
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanisms of SN2 reactions: insights from a nearside/farside analysis
Carsten
Hennig
and
Stefan
Schmatz
*
Institut für Physikalische Chemie, Universität Göttingen, Tammannstr. 6, D-37077 Göttingen, Germany. E-mail: sschmat@gwdg.de
First published on 22nd September 2015
Abstract
A nearside/farside analysis of differential cross sections has been performed for the complex-forming SN2 reaction Cl− + CH3Br → ClCH3 + Br−. It is shown that for low rotational quantum numbers a direct “nearside” reaction mechanism plays an important role and leads to anisotropic differential cross sections. For high rotational quantum numbers, indirect mechanisms via a long-lived intermediate complex are prevalent (independent of a nearside/farside configuration), leading to isotropic cross sections. Quantum mechanical interference can be significant at specific energies or angles. Averaging over energies and angles reveals that the nearside/farside decomposition in a semiclassical interpretation can reasonably account for the analysis of the reaction mechanism.
I. Introduction
State-resolved differential cross sections for chemical reactions, containing information on angular distributions of the scattered products and thus describing a reactive process at a very detailed level, provide the most stringent test for a quantitatively accurate theoretical model for elementary chemical reactions.1–6 Only a few reactive systems could be studied with quantum-state resolution in scattering experiments,7–14 where the flux of the reaction products is measured that go into different scattering angles and final ro-vibrational states for different initial levels. Differential cross sections often exhibit complicated interference patterns1–5,15 which are difficult to analyse. Ion–molecule reactions, studied experimentally employing crossed-beam imaging,15 an approach widely used for reactions between neutral species,3,16 are also employed for the investigation of reactions between neutrals and anions, based on the technique of velocity map imaging.17 A velocity map imaging spectrometer in conjunction with a low-energy ion source has been used successfully to study gas-phase SN2 reactions by Wester and co-workers.18Several reaction mechanisms are possible in the prototypical SN2 reaction X− + CH3Y → XCH3 + Y− with halogen atoms X and Y.19–25 In the direct mechanism, the nucleophile X− directly attacks the cone built by the carbon and the three hydrogen atoms and forms the new X–C bond in a direct collision while simultaneously the other bond, C–Y, is broken. In the indirect mechanism, on the other hand, a relatively long-lived resonance state is produced, corresponding to an intermolecular complex that is bound by long-range ion–dipole forces. This complex carries out rotations before dissociating back into the reactants or finally forming the products. By investigating the importance of different reaction channels, new insight can be gained into this fundamental chemical reaction mechanism.
In our previous quantum-mechanical studies on nucleophilic bimolecular substitutions,26–32 reaction probabilities and total reaction cross sections have been calculated and discussed in detail. In ref. 32, differential cross sections have been computed based on rotationally resolved time-independent quantum-mechanical scattering calculations for Cl− + CH3Br → ClCH3 + Br−. The results show almost isotropic cross sections for reactant molecules with high rotational quantum numbers. Backward scattering is disfavoured for reaction out of states with small rotational excitation, in particular the rovibrational ground state. This is due to a quantum-mechanical effect, the interference of partial waves, that in general can be rationalized by simple classical arguments. In particular for higher vibrational excitation, an umbrella effect that favours the backward direction can be explained by the strong enhancement of the reactivity by opening a direct reaction mechanism. In non-central collisions, or in other words collisions with initial rotational motion, a torque due to the ion–dipole interaction is exerted on the molecule which after a rotation by about 90° completes the reaction.
Connor and co-workers for the first time applied an approximate version of Fuller's nearside/farside theory of angular scattering – originally developed to understand nuclear heavy-ion differential elastic cross sections – to molecular collisions.33 While the nearside/farside theory provides valuable insight into structured angular distributions, a disadvantage of a nearside/farside decomposition is that it is not unique.
In the present paper, we report on a nearside/farside analysis in the prototypical substitution reaction Cl− + CH3Br → ClCH3 + Br−. From a theoretical point of view, it is necessary to extract converged complex S-matrix elements from a quantum scattering calculation, not only the squared moduli in which the phase information is lost. While the latter are sufficient for the calculation of integral scattering cross sections (and can be obtained by averaging without taking into account the phases), differential cross sections are computed by first summing up the S-matrix elements weighted by Legendre polynomials and squaring the moduli of that sums afterwards. The complex S-matrix elements are obtained from the scattering wavefunction after projecting the asymptotic wavefunction onto channel Jacobi coordinates.
The paper is organized as follows: in Section II we report on the theory behind the nearside/farside analysis as applied to a molecular collision, while numerical details are contained in Section III. The results and their discussion are presented in Section IV. Finally, Section V gives our conclusions. Throughout, all energies are quoted in cm−1.
II. Theory
The computation of S-matrix elements for the reaction Cl− + CH3Br (v,j) → ClCH3 (v′,j′) + Br− is described in detail in ref. 31 and 32. In ref. 32, state-resolved differential cross sections at total energy E for vanishing helicity have been computed from S-matrices SJnm(E) employing the usual formula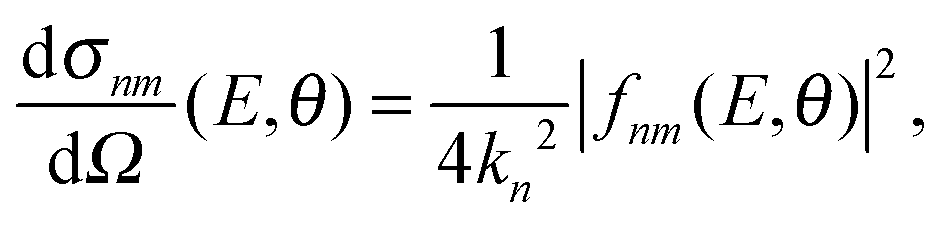 | (1) |
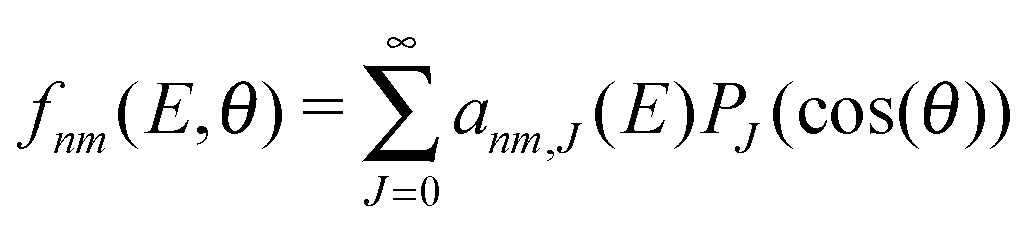 | (2) |
| anm,J(E) = (2J + 1)SJnm(E). | (3) |
The differential cross sections themselves can be directly related to experimental results. To shed more light on the underlying reaction mechanisms, we employed a nearside/farside analysis introduced by Fuller34 which has successfully been applied to reactive scattering.33,35 In a nearside/farside analysis, the scattering amplitude is decomposed into a sum of two subamplitudes moving clockwise (f−(θ)) and counter-clockwise (f+(θ)) around the interaction zone:
| f(θ) = f+(θ) + f−(θ) | (4) |
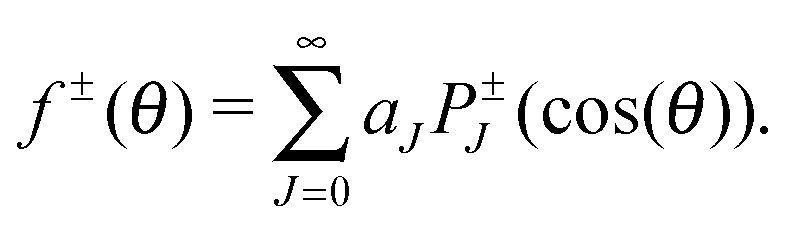 | (5) |
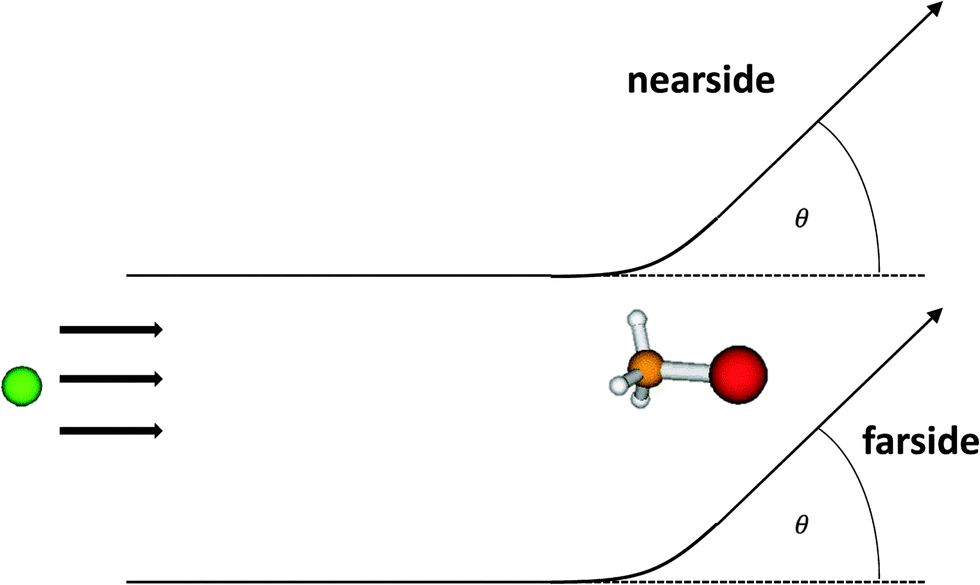 | ||
| Fig. 1 Nearside and farside scattering for the SN2 reaction Cl− + CH3Br → ClCH3 + Br−. Two semiclassical trajectories with the same scattering angle, i.e. equally contributing to the differential cross section are shown. The nearside trajectory runs nearer to the detector which is thought to be positioned at the angle θ. See the text for comments on this simplified picture since for collisions beyond the simple elastic atom–atom case the semiclassical nearside/farside theory is not trivial. | ||
However, the decomposition of the scattering amplitude into subamplitudes is not unique. The final differential cross section is composed of contributions from nearside scattering, farside scattering and interferences of both. In the present analysis, we use the canonical decomposition
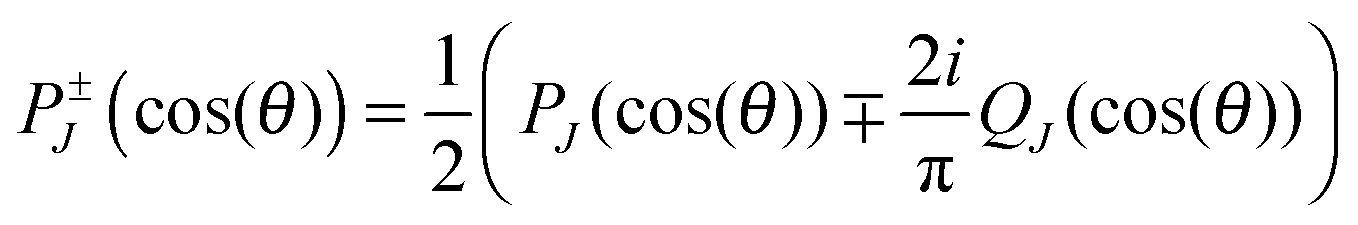 | (6) |
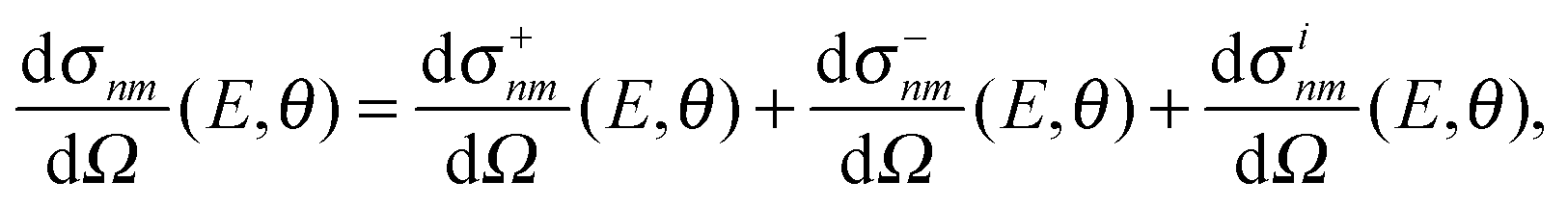 | (7) |
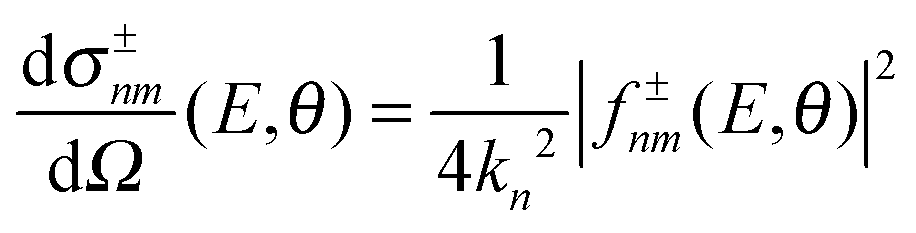 | (8) |
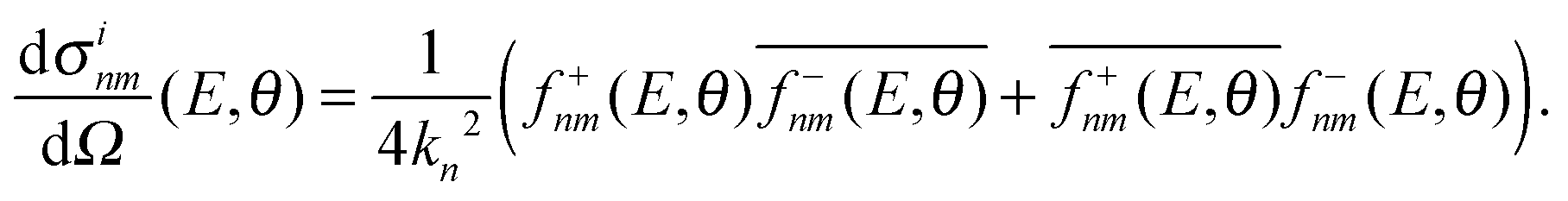 | (9) |
III. Numerical details
The SN2 reaction is treated as a pseudo-triatomic system with the methyl group regarded as a united atom. The internal geometry of this group is optimised for each set of Jacobi coordinates R, r and γ. Starting with the exact Hamiltonian operator in Jacobi coordinates and restricting ourselves to vanishing helicity, we drop the Coriolis coupling term (centrifugal sudden approximation, CS37,38) and obtain the Hamiltonian for total angular momentum J > 0ĤCS = ![[T with combining circumflex]](https://www.rsc.org/images/entities/i_char_0054_0302.gif) J=0 + JCS + J=0 + JCS + ![[V with combining circumflex]](https://www.rsc.org/images/entities/i_char_0056_0302.gif) , , | (10) |
JCS according to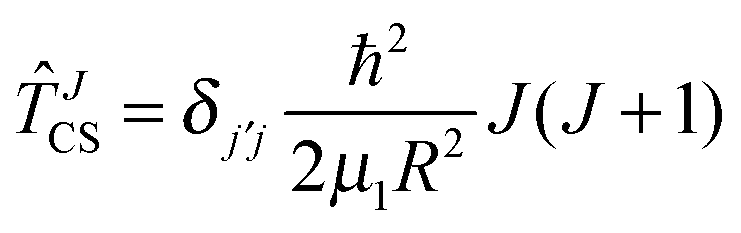 | (11) |
In the framework of time-independent quantum scattering theory the Schrödinger equation for fixed total energy E and fixed J is solved employing an expansion of the wavefunction in products of translational functions and eigenfunctions of the surface Hamiltonian. The numerical solution of the scattering problem makes use of the technique of R-matrix propagation.39 We employ the wave functions and overlap matrices computed for J = 0 and add the above term JCS as an energetic correction to the Hamiltonian in the propagation of the wavefunction along the hyperradius. Asymptotically we impose appropriate boundary conditions and finally obtain the complex scattering matrix.
We made use of the same parameters as employed in ref. 31 and 32. To be able to compare the results, we carry out the calculations at the same energies for nearside/farside state-resolved differential cross sections as in ref. 32: Edif = 350, 850, 1400 and 1900 cm−1; to smoothen many fluctuations that are characteristic of this resonance-driven reaction, for each energy Edif, differential cross sections were actually computed at 21 equidistant values Edif − 1.0 cm−1, Edif − 0.9 cm−1,…, Edif + 1.0 cm−1 and then averaged. In addition, for each angle θdif the differential cross sections have been averaged over intervals of [θdif − θav, θdif + θav], with θav = 5°.
IV. Results and discussion
In Fig. 2, 3, 5 and 6, the initial-state selected differential reaction cross sections and the corresponding nearside/farside decompositions are graphically displayed. They are summed over all product states for total energies Etot of 350, 850, 1400 and 1900 cm−1. sin(θ)(dσ/dΩ)(Etot) is shown, which is the integrand employed in the calculation of the total cross sections σtot(Etot), making the contributions to σtot visible. As a consequence, the data for θ = 0 and θ = 180° are exactly zero, as long as the differential cross sections remain finite. This is not seen in the plots, first because this occurs only at precisely these two angles and second because of data averaging. The data are smoothly averaged over intervals of 10° with the exception of 0 and 180°. Forward scattering with the newly formed Br− ion moving to the left corresponds to θ = 0, while backward scattering with that ion moving to the right is associated with θ = 180°.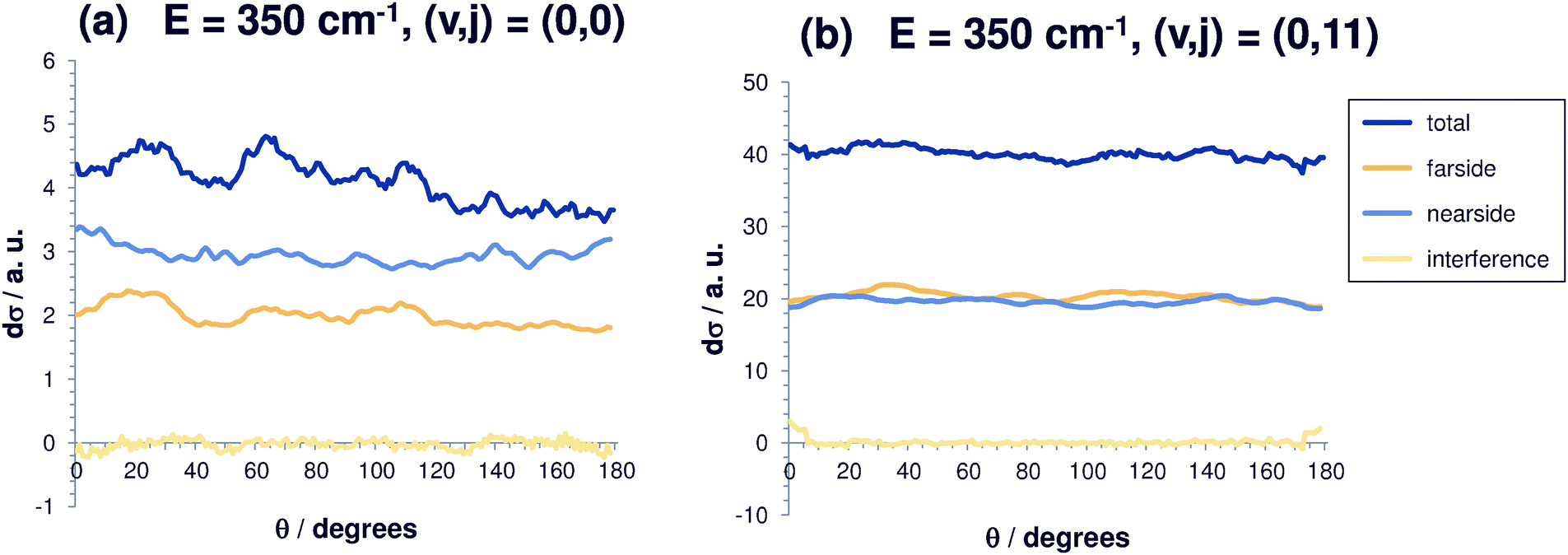 | ||
| Fig. 2 Differential reaction cross sections for Cl− + CH3Br(v,j) → ClCH3 + Br− at E = 350 cm−1 and decomposition in nearside and farside contributions for selected initial rovibrational states (v,j). Also shown is the interference contribution. (a) (v,j) = (0,0); (b) (v,j) = (0,11). | ||
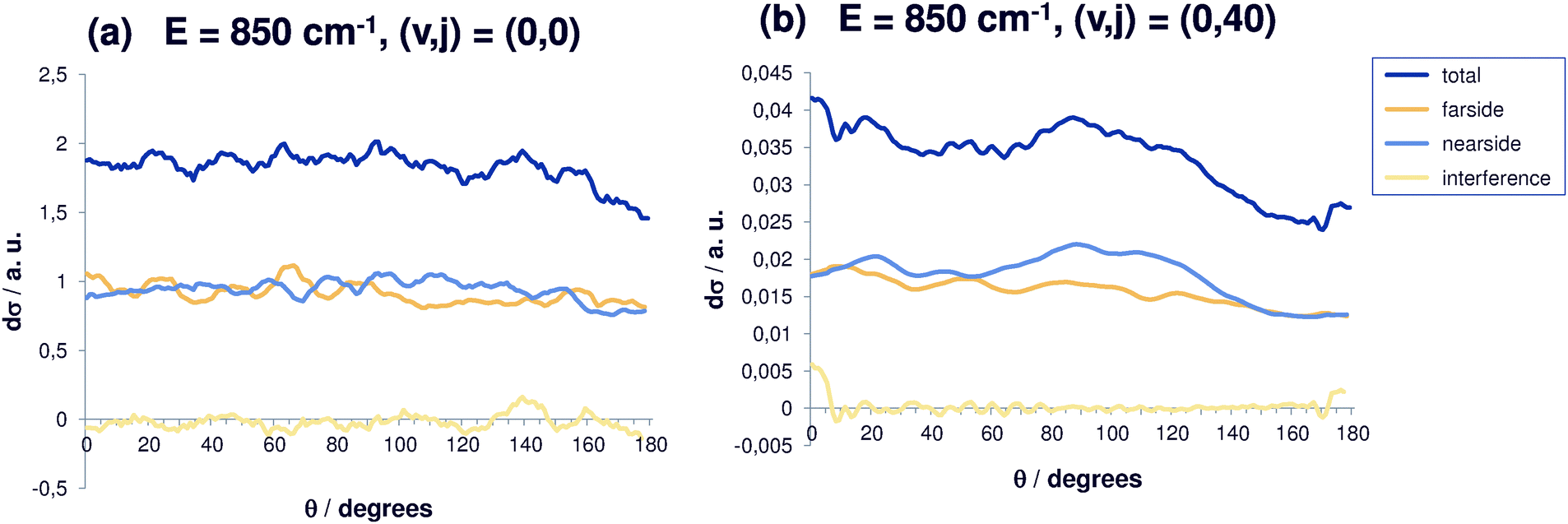 | ||
| Fig. 3 Differential reaction cross sections for Cl− + CH3Br(v,j) → ClCH3 + Br− at E = 850 cm−1 and decomposition in nearside and farside contributions for selected initial rovibrational states (v,j). Also shown is the interference contribution. (a) (v,j) = (0,0); (b) (v,j) = (0,40). | ||
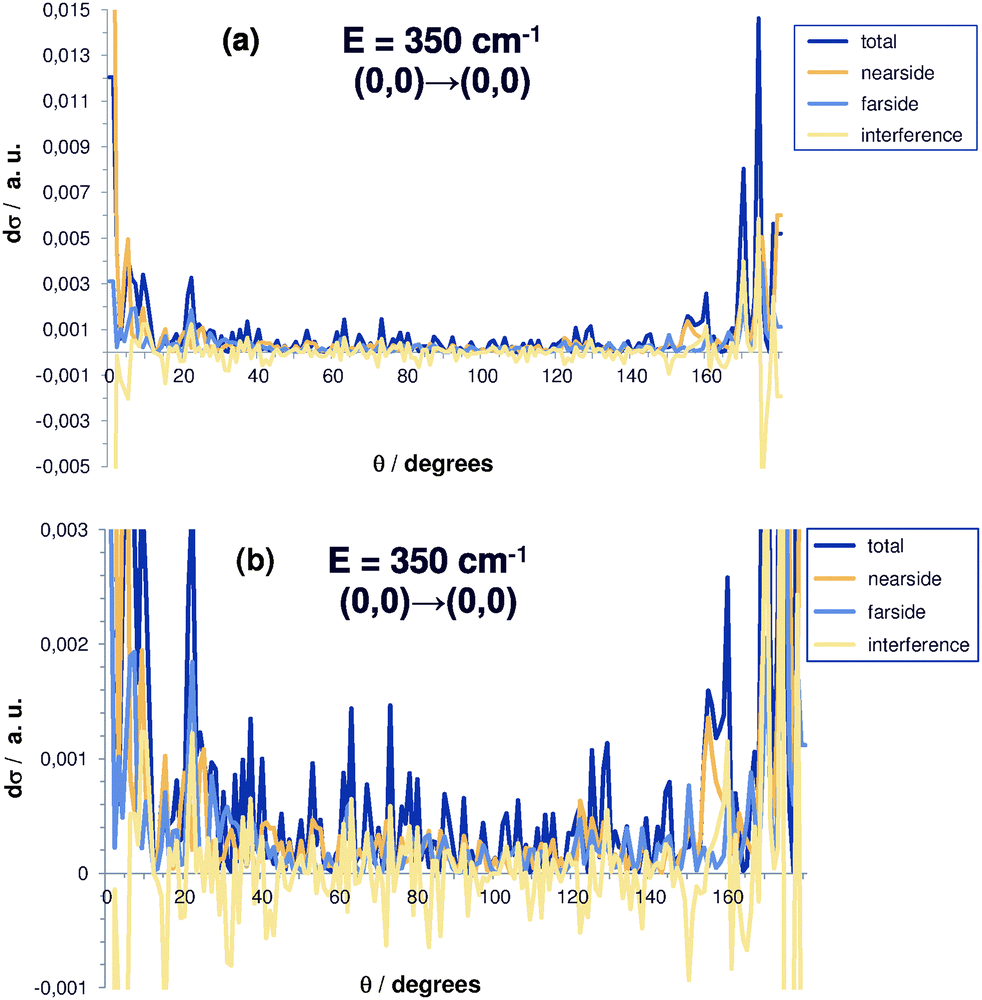 | ||
| Fig. 4 (a) State-to-state specific differential reaction cross section for Cl− + CH3Br (0,0) → ClCH3 (0,0) + Br− at E = 350 cm−1 and decomposition in nearside, farside and interference contributions. (b) Same as (a), but detail with enlarged scale along the vertical coordinate axis. Note the negative and positive parts in the interference contribution. | ||
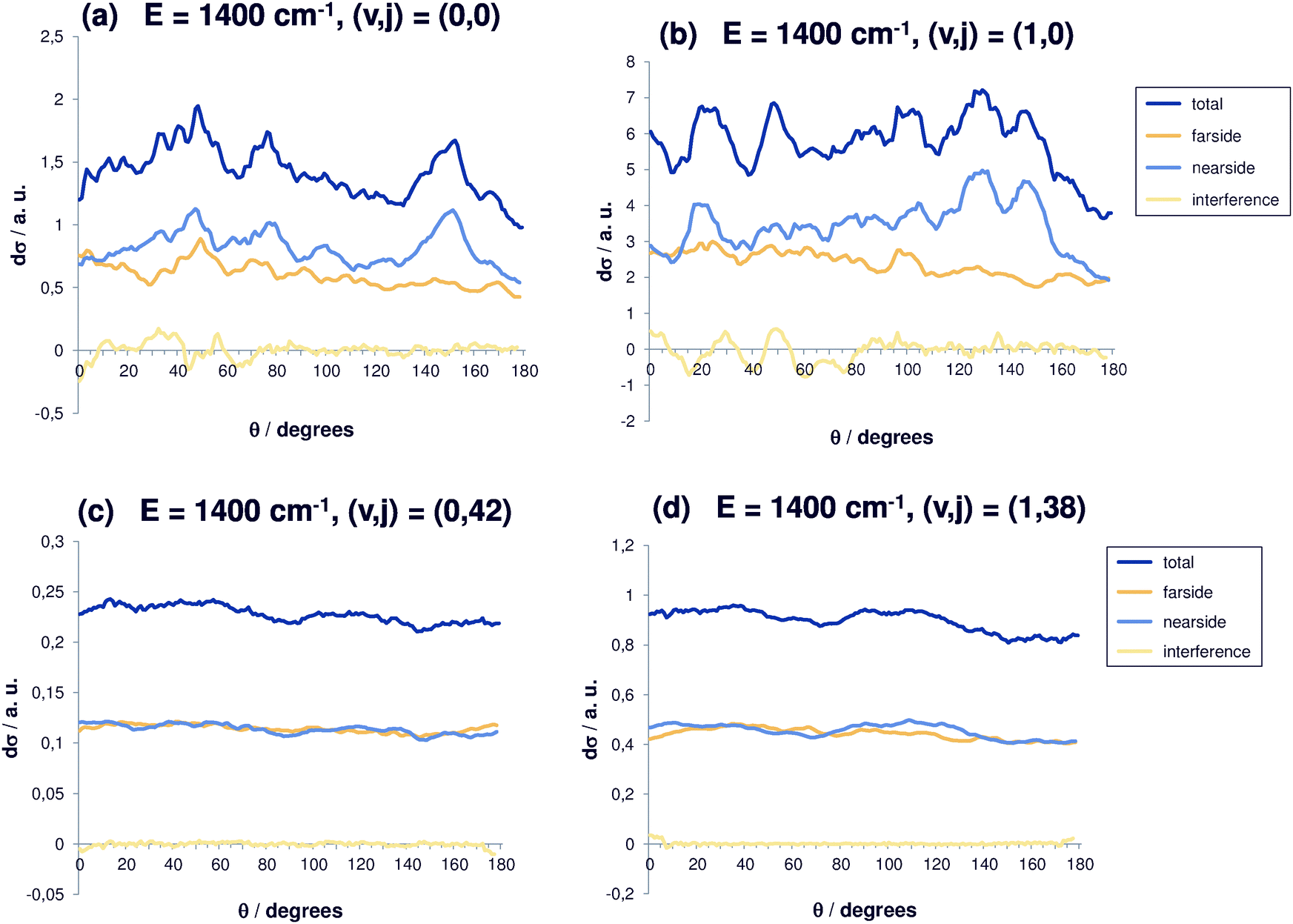 | ||
| Fig. 5 Differential reaction cross sections for Cl− + CH3Br(v,j) → ClCH3 + Br− at E = 1400 cm−1 and decomposition in nearside and farside contributions for selected initial rovibrational states (v,j). Also shown is the interference contribution. (a) (v,j) = (0,0); (b) (v,j) = (1,0); (c) (v,j) = (0,42); (d) (v,j) = (1,38). | ||
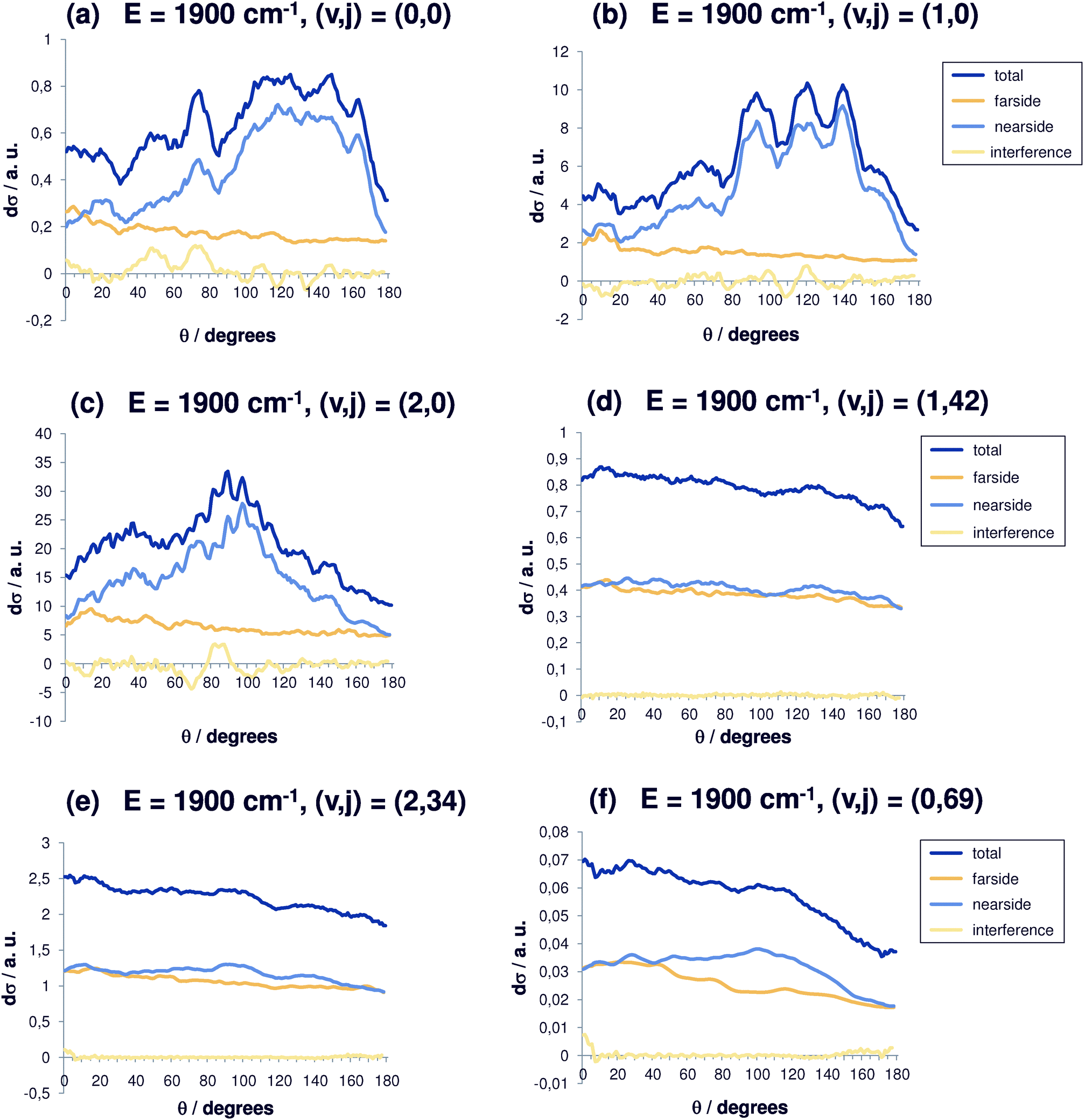 | ||
| Fig. 6 Differential reaction cross sections for Cl− + CH3Br(v,j) → ClCH3 + Br− at E = 1900 cm−1 and decomposition in nearside and farside contributions for selected initial rovibrational states (v,j). Also shown is the interference contribution. (a) (v,j) = (0,0); (b) (v,j) = (1,0); (c) (v,j) = (2,0); (d) (v,j) = (1,42); (e) (v,j) = (2,34); (f) (v,j) = (0,69). | ||
Fig. 2 shows the differential reaction cross sections for a total energy of 350 cm−1 and different initial rotational excitations (v = 0; j = 0, 11). At this relatively small energy, it can be seen clearly that (dσ/dΩ)sin(θ) increases with j (note the scaling) and becomes increasingly more isotropic. For small j, many oscillations typical of resonance features can be observed. Here, the fact that the cross sections become larger with j can be explained mainly by an energetic effect. Rotationally excited initial states are higher in energy, and thus the systems have – at a given total energy – less translational energy available which, however, enters the equation for the calculation of the cross section in the denominator. For the initial state (0,0), back scattering is slightly less effective. For low total energies, the nearside/farside analysis reveals some structure in the differential cross sections with a slight preference of small scattering angles. For initially vibrationless and rotationless states the nearside scattering dominates the farside contribution by 20%, partly by more than 50%, pointing towards a preference of the direct reaction mechanism over complex-formation.
However, if rotation is excited, the relative difference between nearside and farside contribution becomes negligible. The interference contribution between the nearside and the farside amplitude is very small. For higher rotational excitation, this effect becomes even more pronounced, indicating that if the methyl bromide molecule rotates, the reaction occurs mainly via complex-formation and not via the direct reaction pathway. So, the differential cross section becomes isotropic for high initial rotational excitation.
In agreement with our previous results,30 the differential cross sections are largest for small energies. At higher total energies (850 cm−1, Fig. 3), the (0,0) differential cross section shows a similar behavior, i.e. back scattering being slightly less probable, but the average differential cross section has become smaller by roughly a factor of two. Also, the oscillations in the differential cross sections are less pronounced. For initial rotational excitation, the differential cross sections decrease and become more isotropic, so that finally almost no anisotropy can be observed (the cross section for angular momentum quantum number j = 40 is shown in Fig. 3(b)). Isotropic cross sections point to the existence of long-living resonance states which shows that the correlation with the initial angle disappears. For reaction out of the ground state and out of the state with rotational excitation j = 40, the reaction cross sections are anisotropic, the coarse-grained fluctuation is more pronounced for j = 40 while the fine structure is less distinct. The scale of the cross sections in Fig. 3 should be noted. Again, nearside and farside scattering contributions are equally strong and overall the interference contribution obviously is negligible.
However, it should be kept in mind that the curves shown are averaged due to strong oscillating patterns in the cross sections for this resonance-driven reaction. Also in the integral cross sections calculated within a model with the number of degrees of freedom reduced to only two, many extremely narrow resonances are present; an even more pronounced resonance structure is found in the differential cross section. Furthermore, the differential cross sections shown in the present paper are summed over product states, and this summation process smoothens the curves presented in this paper.
The raw data, i.e. state-to-state cross sections before averaging over energies and angles, show prominent interference patterns. This can be seen from Fig. 4 for the state-to-state differential cross section at a fixed energy of 350 cm−1 as a function of the scattering angle (the (0,0) → (0,0) transition is shown). In particular, while the total, nearside and farside contributions are strictly positive due to their definition, the interference term is real, but can be negative as can be clearly seen from Fig. 4. After averaging, the interference contribution becomes negligible. This to a large extent is due to the annihilation of negative and positive components. That justifies the interpretation of this term as an interference type component of the differential cross section and helps to identify the reaction mechanisms in a semiclassical picture and to establish the link to experimental results.
For even higher total energy (Fig. 5(a)), 1400 cm−1, similar features can be observed so that the trend becomes even clearer. However, the coarse oscillations for (0,0) are again more pronounced. Fig. 5(b) shows the same for additional initial vibrational excitation, which is possible at this given total energy. The differential cross sections increase as expected from our previous collinear studies,30 while the behaviour with respect to angular excitation remains as before. The dip for backward scattering becomes more pronounced and the oscillations are coarser. For reaction out of the ground state and the state with one quantum in the C–Br stretching vibration, the differential cross sections show structure, but not a clear preference of a particular angular regime. Nearside scattering dominates, in particular for (v,j) = (1,0) and for wide angles, whereas for small angles the contributions of nearside and farside scattering are very similar. A relatively strong interference contribution is observed. Clearly, the direct mechanism is enhanced when the broken bond is excited (v = 1, Fig. 5(a)). High rotational energy means low translational energy. The cross sections are almost perfectly isotropic and the nearside and farside contributions are very similar.
Again, for energies of 1900 cm−1 (Fig. 6(a–f)), the differential cross sections tend to be more isotropic for larger values of j. The cross section for the initial state (0,0) becomes more and more interesting (Fig. 6(a)), because the peak for backward scattering (θ = 180°) is only one third as high as the maximum value located in the range between 100° and 160°. The structural features become even coarser and the typical resonance structure is lost. Without initial excitation (Fig. 6(a)), the nearside part is strongly dominating. The scattering occurs in the angular range between 100° and 170°. The farside contribution remains almost constant, which points towards a direct reaction mechanism. Additional vibrational excitation (Fig. 6(b)) shifts this range to 90°–140° with backward scattering less than one third of the maximum cross section. An even larger effect is seen in Fig. 6(b) with a pronounced interference pattern. Two quanta in the vibrational mode cause a further shift down to 80°–115°. For two quanta in the C–Br stretching vibration, (v,j) = (2,0) (Fig. 6(c)), an angular maximum at 90° is observed. For even higher initial energies (1900 cm−1) the differential cross sections are shown in Fig. 6(d). For high rotational excitation (v = 42), the same effect as already discussed occurs. The same can be said for Fig. 6(e). From Fig. 6(f), we see that the forward scattering is reduced and the nearside contribution again dominates.
To rationalize the calculated behaviour of the differential reaction cross sections, in particular the isotropy for large j-values, it is useful to go back to simple classical-mechanical arguments. For a fastly rotating target molecule, all directions for dissociation of the intermediate complex are possible and equally probable. A complex that lives long enough (i.e. the resonance of which is narrow enough with respect to energy) can perform several rotations before the actual reaction takes place. So, no direction is favoured, yielding isotropic differential cross sections.
It should be discussed why cross sections for backward scattering are smaller. Remember that in our previous collinear studies28–30 only backward scattering was possible, with the chloride anion attacking the methyl group, forming a bond with the carbon atom, and after the umbrella flip of the methyl group bouncing back to the direction where the chloride ion came from. In the full three-dimensional picture, this direction is strongly disfavoured because the chlorine anion approaches the CH3Br molecule off from the collinear axis. In the classical-mechanical picture, due to the strong ion–dipole interaction a torque is exerted on the molecule, and it begins to carry out a rotation or, when the ion is very close to the molecule, a libration. However, due to the shape of the potential, no full rotational cycle can be performed if the available energy is not high enough, and an angle of about 90° seems most probable. This explains why exact backward scattering is such a disfavoured event. The strong electrostatic forces have a substantial influence on the differential cross sections in the cases without initial rotational excitation. The molecule orients towards the ion in the case of any deviation from the central collision.
V. Conclusions
We have carried out a nearside/farside analysis on the nucleophilic bimolecular substitution reaction Cl− + CH3Br(v,j) → ClCH3(v′,j′) + Br− as it was first introduced in molecular scattering by Connor and co-workers based on an idea of Fuller. It is revealed that in gas-phase SN2 reactions the structure in the differential cross sections at higher energies is related to the nearside scattering contribution, pointing towards a direct reaction mechanism where the attacking ion directly approaches the open cone formed by the carbon and the three connected hydrogen atoms. The indirect mechanism, on the other hand, is described by complex-formation and subsequent dissociation after carrying out rotational motion. The differential cross section becomes isotropic because no angle is favoured. This mechanism clearly is connected with the farside contribution which introduces no structure in the differential reaction cross section.Acknowledgements
This work was financially supported by the Deutsche Forschungsgemeinschaft (DFG), grant SCHM 1651/1-3. Additional support from the Fonds der chemischen Industrie is gratefully acknowledged.References
- D. C. Clary, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12649 CrossRef CAS PubMed.
- D. C. Clary, Science, 2008, 321, 789 CrossRef CAS PubMed.
- K. Liu, Annu. Rev. Phys. Chem., 2001, 52, 139 CrossRef CAS PubMed.
- X. Yang, Annu. Rev. Phys. Chem., 2007, 58, 433 CrossRef CAS PubMed.
- W. Hu and G. C. Schatz, J. Chem. Phys., 2006, 125, 132301 CrossRef PubMed.
- J. Mikosch, M. Wei and G. Nyman, J. Phys. Chem. A, 2010, 114, 9617 Search PubMed.
- P. D. D. Monks, J. N. L. Connor and S. C. Althorpe, J. Phys. Chem. A, 2006, 110, 741 CrossRef CAS PubMed.
- D. Dai, C. C. Wang, S. A. Harich, X. Wang, X. Yang, S. D. Chao and R. T. Skodje, Science, 2003, 300, 1730 CrossRef CAS PubMed.
- F. J. Aoiz, L. Banares, B. Martinez-Haya, J. Castillo, D. E. Manolopoulos, K. Stark and H.-J. Werner, J. Phys. Chem. A, 1997, 101, 6403 CrossRef CAS.
- M. Qiu, Z. Ren, L. Che, D. Dai, S. A. Harich, X. Wang, X. Yang, C. Xu, D. Xie, M. Gustafsson, R. T. Skodje, Z. Sun and D. H. Zhang, Science, 2006, 311, 1440 CrossRef CAS PubMed.
- J. N. L. Connor, Phys. Chem. Chem. Phys., 2004, 6, 377 RSC.
- N. Balucani, D. Skouteris, L. Cartechini, G. Cappozza, E. Segeloni, P. Casavecchia, M. H. Alexander, G. Capecchi and H.-J. Werner, Phys. Rev. Lett., 2003, 91, 13201 CrossRef CAS.
- D. H. Zhang and J. Z. H. Zhang, J. Chem. Phys., 1994, 101, 1146 CrossRef CAS PubMed.
- E. J. Rackham, T. Gonzales-Lezana and D. E. Manolopoulos, J. Chem. Phys., 2003, 119, 12895 CrossRef CAS PubMed.
- J. Mikosch, M. Weidemüller and R. Wester, Int. Rev. Phys. Chem., 2010, 29, 589 CrossRef CAS PubMed.
- P. Casavecchia, Rep. Prog. Phys., 2000, 63, 355 CrossRef CAS.
- A. T. J. B. Eppink and D. H. Parker, Rev. Sci. Instrum., 1997, 68, 3477 CrossRef CAS PubMed.
- J. Mikosch, U. Frühling, S. Trippel, D. Schwalm, M. Weidemüller and R. Wester, Phys. Chem. Chem. Phys., 2006, 8, 2990 RSC.
- W. L. Hase, Science, 1994, 266, 998 CAS.
- M. L. Chabinyc, S. L. Craig, C. K. Regan and J. I. Brauman, Science, 1998, 279, 1882 CrossRef CAS.
- J. K. Laerdahl and E. Uggerud, Int. J. Mass Spectrom. Ion Processes, 2002, 214, 277 CrossRef CAS.
- S. Schmatz, ChemPhysChem, 2004, 5, 600 CrossRef CAS PubMed.
- J.-L. Le Garrec, B. R. Rowe, J. L. Queffelec, J. B. A. Mitchell and D. C. Clary, J. Phys. Chem., 1997, 107, 1021 CrossRef CAS PubMed.
- J. Mikosch, S. Trippel, C. Eichhorn, R. Otto, U. Lourderaj, J. X. Zhang, W. L. Hase, M. Weidemüller and R. Wester, Science, 2008, 319, 183 CrossRef CAS PubMed.
- J. Zhang and W. L. Hase, J. Phys. Chem. A, 2010, 114, 9635 CrossRef CAS PubMed.
- S. Schmatz and D. C. Clary, J. Chem. Phys., 1998, 109, 8200 CrossRef CAS PubMed.
- S. Schmatz and D. C. Clary, J. Chem. Phys., 1999, 110, 9483 CrossRef CAS PubMed.
- C. Hennig and S. Schmatz, J. Chem. Phys., 2004, 121, 220 CrossRef CAS PubMed.
- C. Hennig and S. Schmatz, J. Chem. Phys., 2005, 122, 234307 CrossRef PubMed.
- C. Hennig and S. Schmatz, Chem. Phys. Lett., 2007, 446, 250 CrossRef CAS PubMed.
- C. Hennig and S. Schmatz, J. Chem. Phys., 2009, 121, 224303 CrossRef PubMed.
- C. Hennig and S. Schmatz, Phys. Chem. Chem. Phys., 2012, 14(37), 12982 RSC.
- J. N. L. Connor, P. McCabe, D. Sokolovski and G. C. Schatz, Chem. Phys. Lett., 1993, 206, 119 CrossRef CAS.
- R. C. Fuller, Phys. Rev. C: Nucl. Phys., 1975, 12, 1561 CrossRef CAS.
- A. J. Dobbyn, P. McCabe, J. N. L. Connor and J. F. Castillo, Phys. Chem. Chem. Phys., 1999, 1, 1115 RSC.
- D. Sokolovski and J. N. L. Connor, Chem. Phys. Lett., 1999, 305, 238 CrossRef CAS.
- R. T. Pack, J. Chem. Phys., 1974, 60, 633 CrossRef CAS PubMed.
- P. McGuire and D. J. Kouri, J. Chem. Phys., 1974, 60, 2488 CrossRef CAS PubMed.
- E. B. Stechel, R. B. Walker and J. C. Light, J. Chem. Phys., 1978, 69, 3518 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2015 |