In situ ATR-FTIR study of H2O and D2O adsorption on TiO2 under UV irradiation†
Received
8th July 2015
, Accepted 6th August 2015
First published on 6th August 2015
Abstract
The adsorption of water and deuterium oxide on TiO2 surfaces was investigated in the dark as well as under UV(A) irradiation using in situ ATR-FTIR spectroscopy under oxygen and oxygen free conditions. Adsorption of H2O–D2O mixtures revealed an isotopic exchange reaction occurring onto the surface of TiO2 in the dark. Under UV(A) irradiation, the amount of both OH and OD groups was found to be increased by the presence of molecular oxygen. Furthermore, the photocatalytic formation of hydroperoxide under oxygenated condition has been recorded utilizing Attenuated Total Reflection Fourier Transformed Infrared (ATR-FTIR) spectroscopy which appeared as new band at 3483 cm−1. Different possible mechanisms are discussed in terms of the source of hydroxyl groups formed and/or hydration water on the TiO2 surface for the photocatalytic reaction and photoinduced hydrophilicity.
1. Introduction
Photocatalytic reactions at the surface of titanium dioxide (TiO2) have been attracting much attention, in view of their practical applications including environmental aspects such as water purification, self-cleaning and antifogging.1,2 UV(A) irradiation of the TiO2 surface induces photocatalytic reactions and photoinduced hydrophilicity.3 In general, electrons and holes generated after the band gap excitation can respectively, reduce and oxidize electron acceptor and electron donor species which are already adsorbed on the TiO2 surface. The importance of the photogenerated active oxygen species, such as hydroxyl radicals (˙OH), superoxide radicals (O2˙−) and hydrogen peroxide (H2O2) in photocatalytic applications has been extensively studied.4,5 The mechanism of hydroxyl radical formation has been suggested as a result of the oxidation of water,6 while hydrogen peroxide can be formed by both reduction and oxidation processes, such as dimerization of hydroxyl radicals, as well as disproportionation and reduction of superoxide radical anions.5 On the other hand, the photoinduced hydrophilicity is reported as a result of increasing the number of hydroxyl groups on the TiO2 surface upon irradiation.7 During these processes, the surface OH groups on the TiO2 surface have been assumed to play important roles in both photocatalytic reactions and photoinduced hydrophilicity.8 Adsorbed species such as water and molecular oxygen, however, are expected to affect the hydroxyl group behaviour on the TiO2 surface,9 but their influences have to date not yet been clearly determined.
Several mechanisms regarding the interaction of UV(A) light and hydroxyl group formation on TiO2 have been proposed by different research groups.8 One of the proposed mechanisms for the photoinduced hydrophilicity is based on the formation of oxygen vacancies and the reconstruction of Ti–OH bonds caused by the photogenerated electron–hole pairs.7,10 Another mechanism has been suggested to involve the photocatalytic removal of organic contaminants from the surface.11 Hashimoto and co-workers have followed the idea of photogenerated surface OH groups to introduce their surface reconstruction model of hydroxyl groups as the mechanism for the hydrophilic conversion.12,13 Another mechanism was proposed by Yates et al., who demonstrated that only under UV light illumination the decomposition of organic contaminants can take place leading to the creation of superhydrophilic surfaces.11 In other studies, Munuera et al. and Anpo et al. have reported that H2O desorbs from the TiO2 surface during UV light irradiation, i.e., photodesorption of H2O, due to the heating effect of the light source.14,15 Recently, special attention has been focused on the particle network before and after irradiation and how it changes during illumination.16,17 Wang et al. reported that the metal oxide nanoparticles tend to form 3D networks via a self-aggregation mechanism when suspended in aqueous solution.16 Some investigations indicated that UV light activation of TiO2 leads to an increase in the surface area due to de-aggregation of particles agglomerates which in turn enhances the photonic efficiency.17,18 The behaviours of the water adsorption and the associated hydroxyl groups during irradiation are, however, still not well understood. The relative roles of the active oxygen and the hydroxyl groups for the photocatalytic process and for the photoinduced hydrophilicity, which have been under debate for a long time, still need to be investigated in detail. From this point of view, one of the most important issues of the current investigations concerning photocatalytic reactions and photoinduced hydrophilicity is to understand the fundamental process of water adsorption and the source of hydroxyl groups formed on the TiO2 surface under UV irradiation.
Herein, a detailed investigation of the H2O and/or D2O adsorption on the TiO2 surface is reported using Attenuated Total Reflection Fourier Transformed Infrared spectroscopy (ATR-FTIR). Qualitative and quantitative evaluations of the D2O effect and the isotopic exchange on the TiO2 surface are presented. This work focuses on the analysis of the behaviours of OH and OD stretching groups before and after UV irradiation considering the role of molecular oxygen to provide new insights into the mechanism of photoinduced hydrophilicity during UV illumination.
2. Experimental
2.1 Materials
The photocatalyst, titanium dioxide PC-500 (Cristal Global) consisted of 100% pure anatase with a specific BET-surface area of 340 m2 g−1 and a primary particle size of 5–10 nm. Deuterium oxide (D2O) (99.9 atom% D) was purchased from Sigma Aldrich. Deionized water (H2O) was supplied from a Millipore Mill-Q system with a resistivity equal to 18.2 Ω cm at 25 °C.
2.2 ATR-FTIR spectroscopic measurements
2.2.1 Preparation of TiO2 films on the ATR-FTIR crystal.
An aqueous suspension of TiO2 at a concentration of 5.75 g l−1 was initially prepared and sonicated for 15 min in an ultrasonic cleaning bath. An aliquot of 400 μL of the TiO2 suspension was placed on the surface of the ZnSe ATR crystal. This small volume was simply spread by balancing the unit manually. The suspension was then directly allowed to evaporate to dryness by storing the crystal in a semi-opened desiccator at room temperature. Prior to deposition of the TiO2 films, the ZnSe surface (area = 6.8 mm × 72 mm) was cleaned by polishing with 1 mm diamond paste (Metadi II, polishing grade) and rinsed with methanol and deionised water. The coverage of the final dry layer of particles obtained was 2.3 g m−2 and the layer appeared homogeneous under visual inspection. In the original preparation by Hug et al., AFM measurements of layers with coverage of 2.3 g m−2 yielded a thickness of 1–3 μm.19,20 The final resulting layers of particles remained stable over the entire course of the experiment. Thus, it can be assumed that the effective path lengths at all wavelengths remained unchanged.
2.2.2 ATR-FTIR measurement.
The ATR-FTIR spectra of the TiO2 samples were monitored by a FTIR spectrometer (IFS 66 BRUKER) equipped with an internal reflection element 45° ZnSe crystal and a deuterated triglycine sulfate (DTGS) detector. The interferometer and the infrared light path in the spectrometer were constantly purged with Argon to avoid H2O and CO2 contamination. The spectra were recorded with 350 scans at 4 cm−1 resolution and analysed using OPUS version 6.5 software. The background was subtracted and baseline correction was made in order to eliminate minor fluctuations due to instrumental instability. Irradiation of samples with UV(A) light was carried out using an LED lamp (Model LED-Driver, THORLABS) emitting UV light (365 nm). The distance from the UV lamp to the surface of the test solution was kept at 30 cm on which the intensity of UV(A) light was of 1.0 mW cm−2 as measured by an UV radiometer (Dr Hönle GmbH, Martinsried, Germany). Prior to starting the irradiation experiments, spectra of adsorption of H2O and D2O on the TiO2 were monitored in the dark. When the last spectrum of each experiment had been recorded, the UV(A) lamp was turned on and another sequence of spectra was recorded. These two groups of spectra were considered as the blank reference spectra in the dark and under UV(A) illumination, respectively.
3. Results
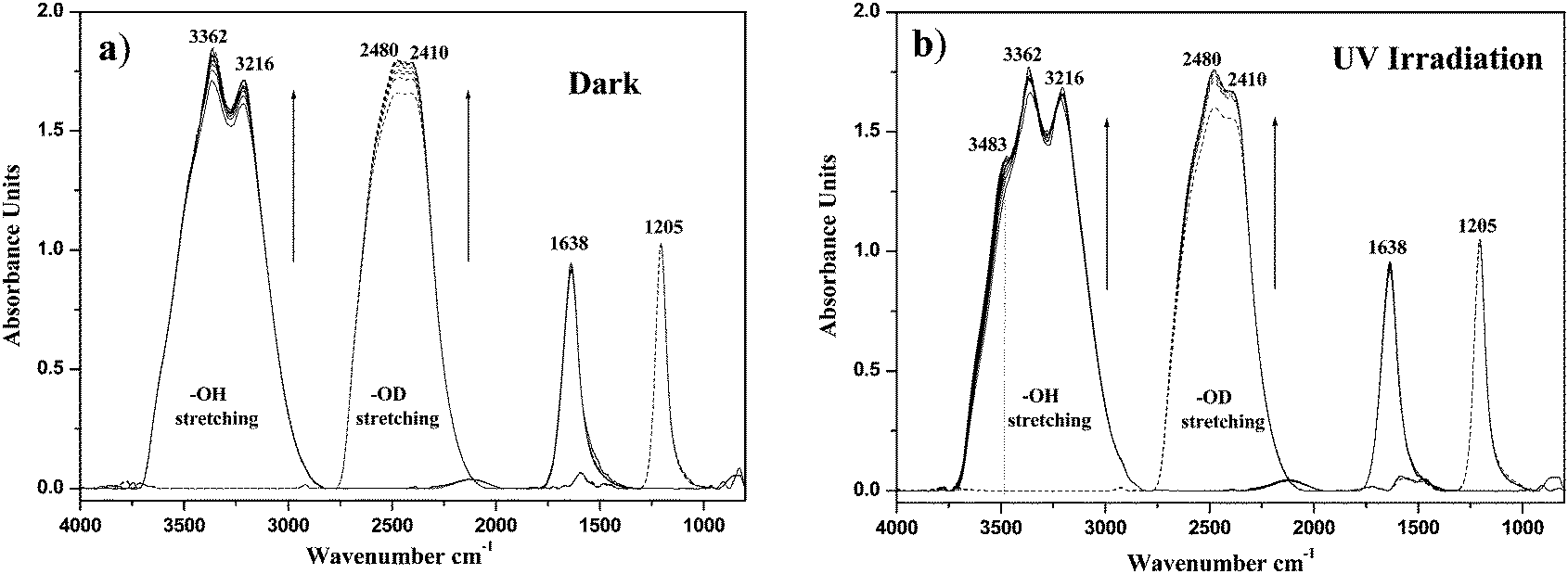
Fig. 1a shows the time evolution of the spectra of adsorbed H2O and D2O on TiO2 in the dark. The signal of the adsorbed water on TiO2 with maxima at 3362, 3216, and 1638 cm−1 can be clearly seen. As is well known, the adsorption of H2O is represented by strong IR absorbances of O–H stretching in the region between 3200 and 3550 cm−1 and δ (H–O–H) bending at 1638 cm−1 which is assigned to undissociated water molecules. When D2O was used instead of H2O, the results showed that all bands of adsorbed water on TiO2 surface were shifted to lower frequency with exchanging H for D. Additionally, an increase in the intensities of OH and OD stretching has been observed by increasing the adsorption time in the dark, which can be explained by increased adsorption of water molecules as well as of deuterium oxide on the TiO2 surface until attaining an equilibrium.
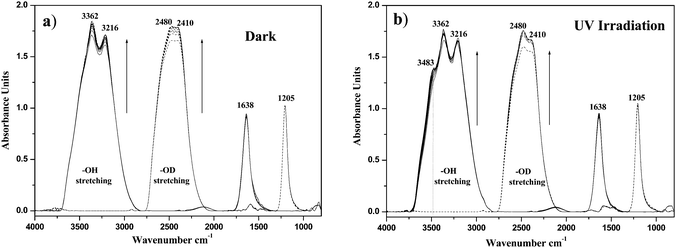 |
| | Fig. 1 Time evolution of the ATR-FTIR spectra of adsorbed pure H2O (solid lines) and D2O (dashed lines) in the presence of O2 on TiO2 (a) in the dark for 4 h, (b) under 8 h of UV(A) illumination. | |
It can be clearly seen from Fig. 1b that subsequent illumination with UV(A) light in the presence of O2, results in the formation of a shoulder in the FTIR spectrum of the adsorbed H2O at 3483 cm−1 as well as an increase in the bands at 2480 cm−1 in the case of D2O (Fig. 1b). The shoulder formed around 3483 cm−1 can be attributed to a photocatalytically generated hydroperoxide. This was confirmed by recording the spectrum of H2O2 added to the water–TiO2 system in the dark in which a similar band (at 3483 cm−1) has been detected (Fig. S1, in ESI†). Furthermore, no formation of such band around 3483 cm−1 was observed in the absence of O2 (Fig. S2, in ESI†).
Fig. 2 shows the spectra of D2O–H2O mixtures adsorbed on TiO2 in the dark. It is obvious from these spectra that the intensity of the band in the OH-stretching region decreased gradually with increasing dosage of D2O. Interestingly, the peak of the isotopologue HDO bending band centred at 1450 cm−1 was formed and increased by increasing the concentration of H2O in D2O until it approached equimolar proportions (Fig. 2 insert). This peak then decreased again with further increases in the amounts of H2O in D2O. The mixture of H2O and D2O reacted and equilibrated via transfer by self-dissociation and recombination to form HDO.21
The equilibrium constant
K of this reaction is given by
| | | K = [HOD]2/[H2O][D2O] Keq = 3.86 at 25 °C | (2) |
To investigate the effect of D
2O on the hydroxyl group behaviour during UV irradiation, a series of experiments for water adsorption with different ratios of D
2O were performed under irradiation in the presence of oxygen.
Fig. 3 shows the time evolution of the intensities of the integrated spectral areas of OH and OD stretching groups that are formed on the surface of TiO
2 before and after UV irradiation. As can be clearly seen, in the dark, at low concentrations of H
2O (H
2O% < D
2O%), a strong decrease in the amount of OH stretching was observed especially in the first hour. Simultaneously, the amount of OD stretching groups adsorbed on TiO
2 increased during this period until the system reached equilibrium. In contrast, at higher concentrations of H
2O (H
2O% > D
2O%), both OH and OD stretching bands increased. Interestingly, at an equimolar mixture of 50% H
2O and D
2O, a decrease in intensity of OH stretching with a simultaneous increase in the OD stretching could be observed in the dark. From these results we suggest that the deuteride ion in the dark shows a stronger adsorption ability than hydroxyl ions at the surface of TiO
2. Thus, the deuteride ions could lead to isotopic exchange by replacing hydroxyl groups adsorbed on the TiO
2 surface (
reaction 3):
| | | Ti − OH + OD− → Ti − OD + OH− | (3) |
It is worth noting that the time required to reach equilibrium for all experiments was nearly constant,
i.e., it took almost 3 hours to approach isotopic exchange equilibrium. Furthermore, this evaluation of OD groups could be associated with the better exchange of the isolated Ti–OH species on TiO
2 at higher concentrations than at lower concentrations of D
2O. Interestingly, upon UV(A) irradiation, an increase in the intensity of the OH stretching could be observed almost immediately as well as an increasing intensity of the OD stretching band,
i.e., Fig. S3 (see ESI
†). These results, however can be largely attributed to either an increase of the amount of OH and OD groups by the formation of new OH and OD groups on the surface or by increasing the amount of H
2O and D
2O molecules chemisorbed on the TiO
2 surface.
 |
| | Fig. 2 ATR-FTIR spectra of D2O–H2O mixtures with different concentrations adsorbed on TiO2 in the dark. Inset: Evolution of the intensity of the integrated spectral areas of H2O, HOD and D2O bending modes. | |
 |
| | Fig. 3 Time evolution of the intensity of the integrated spectral areas of the OH and OD stretching groups before and after UV(A) irradiation with different ratios (H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D2O). :D2O). | |
To determine the effect of molecular oxygen on the behaviour of the H2O and D2O adsorption on the TiO2 surface, the solution was saturated and constantly purged with different types of gases, i.e., oxygen, nitrogen, and argon during the ATR-FTIR measurements. As shown in Fig. 4, the integrated intensity of the OH and OD stretching groups increased significantly upon illumination in the presence of molecular O2. By contrast, when the sample was purged with nitrogen or argon, no increase in the OH and/or OD stretching group was observed. A similar isotopic exchange behaviour, however, was observed during the dark period.
 |
| | Fig. 4 Evolution of the intensity of the integrated spectral areas of OH and OD stretching groups before and after UV irradiation: effect of dissolved O2, N2 and Ar on the adsorption of H2O–D2O on the TiO2 surface. | |
The time evolution of the intensity of the integrated spectral areas of the OH and OD stretching groups before and after UV irradiation at prolonged time periods are shown in Fig. 5. Again as described above under dark conditions, the isotopic exchanges inevitably took place on the TiO2 surface. When the system was subsequently stored again in the dark (after the UV illumination was turned off), however, no more isotopic exchange behaviour was detected and the amount of OH and OD groups remained steady during this period. Additionally, when the system was illuminated again, the amount of OH and OD groups immediately increased.
 |
| | Fig. 5 Time evolution of the intensity of the integrated spectral areas of OH and OD stretching groups before and after UV(A) irradiation with prolonged time (alternately). | |
4. Discussion
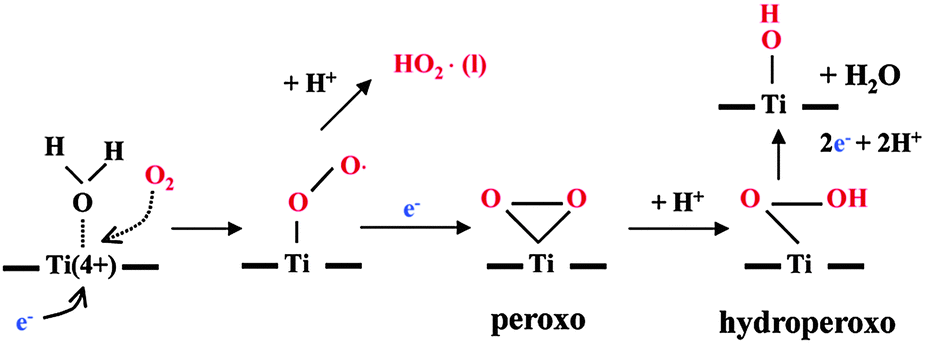
When the TiO2 surface was illuminated with UV light, electron and hole pairs were generated, and they, respectively, reduced and oxidized adsorbates on the surface. As can be seen from the ATR-FTIR spectra (Fig. 1b), a clear shoulder appears at 3483 cm−1 upon irradiation of the system by UV(A) in the presence of molecular oxygen (O2). This peak can most likely be assigned to a Ti–OOH vibration as has been confirmed from the spectrum of H2O2 added to the water–TiO2 system in the dark in which a similar band (at 3483 cm−1) has been detected as shown in Fig. S1 (see ESI†). The formation of this adsorbed hydroperoxy species only in the presence of (O2) can be considered as evidence that it is resulting from the reduction of molecular oxygen adsorbed at the TiO2 surface by the photogenerated electrons rather than from the oxidation of water by the holes. These results are in agreement with Nakamura et al., who revealed and assigned O–O stretching of a surface peroxo species to identify Ti–OOH as a primary intermediate from the O2 photoreduction.22 Robertson et al., however, reported that the formation of hydroxyl radicals for the photocatalytic process could be preferable through the water oxidation at the valence band rather than through the oxygen reduction at the conduction band.6,23 Based on the identification of these intermediates, a mechanism for the photocatalytic formation of hydroperoxide has previously been proposed (Scheme 1).22,24 In this mechanism, the photogenerated conduction band electrons reduce Ti4+ at the surface that adsorbs H2O, then molecular oxygen attacks immediately to form superperoxo TiOO˙. This superperoxo is reduced to peroxo Ti(O2), which is equivalent to the hydroperoxide Ti–OOH when it is protonated. Furthermore, an increase in the intensities of the OH and OD stretching bands is observed after illumination which may be assigned either to the stretching modes of Ti–OH formed upon illumination or to the OH stretching vibration of adsorbed water molecules which in turn increases the hydrophilicity of the TiO2 surface. Hashimoto et al. reported that during UV irradiation not only the photocatalytic reactions take place on the surface but also the photoinduced hydrophilicity resulting from the separation and diffusion of photogenerated electrons and holes occurring on the surface of TiO2.3 As shown in Fig. 3, at low concentrations of H2O (H2O% < D2O%), the reduction of the OH band with the simultaneous increase in the OD stretching band in the dark revealed that the deuteride ions have a stronger adsorption affinity than the hydroxyl ions. Furthermore, it can be clearly observed that the photoinduced hydrophilicity of the TiO2 surfaces which was explained by an increase in the amount of hydroxyl groups is readily induced upon UV light irradiation in the presence of O2. These results can be largely attributed to either an increase in the number of surface OH groups or to an increased adsorption of water molecules on the TiO2 surface.
 |
| | Scheme 1 Proposed mechanism for the photocatalytic formation of hydroperoxides. (Copyright 2003 American Chemical Society.) | |
Several mechanisms suggested that the photoinduced holes play a crucial role in the photoinduced hydrophilicity conversion. It was thus proposed that the photogenerated holes produced in the bulk of TiO2 diffuse to the surface where they are trapped at lattice oxygen sites. The trapped holes are consumed by reaction with the TiO2 itself creating oxygen vacancies,25 or by breaking the bond between the lattice titanium and oxygen ions upon the coordination of water molecules at the titanium site.10 Subsequently the water molecule dissociates in an oxygen vacancy or releases a proton for charge compensation to produce new OH groups. As shown in Fig. 5, however, an isotopic exchange between deuteride ions and hydroxyl groups takes place on the TiO2 surface in the dark according to eqn (3). After 6 h of irradiation even during a prolonged time in the dark, however, no isotopic exchange was detected between hydroxyl groups and deuteride ions. From this result we suggest that the increase of OH and OD stretching groups under irradiation is most likely caused by adsorption of H2O and D2O molecules, respectively, and not by the generation of hydroxyl groups. There have been several reports supporting our suggestion that water molecules strongly adsorb onto the TiO2 surface under UV illumination. Nosaka et al. reported that the increase in the amount of adsorbed water on the TiO2 surface after UV light irradiation was observed by 1H NMR spectroscopy.26 Another study by Uosaki et al. using SFG spectroscopy confirmed that the UV illumination increased not only the amount of adsorbed water but also the order of the adsorbed water on the TiO2 surface.27 Although the mechanism for the H2O/UV light interactions still remains uncertain, it seems that the particle network during UV light irradiation might be responsible for the enhancement of water adsorption on a TiO2 particle film due to a new distribution of the particles. Wang et al. revealed the fact that under UV(A) illumination the total exposed TiO2 surface increases due to the de-aggregation of particles agglomerates which was explained by assuming that part of the absorbed light energy is converted non-adiabatically into heat which is subsequently used to break the bonds between the particles thus producing additional surface area for the photocatalytic process.17,28 One of the first ATR-FTIR studies that focused on the adsorption phenomena in aqueous solution was performed by Mendive et al., who combined experimental data and theoretical calculations to demonstrate that the water molecule can fill the space in between the particles which was demonstrated by the increase in the IR band corresponding to the bending mode of water.29
We propose that UV irradiation leads to an increase in the amount of adsorbed water by thermal processes with hydrophilicity effects simultaneously taking place on the TiO2 surface. Moreover, as shown in Fig. 4, the results clearly indicate that the presence of O2 is necessary to enhance the photoadsorption of H2O and D2O on TiO2 surfaces during UV(A) irradiation. This confirms that the adsorption of water and D2O to TiO2 surfaces is caused by a photoinduced charge transfer process in the presence of molecular oxygen. Takeuchi et al.30 proposed a mechanism which revealed that when TiO2 surfaces were irradiated with UV light in the absence of O2, the electrons trapped on the Ti3+ sites were not scavenged by O2 and the holes trapped on the TiO2 surface were immediately consumed to oxidize the lattice oxygen, resulting in the formation of oxygen vacancies. Such photoreduced TiO2 surfaces can behave as negatively charged surfaces. Thus, water molecules could hardly adsorb on the photoreduced surfaces due to repulsive effects (Scheme 2). On the other hand, when the TiO2 surfaces are irradiated in the presence of O2, the surface trapped electrons are scavenged by O2 and the photoreduced surfaces are immediately oxidized. In this way, the TiO2 surface is very likely interacting with water molecules due to interaction effects resulting in an increase in the amount of adsorbed water. The results of this investigation indicate that the photoinduced hydrophilic effect is achieved by an increase in the amount of adsorbed H2O molecules and this phenomenon occurs not only by a thermal process but also by a photoinduced charge transfer process.
 |
| | Scheme 2 Interaction model of adsorbed water during UV(A) light irradiation in the absence and presence of O2 molecules. | |
5. Conclusions
The behaviour of adsorbed H2O and D2O in photocatalytic processes and in photoinduced hydrophilicity on TiO2 surfaces has been addressed using ATR-FTIR spectroscopy. This study made it possible to investigate adsorption processes at the TiO2/aqueous solution interface quantitatively and in situ. It was shown that the photocatalytic reduction of O2 at the anatase surface leads to the formation of hydroperoxo groups as demonstrated by a FTIR band at 3483 cm−1. Adsorption of H2O with different ratios of D2O on TiO2 revealed that the deuteride ions exhibit stronger adsorption ability than hydroxyl ions resulting in an isotopic exchange reaction which takes place in the dark. Upon illumination with UV light in the presence of O2 both OH and OD groups are formed, which in turn increase the hydrophilicity of the TiO2 surface. In contrast, when the solution was saturated with argon or nitrogen, no formation of OH and OD groups was detected and the hydrophilicity was strongly inhibited. The increase in the amount of OH and OD groups is suggested to be caused by photoadsorption of H2O and D2O due to de-aggregation of the pre-existing network of particles rather than by dissociative water and D2O adsorption at surficial oxygen vacancies. Finally, it is concluded that the role of oxygen during UV irradiation is very crucial for the presence of both the photoinduced hydrophilic as well as the photocatalytic response.
Acknowledgements
Belhadj H. gratefully acknowledges a scholarship from the Deutscher Akademischer Austauschdienst (DAAD) providing the financial support to perform his PhD. studies in Germany. The present study was performed within the Project “Establishment of the Laboratory ‘Photoactive Nanocomposite Materials’” No. 14.Z50.31.0016 supported by a Mega-grant of the Government of the Russian Federation.
References
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- J. Tschirch, R. Dillert, D. Bahnemann, B. Proft, A. Biedermann and B. Goer, Res. Chem. Intermed., 2008, 34, 381–392 CrossRef CAS PubMed.
- M. Miyauchi, A. Nakajima, T. Watanabe and K. Hashimoto, Chem. Mater., 2002, 14, 2812–2816 CrossRef CAS.
- J. Xu, N. Sahai, C. M. Eggleston and M. A. A. Schoonen, Earth Planet. Sci. Lett., 2013, 363, 156–167 CrossRef CAS PubMed.
- T. Hirakawa and Y. Nosaka, Langmuir, 2002, 18, 3247–3254 CrossRef CAS.
- P. K. J. Robertson, D. W. Bahnemann, L. A. Lawton and E. Bellu, Appl. Catal., B, 2011, 108–109, 1–5 CrossRef CAS PubMed.
- R. Wang, K. Hashimoto, A. Fujishima, M. Chikuni, E. Kojima, A. Kitamura, M. Shimohigoshi and T. Watanabe, Adv. Mater., 1998, 10, 135–138 CrossRef CAS.
- L. Zhang, R. Dillert, D. Bahnemann and M. Vormoor, Energy Environ. Sci., 2012, 5, 7491 CAS.
- L. M. Liu, P. Crawford and P. Hu, Prog. Surf. Sci., 2009, 84, 155–176 CrossRef CAS PubMed.
- N. Sakai, A. Fujishima, T. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 1028–1035 CrossRef CAS.
- T. Zubkoy, D. Stahl, T. L. Thompson, D. Panayotov, O. Diwald and J. T. Yates, J. Phys. Chem. B, 2005, 109, 15454–15462 CrossRef PubMed.
-
H. Irie and K. Hashimoto, Environmental Photochemistry Part II, 2005, vol. 2M Search PubMed.
- K. Hashimoto, H. Irie and A. Fujishima, Jpn. J. Appl. Phys., 2005, 44, 8269–8285 CrossRef CAS.
- G. Munuera, A. R. Gonzalez-Elipe, J. Soria and J. Sanz, J. Chem. Soc., Faraday Trans. 1, 1980, 76, 1535 RSC.
- M. Takeuchi, K. Sakamoto, G. Martra, S. Coluccia and M. Anpo, J. Phys. Chem. B, 2005, 109, 15422–15428 CrossRef CAS PubMed.
- C. Wang, C. Böttcher, D. W. Bahnemann and J. K. Dohrmann, J. Mater. Chem., 2003, 13, 2322 RSC.
- C. Wang, R. Pagel, J. K. Dohrmann and D. W. Bahnemann, C. R. Chim., 2006, 9, 761–773 CrossRef CAS PubMed.
- C. Y. Wang, R. Pagel, D. W. Bahnemann and J. K. Dohrmann, J. Phys. Chem. B, 2004, 108, 14082–14092 CrossRef CAS.
- S. J. Hug and B. Sulzberger, Langmuir, 1994, 10, 3587–3597 CrossRef CAS.
- C. B. Mendive, T. Bredow, A. Feldhoff, M. A. Blesa and D. Bahnemann, Phys. Chem. Chem. Phys., 2009, 11, 1794–1808 RSC.
- J. C. Duplan, L. Mahi and J. L. Brunet, Chem. Phys. Lett., 2005, 413, 400–403 CrossRef CAS PubMed.
- R. Nakamura, A. Imanishi, K. Murakoshi and Y. Nakato, J. Am. Chem. Soc., 2003, 125, 7443–7450 CrossRef CAS PubMed.
- L. A. Lawton, P. K. J. Robertson, B. J. P. A. Cornish, I. L. Marr and M. Jaspars, J. Catal., 2003, 213, 109–113 CrossRef CAS.
- G. Mattioli, F. Filippone and A. A. Bonapasta, J. Am. Chem. Soc., 2006, 128, 13772–13780 CrossRef CAS PubMed.
- R. Wang, K. Hashimoto, A. Fujishima, M. Chikuni, E. Kojima, A. Kitamura, M. Shimohigoshi and T. Watanabe, Nature, 1997, 388, 431–432 CrossRef CAS.
- A. Y. Nosaka, E. Kojima, T. Fujiwara, H. Yagi, H. Akutsu and Y. Nosaka, J. Phys. Chem. B, 2003, 107, 12042–12044 CrossRef CAS.
- K. Uosaki, T. Yano and S. Nihonyanagi, J. Phys. Chem. B, 2004, 108, 19086–19088 CrossRef CAS.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- C. B. Mendive, D. Hansmann, T. Bredow and D. Bahnemann, J. Phys. Chem. C, 2011, 115, 19676–19685 CAS.
- M. Takeuchi, G. Martra, S. Coluccia and M. Anpo, J. Phys. Chem. C, 2007, 111, 9811–9817 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp03947a |
|
| This journal is © the Owner Societies 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence