
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic structure and excited state dynamics in a dicyanovinyl-substituted oligothiophene on Au(111)
Lea
Bogner†
a,
Zechao
Yang
a,
Martina
Corso‡
a,
Roland
Fitzner
b,
Peter
Bäuerle
b,
Katharina J.
Franke
a,
José Ignacio
Pascual
*c and
Petra
Tegeder
*d
aFreie Universität Berlin, Fachbereich Physik, Arnimallee 14, D-14195 Berlin, Germany
bUniversität Ulm, Institut für Organische Chemie II und Neue Materialien, Albert-Einstein-Allee 11, 89081 Ulm, Germany
cCIC nanoGune, 20018 Donostia-San Sebastin, Spain. E-mail: ji.pascual@nanogune.eu
dRuprecht-Karls-Universität Heidelberg, Physikalisch-Chemisches Institut, Im Neuenheimer Feld 253, 69120 Heidelberg, Germany. E-mail: tegeder@uni-heidelberg.de
First published on 16th September 2015
Abstract
Dicyanovinyl (DCV)-substituted oligothiophenes are promising donor materials in vacuum-processed small-molecule organic solar cells. Here, we studied the structural and the electronic properties of DCV-dimethyl-pentathiophene (DCV5T-Me2) adsorbed on Au(111) from submonolayer to multilayer coverages. Using a multi-technique experimental approach (low-temperature scanning tunneling microscopy/spectroscopy (STM/STS), atomic force microscopy (AFM), and two-photon photoemission (2PPE) spectroscopy), we determined the energetic position of several affinity levels as well as ionization potentials originating from the lowest unoccupied molecular orbitals (LUMO) and the highest occupied molecular orbitals (HOMO), evidencing a transport gap of 1.4 eV. Proof of an excitonic state was found to be a spectroscopic feature located at 0.6 eV below the LUMO affinity level. With increasing coverage photoemission from excitonic states gains importance. We were able to track the dynamics of several electronically excited states of multilayers by means of femtosecond time-resolved 2PPE. We resolved an intriguing relaxation dynamics involving four processes, ranging from sub-picosecond (ps) to several hundred ps time spans. These show a tendency to increase with increasing coverage. The present study provides important parameters such as energetic positions of transport levels as well as lifetimes of electronically excited states, which are essential for designing organic-molecule-based optoelectronic devices.
Introduction
Semiconducting materials based on organic molecules are being explored as potential active materials in (opto)electronic devices such as organic photovoltaic cells, light emitting diodes, or thin film transistors.1–4 For device performance the properties of interfaces between the inorganic substrate and the organic material as well as between the active layers (donor/acceptor) are crucial. Thereby the orientation of the molecules is of particular importance for the light emission/absorption and charge transport properties. Intermolecular interactions compete with molecule/substrate interactions both defining the adsorption geometry and the arrangement of the molecular film and, accordingly, the electronic structure of the system. The electronic structure of interfaces crucially influences the functionalities of organic films.5–7 The energetic positions of molecular electronic states or bands (transport levels) with respect to the Fermi level of a (metallic) substrate yields quantitative information on the barriers for electron (hole) injection at the molecule–substrate interface. Additionally, the formation and decay dynamics of excitonic states both within the films and at interfaces (e.g. donor/acceptor-interfaces) are key processes governing the optoelectronic functionality of the films.Oligothiophene derivatives, in particular dicyanovinyl (DCV)-substituted oligothiophenes, have been utilized in vacuum-processed small-molecule organic solar cells.8–13 Using these low-band gap donor materials promising efficiencies of up to 8.3% for a single junction and up to 9.7% for a triple junction cell have been obtained.14 The adsorption and electronic properties of bare non-substituted oligothiophenes adsorbed on noble metal surfaces are well-known.15–25 The substituted species DCV5T-Me2 (see Fig. 1(a)) is expected to show different structural properties that explain its larger photovoltaic efficiency. It combines an electron-rich donor backbone and two terminal electron-poor acceptor moieties. On the Au(111) surface, this compound self-assembles in extended chains stabilized by intercalated Au adatoms coordinated via terminal CN-groups resulting in a redistribution of molecular orbitals.26
 | ||
| Fig. 1 (a) STM overview (scale bar = 56 nm; I = 63 pA, V = 0.69 V) of DCV5T-Me2 molecules deposited on the Au(111) surface held at room temperature. Inset shows the chemical structure of DCV5T-Me2. (b) High resolution STM image of the island (scale bar = 1.2 nm; I = 50 pA, V = 0.31 V). The black rhombus shows the unit cell of the island (a = 1.77 nm, b = 3.38 nm, α = 130°). (c) Laplace filtered frequency shift AFM image of the same area as (b) with the DFT-optimized molecular model overlaid. The tip was approached by 1.4 Å to the sample from the current set point of 0.31 V and 50 pA over the molecule. | ||
In the present contribution we utilized complementary experimental techniques such as scanning tunneling microscopy/spectroscopy (STM/STS), atomic force microscopy (AFM), and two-photon photoemission (2PPE) spectroscopy to obtain a comprehensive picture of the adsorption and electronic properties of DCV5T-Me2 on Au(111) from the submonolayer to the multilayer regime. We found that two-dimensional DCV5T-Me2 molecular assemblies appear on the surface at submonolayer coverages with a structure similar to that adopted in multilayer growth. A combination of tunneling and photoemission spectroscopy of the first and second layer provided detailed insights into the energetic positions of affinity levels and ionization potentials. Additionally we utilized femtosecond time-resolved 2PPE to elucidate the dynamics of electronically excited states in DCV5T-Me2 after optical excitation. We show that the submonolayer is strongly affected by the electronic coupling to the metallic states. With increasing coverage, the molecular states are better decoupled and the relaxation dynamics of the excited states slow down. These findings uncover the limit of ultra-thin films as functional building blocks for photovoltaic cells.
Methods
The Au(111) single crystal was prepared by Ar+ or Ne+ sputtering and annealing at 800 K. DCV5T-Me2 molecules were evaporated from a Knudsen cell onto the substrate held at room temperature (around 300 K). STM/STS, AFM and 2PPE experiments were carried out in two different setups under ultrahigh vacuum conditions.STM/AFM experiments
STM/AFM measurements were done on a custom-made low temperature scanning tunneling microscope, at 4.8 K, equipped with a tuning fork force sensor27 for simultaneous non-contact AFM and STM measurements. Tunneling spectroscopy was performed using lock-in techniques to measure the differential conductance of the junction, which allows us to localize the energy alignment of molecular-derived resonances. Spatial maps of the differential conductance at the resonance voltage were drawn with an open feedback loop. Non-contact AFM images were obtained by mapping relative changes in the resonance frequency of the sensor at a constant tip height over the sample. High resolution images with clear intramolecular contrast were obtained with Xe functionalized tips at the onset of repulsive tip-surface forces. AFM images were Laplace filtered to emphasize the intramolecular corrugation over the smooth molecular background.Identification of molecular structures and orbitals from AFM and dI/dV maps was based on density functional theory (DFT) simulations of minimum energy molecular structure and orbital isosurfaces. We employed the Gaussian package, with the B3LYP exchange–correlation functional and the 6-31G basis set. Since DFT methods fail to reproduce level alignment correctly,28 the orbital level was strictly obtained by comparing STS and 2PPE spectra.
2PPE experiments
2PPE allows the investigation of occupied, unoccupied electronic and excitonic states29 in a pump–probe scheme using femtosecond (fs) laser pulses. While the pump pulse hν1 excites an electron from below the Fermi level (EF) to intermediate unoccupied states at energies E − EF = Ekin + Φ − hν2 (with Φ the work function), the probe pulse hν2 photoionizes the sample by lifting the excited electron above the vacuum level (Evac). Photoelectrons are detected using a custom-built time-of-flight (TOF) spectrometer and are analyzed with respect to their kinetic energy (Ekin) (for details see ref. 30). In order to elucidate the origin of peaks in the 2PPE spectrum, viz. identify whether they originate from occupied initial or unoccupied intermediate states in the 2PPE process, the dependence of the electron kinetic energy Ekin on the photon energy was investigated. When an unoccupied intermediate state, such as the LUMO, is probed in one-color 2PPE (pump and probe photon have the same energy) the change in Ekin scales with that in photon energy hν and hν′, i.e., Ekin = 1Δhν (Δhν = hν − hν′). On the other hand, for an occupied initial state, e.g., the HOMO, the kinetic energy of the electron ejected scales with twice the photon energy 2Δhν. In two-color 2PPE (pump and probe photon have different energies, one in the visible the other in the UV regime. The latter is the frequency doubled visible pulse.) the energy of the ejected electron scales with three times the photon energy (i.e., 3Δhν; with respect to the photon in the visible regime) for occupied initial states and twice or once the photon energy (2Δhν or 1Δhν) for unoccupied intermediate states. This analysis is generally not applicable for transitions between bulk bands due to their strong perpendicular dispersion but holds in the case of surface and adsorbate-derived states.31 The assignment of 2PPE features to photoemission from occupied or unoccupied molecular orbitals is carried out via their energetic position with respect to the substrate Fermi level, i.e., the closed state to EF is the HOMO or the LUMO, respectively. Energetically lower and higher lying states are accordingly lower lying HOMOs (HOMO−n) and higher lying LUMOs (LUMO+n). Additionally, the STS results are used for the assignment. The 2PPE spectra are displayed with an energy axis representing the final state of the photoelectron, referenced to the Fermi level (EFinal − EF = Ekin + Φ). The UV-spectra were fitted in two parts: the low energy part was fitted with an exponential function, for the secondary electron background, and a reasonable number of Gaussians, depending on the number of the peaks observed. The high energy part of the spectrum was fitted only with Gaussians since the secondary electron background is rather flat at high energies. The same fitting principle was applied to the correlated spectra. In time-resolved 2PPE, the pump–probe scheme enables us to study the population dynamics of excited electronic states on the time scale of the femtosecond laser pulses by delaying the probe pulse with respect to the pump pulse. For 2PPE experiments the DCV5T-Me2 coverage was determined by temperature-programmed desorption and work function measurements.Results and discussion
The properties of an organic film depend on the degree of electronic mixing between molecular and metal states at the organic–inorganic interface, and on the energy alignment of molecular states. Hence, we start by characterizing the regime of coverage below one monolayer.Sub-monolayer coverage
We grew a sub-monolayer coverage of DCV5T-Me2 molecules on Au(111) at room temperature and cooled down to 4.8 K for STM inspection. We found that DCV5T-Me2 forms both two-dimensional molecular islands and extended chain structures (Fig. 1(a)). The latter were found to consist of coordination-bonded DCV5T-Me2–Au structures in ref. 26. Here we study the two-dimensional assemblies, which dominate the aspect of the sample for coverages closer to one monolayer. We typically find domains with the extension of hundreds of nanometers. STM images show that the islands are composed of molecules adopting a bent configuration with the DCV moieties contacting the center of neighboring molecules (Fig. 1(b)). This molecular structure suggests the absence of coordinating Au adatoms in the layer structure, in opposition to the chains.26In order to precisely determine the bonding geometry within the islands, we performed AFM measurements using a Xe functionalized tip. Fig. 1(c) shows a Laplace-filtered constant height AFM image of a molecular island, where the chemical structure of the DCV5T-Me2 molecules is resolved. The thiophene rings are imaged as pentagonal structures with their sulfur atoms exhibiting the sharpest contrast.
The pentagon alternate their orientation along the molecular axis, as indicated in the molecular models superimposed to Fig. 1(c). The DCV groups are resolved as protrusion with brighter contrast localized at the terminal nitrogen atoms. AFM images further exclude that Au adatoms are forming part of the structure and, hence, mediate the intermolecular bonding, as it is the case for the chain structures. Instead, the DCV endgroups are arranged with a cyano end pointing towards a sulfur atom at the neighbor molecule. In spite of the large conformational flexibility of DCV5T-Me2, all of the molecules appear with this shape. DFT calculations of the isolated molecule revealed that the shape observed here corresponds to the global minimum.26 This minimum energy configuration has a planar structure, thus enhancing its relative stability when forced to lay in a planar adsorption geometry on a metal surface.
Next, we investigated the electronic properties of DCV5T-Me2 molecules in islands by combining STS and 2PPE measurements. Fig. 2(b) compares the STS spectra of unoccupied states (i.e., positive sample bias) acquired at three different positions within one DCV5T-Me2 molecule. All spectra show a peak of differential conductance at 1.3 V with similar intensity at the three sites, but with a clear shoulder at 1.65 V only at the endgroups of the DCV5T-Me2 molecule. Furthermore, the spectrum at the molecular center shows a second peak at 2.35 V, completely absent at the sides. This peculiar distribution of unoccupied molecular states is further confirmed by spatially mapping the corresponding dI/dV signal at each resonance bias position (Fig. 2(c)–(e)). Supported by DFT calculations of the orbital shape (wave-function amplitude isosurfaces) we assign the two lowest resonances to molecular states derived from the LUMO and LUMO+1, respectively. The resonance at 2.35 V is highly localized at the central part of the molecule. We assign it to the LUMO+2 derived resonance, because it is also weighted at the center of the molecule. The resulting spectral structure indicates that the molecules retain their free-molecule character on the surface, and the degree of hybridization with the surface is relatively small.
 | ||
| Fig. 2 Electronic properties of molecular islands. (a) STM image (scale bar = 1.7 nm; I = 81 pA and V = 1.3 V) of a molecular island. (b) dI/dV spectra (solid) acquired with a closed feedback (set point: I = 50 pA and V = 0.5 V) at different locations of one DCV5T-Me2 molecule in the island and the corresponding Gaussian fits (dash). The energetic positions of the resonances in the dI/dV spectra are highlighted by the black dashed vertical lines, and all the spectra are offset for clarity. (c–e) Constant height dI/dV maps of the same island as in (a) recorded at biases of 1.3 V (81 pA), 1.6 V (81 pA) and 2.25 V (39 pA), respectively. In dI/dV channels the red color represents the lower intensity, while the yellow color the higher intensity. Insets show the DFT-calculated orbital shape of the LUMO, LUMO+1, and LUMO+2 of the free molecule, obtained with the Gaussian code32 using the B3LYP exchange–correlation functional and the 6-31G basis set. | ||
We will demonstrate that using 2PPE enables us to also gain further insights into the energetic positions of occupied electronic molecular states as well as excitonic states. Fig. 3 shows 2PPE data of a sub-monolayer DCV5T-Me2/Au(111).
 | ||
| Fig. 3 (a) One-color 2PPE spectrum of 0.3 ± 0.2 ML DCV5T-Me2/Au(111) recorded with a photon energy of 4.78 eV. The spectrum is displayed as a function of final-state energy above the Fermi level, EFin − EF = Ekin − Φ (with Φ the work function). (b) Two-color 2PPE spectrum recorded with 2.39 and 4.78 eV photons. The insets in (a) and (b) show the photon energy dependent peak position in order to determine the energetic position of the observed electronic states with respect to EF. The denoted numbers indicate the respective slope. | ||
The one-color 2PPE spectrum shows several peaks related to either occupied electronic states of the DCVT5T-Me2/Au(111) systems, such as d-bands (located around −2.0 eV and −2.6 eV![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33,34), the Shockley surface state (SS) (at −0.48 eV35), or occupied molecular resonances, or to unoccupied molecular resonances. In order to identify whether the peaks originate from occupied initial or unoccupied intermediate states in the 2PPE process, their energetic position is measured as a function of photon energy (see the inset of Fig. 3(a)). The peaks labeled as A, B, and C shift equally with the change in photon energy, resulting in photoemission from unoccupied electronic states located at 4.0 eV (state A), 3.6 eV (state B) and 3.4 eV (state C) with respect to EF. At EFinal − EF = 8.65 eV we observe a peak which shifts by twice the photon energy. This state is assigned to a low-lying occupied state, namely the HOMO−2 (see below), with a binding energy of −1.0 eV. Note that the work function adopts a value of 5.4 eV in this low-coverage regime, i.e., a reduction by 50 meV compared to the bare Au(111) surface. In the two-color spectrum (Fig. 3(b)) two additional features are found, both shifting with twice the photon energy (hν1, visible pulse) indicating that they originate from the photoemission of unoccupied states. They are located at 1.3 and 1.8 eV with respect to EF. Comparing with the STS data, we assign them to the LUMO and LUMO+1, respectively. However, in STS the LUMO+1 is found to be at 1.65 eV, i.e., 150 meV closer to EF. We admit that the feature overlaps with the very intense photoemission from the surface state leading to a higher uncertainty in the determination of the energetic position.
33,34), the Shockley surface state (SS) (at −0.48 eV35), or occupied molecular resonances, or to unoccupied molecular resonances. In order to identify whether the peaks originate from occupied initial or unoccupied intermediate states in the 2PPE process, their energetic position is measured as a function of photon energy (see the inset of Fig. 3(a)). The peaks labeled as A, B, and C shift equally with the change in photon energy, resulting in photoemission from unoccupied electronic states located at 4.0 eV (state A), 3.6 eV (state B) and 3.4 eV (state C) with respect to EF. At EFinal − EF = 8.65 eV we observe a peak which shifts by twice the photon energy. This state is assigned to a low-lying occupied state, namely the HOMO−2 (see below), with a binding energy of −1.0 eV. Note that the work function adopts a value of 5.4 eV in this low-coverage regime, i.e., a reduction by 50 meV compared to the bare Au(111) surface. In the two-color spectrum (Fig. 3(b)) two additional features are found, both shifting with twice the photon energy (hν1, visible pulse) indicating that they originate from the photoemission of unoccupied states. They are located at 1.3 and 1.8 eV with respect to EF. Comparing with the STS data, we assign them to the LUMO and LUMO+1, respectively. However, in STS the LUMO+1 is found to be at 1.65 eV, i.e., 150 meV closer to EF. We admit that the feature overlaps with the very intense photoemission from the surface state leading to a higher uncertainty in the determination of the energetic position.
Monolayer and higher coverages
The growth of thicker films proceeds in a layer-by-layer fashion: only when the coverage is increased above one monolayer, self-assembled structures appear on top of a fully saturated first layer (Fig. 4(a)). The unit cell of the molecules in the second layer has the same dimension as in the first layer. However, high resolution STM images (Fig. 4(b)) suggest that the upper layer is shifted with respect to the underlying layer and that the molecules adopt a more asymmetric configuration, with their two ends appearing differently in the STM images. We expect that due to their large molecular flexibility, DCV endgroups and thiophene rings of second layer molecules rotate to bond and accommodate their structure over the first layer. However, the layer-by-layer growth indicates that the interaction between layers is weak, which is further confirmed by STS measurements. | ||
| Fig. 4 Structural and electronic properties of the bilayer film. (a) Large scale STM image (scale bar = 10 nm; I = 63 pA and V = 0.81 V) of a bilayer film of DCV5T-Me2 molecules on Au(111) showing a large ordered island located on top of the first layer. (b) High resolution STM image (scale bar = 3.3 nm; I = 24 pA and V = 0.63 V) of the bilayer film with a model for the structure superposed. Every rhombus in the black dash grid surrounds a molecule in the first layer. (c) dI/dV spectra (solid) acquired with an open feedback (set point: I = 70 pA and V = −2 V) at the molecules in the first and second layer and corresponding Gaussian fits (dash). In all the dI/dV spectra, the energetic positions of the states are highlighted by the vertical dash lines. In particular, the black dash lines indicate the electronic states, while the orange dash line indicates the vibronic state. All the spectra are offset for clarity. The FWHM of the peaks are listed in the inset table with the unit of volts. | ||
In order to elucidate the evolution of molecular electronic properties as a function of coverage, we performed tunnel spectroscopy measurements on second layer DCV5T-Me2 molecules (Fig. 4(c)). The spectra now show the effect of a less interacting layer. The broad resonance of the first layer, composed of both the LUMO and the LUMO+1, appears now as a multiple component peak with two main features at 1.78 V (over the whole molecule) and 2.14 V (at the sides), attributing to the LUMO and the LUMO+1. The higher alignment of the peaks (∼0.5 V higher) and the sharper linewidth (∼0.1 V and 0.2 V FWHM) prove that this molecular system is less electronically coupled to the metal surface due to reduced screening and less hybridization, respectively. It is thus a direct consequence of tunneling spectroscopy probing affinity levels instead of ground state unoccupied state alignment.36 Accompanying the LUMO state, a third component could be resolved in the spectra as a peak shifted by 180 meV. The energy separation between the LUMO and this feature coincides with the energy of totally symmetric (anti-symmetric) in-plane phonon of the DCV5T-Me2 backbone, located at 179.4 meV in the Raman (infrared) spectrum.37 We thus attribute it to a vibronic feature excited by electrons during tunneling.38 In fact, the excitation of similar modes of the plain tetrathiophene (4T) molecules has been found to be crucial to explain the polaronic charge mobility in these molecular materials because they are highly coupled to 4T frontier orbitals.39
Fig. 5 displays the 2PPE results obtained for 1 and approx. 3 ML of DCV5T-Me2 on Au(111). The work function for both coverages possesses a value of 5.1 eV. In comparison to the one-color 2PPE spectrum in the sub-monolayer regime, for 1 ML an additional feature close to the Fermi-edge at EFinal − EF = 9.4 eV is observed (see Fig. 5(a)). On the basis of photon energy dependent measurements we can assign this feature to photoemission from an occupied state located at −0.1 eV below EF. This state is also visible in the two-color 2PPE data at EFinal − EF = 7.05 eV. We assign this state to the HOMO of DCV5T-Me2. The two-color 2PPE spectrum is dominated by a broad photoemission between EFinal − EF = 6 eV and 7 eV (see Fig. 5(b)). From photon energy dependent experiments we are able to identify a further occupied state located at −0.6 eV below EF which we assign to the HOMO−1. A striking feature is observed at EFinal − EF = 5.4 eV labeled as Exc in Fig. 5(b). This feature is the result of photoemission from an unoccupied state located 0.7 eV above EF. We assign it to an excitonic state (molecular Frenkel exciton). The photoemission intensity from this excitonic state increases drastically with increasing coverage as can be seen already in the 2PPE spectrum from 3 ML (see Fig. 5(c)). For a higher coverage (>6 ML) the spectrum is dominated by this feature, hiding completely the photoemission features associated with the LUMO and LUMO+1. As discussed below, most likely two excitonic states contribute to this broad photoemission signal. At a coverage of 3 ML we observed (see Fig. 5(d)) two new features located at 1.8 and 1.95 eV above EF in the two-color 2PPE spectrum. According to the STS results (see Fig. 4(c)), we assign them to the LUMO and LUMO+1 of the molecules in the second decoupled layer.
 | ||
| Fig. 5 (a and b) One- and two-color 2PPE spectra of 1 ML DCV5T-Me2, respectively. (c and d) Corresponding data for 3 ML. * indicates that the peaks originate from molecules in the second or higher layer. | ||
Electronic states as a function of coverage
Merging together the STS and 2PPE data, we obtain a comprehensive picture of the energetic positions of the adsorbate-derived unoccupied and occupied electronic states. The energy levels and their assignments as a function of coverage are summarized in Fig. 6. Note that electronic states determined using 2PPE are transport states since in the 2PPE the unoccupied states (e.g. LUMOs) are populated via a metal-to-molecule electron transfer (creating a negative ion resonance) and not through an intramolecular charge transfer. Occupied electronic states are ionized, creating a positive ion. Thus we obtain the ionization potential or the electron affinity of the respective molecular state and for the quasi-particle (the exciton) the ionization potential is measured. The same is true for STS, since electron tunneling into unoccupied states leads to the creation of negative ion resonances; tunneling out of occupied states to the generation of positive ion resonances. Hence, both techniques allow the determination of transport levels and accordingly transport gaps. The lowest exciton state is generated by exciting an electron from the HOMO to the LUMO (the molecule remains neutral). The energy needed for this process is Eopt, the optical gap. Eopt is lower than the difference between the ionization potential and electron affinity of the respective HOMO and LUMO levels (transport gap).40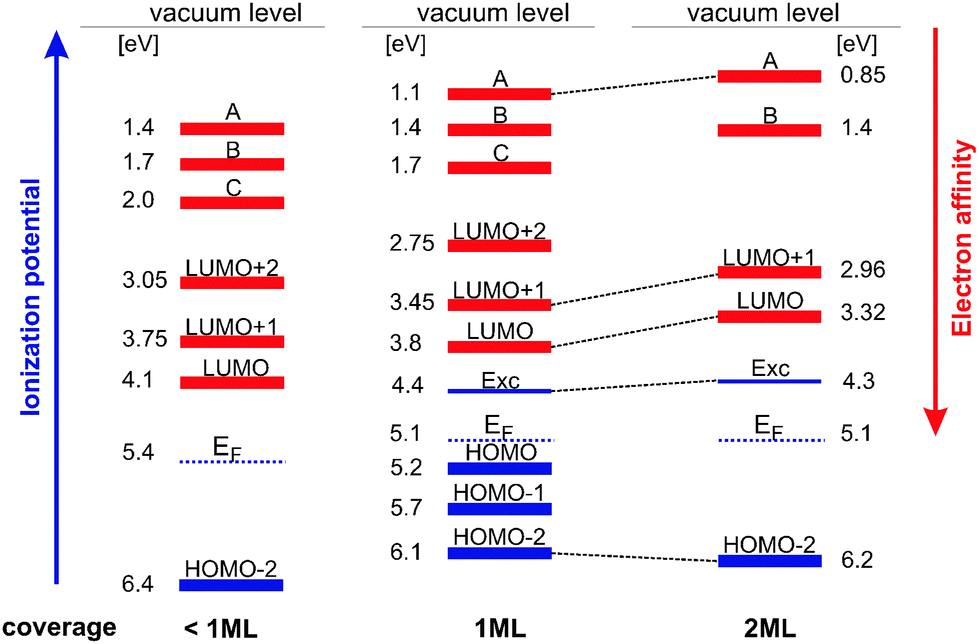 | ||
| Fig. 6 Energy level diagram as a function of DCV5T-Me2 coverage. The blue levels are the ionization potentials (left axis) and the red ones are the electron affinities (right axis); EF denotes the Au(111) Fermi level. | ||
Comparing the sub-monolayer and monolayer coverage, the main difference is a shift of the work function which affects all energy levels equally. Furthermore, the HOMO and HOMO−1 are not observed in 2PPE at sub-MLs, which is most likely due to the weak signal from a very small number of molecules. At 1 ML, with the LUMO level at 3.8 eV and the HOMO at 5.2 eV we obtain a transport gap of 1.4 eV. For comparison, DCV5T-Me2 in solution possesses oxidation and reduction potentials of −3.75 and −5.66 eV measured using an electrochemical technique of cyclic voltammetry corresponding to an energy gap of 1.91 eV,11 0.5 eV larger than the gap measured in our study. This clearly indicates that the metallic substrate leads to a decrease of the gap size due to screening of molecular charge states.36 The excitonic state is located 0.6 eV below the affinity level associated with the LUMO. For comparison, in sexithiophene/Au(111) an energy difference of 0.9 eV has been found.20
To clarify the origin of the higher lying unoccupied states labeled as A, B, and C we performed angular-resolved 2PPE measurements. Both states A and B show no dispersion (the signal of state C was too weak to resolve it). Additionally, they are not pinned to the vacuum level. Thus, we can exclude that they arise from image potential states (IPS), since IPS possess a free-electron like dispersion and are pinned to the vacuum level. Other possibilities are: (i) interface states (IS), (ii) charge-transfer excitons41,42 on the surface of the DCV5T-Me2 with the image band serving as an electron acceptor, or (iii) higher lying molecule-derived states. Considering possibility (i): IS have been observed at organic/metal interfaces, for instance at the interface of an imine-derivative at Au(111).43 There two delocalized unoccupied electronic states have been identified, which have been proposed to originate from a modified image potential at the interface. At the perylene-tetracarboxylic acid dianhydride (PTCDA) on the Ag(111) and Ag(100) interface,44–46 respectively, an interface state generated from a Shockley resonance has been suggested. Thereby the organic overlayer induced energetic shifts of a Shockley resonance. However, interface states disperse and do not reproduce the absence of angular dispersion found for states A and B. In both cases (ii) and (iii) one would expect no dispersion of the respective states, as we find here. Photoemission from an exciton (case ii) does not show dispersion since the photoionization process destroys the exciton (quasiparticle).47 The dispersion of molecular states (case iii) is highly improbable in flat-lying molecular layers. Hence, possibilities (ii) and (iii) have to be taken into account.
Going to higher coverages, i.e., above one ML, we observe pronounced shifts in energy for several molecular states, most dominant for the LUMO and LUMO+1 levels, where a shift of ≈0.5 eV is found. This effect is attributed to electronic decoupling from the metallic substrate. In contrast, for the octithiophene (8T)/Au(111) no energetic shift of the LUMO and LUMO+1 in the first and second layer was found.24 In that case the molecules were arranged in a tilted geometry with respect to the surface plane, which allowed for intermolecular π–π interaction. With such adsorption geometry, already the adsorbate/substrate interaction in the first layer is not strong and accordingly, the electronic coupling. Therefore no significant decoupling effect was observed contrary to the present system, where DCV5T-Me2 adsorbs in a flat configuration. Hence, there is a stronger coupling with metal states. The first layer then acts as an isolating layer resulting in electronically decoupled molecular states in the second layer. The comparison between the two systems DCV5T-Me2/Au(111) and 8T/Au(111) nicely pictures the effect of the adsorption geometry on the electronic structure of the adsorbate/substrate system (structure/property relationship).
Excited state dynamics
In the following, we investigate the excited state dynamics using femtosecond time-resolved 2PPE and show that they are also strongly influenced by the electronic coupling between the adsorbate and the surface at the interface. In the low-coverage regime (1–2 ML), the lifetimes of electrons in the molecular excited states, viz. the LUMO and LUMO+1 as well as the excitonic state possess values that are below the limit of our experimental resolution (τ < 10 fs). These ultrashort lifetimes point towards an efficient back transfer of electrons to the metal substrate. At metallic surfaces such short lifetimes of excited states, i.e., in the range of fs, have been found for several adsorbates.43,48–50 Contrarily, for higher coverages long lifetimes are found as can be seen in Fig. 7(a) for 20 ML DCV5T-Me2 on Au(111). In this false color two-dimensional representation, the 2PPE intensity at a particular energy, EFinal − EF, is displayed as a function of time delay (Δt) between the two laser pulses hν1 = 2.4 eV and hν2 = 4.8 eV. Thereby positive time delays represent delayed UV pulses (hν2, probe pulse). In the high coverage regime the excitation in 2PPE involves an intramolecular process, e.g. a HOMO–LUMO transition. The optical gap in a condensed DCV5T-Me2 film is 1.69 eV as determined using UV/VIS absorption spectroscopy.11 The absorption spectrum shows a broad band between 730 and ≈450 nm,11 which is attributed to the S0 → S1 (HOMO → LUMO) well as the S0 → S2-transition (HOMO → LUMO+1). Thus with a pump pulse photon energy of 2.4 eV (corresponding to 520 nm) we are able to induce both transitions. Therefore we assign the broad 2PPE signal to excitonic levels related to the LUMO and LUMO+1. As mentioned above with increasing coverage the 2PPE spectrum is dominated by this feature, which possesses a peak width much larger than the width of all other observed signals. This also underlines that only one excitonic state does not contribute to the signal. | ||
| Fig. 7 (a) Two-dimensional spectrum of time-resolved 2PPE measurements of 20 ML DCV5T-Me2 on Au(111) recorded with hν1 = 2.4 eV and hν2 = 4.8 eV. Positive pump–probe delay represents a delayed UV pulse, hν2. (b) Cross correlation (XC) trace of the 2PPE intensity integrated over the excited state peak intensities (XC energy range in (a)). The data can be described with an exponential decay using four different time constants τ1–τ4 (black solid line). | ||
The cross correlation (XC) curve for the energy region of the excited states is shown in Fig. 7(b). The solid line corresponds to a fit used to determine lifetimes. The fit model contains a Gaussian function representing the laser pulse duration convoluted with the response function of the intermediate state. The response function is not a single exponential due to different excitonic states involved in the excited state dynamics. A superposition of four exponential decays with different time constants τ1–τ4 results into agreement with the data. For a coverage of 20 ML the time constants are τ1 = 130 ± 13 fs, τ2 = 1 ± 0.1 ps, τ3 = 11 ± 1.1 ps, and τ4 = 390 ± 39 ps. Coverage-dependent measurements showed similar excited state dynamics. In all cases an exponential decay using four different time constants is needed to describe the data and the lifetimes increase with rising coverage. A coverage-dependent decay time indicates that the excited state decays via two relaxation channels, an intrinsic and a distance-dependent channel.21,49 The intrinsic decay rate corresponds to the decay in the bulk material and the distance-dependent (external) rate to quenching by the metal substrate.
So far neither time-dependent measurements on excited state dynamics of DCV5T-Me2 both in the liquid or condensed phase nor electronic structure calculations are available in the literature. However we propose that the following processes may play a role. An initial hot exciton population which is highly delocalized within the first and second excitonic band structure localizes on an ultrafast time-scale (τ1 = 130 ± 13 fs). The relaxation and localization of higher non-relaxed delocalized excitonic states on these short time-scales have also been observed in other organic semiconductors.51–54 Strong coupling of excited electrons to phonons within the film can explain the very effective energetic relaxation within the first picosecond. STS on the second decoupled DCV5T-Me2 layer showed vibronic states (see Fig. 4(c)) indicating strong electron–vibrational coupling with vibrational states of around 180 meV.37,55 The longer lived components (τ3 = 11 ± 1.1 ps and τ4 = 390 ± 39 ps) may be associated with the lifetime of the lowest lying excitonic state as well as polarons or electrons trapped into defect sites.
Conclusions
In summary, we have determined the electronic structure, i.e., the energetic position of affinity levels and ionization potentials of DCV5T-Me2 adsorbed on a Au(111) surface as a function of coverage using scanning tunneling spectroscopy and two-photon photoemission (2PPE). Several unoccupied molecular electronic states shift to higher energies with respect to the Fermi level of Au(111) in the second adsorbate layer, indicating an efficient electronic decoupling from the metallic substrate. For molecular multilayers, we identified the presence of excitonic states, becoming stronger with the film thickness. Femtosecond time-resolved 2PPE pictured the excited state dynamics of the two lowest exciton bands as composed of four processes with different time scales. The quantitative determination of the energetic position of molecular electronic levels with respect to the Fermi level of an electrode and the elucidation of the charge carrier and exciton dynamics are important for the improvement and optimization of molecule-based device performance.Acknowledgements
Funding by the Deutsche Forschungsgemeinschaft through the priority program SPP 1355 and the collaborative research center Sfb 658 is gratefully acknowledge. Z.Y. acknowledges the Chinese CSC program for his grant. M.C. acknowledges for her research grant the Alexander von Humboldt Foundation. J.I.P. acknowledges financial support from Spanish MINECO (MAT2013-46593-C6-01).References
- H. Katz and J. Huang, Thin-Film Organic Electronic Devices, Annu. Rev. Mater. Res., 2009, 39, 1–92 CrossRef.
- N. Koch, Organic Electronic Devices and Their Functional Interfaces, ChemPhysChem, 2007, 8, 1438–1455 CrossRef CAS PubMed.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Conjugated Polymer-Based Organic Solar Cells, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- K. Walzer, B. Maennig, M. Pfeiffer and K. Leo, Organic Devices Based on Electrically Doped Transport Layers, Chem. Rev., 2007, 107, 1233–1271 CrossRef CAS PubMed.
- H. Ishii, K. Sugiyama, E. Ito and K. Seki, Energy Level Alignment and Interfacial Electronic Structures at Organic/Metal and Organic/Organic Interfaces, Adv. Mater., 1999, 11, 605–625 CrossRef CAS.
- S. Braun, W. R. Salaneck and M. Fahlman, Energy-Level Alignment at Organic/Metal and Organic/Organic Interfaces, Adv. Mater., 2009, 21, 1450–1472 CrossRef CAS PubMed.
- M. Oehzelt, N. Koch and G. Heimel, Organic Semiconductor Density of States Controls the Energy Level Alignment at Electrode Interfaces, Nat. Commun., 2014, 5, 4174 CAS.
- A. Mishra, C. Uhrich, E. Reinold, M. Pfeiffer and P. Bäuerle, Synthesis and Characterization of Acceptor-substituted Oligothiophenes for Solar Cell Applications, Adv. Energy Mater., 2011, 1, 265–273 CrossRef CAS PubMed.
- H. Ziehlke, R. Fitzner, C. Körner, R. Gresser, E. Reinold, P. Bäuerle, K. Leo and M. Riede, Side Chain Variations on a Series of Dicyanovinyl-Terthiophenes: A Photoinduced Absorption Study, J. Phys. Chem. A, 2011, 115, 8437–8446 CrossRef CAS PubMed.
- R. Fitzner, E. Reinold, A. Mishra, E. Mena-Osteritz, H. Ziehlke, C. Körner, K. Leo, M. Riede, M. Weil, O. Tsaryova, A. Weiß, C. Uhrich, M. Pfeiffer and P. Bäuerle, Dicyanovinyl-substituted Oligothiophenes: Structure-Property Relationships and Application in Vacuum-Processed Small-Molecule Organic Solar Cells, Adv. Funct. Mater., 2011, 21, 897–910 CrossRef CAS PubMed.
- R. Fitzner, E. Mena-Osteritz, A. Mishra, G. Schulz, E. Reinold, M. Weil, C. Körner, H. Ziehlke, C. Elschner, K. Leo, M. Riede, M. Pfeiffer, C. Uhrich and P. Bäuerle, Correlation of π-Conjugated Oligomer Structure with Film Morphology and Organic Solar Cell Performance, J. Am. Chem. Soc., 2012, 134, 11064–11067 CrossRef CAS PubMed.
- B. Baumeier, D. Andrienko, Y. Ma and M. Rohlfing, Excited States of Dicyanovinyl-Substituted Oligothiophenes from Many-Body Green's Functions Theory, J. Chem. Theory Comput., 2012, 8, 997–1002 CrossRef CAS.
- M. M. Bader, P.-T. T. Pham and E. H. Elandaloussi, Dicyanovinyl-Substituted Oligothiophenes, Cryst. Growth Des., 2010, 10, 5027–5030 CAS.
- R. Meerheim, C. Körner and K. Leo, Highly Efficient Organic Multi-Junction Solar Cells with a Thiophene Based Donor Material, Appl. Phys. Lett., 2014, 105, 063306 CrossRef PubMed.
- I. Hill, A. Kahn, Z. Soosb and J. R. A. Pascal, Charge-Separation Energy in Films of π-Conjugated Organic MSolecules, Chem. Phys. Lett., 2000, 327, 181–188 CrossRef CAS.
- M. Kiguchi, S. Entani, K. Saikil and G. Yoshikawa, One-Dimensional Ordered Structure of α-Sexithienyl on Cu(110), Appl. Phys. Lett., 2004, 84, 3444–3446 CrossRef CAS PubMed.
- N. Koch, G. Heimel, J. Wu, E. Zojer, R. L. Johnson, J.-L. Brédas, K. Müllen and J. P. Rabe, Influence of Molecular Conformation on Organic/Metal Interface Energetics, Chem. Phys. Lett., 2005, 413, 390–395 CrossRef CAS PubMed.
- M. Kiel, K. Duncker, C. Hagendorf and W. Widdra, Molecular Structure and Chiral Separation in α-sexithiophene Ultrathin Films on Au(111): Low-energy Electron Diffraction and Scanning Tunneling Microscopy, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 195439 CrossRef.
- M. Grobosch and M. Knupfer, Charge-Injection Barriers at Realistic Metal/Organic Interfaces: Metals Become Faceless, Adv. Mater., 2007, 19, 754 CrossRef CAS PubMed.
- E. Varene, I. Martin and P. Tegeder, Optically Induced Inter- and Intrafacial Electron Transfer Probed by Two-Photon Photoemission: Electronic States of Sexithiophene on Au(111), J. Phys. Chem. Lett., 2011, 2, 252–256 CrossRef CAS.
- E. Varene, L. Bogner, C. Bronner and P. Tegeder, Ultrafast Exciton Population, Relaxation, and Decay Dynamics in Thin Oligothiophene Films, Phys. Rev. Lett., 2012, 109, 7601 CrossRef.
- T. Yokoyama, S. Kurata and S. Tanaka, Direct Identification of Conformational Isomers of Adsorbed Oligothiophene on Cu(100), J. Phys. Chem. B, 2006, 110, 18130 CrossRef CAS PubMed.
- T. Kakudate, S. Tsukamoto, M. Nakaya and T. Nakayama, Initial Stage of Adsorption of Octithiophene Molecules on Cu(111), Surf. Sci., 2006, 605, 1021–1026 CrossRef PubMed.
- E. Varene, Y. Pennec and P. Tegeder, Assembly and Electronic Structure of Octithiophene on Au(111), Chem. Phys. Lett., 2011, 515, 141–145 CrossRef CAS PubMed.
- E. Varene, L. Bogner, S. Meyer, Y. Pennec and P. Tegeder, Coverage-Dependent Adsorption Geometry of Octithiophene on Au(111), Phys. Chem. Chem. Phys., 1012, 14, 691–696 RSC.
- Z. Yang, M. Corso, R. Robles, C. Lotze, R. Fitzner, E. Mena-Osteritz, P. Bäuerle, K. Franke and J. Pascual, Orbital Redistribution in Molecular Nanostructures Mediated by Metal–Organic Bonds, ACS Nano, 2014, 8, 10715–10722 CrossRef CAS PubMed.
- F. J. Giessibl, High-Speed Force Sensor for Force Microscopy and Profilometry Utilizing a Quartz Tuning Fork, Appl. Phys. Lett., 1998, 73, 3956–3958 CrossRef CAS PubMed.
- Note that we utilized the DFT calculations just for visualizing of the molecular orbital shapes since they are used for assignment of the dI/dV maps and not for their energy level alignment. In the latter case special types of functionals (range-separated DFT) have to be used as demonstrated e.g. for various thiophenes [ B. M. Wong, et al. , Phys. Chem. Chem. Phys., 2009, 11, 4498 RSC; T. Körzdörfer, et al. , J. Chem. Phys., 2011, 135, 204107 CrossRef CAS PubMed ].
- X.-Y. Zhu, Electronic Structure and Electron Dynamics at Molecule-Metal Interfaces: Implications for Molecule-Based Electronics, Surf. Sci. Rep., 2004, 56, 1–83 CrossRef CAS PubMed.
- P. Tegeder, Optically and Thermally Induced Molecular Switching Processes at Metal Surfaces, J. Phys.: Condens. Matter, 2012, 24, 394001 CrossRef PubMed (34 pp).
- C. Bronner, G. Schulze, K. Franke, J. I. Pascual and P. Tegeder, Switching Ability of Nitro-Spiropyran on Au(111): Electronic Structure Changes as a Sensitive Probe During a Ring-Opening Reaction, J. Phys.: Condens. Matter, 2011, 23, 484005 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr, T. Vreven, K. N. Kudin and J. C. Burant, et al., Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- H. Eckardt, L. Fritsche and J. Noffke, Self-Consistent Relativistic Band Structure of the Noble Metals, J. Phys. F: Met. Phys., 1984, 14, 97–112 CrossRef CAS.
- R. Courths, H. G. Zimmer, A. Goldmann and H. Saalfeld, Electronic Structure of Gold: An Angle-Resolved Photoemission Study Along the Γ Line, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 3577 CrossRef CAS.
- G. Nicolay, F. Reinert, S. Hüfner and P. Blaha, Spin-Orbit Splitting of the L-Gap Surface State on Au(111) and Ag(111), Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 65, 033407 CrossRef.
- I. F. Torrente, K. J. Franke and J. I. Pascual, Spectroscopy of C60 Single Molecules: The Role of Screening on Energy Level Alignment, J. Phys.: Condens. Matter, 2008, 20, 184001 CrossRef.
- Z. Yang, Structural and Electronic Properties of Thiophene-based Supramolecular Architectures on Metal Surfaces, PhD thesis, Freie Universität Berlin, 2014 Search PubMed.
- K. J. Franke and J. I. Pascual, Effects of Electron-Vibration Coupling in Transport Through Single Molecules, J. Phys.: Condens. Matter, 2012, 24, 394002 CrossRef PubMed (15 pp).
- S. M. Falke, C. A. Rozzi, D. Brida, M. Maiuri, M. Amato, E. Sommer, A. D. Sio, A. Rubio, G. Cerullo, E. Molinari and C. Lienau, Coherent Ultrafast Charge Transfer in an Organic Photovoltaic Blend, Science, 2014, 344, 1001–1005 CrossRef CAS PubMed.
- M. Muntwiler and X.-Y. Zhu, in Dynamics at Solid State Surfaces and Interfaces, ed. U. Bovensiepen, H. Petek and M. Wolf, Wiley-VCH, 2010 Search PubMed.
- M. Muntwiler, Q. Yang, W. Tisdale and X.-Y. Zhu, Coulomb Barrier for Charge Separation at an Organic Semionductor Interface, Phys. Rev. Lett., 2008, 101, 196403 CrossRef.
- X.-Y. Zhu, Q. Yang and M. Muntwiler, Charge-Transfer Excitons at Organic Semiconductor Surfaces and Interfaces, Acc. Chem. Res., 2009, 42, 1779–1787 CrossRef CAS PubMed.
- S. Hagen, Y. Luo, R. Haag, M. Wolf and P. Tegeder, Electronic Structure and Electron Dynamics at an Organic Molecule/Metal Interface: Interface States of Tetra-tert-butyl-imine/Au(111), New J. Phys., 2010, 12, 125022 CrossRef (17 pp).
- C. H. Schwalb, S. Sachs, M. Marks, A. Schöll, F. Reinert, E. Umbach and U. Höfer, Electron Lifetime in a Shockley-Type Metal-Organic Interface State, Phys. Rev. Lett., 2008, 101, 146801 CrossRef CAS.
- M. Galbraith, M. Marks, R. Tonner and U. Höfer, Formation of an Organic/Metal Interface State from a Shockley Resonance, J. Phys. Chem. Lett., 2014, 5, 50–55 CrossRef CAS PubMed.
- M. Marks, A. Schöll and U. Höfer, Formation of Metal-Organic Interface States Studied with 2PPE, J. Electron Spectrosc. Relat. Phenom., 2014, 195, 263–271 CrossRef CAS PubMed.
- M. Weinelt, M. Kutschera, T. Fauster and M. Rohlfing, Dynamics of Exciton Formation at the Si(100) c(4 × 2) Surface, Phys. Rev. Lett., 2004, 92, 126801 CrossRef.
- A. Yang, S. T. Shipman, S. Garrett-Roe, J. Johns, M. Strader, P. Szymanski, E. Muller and C. B. Harris, Two-Photon Photoemission of Ultrathin Film PTCDA Morphologies on Ag(111), J. Phys. Chem. C, 2008, 112, 2506 CAS.
- G. Dutton, D. P. Quinn, C. D. Lindstrom and X.-Y. Zhu, Exciton Dynamics at Molecule-Metal Interfaces: C60/Au(111), Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 045441 CrossRef.
- C. Bronner, M. Schulze, S. Hagen and P. Tegeder, The Influence of the Electronic Structure of Adsorbate-Substrate Complexes on the Photoisomerization Ability, New J. Phys., 2012, 14, 043032 CrossRef.
- M. Marks, S. Sachs, C. H. Schwalb, A. Schöll and U. Höfer, Electronic Structure and Excited State Dynamics in Optcally Excited PTCDA Films Ivestigated with Two-Photon Photoemission, J. Chem. Phys., 2013, 139, 124701 CrossRef CAS PubMed.
- E. Engel, M. Koschorreck, K. Leo and M. Hoffmann, Ultrafast Relaxation in Quasi-One-Dimensional Organic Molecular Crystals, Phys. Rev. Lett., 2005, 95, 157403 CrossRef CAS.
- J. Guo, H. Ohkita, H. Benten and S. Ito, Near-IR Femtosecond Transient Absorption Spectroscopy of Ultrafast Polaron and Triplet Exciton Formation in Polythiophene Films with Different Regioregularities, J. Am. Chem. Soc., 2009, 131, 16869–16880 CrossRef CAS PubMed.
- K. Chen, A. Barker, M. Reish, K. Gordon and J. M. Hodgkiss, Broadband Ultrafast Photoluminescence Spectroscopy Resolves Charge Photogeneration via Delocalized Hot Excitons in Polymer: Fullerene Photovoltaic Blends, J. Am. Chem. Soc., 2013, 135, 18502–18512 CrossRef CAS PubMed.
- J. Repp, P. Liljeroth and G. Meyer, Coherent Electron-Nuclear Coupling in Oligothiophene Molecular Wires, Nat. Phys., 2010, 6, 975–979 CrossRef CAS PubMed.
Footnotes |
| † Current address: Fritz-Haber-Institut der MPG, Germany. |
| ‡ Current address: Centro de Fisica de Materiales, San Sebastian, Spain. |
| This journal is © the Owner Societies 2015 |