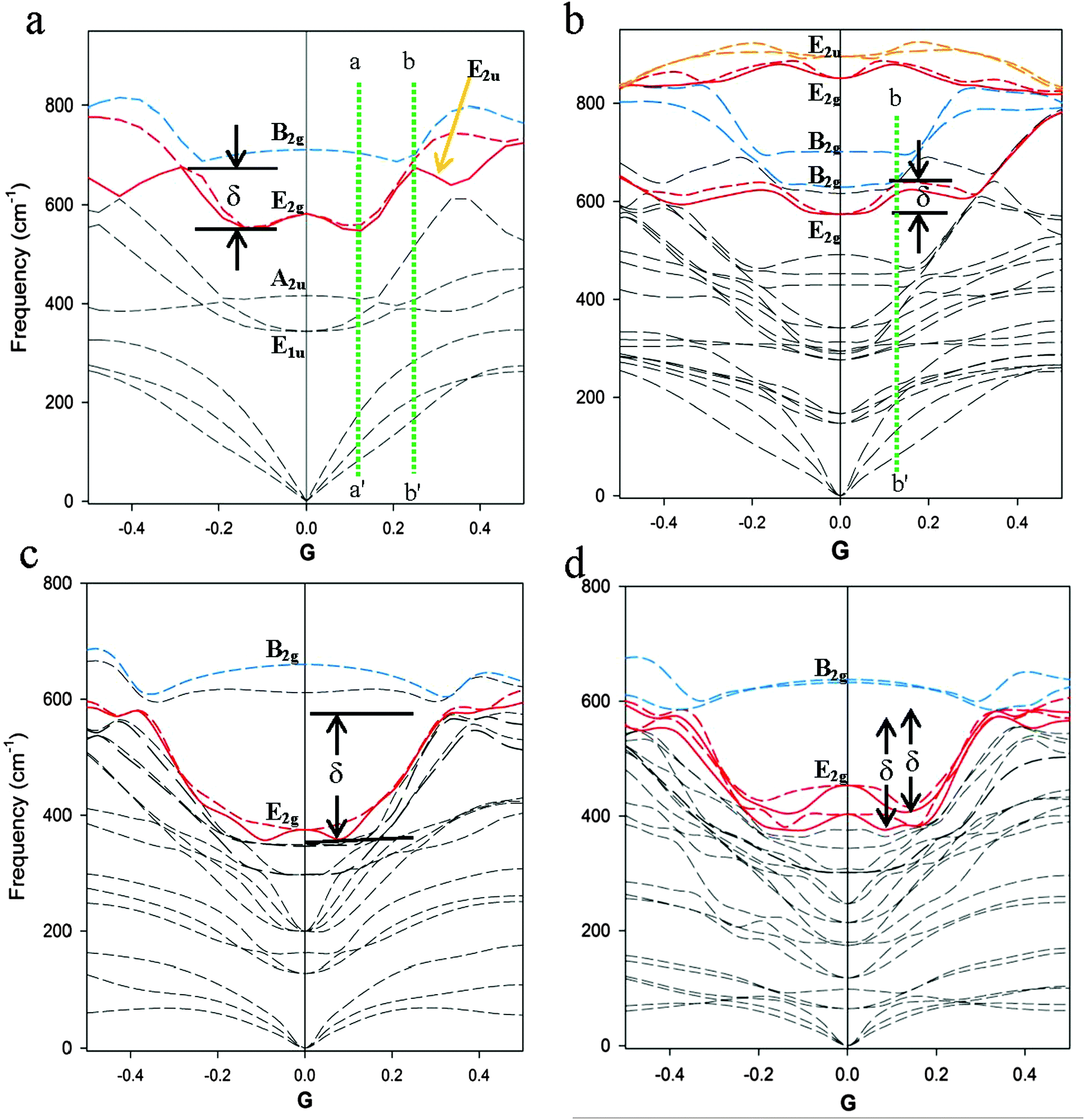 | ||
| Fig. 2 Partial PD plots based on DFT models with k = 0.02 Å−1 along the G–M and G–K directions of reciprocal space: (a) for MgB2 and (b) for Mg2AlB6 using the LDA functional; for the predicted compositions (c) MgBaB4 using the GGA functional and (d) for MgBa2B6 using the LDA functional. The phonon anomalies for the E2g mode around the G-point are highlighted in red and the magnitude of the anomaly, δ, is shown. The dotted red lines show regions with non-degenerate E2g modes. The green dotted line b–b′ corresponds to reciprocal lattice dimensions equivalent to the average diameter of the Fermi surface for the specific composition. The distance between a–a′ and b–b′ along the G–M direction is an indicator of coherence length in the a–b plane of an AlB2-type structure. | ||
In the vicinity of the G-point, the E2g PD bands are degenerate and extend along the basal plane directions (i.e., G–K and G–M) with a characteristic inflection along these directions that is limited, or defined, by the B2g mode. Note that outside the anomaly, along the G–K and G–M directions, the E2g mode changes symmetry to E2u for MgB2. The magnitude of the anomaly, δ, is measured in frequency units (cm−1), as shown in Fig. 2. This anomaly is evident in other publications9,21,28 when there is sufficient resolution of the k-grid and is referred to as a Kohn anomaly in earlier work.5,29
For high-Al compositions such as MgAl2B6, the frequency difference between E2g and E2u bands at the G-point is minimal or zero, and no anomaly occurs. In addition, for these high-Al compositions, the E2g and E2u bands do not show the inflection along the G–M and G–K directions as we show for MgB2.9 For AlB2, the E2g mode is the highest frequency optical phonon and does not show a phonon anomaly.21 Similar PD plots for end-member compositions such as MgB2 and AlB2 are described in earlier work9,21 and have been calculated for the compositions shown in Fig. 3 below (open symbols).
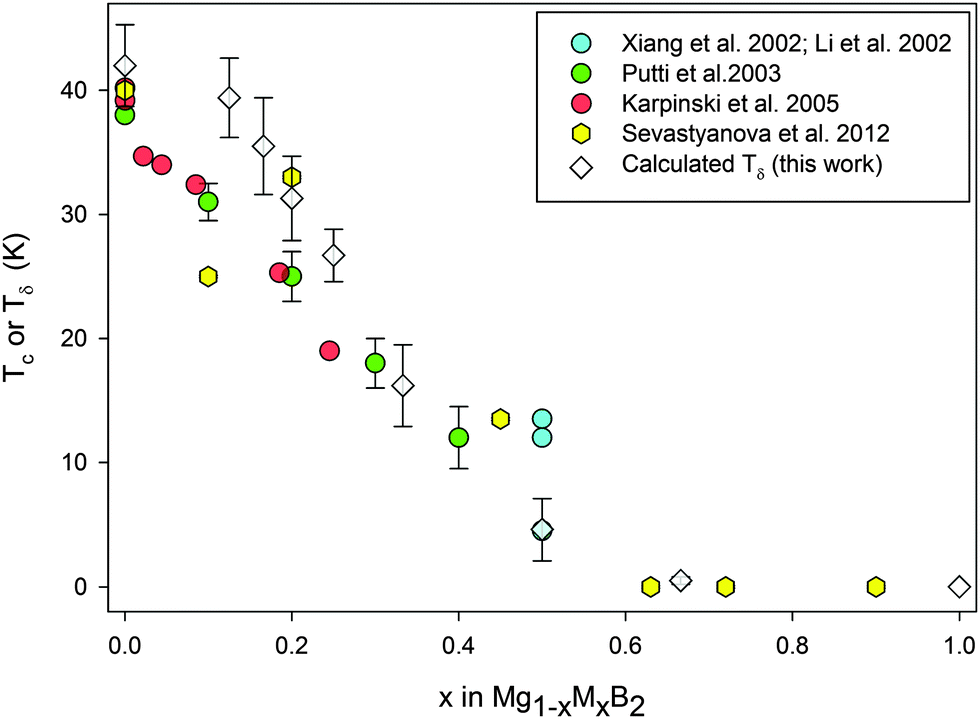 | ||
| Fig. 3 Plot of experimental Tc values for Mg1−xAlxB2 (filled symbols) and calculated Tδ values (open symbols) determined by the method outlined in the text. Error bars (this work) represent one standard deviation and are as reported by Putti et al.31 | ||
Compositional trends
A summary of the modelled parameters for end-member and intermediate compositions of the Mg1−xAlxB2 suite is shown in Table 1. The E2g mode for non-end-member compositions in the Mg–Al series increases in energy with increase in Al content, as noted previously4 and, in each case, a higher phonon energy (e.g. B2g or E2u) occurs until x = 1 where E2g is the highest frequency mode.21 These trends are evident with either the LDA or GGA model in the DFT calculation and are consistent with data calculated at sufficient k-grid mesh density.9 The calculated values for E2g modes at G shown in Table 1 correspond with experimental values determined by spectroscopic techniques.9,30 Calculated partial densities of phonon states (data not shown) show that Mg and Al contribute almost exclusively to modes below 350 cm−1. As noted in earlier work,4 B atoms contribute to modes above 350 cm−1 with minor contributions in a small overlap region at 100–350 cm−1.The E2g band is doubly degenerate around the G-point along the basal plane directions for non-end-member compositions in the Mg1−xAlxB2 series. Fig. 2b shows a portion of the PD around the G-point for x = 0.33 calculated using the LDA with a k-grid mesh value of 0.02 Å−1. The two E2g modes, which are degenerate at G, are consistent with ordered alternation of boron layers with a metal layer (e.g. either Mg or Al) in the AlB2-type structure. In Fig. 2b, the E2g modes at ∼570 cm−1 and at ∼851 cm−1 are highlighted in red, and the higher energy E2u mode is indicated in orange. For this composition, measurement of the anomaly δ for the lower energy E2g mode shows that it is significantly less (see Table 1) than the anomaly in Fig. 2a. This lower value of δ is consistent with a dampening of the dominant mode that influences superconductivity in Mg1−xAlxB2.
Phonon thermal energy and Tc
We have calculated a thermal energy, Tδ, for each composition in Table 1 based on the equation:| δ = (nN/Z)·(kBTδ/2) | (1) |
Fig. 3 shows the calculated temperature (open symbols), Tδ, associated with the phonon anomaly compared with the experimentally determined Tc (solid symbols) for Al content in Mg1−xAlxB2. Experimental data for Fig. 3 have been collated from studies that used structure refinements22 and/or microstructural and compositional analyses26,31 to define stoichiometry and to account for the presence of second phases (e.g. MgB4).26,32 These experimental Tc values are for compositions determined on the as-synthesized product(s) made by an internally consistent method. The trend towards a lower Tδ with increased x value at intermediate compositions for 0.125 < x < 0.5 is consistent with experimental data22,26,31 on well-characterized Mg1−xAlxB2. For reference, experimental Tc values for x = 0.5 range from ∼4 K33 to ∼13.5 K.10,26,34
We have used this method to estimate Tc on other AlB2-type structures. In these cases, the experimentally determined values for Tc are much lower (∼10 K) and thus, features ascribed to a phonon anomaly will be more difficult to detect. The base case for low Tc compounds is provided for the high-Al compositions listed in Table 1 (e.g. Mg0.5Al0.5B2). However, compositions that do not contain boron atoms may also form in the AlB2-type structure and, in some cases, show superconducting properties.35–37 We use the approach outlined above to estimate the Tc of the disilicide compounds BaSi2 and Ca(Al0.5Si0.5)2. For these disilicides, our estimate for BaSi2 of Tc = 9.3 ± 0.5 K compares with an experimental determination36 of 8.9 K. An estimated Tc = 7.5 ± 0.5 K for Ca(Al0.5Si0.5)2 is similar to the experimental value35 of 7.8 K.
Phonon anomaly for Mg1−xBaxB2
We show in Fig. 2c and d partial PD plots for MgBaB4 and MgBa2B6. For these DFT calculations, both compositions are constructed as AlB2-type structures with P6/mmm symmetry. The extent of the phonon anomaly, δ, is shown for MgBaB4 in Fig. 2c and is similar in form to MgB2 shown in Fig. 2a. Determinations of δ, as shown in Table 2 show that the magnitude of the anomaly for MgBaB4 is significantly higher than for MgB2. Table 2 summarises the calculated parameters for compositions of the Mg1−xBaxB2 series similar to that shown in Table 1. We have calculated fewer compositions for this series compared with the Mg1−xAlxB2 series because similar conclusions are evident from these calculations.| Ba | Grid | Optimised unit cell parameters | DFT type | Fermi energy | DFT modelled values | Average valuesb | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E2g at G | δ | T δ | δ av | T δ (av) | σ T(error) | |||||||
| x | k | a (Å) | c (Å) | (eV) | (cm−1) | (cm−1) | (meV) | (K) | (meV) | (K) | (K) | |
| a DFT calculation undertaken at equivalent to 16 GPa hydrostatic pressure. b Average for DFT models: both LDA and GGA models. | ||||||||||||
| 0.333 | 0.020 | 3.0896 | 11.8631 | LDA | 8.0111 | 420.9 | 207.4 | 25.7 | 66.3 | 25.0 | 64.4 | 2.2 |
| 0.333 | 0.020 | 3.1390 | 12.1094 | GGA | 7.6371 | 431.7 | 195.2 | 24.2 | 62.4 | |||
| 0.500 | 0.020 | 3.1186 | 8.3362 | LDA | 7.8668 | 429.1 | 181.6 | 22.5 | 58.1 | 24.7 | 63.6 | 6.6 |
| 0.500 | 0.020 | 3.1727 | 8.5178 | GGA | 7.4234 | 374.6 | 216.3 | 26.8 | 69.2 | |||
| 0.666 | 0.020 | 3.1474 | 13.1914 | LDA | 7.6721 | 403.2, 453.6 | 179.3 | 22.2 | 57.3 | 24.1 | 62.1 | 5.8 |
| 0.666 | 0.020 | 3.2072 | 13.4716 | GGA | 7.1948 | 334.4, 364.8 | 208.9 | 25.9 | 66.8 | |||
| 1.00a | 0.015 | 3.0840 | 4.6612 | LDA | 8.4699 | 376.4 | 175.7 | 21.8 | 56.2 | 23.1 | 59.7 | 6.2 |
| 1.00a | 0.015 | 3.1424 | 4.7314 | GGA | 7.9646 | 415.8 | 197.5 | 24.5 | 63.1 | |||
Comparison of Tables 1 and 2 shows that the Fermi energies for Mg1−xBaxB2 are lower than the Mg1−xAlxB2 series by approximately 0.7 eV to 1.0 eV across the compositional range. In addition, the optical phonons for Mg1−xBaxB2 compositions are typically at lower frequencies than Mg1−xAlxB2 with the critical E2g modes at lower frequencies at the G point for equivalent values of x.
Discussion
The Mg–Al diboride system is characterized by a sharp superconducting transition at x = 0 that decreases in value and gradually broadens with higher Al content.12 Superconductivity in Mg1−xAlxB2 is moderated, but not extinguished, by other influences such as order–disorder,38 superlattice(s)10–12 and the presence of other phases32,33 for 0.0 < x < 0.5. Band structure calculations,2,39,40 experimental data41 and phonon calculations8 have established that the vibrational properties of the E2g mode for MgB2 influence electron–phonon interactions and superconductivity.4,7Ordering and superlattice models
Superlattices are observed in Mg1−xAlxB2 systems10,11,26,34 with prominent diffraction spots at the reciprocal lattice dimension c*/2, which demonstrate a 2x c-axis superlattice34 for Mg0.5Al0.5B2. Superlattice structures are also predicted42 for vacancy-ordered MgB2 but are not considered in this analysis. Detailed analysis of Al-substituted MgB2 shows that superstructures occur for a range of compositions (x = 0.17;34x = 0.25;10x = 0.45, 0.5 and 0.5511) and microstructures.10,11 Microstructural studies10 suggest that intimate mixtures of MgB2 and Mg0.5Al0.5B2 may also occur for 0.1 < x < 0.5. However, the dominant motif involves alternation of Mg and Al layers for x = 0.5.12 Long range ordering of Al and Mg is observed in the a–b plane12 with an extent ∼10 nm. Representation of this periodicity in the a–b plane is non-trivial for DFT calculations and is not considered here. Brutti and Gigli38 used DFT calculations to show that for x > 0.31, the formation of an Al-rich phase leads to formation of thermodynamically stable Mg0.5Al0.5B2 at 50% Al concentration in MgB2.Raman spectroscopy and inelastic neutron scattering studies43 on Mg1−xAlxB2 samples show evidence for alternate stacking of Mg and Al layers for x ∼ 0.5. In our earlier work, we note that additional Raman and IR peaks not predicted by P6/mmm symmetry for MgB2 are due to super-lattice modes that approximate a dynamic, phonon-distorted lower-symmetry crystal.9 We show that a 2x super-lattice in the c-direction allows a simple correlation of the pair breaking energy and the superconducting gap.9
We have evaluated other c-axis ordered structures in which multiple adjacent Al-layers alternate with Mg-layers for key compositions (e.g. Mg–Mg–Al–Al for x = 0.5 or Mg–Mg–Mg–Mg–Al–Al for x = 0.33). In these cases, while there are differences of ∼0.15 eV in calculated enthalpies favouring the stability of single Al-layer stacking, ordered motifs with adjacent Al-layers also show a phonon anomaly similar to that in Fig. 2a and b. Thus, a superlattice repeat along the c-axis utilising a simple alternation of Mg and Al layers to minimise adjacent Al layers is an optimal configuration. For simplicity of computation, we follow a superlattice formalism confirmed by detailed experimental studies11,43 and implied by DFT models38
Phonon anomalies
The magnitude of the phonon anomaly varies with composition, as do experimentally determined Tc values. The average value of δ for MgB2 shown in Table 1 is comparable to the gap energy,5 2δ, of ∼15 meV. The variation in magnitude of the phonon anomaly reflects the predominant role of boron layer vibrations in many AlB2-type structures44,45 and, in this case, the influence of Al substitution on these vibrations. For the AlB2-type structure, the D6h point group symmetry results in two E2g modes at the G-point and equivalent displacement modes, E2u, at the A point of the hexagonal Brillouin zone.8The doubly degenerate E2g(G) band describes two distinct displacement patterns that are equivalent within the harmonic approximation.8 The E2g and E2u modes have the same movement pattern, albeit with different parity (gerade or ungerade), via a difference in relative phase.8,46 One of the E2g modes reflects in-plane B–B bond stretching modes that are strongly coupled to the sigma bonded Fermi surfaces related to the px and py in-plane orbitals.46
Table 1 summarizes the difference in frequency, or the phonon anomaly, δ, between the low and high points of the E2g mode inflection for both G–M and G–K directions for each calculated composition using both LDA and GGA in the Mg1−xAlxB2 series. For both DFT calculation methods, the range of values for the phonon anomaly, δ, of each composition is similar, but not equal, because of different assumptions in the methods to calculate the charge distribution in the LDA and GGA methods,4 differences in optimized lattice parameters and the k-grid value required to attain convergence.
For each calculated composition in the Mg–Al series, the value of Tδ is slightly higher than the experimentally determined values for Tc as shown in Fig. 3. A difference of 1.0 K to 2.5 K is evident for models with x < 0.25 and probably relates to factors such as (a) a higher k-grid value (particularly for x = 0.125) that allows convergence of the PD calculation, (b) systematic errors associated with extended superlattice construction and (c) our DFT calculations are for absolute zero, ground state properties without correction for higher temperatures. Nevertheless, this ab initio determination of Tδ is internally consistent and in close agreement with the experimentally determined Tc trends22,26,31 for Al-substituted MgB2.
The phonon anomaly can be described in terms of interconnected hyperboloid surfaces in different proportions showing origins at different energies and inverse directions along a fixed axis that intercepts G. We can describe this hyperboloid for the E2g mode(s) as follows. For the E2g dispersion shown in Fig. 1 and 2, the phonon band is in the kx − ky plane for which kz = 0. For one hyperboloid, the equation for kz = 0 becomes
| ω2 = ω02 + a2(kx2 + ky2) | (2) |
| ω2 = ω02 + a2kx2 or (ω/ω0)2 − (akx/ω0)2 = 1 | (3) |
| ω = ω0(1 + a2(kx/ω0)2)1/2 ∼ ω0(1 + (a2/2)(kx/ω0)2) |
| ω0 + (a2/2ω0)kx2. | (4) |
2ω![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∂ω/∂kx = 2a2kx. ∂ω/∂kx = 2a2kx. | (5) |
| νg = a2/νp. | (6) |
In the asymptotic region of the E2g band, where the phonon dispersion changes abruptly from the lowest point of the anomaly towards the higher E2u phonon band, the curve displays an approximate linear behaviour (in Fig. 2, the distance along the G–M direction between the green dotted lines a–a′ and b–b′). This behaviour indicates that the group velocity, νg, is constant and, as shown above, the phase velocity, νp, is also constant. Therefore, there is an interval of k-vectors, Δk, relating to phonon waves for which the wave is non-dispersive. That is, the group of waves moves at constant group velocity and each component of the interval also moves at constant phase velocity. In this instance, the wave packet retains shape and can be viewed as a coherent wave. The slope of the E2g band in the linear section of the anomaly appears approximately parallel to the acoustic band of highest energy. This relationship suggests that the group velocity of the corresponding optical waves matches the sound velocity.
Phonons and Fermi surfaces
Comparison of electronic bands with PDs shows that the phonon anomalies in the Mg1−xAlxB2 system originate from cusps of paraboloid bands across the Fermi level in the electronic band structure at the G-point. These anomalies are effected by a transfer of electronic charge from the vicinity of the cusps to and from adjacent unit cells and the flat bands in the G–A direction.39,48 Our calculations show that cusp size is directly proportional to the PD anomaly. For example, the PD anomaly is deep when the cusp size in the electronic band is large. If the cusp dips below the Fermi level, for example as with AlB2, the phonon anomaly does not occur; this is consistent with experimental data49 that show no superconductivity for this composition. In addition, for superlattice constructs, multiple parallel cusps that intersect with the Fermi level occur. These intersections are reflected in a multiplicity of E2g modes and of tubular sections in Fermi surface models.Calculations of Fermi energies link electrons on or near the Fermi surface to strongly coupled phonons in MgB2.39 For example, Fig. 4 shows the Fermi surface for MgB2 calculated with the GGA model for k = 0.02 Å−1. For this model, the Fermi energy is 8.1087 eV. In a free electron approximation, the Fermi wave vector, kF, is determined from the equation
| EF = ℏ2kF2/2m | (7) |
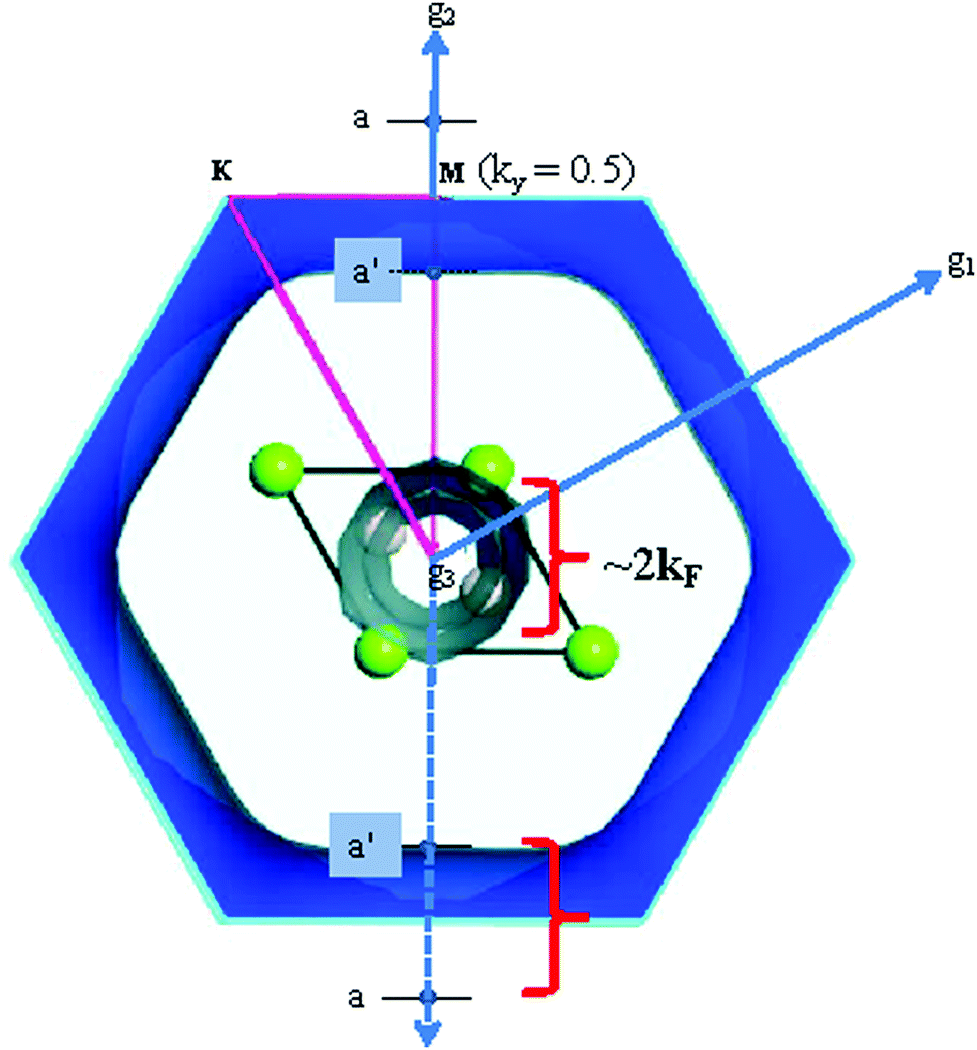 | ||
| Fig. 4 Fermi surface projection along g3 calculated for MgB2 using the GGA model with k = 0.02 Å−1. The projection shows the equivalent magnitude of the Fermi vector, 2kF, along the ky direction and tubular sections on the Fermi surface. Green spheres are Mg atoms; off-white spheres (partially obscured) are B atoms. | ||
For two electrons to interact through a phonon, conservation of energy and momentum give the equation:
| ℏ(ke1 − ℏke2) = ℏ(Kph + G) | (8) |
For Al-substituted compositions, this point shifts closer to the G-point in a PD plot, as shown in Fig. 2b (green dotted line denoted b–b′; ∼0.13 along G–M; equivalent to 0.31 Å−1). This shift implies a reduction in size of the sigma sheets coupled to the E2g phonon and is consistent with de Haas van Alfen effect measurements50 of Al-substituted MgB2. Our calculations for other compositions (data not shown) also show a size reduction of the cylindrical sigma sheets in Fermi surface projections with increased Al substitution. Al substitution in MgB2 results in a commensurate change in the number of tubular sections in Fermi surface projections in proportion to the multiplicity of E2g modes using superlattice models.
Predicted superconducting compositions
Extrapolation of the methods described above for Mg1−xAlxB2 to other compositions12,51 of MgB2 and a consideration of diboride thermodynamics52 suggest particular atom substitutions to the type structure may also result in superconducting behaviour. For example, our LDA calculations on BaB2 reveal an electronic band structure similar to MgB2 but with a PD that shows unusual frequency variation and negative frequency values for modes in the A–H direction and around G. Subsequent calculations constrained to include hydrostatic pressure reduced the number and range of negative frequency values. At an applied hydrostatic pressure of 16 GPa convergence of the LDA and GGA models is achieved with phonon anomalies similar to that shown for MgB2 in Fig. 2. The E2g modes show a strong anisotropy in the PD plot particularly in the G–K direction. Nevertheless, these calculations indicate that a BaB2 structure with an applied stress at 16 GPa is likely to show a phonon anomaly, and by inference, superconductivity.BaB2 is not a well-known compound and may be structurally unstable due to a larger Ba+2 ionic radius compared with Mg+2. Cava et al.12 notes that a variation of approximately 50% of the metal atom size can be accommodated by the diboride structure. However, the existence of BaB2 is not readily confirmed. Early literature53 on the synthesis of BaB2 reports cell dimensions that are inconsistent with an AlB2-type structure and suggests that this compound is yet to be synthesised. In addition, the geometry optimised cell dimensions for BaB2 shown in Table 2 are comparable to MgB2 for the a axis. The c-axis dimension is ∼33% greater than the calculated values for MgB2. These attributes, and the unstable nature of DFT models without a hydrostatic pressure constraint, suggest that BaB2 may not be thermodynamically stable except at high pressure.
Our computational method utilises a linear response for both LDA and GGA models and is effective for structures with low anharmonicity. Outcomes from these linear calculations as well as structural considerations (e.g. Ba+2 ionic radius), infer that anharmonicity is an important factor for BaB2 PD calculations. An alternative calculation for BaB2 using the Finite Displacement (FD) method at similar hydrostatic pressure shows a PD anomaly very similar to Fig. 2a. In this case, the extent of anomaly is significant (∼250 cm−1) and, by similar analysis to that for DFPT calculations, suggests a Tc ∼ 79.1 ± 10.2 K.
The result from DFT calculations on BaB2 under an applied stress indicates that substitution of an appropriate valence atom for Ba may also induce a similar shift in structural parameters or an improvement in the calculated PD. DFT calculations for compositions of Mg1−xBaxB2, where 0 < x < 1, display phonon anomalies of varying magnitude as shown in Fig. 2c and d. Table 2 lists phonon anomalies for three compositions (x = 0.333; x = 0.5 and x = 0.666) and for the end-member BaB2 at a hydrostatic pressure of 16 GPa. In all cases, calculated PDs for Mg1−xBaxB2 show a phonon anomaly with an extent significantly greater than calculated for MgB2. This analysis predicts that Mg1−xBaxB2 will show superconductivity at Tc > 60 K over a wide compositional range. We infer from the smaller average c-axis cell dimensions shown in Table 2 that Mg0.66Ba0.33B2 is more likely to be a stable phase in this compositional series. This Tc prediction for Mg1−xBaxB2 is ∼20 K higher than that for MgB2, currently the stand-out material in the diboride suite.54
Coherence lengths
Using the width Δk in reciprocal space, we can derive a width Δx in real space that may be associated with the coherence length of the superconductor. Using the calculated PD for MgB2 shown in Fig. 2a, we estimate the coherence length in the a–b plane as the distance along the G–M direction (i.e. the reciprocal a axis) between the approximately linear sections of the phonon anomaly manifest in the E2g modes. This distance, measured in reciprocal lattice dimensions, can be converted to real space dimensions for the symmetry conditions of the unit cell. For MgB2, our estimate of the coherence length is ∼50 Å ± 5 Å. This value compares favourably with experimentally determined values54 between 61 Å and 65 Å for coherence length in the a–b plane for single crystals; noting that experimental determination of coherence length is dependent on the applied magnetic field55 and temperature.56 Similar estimates of coherence length for substituted MgB2 can be inferred from these PD calculations. For Al-substituted MgB2, these estimates of coherence length range from ∼70 Å to ∼80 Å with increasing Al content. For Mg1−xBaxB2, estimates of coherence length range from ∼30 Å to ∼50 Å in the a–b plane.Conclusions
We have utilised ab initio DFT calculations to show that PD plots of AlB2-type structures including Mg1−xMxB2 (where M = Al or Ba) and MSi2 (where M = Ca, Al or Ba) are key indicators of physical properties. For known compositions, the phonon anomaly predicts physical properties such as the presence (or absence) of superconductivity in this structure type. The extent, or size (in frequency units), of the anomaly provides an estimate of Tc by the well-known relationship between thermal energy and degrees of freedom for a particular structure. Agreement between theory and experiment is strong even though DFT models ground state properties at absolute zero temperature. In addition, this approach does not use modified functionals or post facto corrections and is unable, at this time, to account for time dependent phenomena. The phonon anomaly for the AlB2-type structure is also known as a Kohn anomaly. If present, the Kohn anomaly provides a means to predict Tc of unknown materials and to estimate other key parameters such as coherence length and Fermi surface structure that link electronic and magnetic properties of these materials. This work predicts that Mg–Ba compounds with the AlB2-type structure and BaB2 will show superconducting properties. The approach used in this work amplifies the value of DFT computations as a predictive tool.Acknowledgements
We acknowledge financial assistance from QUT and assistance of Ashley Wright, Adam Siliato and Mark Barry with the High Performance Computing facilities.References
- J. Nagamatsu, N. Nakagawa, T. Muranaka, Y. Zenitani and J. Akimitsu, Nature, 2001, 410, 63–64 CrossRef CAS PubMed.
- J. Kortus, Phys. C, 2007, 456, 54–62 CrossRef CAS PubMed.
- I. I. Mazin and V. P. Antropov, Phys. C, 2003, 385, 49–65 CrossRef CAS.
- R. Heid, K.-P. Bohnen and B. Renker, Adv. Solid State Phys., 2002, 42, 293–305 CAS.
- A. Q. R. Baron, H. Uchiyama, Y. Tanaka, S. Tsutsui, D. Ishikawa, S. Lee, R. Heid, K.-P. Bohnen, S. Tajima and T. Ishikawa, Phys. Rev. Lett., 2004, 92, 197004 CrossRef CAS.
- H. Kotegawa, K. Ishida, Y. Kitaoka, T. Muranaka, N. Nakagawa, H. Takagiwa and J. Akimitsu, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 064516 CrossRef.
- A. Y. Liu, I. I. Mazin and J. Kortus, Phys. Rev. Lett., 2001, 87, 087005 CrossRef CAS.
- K. Kunc, I. Loa, K. Syassen, R. K. Kremer and K. Ahn, J. Phys.: Condens. Matter, 2001, 13, 9945–9962 CrossRef CAS.
- J. A. Alarco, A. Chou, P. C. Talbot and I. D. R. Mackinnon, Phys. Chem. Chem. Phys., 2014, 16, 24443–24456 RSC.
- J. Y. Xiang, D. N. Zheng, J. Q. Li, L. Li, L. P. L. Lang, H. Chen, C. Dong, G. C. Che, Z. A. Ren, H. H. Qi, H. Y. Tian, Y. M. Ni and Z. X. Zhao, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 214536 CrossRef.
- H. W. Zandbergen, M. Y. Wu, H. Jiang, M. A. Hayward, M. K. Haas and R. J. Cava, Phys. C, 2002, 366, 221–228 CrossRef CAS.
- R. J. Cava, H. W. Zandbergen and K. Inumaru, Phys. C, 2003, 385, 8–15 CrossRef CAS.
- L. N. Oliveira, E. K. U. Gross and W. Kohn, Phys. Rev. Lett., 1988, 60, 2430–2433 CrossRef.
- W. Kohn, Rev. Mod. Phys., 1999, 71, 1253–1266 CrossRef CAS.
- A. J. Cohen, P. Mori-Sanchez and W. Yang, Chem. Rev., 2012, 112, 289–320 CrossRef CAS PubMed.
- A. Floris, A. Sanna, M. Luders, G. Profeta, N. N. Lathiotakis, M. A. L. Marques, C. Franchini, E. K. U. Gross, A. Continenza and S. Massidda, Phys. C, 2007, 456, 45–53 CrossRef CAS PubMed.
- J. C. Phillips, Adv. Condens. Matter Phys., 2010, 2010, 1–13 CrossRef PubMed.
- N. N. Lathiotakis, M. A. L. Marques, M. Luders, L. Fast and E. K. U. Gross, Int. J. Quantum Chem., 2004, 99, 790–797 CrossRef CAS PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CrossRef CAS.
- K. Refson, S. J. Clark and P. R. Tulip, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 155114 CrossRef.
- J. A. Alarco, P. C. Talbot and I. D. R. Mackinnon, Phys. Chem. Chem. Phys., 2014, 16, 25386–25392 RSC.
- J. Karpinski, N. D. Zhigadlo, S. Katrych, R. Puzniak, K. Rogacki and R. Gonnelli, Phys. C, 2007, 456, 3–13 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- K.-P. Bohnen, R. Heid and B. Renker, Phys. Rev. Lett., 2001, 86, 5771–5774 CrossRef CAS.
- A. J. Cohen, P. Mori-Sanchez and W. Yang, Science, 2008, 321, 792–794 CrossRef CAS PubMed.
- L. G. Sevastyanova, O. K. Gulish, V. A. Stupnikov, V. K. Genchel, O. V. Kravchenko, B. M. Bulychev, R. A. Lunin and V. P. Tarasov, Cent. Eur. J. Phys., 2012, 10, 189–196 CAS.
- L. He, F. Liu, G. Hautier, M. J. T. Oliveira, M. A. L. Marques, F. D. Vila, J. J. Rehr, G.-M. Rignanese and A. Zhou, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 064305 CrossRef.
- T. Morshedloo, M. R. Roknabadi and M. Behdani, Phys. C, 2015, 509, 1–4 CrossRef CAS PubMed.
- A. Q. R. Baron, H. Uchiyama, R. Heid, K. P. Bohnen, Y. Tanaka, S. Tsutsui, D. Ishikawa, S. Lee and S. Tajima, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 020505 CrossRef.
- V. P. S. Awana, A. Vajpayee, M. Mudgel and H. Kishan, Supercond. Sci. Technol., 2009, 22, 034015 CrossRef.
- M. Putti, M. Affronte, P. Manfrinetti and A. Palenzona, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 1–6 CrossRef.
- R. A. Ribeiro, S. L. Bud'ko, C. Petrovic and P. C. Canfield, Phys. C, 2003, 385, 16–23 CrossRef CAS.
- S. Agrestini, A. Bianconi, S. De Negri, M. Giovannini and A. Saccone, Intermetallics, 2003, 11, 1339–1344 CrossRef CAS.
- J. Q. Li, L. Li, F. M. Liu, C. Dong, J. Y. Xiang and Z. X. Zhao, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 132505 CrossRef.
- M. Imai, K. Nishida, T. Kimura and H. Abe, Appl. Phys. Lett., 2002, 80, 1019–1021 CrossRef CAS PubMed.
- J. A. Flores-Livas, R. Debord, S. Botti, A. San Miguel, S. Pailhes and M. A. L. Marques, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 184503 CrossRef.
- M. Imai, E. H. S. Sadki, H. Abe, K. Nishida, T. Kimura, T. Sato, K. Hirata and H. Kitazawa, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 064512 CrossRef.
- S. Brutti and G. Gigli, J. Chem. Theory Comput., 2009, 5, 1858–1864 CrossRef CAS.
- J. M. An and W. E. Pickett, Phys. Rev. Lett., 2001, 86, 4366–4369 CrossRef CAS.
- O. de la Pena, A. Aguayo and R. de Coss, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 01251 CrossRef.
- G. Campi, T. Proffen, X. Qiu, E. S. Bozin, S. J. L. Billinge, S. Agrestini, N. L. Saini and A. Bianconi, J. Supercond. Novel Magn., 2007, 20, 505–510 CrossRef CAS.
- Y. Zhao, C. Ban, Q. Xu, S.-H. Wei and A. C. Dillon, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 1–5 Search PubMed.
- B. Renker, K. B. Bohnen, R. Heid, D. Ernst, H. Schober, M. Koza, P. Adelmann, P. Schweiss and T. Wolf, Phys. Rev. Lett., 2002, 88, 067001 CrossRef CAS.
- X. K. Chen, M. J. Konstantinovic, J. C. Irwin, D. D. Lawrie and J. P. Franck, Phys. Rev. Lett., 2001, 87, 157002 CrossRef CAS.
- A. Pallas and K. Larsson, J. Phys. Chem. B, 2006, 110, 5367–5371 CrossRef CAS PubMed.
- A. Q. R. Baron, H. Uchiyama, S. Tsutsui, Y. Tanaka, D. Ishikawa, J. P. Sutter, S. Lee, S. Tajima, R. Heid and K.-P. Bohnen, Phys. C, 2007, 456, 83–91 CrossRef CAS PubMed.
- R. Englman, The Jahn-Teller Effect in Molecules and Crystals, Wiley-Interscience, 1972 Search PubMed.
- J. Kortus, I. I. Mazin, K. D. Belashchenko, V. P. Antropov and L. L. Boyer, Phys. Rev. Lett., 2001, 86, 4656–4659 CrossRef CAS.
- D. Sharma, J. Kumar, A. Vajpayee, R. Kumar, P. K. Ahluwalia and V. P. S. Awana, J. Supercond. Novel Magn., 2011, 24, 1925–1931 CrossRef CAS.
- A. Carrington, J. D. Fletcher, J. R. Cooper, O. J. Taylor, L. Balicas, N. D. Zhigadlo, S. M. Kazakov, J. Karpinski, J. P. H. Charmant and J. Kortus, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 060507(R) CrossRef.
- D. G. Hinks, J. D. Jorgensen, H. Zheng and S. Short, Phys. C, 2002, 382, 166–176 CrossRef CAS.
- A. Saengdeejing, PhD dissertation, Pennsylvania State University, 2011.
- K. Torkar, H. Krisehner and E. Hitsch, Monatsh. Chem., 1972, 103, 744–750 CrossRef CAS.
- C. Buzea and T. Yamashita, Supercond. Sci. Technol., 2001, 14, R115–R146 CrossRef CAS.
- T. Klein, L. Lyard, J. Marcus, Z. Holanova and C. Marcenat, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 184513 CrossRef.
- J. D. Fletcher, A. Carrington, O. J. Taylor, S. M. Kazakov and J. Karpinski, Phys. Rev. Lett., 2005, 95, 079005 CrossRef.
| This journal is © the Owner Societies 2015 |