
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Building solids inside nano-space: from confined amorphous through confined solvate to confined ‘metastable’ polymorph†
K. P.
Nartowski
a,
J.
Tedder
a,
D. E.
Braun
b,
L.
Fábián
a and
Y. Z.
Khimyak
*a
aSchool of Pharmacy, University of East Anglia, Norwich, Norfolk NR4 7TJ, UK. E-mail: y.khimyak@uea.ac.uk
bInstitute of Pharmacy, University of Innsbruck, Innrain 52c, 6020 Innsbruck, Austria
First published on 17th August 2015
Abstract
The nanocrystallisation of complex molecules inside mesoporous hosts and control over the resulting structure is a significant challenge. To date the largest organic molecule crystallised inside the nano-pores is a known pharmaceutical intermediate – ROY (259.3 g mol−1). In this work we demonstrate smart manipulation of the phase of a larger confined pharmaceutical – indomethacin (IMC, 357.8 g mol−1), a substance with known conformational flexibility and complex polymorphic behaviour. We show the detailed structural analysis and the control of solid state transformations of encapsulated molecules inside the pores of mesoscopic cellular foam (MCF, pore size ca. 29 nm) and controlled pore glass (CPG, pore size ca. 55 nm). Starting from confined amorphous IMC we drive crystallisation into a confined methanol solvate, which upon vacuum drying leads to the stabilised rare form V of IMC inside the MCF host. In contrast to the pure form, encapsulated form V does not transform into a more stable polymorph upon heating. The size of the constraining pores and the drug concentration within the pores determine whether the amorphous state of the drug is stabilised or it recrystallises into confined nanocrystals. The work presents, in a critical manner, an application of complementary techniques (DSC, PXRD, solid-state NMR, N2 adsorption) to confirm unambiguously the phase transitions under confinement and offers a comprehensive strategy towards the formation and control of nano-crystalline encapsulated organic solids.
Introduction
The discovery of the M41S (MCM, Mobile Composition of Matter) family of mesoporous silica materials extended the field of possible applications of porous solids in catalysis, gas storage, size exclusion chromatography and enzyme immobilisation in silica scaffolds.1–7 Furthermore, some unique features of mesoporous composites, including an ordered, size tailored pore network, high pore volume and high surface area drew the attention of the pharmaceutical world to these materials as promising, multifunctional candidates for drug delivery systems (DDS).8–10Research on mesoporous silica materials for drug delivery has focused mainly on two hosts, namely MCM-41 and SBA-15, with pore sizes of ca. 4 and 8 nm, respectively. Van Speybroeck et al. showed that SBA-15 may stabilise the amorphous state of 10 pharmaceutical molecules (including IMC) for over a period of 6 months when entrapped inside the silica scaffold and demonstrated their increased dissolution rates.11 Although the volume of research published on the possible applications of mesoporous silicas as crystallisation chambers is increasing, molecular understanding of the phenomena which govern the aggregation and self-assembly of molecules in porous media is still to be fully achieved. One of the possible reasons for this is the complexity of the system, in which the impact of the size of confined clusters and the additional host–guest surface interactions cannot be ignored. An attempt to understand glass transition, vitrification and solidification of organic liquids entrapped in nanometer scale pores was made by Jackson, Alcoutlabi and McKenna.12–14 Unfortunately, all these findings are concerned mainly with metals, small organic molecules, liquids or polymers, which cannot be translated directly into phase transitions of medium size complex molecules (including pharmaceuticals), which may adopt more than one molecular arrangement in the crystalline state (namely polymorphism).
Engineering of nano-crystals within controlled pore glasses (CPG) and polymeric monoliths has been investigated during the last decade. Polymorph selectivity in the crystallisation of metastable forms of acetaminophen was reported by Beiner et al. with further discussion of the impact of pore size on this phenomenon by Rengarajan et al.15,16 Hamilton et al. showed stabilisation of the metastable β-form of glycine inside porous polystyrene-poly(dimethyl acrylamide) (p-PS-PDMA) monoliths and in CPG hosts with pore diameters less than 24 nm. Furthermore, control of the orientation of the nanocrystalline glycine or α,γ-alkanedicarboxylic acids (HO2C(CH2)n−2CO2H, n = 3–13, odd) was achieved either by stereochemical inhibition or chain length of the confined dicarboxylic acids.17–19 Despite years of studies, to date the largest molecule with fully proven crystallisation within mesoporous hosts is a pharmaceutical intermediate, 5-methyl-2-[(2-nitrophenyl)-amino]-3-thiophenecarbonitrile (ROY), which exhibits conformational flexibility and a molecular mass of 259.3 g mol−1.20
In this work we have studied nano-crystallisation of a much larger (molecular mass 357.8 g mol−1) model pharmaceutical indomethacin (IMC) encapsulated within MCF (mesoscopic cellular foam) and CPG porous solids via careful control of the confined structures from amorphous to crystalline states. With its well described polymorphism and phase transitions IMC is a very good model compound for nano-crystallisation research. SBA-15 mesoporous silica has been demonstrated to be a suitable host for the stabilisation of amorphous IMC loaded via incipient wetness method.11 An attempt to crystallise IMC ‘inside’ the pores of MCM-41 and SBA-15 materials were recently made by Ukmar et al.21,22 Although the authors suggested selective crystallisation of one of the IMC polymorphs under mesoscopic confinement, the presented data show typical bulk fusion of IMC polymorphs and cannot be described as confined crystallisation.22 Moreover, the spectroscopic data are consistent with the presence of amorphous IMC in the pores, in agreement with Van Speybroeck et al.11
In this work, for the first time we show multiple phase transformations of the confined solids assuring in a critical and comprehensive way that all transitions take place inside the nanopores. Starting from confined amorphous IMC, through a confined IMC methanol (MeOH) solvate, we show a novel method to crystallise and stabilise the rare and highly metastable IMC form V*, a desolvation product.23,24 (*we expanded the nomenclature introduced by Borka,25i.e. Roman numerals for anhydrate polymorphs). We further demonstrate methods to control the drug phase through the effect of the pore size, loading procedure and guest content, which is of paramount importance for further industrial development.
Results and discussion
Indomethacin polymorphs – introducing solid-state NMR studies on IMC methanol solvate and IMC form V
Indomethacin is a widely studied model pharmaceutical with well recognised polymorphism and complex phase behaviour.25–43 Recent studies by Surwase et al. reported new crystalline forms of IMC and water driven phase transitions from an amorphous phase, in spite of years of studies.23The thermodynamically most stable form of indomethacin (γ-IMC) has the space group symmetry P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with Z′ = 1. The γ-IMC crystal structure is stabilised by centrosymmetric R22(8) hydrogen bonded ring motifs,44 formed between carboxylic acid groups. γ-IMC shows a melting point at 159.1 to 161.3 °C and an enthalpy of fusion of 110 J g−1.30,45,46 Metastable α-IMC is another frequently occurring polymorph, which crystallises in the monoclinic space group P21 with Z′ = 3 and very different conformations compared to γ-IMC. α-IMC exhibits a melting point in the range from 153.0 to 155.0 °C with an enthalpy of fusion of 97.5 J g−1.30,38
with Z′ = 1. The γ-IMC crystal structure is stabilised by centrosymmetric R22(8) hydrogen bonded ring motifs,44 formed between carboxylic acid groups. γ-IMC shows a melting point at 159.1 to 161.3 °C and an enthalpy of fusion of 110 J g−1.30,45,46 Metastable α-IMC is another frequently occurring polymorph, which crystallises in the monoclinic space group P21 with Z′ = 3 and very different conformations compared to γ-IMC. α-IMC exhibits a melting point in the range from 153.0 to 155.0 °C with an enthalpy of fusion of 97.5 J g−1.30,38
The methanol solvate crystallises in the monoclinic space group P21/n (Z′ = 1), with a stoichiometric ratio of IMC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH of 1:1 (BANMUZ24). The solvate structure is stabilised by hydrogen bonds between the carboxylic acid groups of indomethacin and the alcohol groups of the methanol molecules, which form a R44(12) ring motif. The solvent molecules are located in channels along the crystallographic b axis, from which they may escape easily during desolvation.47 The DSC curve (open pan) of IMC methanol solvate shows an endothermic peak at an onset temperature of 84 °C, which is in agreement with previous studies and corresponds to the desolvation of the solvate to IMC form V.35,47 A second endothermic event at 131.4 °C is related to the melting of form V and followed by recrystallisation of the two more stable forms of IMC (α and γ). Thus, IMC form V, often incorrectly called δ or form IV, can be obtained via slow desolvation of the methanol solvate at room or elevated temperatures or under vacuum.23,47 Based on our experience, a slow desolvation process at room temperature may take up to 3 months, but leads to pure form V, while much faster desolvation at elevated temperatures often leads to a mixture of polymorphs (V, γ, α).
:MeOH of 1:1 (BANMUZ24). The solvate structure is stabilised by hydrogen bonds between the carboxylic acid groups of indomethacin and the alcohol groups of the methanol molecules, which form a R44(12) ring motif. The solvent molecules are located in channels along the crystallographic b axis, from which they may escape easily during desolvation.47 The DSC curve (open pan) of IMC methanol solvate shows an endothermic peak at an onset temperature of 84 °C, which is in agreement with previous studies and corresponds to the desolvation of the solvate to IMC form V.35,47 A second endothermic event at 131.4 °C is related to the melting of form V and followed by recrystallisation of the two more stable forms of IMC (α and γ). Thus, IMC form V, often incorrectly called δ or form IV, can be obtained via slow desolvation of the methanol solvate at room or elevated temperatures or under vacuum.23,47 Based on our experience, a slow desolvation process at room temperature may take up to 3 months, but leads to pure form V, while much faster desolvation at elevated temperatures often leads to a mixture of polymorphs (V, γ, α).
The crystal structure of IMC form V is yet to be determined, as this phase is a desolvation product showing low crystallinity. The diffraction pattern of form V indexed to a monoclinic unit cell (25 °C: a = 18.5450 Å, b = 5.4032 Å, c = 18.2967 Å, β = 95.642°) using the first twenty peaks with DICVOL04 and the space group was determined to be P21 based on a statistical assessment of systematic absences, as implemented in the DASH structure solution package.48,49
From the cell volume it was derived that there are two IMC molecules in the asymmetric unit (Z′ = 2). IMC form V shows small needles which melt between 130–131 °C followed by a recrystallisation directly into γ-IMC or a mixture of both α and γ-IMC.47 All DSC data of the reference samples are summarised in Fig. 1B. The PXRD patterns of the reference samples (Fig. 1A) are in good agreement with the simulated patterns for the IMC structures published in the CSD (Cambridge Structural Database) for structures INDMET (γ-IMC, form I), INDMET02 (α-IMC, form II) and BANMUZ (IMC methanol solvate).24,36,45,50 The PXRD pattern of IMC:MeOH (1:1) solvate even after careful grinding shows two strong intensity peaks with d(002) = 10.27 Å and d(004) = 5.15 Å, which may be related to preferred orientation of the fine needles. Furthermore, the PXRD pattern of form V IMC is in a good agreement with those published previously.23,35 Indomethacin has been studied widely using solid-state NMR spectroscopy, complementing the single crystal structure determinations of both the α- and γ-polymorphs. The advanced solid-state NMR methods focused on detailed analysis of hydrogen bonding patterns and intramolecular distances and were supported by first principles calculations.28,38,43,46,51
 | ||
| Fig. 1 PXRD patterns (A) and DSC thermograms (B) of IMC polymorphs and MeOH solvate. | ||
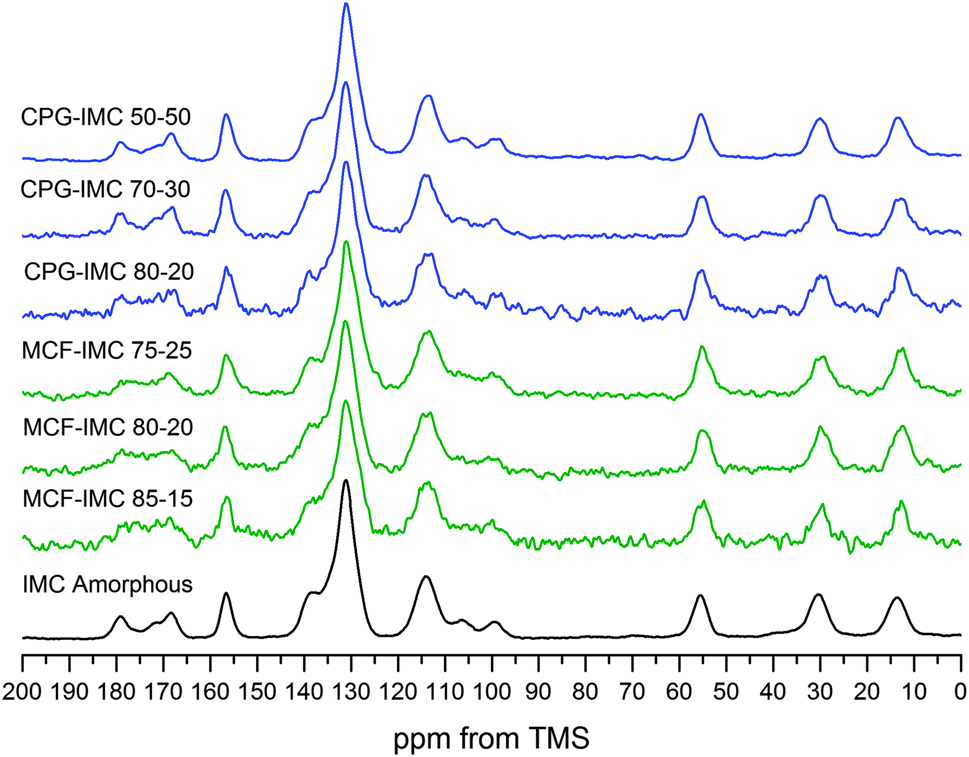
We report for the first time solid-state NMR spectra of IMC methanol solvate and form V. Due to the fact that IMC form V is formed via desolvation of the IMC methanol solvate, we focus on the changes in the peak positions and intensities for both forms and compare them to the previously assigned peaks of the spectrum of γ-IMC (Fig. 2). Further, more detailed structural, spectroscopic and computational studies of this indomethacin polymorph V are in progress and will be presented elsewhere.
 | ||
| Fig. 2 (A) Hydrogen bonded R44(12) ring motif formed between IMC and methanol extracted from the IMC methanol solvate crystal structure (BANMUZ); (B) Molecular diagram of IMC with labelled carbons; (C) 1H–13C CP/MAS NMR spectra of IMC polymorphs and methanol solvate (spinning sidebands are labelled with asterisks). | ||
Similarly to γ-IMC, the IMC methanol solvate has only one indomethacin molecule in the asymmetric unit (and an additional methanol molecule). This leads to a similar number of the peaks in 1H–13C CP/MAS NMR spectra as in the spectrum of γ-IMC with one additional peak at 50.1 ppm arising from the methanol carbon.
The majority of the aromatic peaks, the phenyl group and carbons C-2, C-8, C-9 of the indole ring, undergo slight changes when compared to γ-IMC. The carboxylic acid carbon (C-11) peak of the solvate undergoes three distinct changes, i.e. up-field shift from 179.3 to 176.3 ppm, broadening and decrease in intensity. The latter changes might be related to the close proximity of the methanol molecules in the solvate, forming the R44(12) ring motif, as shown in Fig. 2A.
The spectrum of IMC form V shows several differences as compared to the IMC methanol solvate spectrum. Firstly, the methanol carbon peak disappears and the C-12 peak undergoes downfield shift from 54.1 to 56.2 ppm. Secondly, splitting and decrease of the intensity of the peaks from carbons C-10 and C-4 indicates the presence of two molecules in the asymmetric unit, in agreement with the space group and unit cell parameters derived from PXRD data. The carbon C-11 involved in the hydrogen bonding in the solvate undergoes a downfield shift, splitting and decrease in intensity after desolvation, indicating the presence of two magnetically non-equivalent carboxylic acid motifs.
Loading efficiency of silica hosts
Several methods have been applied for loading drug molecules into the porous silica hosts, including impregnation, incipient wetness, rotary evaporation, immersion, spray drying and melt loading.8,52–56 The studies of crystallisation in the confined space require accurate control of experimental protocols to prevent the loading of the drug outside the pores. Furthermore, it is important to note that distinguishing between the confined drug species and the bulk phase outside the pore space often presents a significant characterisation challenge.We used two loading methods, i.e. incipient wetness and melt loading method, for the encapsulation of indomethacin and two different types of the silica hosts. The first one was the spherical MCF with ink-bottle like pores of 29.6 nm in diameter interconnected by smaller windows (necks) of 11 nm as determined using nitrogen adsorption analysis. The second material was commercially available unstructured CPG host with a mean pore size diameter of 55 nm.
When the MCF silica host is loaded from the melt a gradual decrease of the total pore volume and surface area as a function of an increasing drug content are observed from nitrogen adsorption–desorption isotherms, indicating loading of the drug inside the pores. When the concentration of the embedded drug reaches 50 wt% (MCF–IMC 50–50) the pores seem to be fully filled with the guest molecules.
A substantial increase in the total pore volume and the surface area of the products loaded with high concentration of the drug (MCF–IMC 70–30 and MCF–IMC 50–50) was observed after addition of the solvent (Fig. 3 and 4, Table 1). This may indicate dissolution and rearrangement of the confined amorphous phase prior to further crystallisation or some leakage of the drug from the pores as melting of bulk crystalline IMC was detected in DSC studies of the composites loaded with 30 and 50 wt% IMC (ESI:† S3, S6 and related figures). It is important to mention that careful control over the solvent volume (lower than Vtotal) and total content of the drug within the pores is crucial to avoid leakage of the drug. Large volumes of solvent and high drug content increase the risk of the drug leakage leading to inhomogeneous composites (see ESI,† Fig. S2.4).
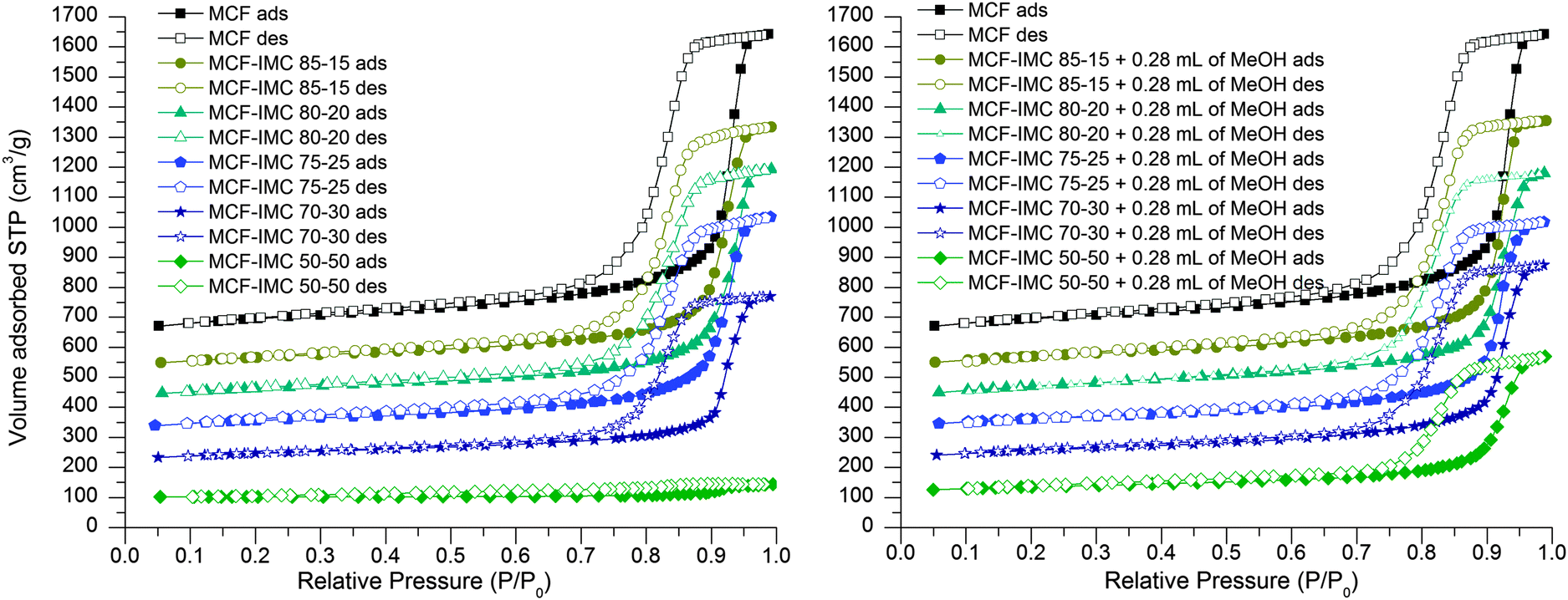 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherms of MCF host loaded with different ratios of IMC using the melting method (left) and MCF host loaded with different ratios of IMC using the melting method and subsequently treated with MeOH (right). The isotherms are offset by 100 cm3 g−1. | ||
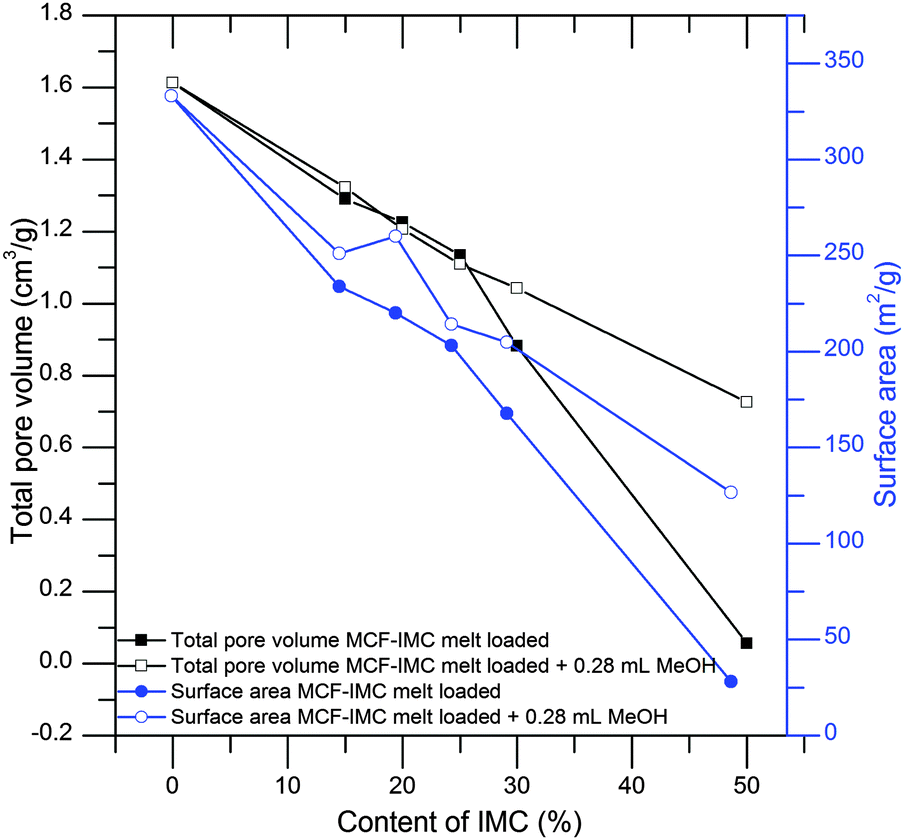 | ||
| Fig. 4 Total pore volume and BET surface area of MCF–IMC melt loaded samples before and after addition of the solvent. The total amount of IMC within the materials was determined using TG analysis. | ||
| Host–guest composite | IMC (g g−1) | V total [cm3 g−1] | d pore [nm] | S BET [m2 g−1] |
|---|---|---|---|---|
| a V total – total pore volume calculated at P/P0 of 0.99, dpore – pore size diameter calculated from the adsorption branch using the BJH (Barrett–Jotyner–Halenda) method, SBET – the BET specific surface area. b Loaded using IW method. | ||||
| MCF | N/A | 1.613 | 29.6 | 333.2 |
| MCF–IMC 85–15 | 0.151 ± 0.003 | 1.290 | 26.2 | 233.9 |
| MCF–IMC 80–20 | 0.199 ± 0.010 | 1.225 | 30.0 | 220.1 |
| MCF–IMC 75–25 | 0.243 ± 0.016 | 1.134 | 23.3 | 203.0 |
| MCF–IMC 70–30 | 0.294 ± 0.010 | 0.880 | 25.8 | 167.8 |
| MCF–IMC 50–50 | 0.494 ± 0.013 | 0.056 | N/A | 28.1 |
| MCF–IMC 85–15 + 0.28 mL MeOH | 0.161 ± 0.012 | 1.322 | 26.1 | 251.2 |
| MCF–IMC 80–20 + 0.28 mL MeOH | 0.191 ± 0.070 | 1.206 | 23.1 | 260.0 |
| MCF–IMC 75–25 + 0.28 mL MeOH | 0.245 ± 0.041 | 1.109 | 26.1 | 214.3 |
| MCF–IMC 70–30 + 0.28 mL MeOH | 0.261 ± 0.027 | 1.042 | 26.0 | 204.8 |
| MCF–IMC 50–50 + 0.28 mL MeOH | 0.471 ± 0.073 | 0.725 | 23.1 | 126.7 |
| MCF 150 mg mL−1 MeOH (0.8 mL g−1)b | 0.109 ± 0.001 | 1.228 | 23.2 | 241.9 |
| MCF 200 mg mL−1 MeOH (0.8 mL g−1)b | 0.146 ± 0.001 | 0.910 | 21.2 | 178.3 |
| MCF 250 mg mL−1 MeOH (0.8 mL g−1)b | 0.179 ± 0.003 | 1.083 | 23.3 | 222.1 |
The CPG host with large pores of 55 nm in diameter can be classified as being on the boundary between mesoporous and macroporous silica materials (Table 2).
| CPG host loaded using melting method | IMC (g g−1) | CPG host loaded using melting method after addition of methanol | IMC (g g−1) |
|---|---|---|---|
| CPG–IMC 85:15 |
0.169 ± 0.010 | CPG–IMC 85:15 + 0.28 mL MeOH |
0.150 ± 0.001 |
| CPG–IMC 80:20 |
0.218 ± 0.008 | CPG–IMC 80:20 + 0.28 mL MeOH |
0.198 ± 0.003 |
| CPG–IMC 75:25 |
0.267 ± 0.018 | CPG–IMC 75:25 + 0.28 mL MeOH |
0.231 ± 0.002 |
| CPG–IMC 70:30 |
0.285 ± 0.024 | CPG–IMC 70:30 + 0.28 mL MeOH |
0.291 ± 0.003 |
| CPG–IMC 50:50 |
0.482 ± 0.032 | CPG–IMC 50:50 + 0.28 mL MeOH |
0.484 ± 0.001 |
| CPG host loaded using IW method with 0.6 mL g−1 of material | IMC (g g−1) | CPG host loaded using IW method with 0.75 mL g−1 of material | IMC (g g−1) |
|---|---|---|---|
| CPG 150 mg mL−1 MeOH | 0.069 ± 0.018 | CPG 150 mg mL−1 MeOH | 0.088 ± 0.001 |
| CPG 200 mg mL−1 MeOH | 0.112 ± 0.004 | CPG 200 mg mL−1 MeOH | 0.135 ± 0.006 |
| CPG 250 mg mL−1 MeOH | 0.133 ± 0.007 | CPG 250 mg mL−1 MeOH | 0.153 ± 0.002 |
The combination of melting point depression (onset and heat of fusion) and changes in N2 adsorption properties serve as a proof of the guest confinement. The loaded CPG silicas show some decrease of the BET specific surface area compared to the pure host. However, the correlation between the drug content and decrease of surface area is not straightforward due to the relatively low value of the BET surface area of the host (i.e. for the CPG material it is ca. 49.5 m2 g−1 compared to 333.2 m2 g−1 for the MCF host (see the ESI,† S4, Table S4.1)). The presence of crystalline guest embedded within the CPG host can be illustrated by the broadening and decrease of the IMC melting point, which is indicative of small crystals.14,57,58
Identification of the phase of the drug under mesoscopic confinement
 | ||
| Fig. 5
1H–13C CP MAS NMR spectra of MCF and CPG hosts loaded with molten IMC at different host:drug ratios. | ||
Although thermal properties and phase transitions of confined gases and liquids have been studied extensively, the effect of confinement on solids is still not understood fully. It is known that nano-size crystals show a decrease and broadening of the melting point and the melting enthalpy of confined crystals is smaller in comparison to the bulk phase.14,57,58
Upon heating bulk amorphous indomethacin shows a glass transition (Tg) at ca. 40 °C followed by recrystallization of α- and γ-IMC at 100–120 °C and the melting of the latter two polymorphs.26 DSC thermograms of the MCF host loaded with indomethacin using the melting method do not show any crystallisation and melting processes, proving it is in an amorphous state (ESI,† S5, Fig. S5.3) and the size of the pores of MCF (ca. 30 nm) prevents the thermally induced recrystallisation of indomethacin. Interestingly, indomethacin confined within the larger pores of the CPG host (ca. 55 nm) shows concentration dependent thermally induced crystallisation (Fig. 6). No thermal transitions were observed at low concentrations of IMC (up to 25% w/w). However, when the drug content reached 30 wt% recrystallisation of the amorphous indomethacin into both α- and γ-IMC is observed. Such recrystallisation occurs inside the pores, as confirmed by a decrease of the melting temperature and broadening of the melting peak (for CPG–IMC 70–30 Tm is 147.5 °C, for CPG–IMC 50–50 Tm is 146.8 °C). Nevertheless, the two other peaks present on the DSC thermogram for the materials loaded with IMC at concentrations of 30 and 50% indicate the presence of the bulk crystalline α- and γ-IMC, which again signifies the importance of the loading protocol on the crystallisation outcome.
 | ||
| Fig. 6 DSC thermograms of CPG host loaded with IMC from the melt at different host:drug ratios. | ||
:methanol (1:1) solvate from hot methanol at a low supersaturation ratio.35 As demonstrated by Joshi et al. and later by Surwase et al. this solvate may yield the metastable form V of IMC under careful desolvation. This form was reported to recrystallise subsequently into more stable polymorphs.23,47
The amorphous drug loaded within the MCF host at the concentration of 30 and 50 wt% crystallises into a mixture of IMC phases after addition of methanol, which are partly loaded outside the pores. The DSC thermograms of both composites with the highest loadings show the melting peaks of bulk γ-IMC. This can be related to a leakage of the dissolved drug from the pores after addition of the solvent and recrystallisation on the external surface of the host, since the presence of the bulk crystalline phase in the highly concentrated samples could not be detected before the addition of the solvent (ESI,† S6 and related figures). The DSC thermograms of IMC confined within the MCF pores show a decrease of the melting temperature and broadening of the melting peak indicating the presence of nano-size crystals (Fig. 7). The host loaded with 15% of IMC shows no recrystallisation in the temperature range from 20 to 180 °C confirming the presence of an amorphous phase, which is in agreement with the PXRD data (Fig. 8). Increasing the host load to 20% results in a mixture of amorphous and IMC form V. The composites loaded with 20 and 25% of IMC, after treatment with methanol, show a single melting point of form V IMC. This may indicate that spatial constrains of ca. 30 nm prevent the concomitant crystallisation of form V and other phase impurities (α- and γ-IMC). Thus, phase pure form V can be obtained.
 | ||
| Fig. 7 DSC thermograms of MCF and CPG hosts loaded with IMC from the melt after addition of methanol. | ||
 | ||
| Fig. 8 PXRD patterns of MCF and CPG hosts loaded with molten indomethacin after treatment with methanol (peaks showing the presence of γ-IMC are labelled with asterisks). | ||
Using CPG host loaded with molten IMC at concentrations 15–25% we were able to obtain exclusively γ-IMC after the addition of ethanol or acetonitrile as a crystallisation driving solvent (ESI,† S2). When methanol was used to drive IMC crystallisation either pure IMC form V, with the methanol solvate as an intermediate, or a mixture of both γ-IMC and IMC form V were obtained. The CPG–IMC composite loaded with 15% of indomethacin shows the PXRD pattern and solid-state NMR spectrum of the pure IMC form V (Fig. 8 and 9). At host–guest ratio of the CPG–IMC 80–20 and the CPG–IMC 75–25, predominately indomethacin form V with a small amount of γ-IMC is formed (confirmed by the PXRD and solid-state NMR). Further increase of the IMC content to 30 and 50% leads to the increase of the proportion of the drug loaded outside the pores.
 | ||
| Fig. 9
1H–13C CP/MAS solid-state NMR spectra of MCF and CPG hosts loaded with molten IMC at different host:drug ratios subsequently treated with methanol (peaks assigned to γ-IMC are labelled with asterisks). | ||
The DSC thermograms of the CPG host loaded with IMC from the melt and subsequently treated with methanol show several thermal events, which may be assigned to the melting of IMC form V (below 125 °C), recrystallisation of α- and γ-IMC, corresponding to the only exothermic event following the form V melting process, and finally the melting of α- (shoulder) and γ-IMC (<157 °C). Broadening and decrease of the melting peaks of form V, as well as α- and γ-IMC, indicates that all observed phases are not bulk material, but are in the confined space (Fig. 7, Table 3).
| Host–guest composite | Melting onset (°C) | ΔHfusion (J g−1) |
|---|---|---|
| a The lower measured heat of fusion value, compared to ‘MCF–IMC 75–25 + 0.28 mL MeOH’, indicates the presence of amorphous IMC. b Melting of form V and recrystallisation of α- and γ-IMC overlap, thus, the stated ΔHfusion values are too low. | ||
| MCF–IMC 85–15 + 0.28 mL MeOH | — | — |
| MCF–IMC 80–20 + 0.28 mL MeOH | 110.0 (form V) | 18.3 (form V)a |
| MCF–IMC 75–25 + 0.28 mL MeOH | 111.9 (form V) | 35.8 (form V) |
| CPG–IMC 85–15 + 0.28 mL MeOH | 122.6 (form V) 152.1 (γ-IMC) | 31.6 (form V) 18.6 (γ-IMC) |
| CPG–IMC 80–20 + 0.28 mL MeOH | 112.3 (form V) 150.1 (γ-IMC) | 25.6 (form V)b 26.5 (γ-IMC) |
| CPG–IMC 75–25 + 0.28 mL MeOH | 110.5 (form V) 149.6 (γ-IMC) | 24.1 (form V)b 25.9 (γ-IMC) |
Neither solid-state NMR nor PXRD indicated the formation of γ- or α-IMC within the CPG–IMC 85–15 composite after addition of methanol, suggesting that the presence of the melting peaks at 146.5 °C (shoulder) and 152.1 °C are due to recrystallisation of the α and γ-IMC forms above the form V melting point. Seeds of γ and α-IMC may be present below the PXRD and NMR detection limit and trigger the recrystallisation. A decrease of the melting peak onset temperature of IMC form V with increasing drug concentration in the CPG host is evident, indicating a decrease of the size of the crystals. Full understanding of the mechanism of thermal events in silica host loaded with large amounts of IMC is beyond the scope of this work.
To investigate the impact of the added volume of the loading solution on the final composition of the loaded hosts we used 0.6 and 0.75 mL g−1 of a loading solution for the CPG and 0.8 mL g−1 for the MCF material. These values were chosen taking into account the total pore volume of the CPG and the MCF hosts used in the study (1.18 and 1.61 cm3 g−1 respectively).
PXRD patterns of the composites loaded with solutions of different indomethacin concentrations show peaks characteristic for IMC form V (and γ) (Fig. 10).
 | ||
| Fig. 10 PXRD patterns of CPG host and MCF materials loaded with IMC using IW method with 0.6 mL g−1 of the solvent for CPG and 0.8 mL g−1 of the solvent for MCF. | ||
Due to overlapping and broadening of the peaks only traces of γ-IMC within the MCF composites can be detected, which is corroborated by DSC results. CPG hosts loaded with a higher volume of the loading solution (0.75 mL g−1) show distinct peaks of both crystalline phases (ESI,† S7, Fig. S7.1). This is corroborated by DSC analysis, where thermograms show peaks of bulk crystalline phases of IMC (ESI,† Fig. S7.2, and Table S7.1).
The DSC thermograms of CPG composites loaded using lower volumes (150 and 200 mL mg−1) of the loading solution show a slightly broader melting endotherm of IMC form V and traces (0.05–1.0% as determined from melting point onset temperatures and heat of fusion) of bulk α- and γ-polymorphs of IMC. Furthermore, there is no exothermic recrystallisation event after the melting of IMC form V. This indicates the formation of confined IMC form V and the importance of the spatial constrains of silica walls in preventing the crystallisation of other IMC polymorphs in the temperature range 20 to 170 °C. The 250 mg mL−1 sample shows a distinct γ-IMC melting peak. Loading from the solution with the highest concentration of IMC in methanol gives sharper form V melting peaks and a slightly lower decrease of the melting onset which may indicate that IMC form V is partly loaded outside the pores (Fig. 11, Table 4).
 | ||
| Fig. 11 DSC thermograms of CPG and MCF hosts loaded with IMC using IW method. | ||
| Host–guest composite | Melting onset (°C) | Melting peak (°C) |
|---|---|---|
| CPG 150 mg mL−1 MeOH | 126.9 (V) | 130.6 |
| CPG 200 mg mL−1 MeOH | 126.7 (V) | 130.4 |
| CPG 250 mg mL−1 MeOH | 129.9 (V) | 132.1 |
| MCF 150 mg mL−1 MeOH | 128.3 (V) | 131.4 |
| 159.7 (γ) | 160.1 | |
| MCF 200 mg mL−1 MeOH | 127.6 (V) | 132.1 |
| 159.5 (γ) | 160.1 | |
| MCF 250 mg mL−1 MeOH | 127.5 (V) | 131.7 |
| 159.5 (γ) | 160.1 |
The MCF host loaded with IMC from methanol using the IW method shows the presence of confined form V IMC, bulk γ-IMC and small amounts of α-IMC. Although the loading volume was much smaller than the total pore volume of the MCF host, crystallisation outside the pores could not be avoided. Similarly to the CPG hosts loaded with higher volumes of loading solution (ESI,† Fig. S7.2, Table S7.1), the DSC thermograms of MCF composites loaded with higher concentrations of IMC loading solution (200 and 250 mg mL−1 MeOH) show melting of form V IMC preceded by a small endothermic event at ca. 85 °C. This may be related to the presence of IMC methanol solvate (Fig. 11). The endotherm below 80 °C is related to the evaporation of water from the silica surface, as water sorption from the air may happen during sample preparation.
Solid-state NMR spectra of both hosts loaded using the IW method at IMC concentration of 200 mg mL−1 in methanol are consistent with the presence of pure IMC form V within the composites. No peaks of other IMC crystalline phases could be detected, indicating that the presence of the γ-IMC peak at high loading concentrations of the MCF material is most likely due to temperature induced recrystallisation of the bulk drug phase loaded on the external surface of the host (Fig. 11 and 12).
 | ||
| Fig. 12 1H–13C CP/MAS solid-state NMR spectra of CPG and MCF hosts loaded using IW method at indomethacin concentration in methanol of 200 mg mL−1. | ||
The outcome of the deposition of the drug on the external surface of the material is related to the concentration of the loading solution. A high concentration of the guest molecules in the loading solution (250 mg mL−1) allows reaching reasonable API content within the composite. This, however, leads to the risk for some of the drug to be loaded outside the pores, as crystallisation of the supersaturated solution may start in contact with the external surface of silica.
 | ||
| Scheme 1 Summary of possible amorphous to crystalline transformations of melt loaded indomethacin under confinement of MCF and CPG hosts. | ||
Thermally induced recrystallisation of indomethacin confined within the CPG host was prevented at up to the 30% drug content within the pores. This indicates an increased possibility for molecules to form different supramolecular assemblies within the pores, which can lead to polycrystalline composites.22
Addition of methanol to the melt loaded MCF composites drives the crystallisation of the methanol solvate and upon vacuum drying IMC form V is obtained (at drug concentrations within the silica pores from 20 to 25 wt%). Interestingly, confined form V does not show any recrystallisation upon heating, indicating that spatial constrains and the crystallisation method allowed us to readily produce phase pure methanol solvate and phase pure form V. Phase purity of the intermediate solvate and form V is crucial if one aims at preventing/slowing down the transformation to any of the other more stable IMC polymorphs.
The addition of the solvent to the MCF composite loaded with IMC at a concentration of 15% does not drive crystallisation into any of crystalline forms of IMC. This may indicate both strong interactions between the drug and the silica walls and/or the insufficient number of drug molecules to form nuclei large enough to form a crystal.
Methanol driven crystallisation of amorphous indomethacin inside the larger pores of the CPG host shows the formation of a mixture of forms within the whole range of investigated host–guest ratios. However, a proportional preference towards the most stable γ-IMC with an increase in drug content is observed. The addition of either ethanol or acetonitrile to the melt loaded CPG composites leads to an exclusive recrystallisation into the most stable γ-IMC (see the ESI,† S2). This highlights the importance of both the nature of the confinement medium and the solvent on the formation of nanoscale confined crystals. The amount of guest molecules that can be successfully embedded within the silica hosts depends on a careful design of the loading procedure. The maximum loading capacity of the host might be overestimated due to the obstruction of the pores, especially when high drug loading is needed. A uniform distribution of the drug within the host material during mixing helps to avoid the presence of drug rich domains which cannot be loaded fully into the pores by a capillary action and may lead to some drug being left on the external surface. Finally, the heating temperature needs to be sufficiently high to melt the guest, while avoiding its decomposition.
The incipient wetness method is one of the solvent mediated methods for drug loading into the mesoporous silica hosts. However, it is much more difficult to control as increasing drug concentration within the loading solution increases risk of the spontaneous crystallisation of the supersaturated solution outside the pores. In case of indomethacin it can even lead to the formation of an elusive polymorph, IMC form V.
Conclusions
Mesoporous silica materials with pore diameters of up to 8 nm (MCM-41 and SBA-15) have been widely studied as drug delivery systems to stabilise amorphous phases. We have demonstrated that an increase in the pore size diameter of mesoporous silicas may lead to exciting changes in the crystallisation behaviour of a widely studied pharmaceutical compound indomethacin. Amorphous IMC can be stabilised when embedded within pores of ca. 30 nm using the melting method, whereas the CPG host with pores of ca. 55 nm can stabilise the amorphous phase only at low drug content within the composite. Furthermore, addition of a solvent to the melt loaded systems leads to the formation of nano-scale crystals inside the pores. The polymorphic phase formed under confinement depends on the pore size, embedded drug content and crystallisation solvent. Guest loading using the IW method leads directly to the formation of a crystalline phase. Thus, by choosing the right conditions it is possible to increase the solubility, which might be crucial for a poorly water soluble compound as indomethacin.In this study, we have used a novel approach to investigate phase transitions inside silica based, nano-scale crystallisation chambers with well organised systems of pores, i.e. MCF hosts. Combined application of nitrogen adsorption–desorption analysis, thermal and diffraction methods and solid-state NMR enabled us to gain a much better control over the phase and transformations of the guest in the final products, which may lead to a better understanding of host guest interactions within drug delivery systems based on mesoporous silica composites. Furthermore, we have demonstrated a new method for the formation and stabilisation of IMC form V using mesopores to prevent recrystallisation into more stable IMC polymorphs. This should enable further studies of MCF based nano-crystalline drug delivery systems and highlights the importance of loading methods and laboratory procedures on the final product, which is of the interest for the pharmaceutical industry. 13C solid-state NMR spectra of IMC methanol solvate and IMC form V were presented for the first time, highlighting the main structural changes related to the desolvation.
Mesoporous silicas with large pores are not only promising systems for nano-crystalline drug delivery systems, but are also well defined model materials for the investigation of early stages of crystallisation.
Possible tailoring of the pore size diameter may enable effective control of the maximum size of critical nucleus during crystal formation. Furthermore, functionalisation of the internal surface of the silica host may target a particular hydrogen bonding motif on crystallisation outcome.
Experimental
Indomethacin, Pluronic P123, 1,2,3-trimethylbenzene, tetraethyl orthosilicate (TEOS), ammonium fluoride, 37 wt% hydrochloric acid, methanol, ethanol and acetonitrile were purchased from Sigma Aldrich and used without further purification.Mesoporous host synthesis
Spherical particles of Mesostructured Cellular Foam (MCF) were synthesised using the method described by Han et al.61 Briefly, 4 g of tribloc co-polymer Pluronic P123 were dissolved in 75 mL of 1.5 M HCl. After that 4 g of swelling agent (1,3,5-trimethylbenzene) was added and the resulting solution was heated to 40 °C in a water bath and stirred vigorously for 2 hours. Subsequently, 9.2 mL of TEOS as a source of silica was added dropwise to the polymer solution and stirred for another 5 min. The mixture was then transferred to a PTFE bottle and aged at 40 °C for 20 hours without stirring. After that time 46 mg of NH4F was added to the mixture and aged at 100 °C for another 24 hours. The resulting white solid was filtered, washed with MiliQ-quality water and ethanol and dried in an oven at 40 °C for 24 hours. The dried powder was then calcined in air at 650 °C for 10 hours.Controlled Pore Glass (CPG), developed by Haller, is a rigid glass containing a network of interconnected pores.62 The glass is formed by a phase rearrangement process of alkali borosilicate glasses at elevated temperatures.63,64 Control of the time and the temperature of the process enables tailoring of the pore size diameter and leads to a narrow distribution of the pores. The product obtained after leaching forms a continuous, silica rich glass with a porous network of specified pore dimensions. The CPG material is used mainly in the exclusion and absorptive chromatography of biomolecules and, due to the possible organofunctionalisation of its internal surfaces, as a support for the immobilisation of bio-reactive molecules.31 In our study commercially available (Merck Millipore) CPG material with a pore size of 550 Å served as a nano-size crystallisation chamber for the investigation of IMC phase changes under confinement. Scanning electron microscopy images of the synthesised MCF spherical particles and the CPG particles are shown in ESI† (Fig. S1.1).
Indomethacin recrystallisation
The indomethacin purchased from Sigma Aldrich was γ-IMC (form I45). α-IMC (form II36) was formed by dissolving 300 mg of the drug in 40 mL of warm ethanol in a round bottom flask and evaporation of the solvent using a rotary evaporator at 50 °C. The resulting powder was then ground in a mortar and analysed using DSC (Differential Scanning Calorimetry), PXRD (Powder X-ray Diffraction) and solid state NMR. An IMC:MeOH (1:1) solvate was obtained using the method described by Joshi et al.47 Briefly, 500 mg of IMC was dissolved in 8 mL of methanol at 80 °C and left on a bench at room temperature in a vial covered by parafilm to reduce evaporation. Fine needles were collected after two days and dried using a filtering paper. The stoichiometric ratio between the solvent and IMC was confirmed using thermogravimetric analysis. A reference sample of IMC form V was obtained by the slow desolvation of the IMC:MeOH (1:1) solvate at room temperature for two months under a perforated parafilm cover.
Drug loading methods
:drug weight ratios of 85:15, 80:20, 75:25, 70:30, 50:50 and put into a vacuum oven (200 mbar) at 165 °C for 2 hours (i.e. a temperature above the melting point of γ-IMC). The loaded composites were then cooled to an ambient temperature and stored in a desiccator at 4 °C. The presence of an amorphous phase was confirmed using DSC, PXRD and solid-state NMR. Composites loaded from the melt were labelled as follows: host–IMC (host is CPG or MCF) and host–guest ratio (e.g. CPG–IMC 85–15).
Characterization methods
Acknowledgements
The financial support from the University of East Anglia and EPSRC Directed assembly network is gratefully acknowledged. The authors would also like to express their gratitude to Professor Sally L. Price (UCL, UK) for many useful discussions.References
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson and E. W. Sheppard, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- K. K. Sharma and T. Asefa, Angew. Chem., Int. Ed., 2007, 46, 2879–2882 CrossRef CAS PubMed.
- A.-H. Lu, J.-J. Nitz, M. Comotti, C. Weidenthaler, K. Schlichte, C. W. Lehmann, O. Terasaki and F. Schuth, J. Am. Chem. Soc., 2010, 132, 14152–14162 CrossRef CAS PubMed.
- T. Nassivera, A. G. Eklund and C. C. Landry, J. Chromatogr. A, 2002, 973, 97–101 CrossRef CAS.
- W.-J. Son, J.-S. Choi and W.-S. Ahn, Microporous Mesoporous Mater., 2008, 113, 31–40 CrossRef CAS PubMed.
- Y. J. Wang and F. Caruso, Chem. Mater., 2005, 17, 953–961 CrossRef CAS.
- Z. Y. Zhao, J. Liu, M. Hahn, S. Qiao, A. P. J. Middelberg and L. He, RSC Adv., 2013, 3, 22008–22013 RSC.
- M. Vallet-Regi, F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548–7558 CrossRef CAS PubMed.
- M. Manzano and M. Vallet-Regí, J. Mater. Chem., 2010, 20, 5593 RSC.
- C. Argyo, V. Weiss, C. Braeuchle and T. Bein, Chem. Mater., 2014, 26, 435–451 CrossRef CAS.
- M. Van Speybroeck, V. Barillaro, T. Do Thi, R. Mellaerts, J. Martens, J. Van Humbeeck, J. Vermant, P. Annaert, G. Van Den Mooter and P. Augustijns, J. Pharm. Sci., 2009, 98, 2648–2658 CrossRef CAS PubMed.
- C. L. Jackson and G. B. McKenna, J. Non-Cryst. Solids, 1991, 131, 221–224 CrossRef.
- C. L. Jackson and G. B. McKenna, Chem. Mater., 1996, 8, 2128–2137 CrossRef CAS.
- M. Alcoutlabi and G. B. McKenna, J. Phys.: Condens. Matter, 2005, 17, R461–R524 CrossRef CAS.
- M. Beiner, G. T. Rengarajan, S. Pankaj, D. Enke and M. Steinhart, Nano Lett., 2007, 7, 1381–1385 CrossRef CAS PubMed.
- G. T. Rengarajan, D. Enke, M. Steinhart and M. Beiner, Phys. Chem. Chem. Phys., 2011, 13, 21367–21374 RSC.
- B. D. Hamilton, M. A. Hillmyer and M. D. Ward, Cryst. Growth Des., 2008, 8, 3368–3375 CAS.
- B. D. Hamilton, I. Weissbuch, M. Lahav, M. A. Hillmyer and M. D. Ward, J. Am. Chem. Soc., 2009, 131, 2588–2596 CrossRef CAS PubMed.
- J.-M. Ha, B. D. Hamilton, M. A. Hillmyer and M. D. Ward, Cryst. Growth Des., 2012, 12, 4494–4504 CAS.
- J. M. Ha, J. H. Wolf, M. A. Hillmyer and M. D. Ward, J. Am. Chem. Soc., 2004, 126, 3382–3383 CrossRef CAS PubMed.
- T. Ukmar, T. Cendak, M. Mazaj, V. Kaucic and G. Mali, J. Phys. Chem. C, 2012, 116, 2662–2671 CAS.
- T. Ukmar, A. Godec, O. Planinsek, V. Kaucic, G. Mali and M. Gaberscek, Phys. Chem. Chem. Phys., 2011, 13, 16046–16054 RSC.
- S. A. Surwase, J. P. Boetker, D. Saville, B. J. Boyd, K. C. Gordon, L. Peltonen and C. J. Strachan, Mol. Pharmaceutics, 2013, 10, 4472–4480 CrossRef CAS PubMed.
- J. G. Stowell, S. R. Byrn, G. Zografi and M. Yoshioka, Private communication to the Cambridge Structural Database. Refcode BANMUZ, Crystallographic Data Centre, 12 Union Road, Cambridge, England, 2002.
- L. Borka, Acta Pharm. Suec., 1974, 11, 295–303 CAS.
- M. Yoshioka, B. C. Hancock and G. Zografi, J. Pharm. Sci., 1994, 83, 1700–1705 CrossRef CAS PubMed.
- M. Otsuka, N. Kaneniwa and T. Matsumoto, J. Pharmacobio-Dyn., 1986, 9, S2 Search PubMed.
- K. Masuda, S. Tabata, H. Kono, Y. Sakata, T. Hayase, E. Yonemochi and K. Terada, Int. J. Pharm., 2006, 318, 146–153 CrossRef CAS PubMed.
- S. Lohani, I. V. Nesmelova, R. Suryanarayanan and D. J. W. Grant, Cryst. Growth Des., 2011, 11, 2368–2378 CAS.
- B. Legendre and Y. Feutelais, J. Therm. Anal. Calorim., 2004, 76, 255–264 CrossRef CAS.
- P. Karmwar, J. P. Boetker, K. A. Graeser, C. J. Strachan, J. Rantanen and T. Rades, Eur. J. Pharm. Sci., 2011, 44, 341–350 CrossRef CAS PubMed.
- N. Hamdi, Y. Feutelais, N. Yagoubi, D. de Girolamo and B. Legendre, J. Therm. Anal. Calorim., 2004, 76, 985–1001 CrossRef CAS.
- J.-B. Guilbaud, L. Cummings and Y. Z. Khimyak, Macromol. Symp., 2007, 251, 41–46 CrossRef CAS PubMed.
- S. S. Dalal and M. D. Ediger, J. Phys. Chem. Lett., 2012, 3, 1229–1233 CrossRef CAS.
- K. J. Crowley and G. Zografi, J. Pharm. Sci., 2002, 91, 492–507 CrossRef CAS PubMed.
- X. M. Chen, K. R. Morris, U. J. Griesser, S. R. Byrn and J. G. Stowell, J. Am. Chem. Soc., 2002, 124, 15012–15019 CrossRef CAS PubMed.
- V. Andronis and G. Zografi, Pharm. Res., 1998, 15, 835–842 CrossRef CAS.
- J. M. Aceves-Hernandez, I. Nicolas-Vazquez, F. J. Aceves, J. Hinojosa-Torres, M. Paz and V. M. Castanoz, J. Pharm. Sci., 2009, 98, 2448–2463 CrossRef CAS PubMed.
- V. Andronis, M. Yoshioka and G. Zografi, Pharm. Res., 1996, 13, S353 Search PubMed.
- V. Andronis, M. Yoshioka and G. Zografi, J. Pharm. Sci., 1997, 86, 346–351 CrossRef CAS PubMed.
- V. Andronis and G. Zografi, J. Non-Cryst. Solids, 2000, 271, 236–248 CrossRef CAS.
- M. Otsuka and N. Kaneniwa, Chem. Pharm. Bull., 1988, 36, 4026–4032 CrossRef CAS.
- J. P. Bradley, S. P. Velaga, O. N. Antzutkin and S. P. Brown, Cryst. Growth Des., 2011, 11, 3463–3471 CAS.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef.
- T. J. Kistenmacher and R. E. Marsh, J. Am. Chem. Soc., 1972, 94, 1340–1345 CrossRef CAS.
- L. Carpentier, L. Bourgeois and M. Descamps, J. Therm. Anal. Calorim., 2002, 68, 727–739 CrossRef CAS.
- V. Joshi, K. R. Morris, S. R. Byrn and M. T. Carvajal, Cryst. Growth Des., 2009, 9, 3359–3366 CAS.
- A. J. Markvardsen, W. I. F. David, J. C. Johnson and K. Shankland, Acta Crystallogr., Sect. A: Found. Crystallogr., 2001, 57, 47–54 CrossRef CAS PubMed.
- W. I. F. David, K. Shankland, J. van de Streek, E. Pidcock, W. D. S. Motherwell and J. C. Cole, J. Appl. Crystallogr., 2006, 39, 910–915 CrossRef CAS.
- F. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
- T. Ukmar, V. Kaucic and G. Mali, Acta Chim. Slov., 2011, 58, 425–433 CAS.
- T. Limnell, H. A. Santos, E. Makila, T. Heikkila, J. Salonen, D. Y. Murzin, N. Kumar, T. Laaksonen, L. Peltonen and J. Hirvonen, J. Pharm. Sci., 2011, 100, 3294–3306 CrossRef CAS PubMed.
- C. Charnay, S. Begu, C. Tourne-Peteilh, L. Nicole, D. A. Lerner and J. M. Devoisselle, Eur. J. Pharm. Biopharm., 2004, 57, 533–540 CrossRef CAS PubMed.
- A. Kiwilsza, J. Mielcarek, A. Pajzderska and J. Wasicki, J. Microencapsulation, 2013, 30, 21–27 CrossRef CAS PubMed.
- Y. Zhang, T. Jiang, Q. Zhang and S. Wang, Eur. J. Pharm. Biopharm., 2010, 76, 17–23 CrossRef CAS PubMed.
- M. Manzano, M. Colilla and M. Vallet-Regi, Expert Opin. Drug Delivery, 2009, 6, 1383–1400 CrossRef CAS PubMed.
- B. D. Hamilton, J.-M. Ha, M. A. Hillmyer and M. D. Ward, Acc. Chem. Res., 2012, 45, 414–423 CrossRef CAS PubMed.
- J.-M. Ha, B. D. Hamilton, M. A. Hillmyer and M. D. Ward, Cryst. Growth Des., 2009, 9, 4766–4777 CAS.
- F. Babonneau, L. Camus, N. Steunou, A. Ramila and M. Vallet-Regi, in Self-Assembled Nanostructured Materials, ed. Y. Lu, C. J. Brinker, M. Antonietti and C. Bai, 2003, vol. 775, pp. 77–82 Search PubMed.
- W. Kolodziejski and J. Klinowski, Chem. Rev., 2002, 102, 613–628 CrossRef CAS PubMed.
- Y. Han, S. S. Lee and J. Y. Ying, Chem. Mater., 2007, 19, 2292–2298 CrossRef CAS.
- T. H. Elmer, Engineered Materials Handbook, ASM International, Materials Park, Ohio, 1992, pp. 427–432 Search PubMed.
- W. Haller, Nature, 1965, 206, 693–696 CrossRef CAS PubMed.
- W. Haller, J. Chem. Phys., 1965, 42, 686–693 CrossRef CAS PubMed.
- W. W. Lukens, P. Schmidt-Winkel, D. Y. Zhao, J. L. Feng and G. D. Stucky, Langmuir, 1999, 15, 5403–5409 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp03880d |
| This journal is © the Owner Societies 2015 |