
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of lithium salt doping on the nanostructure of ionic liquids†
Luis
Aguilera
a,
Johannes
Völkner
a,
Ana
Labrador
b and
Aleksandar
Matic
*a
aDepartment of Applied Physics, Chalmers University of Technology, Göteborg, Sweden. E-mail: matic@chalmers.se
bMAX IV Laboratory, Lund, Sweden
First published on 18th September 2015
Abstract
In this work we report on the evolution of the structure of two model ionic liquid families, N-alkyl-N-methylpyrrolidinium (Pyr1n-TFSI) and 1-alkyl-3-methylimidazolium (CnMIm-TFSI) (n = 3, 4, 6 and 8) both containing the bis(trifluoromethanesulfonyl)imide (TFSI) anion, upon the addition of LiTFSI using small angle X-ray scattering (SAXS). The introduction of a lithium salt (Li-salt) tunes the interactions through the substitution of the large cation in the ionic liquid with the small and charge localized lithium ion, thus increasing the coulombic contribution from ion–ion interactions. We find that the introduction of lithium ions results in a restructuring of the polar groups in the ionic liquids. These changes are manifested as an increase in the correlation lengths related to charge alternation of the ions and a more disordered structure. This restructuring is interpreted as a reconfiguration of the anions as they coordinate to the small and ionic lithium. In contrast, the length scale of the mesoscopic heterogeneities related to the clustering of alkyl chains is virtually unchanged with lithium doping. Moreover, the correlation corresponding to alkyl chain domains becomes more well defined with increasing salt concentration, suggesting that Li-salt doping, i.e. an increased columbic interaction in the system, promotes clustering of the alkyl tails.
Introduction
Ionic liquids (ILs) are low temperature molten salts (Tm < 100 °C).1 They typically consist of a large organic cation (imidazolium, pyridinium, pyrrolidinium) and an inorganic anion (PF6−, BF4−, [N(CF3SO2)2]−) weakly coordinated to each other. The bulky and asymmetrical configuration of the ions reduces the electrostatic interaction and prevents efficient packing in a lattice, thus resulting in low melting points. ILs currently attract considerable attention as key components for a range of different applications including electrochemistry,2 tribology,3 catalysis,4 and bio-preservation5 thanks to the flexibility in the design of the anions and cations which enables the optimisation of the properties. For instance, ILs can be designed to have wide thermal and electrochemical windows, low vapour pressure, low flammability, and high ionic conductivity. These properties have led to increased interest in using ionic liquids as replacement of volatile organic solvents to increase the safety of Li-ion batteries.6,7 However, this application requires the addition of a Li-salt to the ionic liquid, where the small and ionic lithium increases the coulombic interactions in the system resulting in a change in the phase behaviour and an increase in the glass transition temperature.8–10Tailoring the properties of ionic liquids is typically achieved by the synthesis of new anions and cations or by the addition of functional groups to the base ions. While these changes introduce the desired functionality, they also modify the interactions in the system and thus influence its structure and physical properties. For ionic liquids composed of small anions/cations coulombic interactions dominate. On the other hand with larger anions/cations, where side groups such as alkyl chains of different lengths are introduced, the contribution from van der Waals interactions is increased. Furthermore, addition of other side groups can introduce extra components into the interactions, for instance the presence of –OH or –NH groups can enable hydrogen bonding. In contrast to the synthesis of new ionic liquids as a way of changing the interactions mixtures of ionic liquids or the addition of an inorganic salt, which is not liquid at room temperature but can be dissolved in the IL, effectively creating a “new” mixed ionic liquid, offers a systematic and continuous way to tune interactions.
An intriguing feature of ionic liquids, closely related to the introduction of large side groups on the ions and thus changes in the interactions, is the occurrence of structural heterogeneities on mesoscopic length scales.11–23 The hallmark of this is a well-defined peak, typically found in the range of a few nm−1, in the static structure factor, S(Q), determined by for instance small angle X-ray scattering (SAXS) experiments and also found in MD simulations.11–14,21,23 Translating this peak into real space gives characteristic correlation distances of about 10–30 Å, which is very large for a liquid. The heterogeneities are suggested to originate from a segregation of the alkyl chains, due to van der Waals interactions, into domains embedded into a charged matrix. The size of these domains has been shown to depend linearly on the cation alkyl chain length;12,14 however, recent studies on relatively long alkyl chains (n > 12) suggest a departure from this linear dependence when the size of the side chains becomes very large.23 Interestingly, the heterogeneities are suppressed by substitution of carbon atoms in the alkyl chain with oxygen, i.e. when the difference in polarity between chains and charged parts of the cation decreases, hence lowering the driving force for segregation of the chains.13 Thus, the presence of the mesoscopic heterogeneities in ionic liquids is directly influenced by the interactions of the constituent ions.
In this work we report on the influence of Li-salt doping on the structural heterogeneities in two model ionic liquids based on pyrrolidinium (Pyr) and imidazolium (Im) cations and the TFSI− ([N(CF3SO2)2]−) anion by SAXS experiments as a function of salt concentration (x[LiTFSI]/(1−x)[IL] 0 < x < 0.4 x = mole fraction) and length of the alkyl chains on the cations (CnMIm-TFSI and Pyr1n-TFSI with n = 3, 4, 6 and 8). The aim is to investigate the role of interactions on the length scales and the stability of the structural heterogeneities. By using the same anion in the Li-salt as in the ionic liquid (i.e. TFSI−) this doping only exchanges a large charge delocalized cation, with a considerable contribution from van der Waals interactions, with the small ionic lithium, thus increasing the coulombic part of the interactions in the system. We show that despite a large change in the overall interactions of the system, by introduction of up to 40 mole% of Li-salt in the ionic liquid, the mesoscopic heterogeneities related to the segregation of alkyl chains are preserved. In contrast, there is a restructuring of the charge alternation domains with an increase in the characteristic length scale and increased disorder.
Experimental
Materials
Li-salt, lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, Sigma-Aldrich, >99.95%), and ionic liquids N-alkyl-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide (Pyr1n-TFSI) and 1-alkyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide (CnMIm-TFSI), with n = 3, 4, 6, and 8 (Solvionic >99.9%) were used as received. All materials were kept inside an argon filled glovebox where all the sample preparation was performed. Samples were prepared by mixing the corresponding mole fraction of the Li-salt (x = 0.01, 0.02, 0.05, 0.1, 0.2 and 0.4) with the desired IL. The samples were stirred overnight in glass vials to ensure homogeneous mixing. Prior to measurements, the samples were filled in 2 mm diameter glass capillaries, which were sealed with an epoxy resin.Small-angle X-ray scattering
Small-angle X-ray scattering (SAXS) experiments were performed at the I911-4 SAXS beamline at the MAX IV Laboratory, Lund, Sweden. With an incident wavelength of 0.91 Å the instrumental setup allowed the coverage of the momentum transfer range, Q = 1–12 nm−1 in one setting. Measurements were performed at room temperature. Data were normalized to the incoming beam intensity and the corresponding empty cell contribution was subtracted. Calibration to absolute units was not performed. In order to determine peak positions in the SAXS patterns, the data were fitted with a number of Lorentzian functions and a linear baseline.Results and discussion
The small angle X-ray diffraction patterns of the two series of ionic liquids are shown in Fig. 1 and 2. The diffraction patterns for the neat ionic liquids, Fig. 1, are in good agreement with those previously reported.18,20,23–25 As seen in Fig. 1, a pre-peak, or first sharp diffraction peak (FSDP) as it is sometimes referred to in the literature, clearly appears in the static structure factor S(Q) in the low Q region for the longer alkyl side chains, n ≥ 6, but already for n = 4 some excess scattering is observed in the imidazolium based ionic liquid. As expected, the pre-peak is found to move to smaller Q values for longer side chains.12–14 In addition to the pre-peak, a second peak, around Q = 0.85 Å−1, is found in the SAXS patterns of the TFSI-based ionic liquids.11,13,17,23 This peak is commonly associated with distances between ions of similar charge, i.e. it accounts for the charge alternation of the ions in the system.17,26 Due to the relatively large size of the TFSI− anion, the charge alternation peak appears in-between the pre-peak and a peak located around Q = 1.3 Å−1, related to inter- and intra-molecular contributions.17,20 From the data in Fig. 1 it is clear that this charge alternation peak has a much weaker dependence on the alkyl chain length and that it is found at slightly lower Q-values in the pyrrolidinium based ionic liquids compared to those of the imidazolium cation. | ||
| Fig. 1 Left: SAXS patterns of the two ILs families Pyr1n-TFSI and CnMIm-TFSI. Right: Schematic representation of different models to interpret the origin of the peaks in the SAXS diffraction patterns of ionic liquids, where (a) is the pre-peak and (b) the charge alternation peak. | ||
Several configuration models have been proposed in order to explain the structural heterogeneities in ILs. In Fig. 1 some of the commonly proposed configurations of such domains are presented. For instance, in a solvation model27 the presence of a pre-peak (a) is attributed to the second solvation shell, whereas the charge alternation peak (b) is assigned to the first solvation shell, when considering ions of similar charge. In a micelle-like model28 the pre-peak is associated with the size of the micelles, while the charge alternation peak corresponds to the distance between ions of a similar charge in the shell of the micelles. Finally, in a liquid-crystal like model, the hydrophobic tails aggregate in lamella-type structures giving rise to the pre-peak, whereas the charge alternation peak is again associated with the distance between the ions on the surface of the lamellas. It is worth mentioning that these are only schematic models that can be used to provide a basic understanding of possible structures present in the systems studied here. However, the actual structuring in ILs is certainly much more complex and most likely a combination of the different configurations is present.
In Fig. 2 the influence of the addition of Li-salt on the SAXS pattern is presented for the extreme cases of the shortest (n = 3) and longest (n = 8) alkyl chains for Pyr1n-TFSI and CnMIm-TFSI. From the SAXS patterns of the ionic liquids with the longest alkyl chains, Fig. 2b, it is clear that the addition of the salt has virtually no effect on the pre-peak position. To make this more evident the SAXS patterns of the undoped ILs have been superimposed on the patterns of the doped ones (dashed lines). In contrast, there is a rather strong effect of the addition of Li-salt on the charge alternation peak, the peak around Q = 0.85 Å−1 in the undoped ionic liquids. It shifts to lower Q-values and decreases considerably in intensity as the Li-salt concentration is increased. This effect is even more evident in the case of the ionic liquids with shorter side chains, Fig. 2a, where there is no interference from the pre-peak as in the case of long alkyl chains. A similar behaviour was previously reported for Pyr1n-TFSI and CnMIm-TFSI with n = 3 and 2 respectively.29 These findings are also in line with recent computational studies performed on a ethylammonium nitrate ionic liquid doped with a lithium salt of the same anion, where the mesoscopic structure of the ionic liquid is not disrupted by the addition of salt, but reorganized around the polar groups.30 However, to our knowledge, no experimental studies of this kind have been performed for ionic liquids while systematically changing the alkyl chain length.
 | ||
| Fig. 2 SAXS patterns of the two ILs families Pyr1n-TFSI (top) and CnMIm-TFSI (bottom), (a) as a function of Li-salt doping for n = 3, and (b) as a function of Li-salt doping for n = 8. Dashed curves represent the undoped ILs (x = 0), while the dashed vertical lines show the peak positions of the undoped ILs. | ||
The effects of Li-salt doping observed for the two extremes (n = 3 and n = 8) in Fig. 2 suggest that the mesoscopic domains of the ionic liquids, originating from the ordering of the hydrophobic tails of the cations, are barely affected by the addition of lithium salt. On the other hand, the presence of the lithium ions clearly influences the charge alternation. It has previously been shown that lithium ions form rather stable Li[TFSI]2 triplets in LiTFSI doped ionic liquids at concentrations x > 0.1.31,32 We envisage that these complexes effectively can act as one negative charge, which results in an effective longer separation between similar charges (see Fig. 3a). Thus, the changes observed in the SAXS patterns are a result of the re-organization of the polar groups, in particular the TFSI−. However, one should be cautious when interpreting changes in the SAXS patterns as there can be several different partial correlations contributing to one peak in S(Q). It has been shown that the charge alternation peak has contributions from both anion–anion and cation–cation correlations.18 In our experiment the addition of the Li-salt to the ionic liquid reduces the number of Pyr1n or CnMIm cations by the substitution of lithium. Furthermore, in the case of the formation of Li[TFSI]2− clusters, that act as a single larger negative charge, the effective number of anions, or anions complexes Li[TFSI]2−, is also reduced. Thus, we expect that the contribution from both partial structure factors (from cations and anions) should decrease in this scenario, which is in fact observed in our results as a reduced intensity of the charge alternation peak further supporting this interpretation.
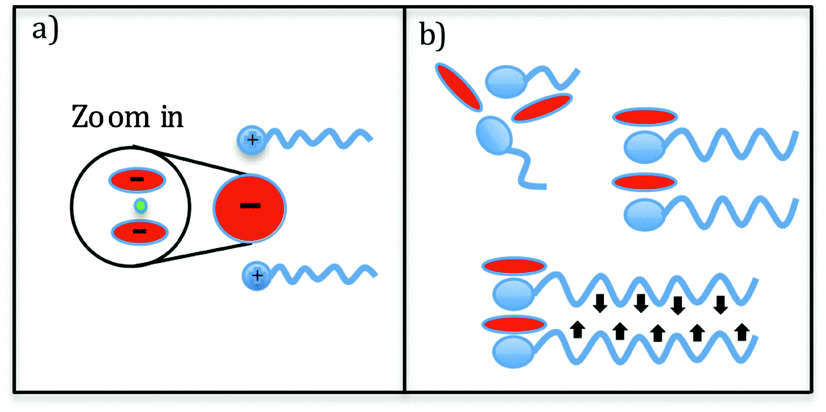 | ||
| Fig. 3 (a) Schematic of Li[TFSI]2 complexes where the cluster can effectively act as a single negative charge (lithium ion in green). (b) Schematic of the effect of the alkyl chain length on the distance between the polar groups. | ||
In order to better quantify the effects of the addition of the Li-salt on the structure of the ionic liquids, the SAXS patterns were fitted with 3 Lorentzian functions accounting for the pre-peak (when present), the charge alternation peak, and the peak associated with inter- and intra-molecular distances, just entering the measured window at high Q, and a linear baseline to account for the background. An example of a fitted SAXS pattern is shown in Fig. S1 in the ESI.† From the fits we obtain the position and the shape (width) of the pre-peak and charge alternation peak in S(Q). From the peak positions we can calculate the real space characteristic distances for the spatial correlations using the relation d = 2π/Qmax12 whereas the width will provide information regarding the extent of ordering in the system.23
The dependence of the characteristic distances corresponding to the pre- and charge alternation peaks, as a function of alkyl chain length and salt concentration are shown in Fig. 4. In agreement with previous results for neat ionic liquids we find that the correlation length derived from the pre-peak position varies linearly as a function of alkyl tail length (n).14 The resulting slopes obtained from a linear fit of the peak positions as a function of alkyl tail length are around 2, in excellent agreement with previous results reported for pyrrolidinium and imidazolium based ionic liquids with the TFSI anion.14,24 The data obtained for the different salt concentrations overlap with each other as well as with the data from the neat ionic liquids, underlining that the length scale of the structural heterogeneities related to the non-polar domains, are not changed by the introduction of the Li-salt. However, it is worth noting that a small deviation is observed for the case of the pyrrolidinium family for the shortest alkyl chain lengths (n = 4). This suggests that a different kind of structural arrangement might be present in this specific system due to the relatively short alkyl tail and the non-planar nature of the pyrrolidinium cation. Hence, the solvation model, see Fig. 1, is most likely the most relevant in this case, short side chains, since an increase in the distance of the second solvation shell is expected as a result of lithium salt doping, in contrast to the other two models proposed in Fig. 1 where the size of the micelles and lamellae should not be affected by lithium salt doping.
 | ||
| Fig. 4 Characteristic distances of structural heterogeneities related to the pre-peak (top panels) and the charge alternation peak (bottom panels) in the static structure factor as a function of alkyl chain length n and Li-salt concentration for Pyr1n-TFSI (top) and CnMIm-TFSI (bottom). Insets show a close-up of the distances related to the charge alternation peak in the low salt concentration region. | ||
The results for the characteristic distances corresponding to the charge alternation peak show a different behaviour compared to that of the pre-peak, as also expected from the first visual analysis of the SAXS data in Fig. 2. For the neat ionic liquids the characteristic charge alternation distance increases slightly as a function of alkyl chain length, reaching a maximum for n = 6 in the specific case of the imidazolium family. This behaviour has previously not been reported in this detail and based on our data we can offer a viable scenario. Under the assumption that this peak is a result of contributions of ions with the same charge, the introduction of longer alkyl tails effectively reduces the possibility of anions to access certain positions around the cation polar head due to steric hindrance, which results in slightly longer correlation distances. However, as the length of the alkyl tail increases van der Waals interactions start to play a dominant role, pulling the cations closer to each other due to the attractive interactions of the hydrophobic tails, thus limiting the distance between the ions, or even decreasing it (see Fig. 3b). In our data this effect is more accentuated in the imidazolium family, possibly due to their planar architecture that is more favourable when it comes to packing of the polar heads. Turning to the effect of the salt doping, it is evident that the charge alternation distance increases as the concentration of Li-salt increases. This effect is present already at low concentrations, as shown in the insets in Fig. 4, but the change in the characteristic distance is more pronounced with increased salt concentration. One can also note that the non-monotonic behaviour of the correlation distance as a function of chain length is also observed in the case of the Li-salt doped ionic liquids.
Further information on the structure of the heterogeneities can be obtained by considering the width of the peaks in the SAXS pattern. The Full Width at Half Maximum (FWHM) of the peaks can be related to how well-defined the structural heterogeneities are.23 The results of the FWHM obtained from the fitting of the diffraction patterns are summarized in Fig. S2 in the ESI.† In Fig. 5 we instead plot the FWHM differences as a function of Li-salt concentration, i.e. the difference between the width of the peaks in the Li-salt doped and the undoped IL SAXS patterns, which highlights the evolution of the structural heterogeneities with salt doping. Focusing first on the width of the pre-peak we find that the FWHM difference becomes increasingly more negative as the Li-salt concentration is increased, i.e. the width of the peak decreases. This is a clear indication of more-well defined structural correlations as the Li-salt concentration increases and the addition of Li-salt results in a more favourable clustering of the alkyl tails. Previous studies, providing indirect evidence by NMR and Raman spectroscopy, on Pyr1n-TFSI have suggested that the addition of lithium salt would increase the IL cations clustering even for relatively short alkyl chains.33 This is fully in line with our findings from the direct observation of the clustering in the SAXS data. However, one should note that there is virtually no effect on the width of the pre-peak for the longest alkyl chains (n = 8). These domains are already well defined as can be seen from the much lower FWHM of the pre-peak in the neat ionic liquids with long side chains, see Fig. S2 (ESI†). In contrast, the width of the charge alternation peak clearly broadens as the Li-salt is added. This is a sign of a more disordered structure at the level of charge alternation. This result is not unexpected since by introducing the Li-salt we are now dealing with a system that has three different components and the IL cations can alternate with either a single TFSI− anion or with a Li[TFSI]2− triplet (see Fig. 3a). Hence, the presence of two different characteristic lengths could give rise to a wider spread of the length scales characteristic of the charge alternation domains.
 | ||
| Fig. 5 FWHM differences, see text, as a function of Li-salt doping x and alkyl chain length n for Pyr1n-TFSI (top) and CnMIm-TFSI (bottom). | ||
Conclusions
We have investigated the effect of lithium salt doping on the nanostructure of two families of ionic liquids with different alkyl chain lengths. Our results provide clear evidence of changes on the characteristic length scale corresponding to the charge alternation domains due to the introduction of Li-salt into the systems. The changes are reflected as an increase in the characteristic distances as well as an increased disorder in the correlation as a result of the reorganization of the polar groups around the lithium ions. Furthermore, our results show that virtually no changes were introduced in the length scale associated with the polar/non-polar domains, apart from a narrowing of the pre-peak in the SAXS patterns that points towards better defined non-polar domains as the Li-salt concentration is increased. Finally, the non-monotonic variation of the charge alternation distances as a function of alkyl tail lengths, found with and without the addition of Li-salt, underlines the importance of the role played by the competing mechanism of steric hindrance and van der Waals interactions of the alkyl tails.Since the main application of Li-salt doped ionic liquids is as electrolytes it is of interest to correlate our findings on the meso-structure with the dynamics and the transport properties. Our results are well in line with previous reports of ionic liquids based electrolytes where the coordination of the anions to the lithium results in an increased viscosity, which hinders the efficient transport of lithium through the electrolyte due to the formation of larger aggregates.34 From NMR-experiments33 it has been shown that the same overall trend of a slowing down of the motion of both anions and cations is observed for both neat ionic liquids and Li-salt doped ionic liquids. This is well in agreement with our findings that the heterogeneous structure caused by the aggregation of alkyl chains is preserved. However, one has also found that the slowing down of the anions is stronger when a Li-salt is added, in agreement with our result of a restructuring of the charge alternation correlation, involving the lithium ions and the TFSI anions.
References
-
S. Zhang, X. Lu, Q. Zhou, X. Li and X. Zhang, Ionic liquids, Elsevier Science, 2009 Search PubMed
.
- H. Sakaebe, H. Matsumoto and K. Tatsumi, Electrochim. Acta, 2007, 53, 1048–1054 CrossRef CAS PubMed
- C. Ye, W. Liu, Y. Chen and L. Yu, Chem. Commun., 2001, 2244–2245 RSC
- H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef CAS PubMed
- K. Fujita, D. R. MacFarlane and M. Forsyth, Chem. Commun., 2005, 4804–4806 RSC
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed
- A. Matic and B. Scrosati, MRS Bull., 2013, 38, 533–537 CrossRef CAS
- J. Pitawala, J.-K. Kim, P. Jacobsson, V. Koch, F. Croce and A. Matic, Faraday Discuss., 2011, 154, 71–80 RSC
- A. Martinelli, A. Matic, P. Jacobsson, L. Börjesson, A. Fernicola and B. Scrosati, J. Phys. Chem. B, 2009, 113, 11247–11251 CrossRef CAS PubMed
- J.-K. Kim, A. Matic, J.-H. Ahn and P. Jacobsson, J. Power Sources, 2010, 195, 7639–7643 CrossRef CAS PubMed
- A. Triolo, O. Russina, B. Fazio, G. B. Appetecchi, M. Carewska and S. Passerini, J. Chem. Phys., 2009, 130, 164521 CrossRef PubMed
- A. Triolo, O. Russina, H.-J. Bleif and E. Di Cola, J. Phys. Chem. B, 2007, 111, 4641–4644 CrossRef CAS PubMed
- O. Russina and A. Triolo, Faraday Discuss., 2011, 154, 97–109 RSC
- O. Russina, A. Triolo, L. Gontrani and R. Caminiti, J. Phys. Chem. Lett., 2012, 3, 27–33 CrossRef CAS
- S. Patra and A. Samanta, J. Phys. Chem. B, 2012, 116, 12275–12283 CrossRef CAS PubMed
- H. K. Kashyap, C. S. Santos, R. P. Daly, J. J. Hettige, N. S. Murthy, H. Shirota, E. W. Castner and C. J. Margulis, J. Phys. Chem. B, 2013, 117, 1130–1135 CrossRef CAS PubMed
- H. K. Kashyap, J. J. Hettige, H. V. R. Annapureddy and C. J. Margulis, Chem. Commun., 2012, 48, 5103–5105 RSC
- H. K. Kashyap, C. S. Santos, N. S. Murthy, J. J. Hettige, K. Kerr, S. Ramati, J. Gwon, M. Gohdo, S. I. Lall-Ramnarine, J. F. Wishart, C. J. Margulis and E. W. Castner, J. Phys. Chem. B, 2013, 117, 15328–15337 CrossRef CAS PubMed
- H. V. R. Annapureddy, H. K. Kashyap, P. M. De Biase and C. J. Margulis, J. Phys. Chem. B, 2010, 114, 16838–16846 CrossRef CAS PubMed
- S. Li, J. L. Bañuelos, J. Guo, L. Anovitz, G. Rother, R. W. Shaw, P. C. Hillesheim, S. Dai, G. A. Baker and P. T. Cummings, J. Phys. Chem. Lett., 2011, 3, 125–130 CrossRef
- S. M. Urahata and M. C. C. Ribeiro, J. Phys. Chem. Lett., 2010, 1, 1738–1742 CrossRef CAS
- B. L. Bhargava, R. Devane, M. L. Klein and S. Balasubramanian, Soft Matter, 2007, 3, 1395–1400 RSC
- A. Martinelli, M. Maréchal, Å. Östlund and J. Cambedouzou, Phys. Chem. Chem. Phys., 2013, 15, 5510–5517 RSC
- C. S. Santos, N. S. Murthy, G. A. Baker and E. W. Castner, J. Chem. Phys., 2011, 134, 121101 CrossRef PubMed
- O. Russina, A. Triolo, L. Gontrani, R. Caminiti, D. Xiao, L. G. Hines Jr, R. A. Bartsch, E. L. Quitevis, N. Plechkova and K. R. Seddon, J. Phys.: Condens. Matter, 2009, 21, 424121 CrossRef
- J. J. Hettige, H. K. Kashyap, H. V. R. Annapureddy and C. J. Margulis, J. Phys. Chem. Lett., 2013, 4, 105–110 CrossRef CAS PubMed
- C. Hardacre, J. D. Holbrey, C. L. Mullan, T. G. A. Youngs and D. T. Bowron, J. Chem. Phys., 2010, 133, 074510 CrossRef PubMed
- A. Triolo, O. Russina, B. Fazio, R. Triolo and E. Di Cola, Chem. Phys. Lett., 1999, 457, 362–365 CrossRef PubMed
- Y. Umebayashi, H. Hamano, S. Seki, B. Minofar, K. Fujii, K. Hayamizu, S. Tsuzuki, Y. Kameda, S. Kohara and M. Watanabe, J. Phys. Chem. B, 2011, 115, 12179–12191 CrossRef CAS PubMed
- T. Mendez-Morales, J. Carrete, O. Cabeza, O. Russina, A. Triolo, L. J. Gallego and L. M. Varela, J. Phys. Chem. B, 2014, 118, 761–770 CrossRef CAS PubMed
- J. Pitawala, A. Martinelli, P. Johansson, P. Jacobsson and A. Matic, J. Non-Cryst. Solids, 2015, 407, 318–323 CrossRef CAS PubMed
- J.-C. Lassègues, J. Grondin, C. Aupetit and P. Johansson, J. Phys. Chem. A, 2009, 113, 305–314 CrossRef PubMed
- M. Kunze, S. Jeong, E. Paillard, M. Schönhoff, M. Winter and S. Passerini, Adv. Energy Mater., 2011, 1, 274–281 CrossRef CAS PubMed
- C. J. F. Solano, S. Jeremias, E. Paillard, D. Beljonne and R. Lazzaroni, J. Chem. Phys., 2013, 139, 034502 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp03825a |
| This journal is © the Owner Societies 2015 |