
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interaction of ionic liquids with noble metal surfaces: structure formation and stability of [OMIM][TFSA] and [EMIM][TFSA] on Au(111) and Ag(111)†
Benedikt
Uhl
ab,
Hsinhui
Huang
ab,
Dorothea
Alwast
ab,
Florian
Buchner
ab and
R. Jürgen
Behm
*ab
aInstitute of Surface Chemistry and Catalysis, Ulm University, Albert-Einstein-Allee 47, D-89081 Ulm, Germany. E-mail: juergen.behm@uni-ulm.de; Fax: +49-731-50-25452
bHelmholtz-Institute Ulm (HIU) Electrochemical Energy Storage, Helmholtzstr. 11, D-89081 Ulm, Germany
First published on 7th August 2015
Abstract
Aiming at a comprehensive understanding of the interaction of ionic liquids (ILs) with metal surfaces we have investigated the adsorption of two closely related ILs, 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide [EMIM][TFSA] and 1-methyl-3-octylimidazolium bis(trifluoromethylsulfonyl)imide [OMIM][TFSA], with two noble metal surfaces, Au(111) and Ag(111), under ultrahigh vacuum (UHV) conditions using scanning tunneling microscopy (STM). At room temperature, the ILs form a 2D liquid on either of the two surfaces, while at lower temperatures they condense into two-dimensional (2D) islands which exhibit ordered structures or a short-range ordered 2D glass structure. Comparison of the adlayer structures formed in the different adsorption systems and also with those determined recently for n-butyl-n-methylpyrrolidinium [TFSA]− adlayers on Ag(111) and Au(111) (B. Uhl et al., Beilstein J. Nanotechnol., 2013, 4, 903) gains detailed insight into the adsorption geometry of the IL ions on the surface. The close similarity of the adlayer structures indicates that (i) the structure formation is dominated by the tendency to optimize the anion adsorption geometry, and that (ii) also in the present systems the cation adsorbs with the alkyl chain pointing up from the surface.
Introduction
The interaction of ionic liquids (ILs) with solid surfaces has attracted increasing interest in recent years because of various possible applications of ILs as solvents, e.g., for the deposition of metals that cannot be deposited from aqueous solutions (e.g. Li or Al),1–4 in catalysis5–9 or in battery electrolytes, in particular in Li ion batteries, exploiting the very high electrochemical stability of these species.10–17 In all cases, a fundamental understanding of the processes at the solid|IL interface is urgently needed for a systematic improvement of the respective systems. A comprehensive account of the current literature on the structure and nanostructure in bulk ILs and at the IL|solid interface was recently published by Hayes et al.18Since the complexity of realistic systems renders it essentially impossible to gain a clear understanding of the ongoing processes on a molecular scale, these processes have been studied using well-defined model systems and simplified conditions. Focussing on applications of ILs as solvent in electrochemistry, the potential dependent interaction of bulk ILs with a single crystal metal surface was investigated combining in situ scanning tunneling microscopy (STM) and cyclic voltammetry (CV).19–21 Another, even more idealized approach involves studies of the interaction of ILs with solid surface under ultrahigh vacuum conditions, which allows the use of a whole arsenal of surface sensitive techniques. Due to their extremely low vapour pressure, even thicker IL films can be investigated this way. The detailed information accessible from such kind of studies, employing angle resolved X-ray photoelectron spectroscopy (ARXPS) as well as infrared reflection absorption spectroscopy (IRRAS), was illustrated by the Steinrück group.22–27
Following these lines, we have started to systematically investigate the interaction of battery relevant ILs with solids, focusing on the structure formation at the solid|IL interface as well as on the possible reactive decomposition of the respective ILs.28–32 In that work it was possible to gain insight into the structure formation on a molecular scale, combining high resolution STM imaging and XPS, which in combination with density functional calculations allowed us to derive detailed information on the nature and order of magnitude of substrate–adsorbate interactions and the interactions between adjacent adsorbed IL species (adsorbate–adsorbate interactions). In the present paper we report results of a comparative STM study on the interaction of two ILs with the same bis(trifluoromethylsulfonyl)imide [TFSA]− anion, [EMIM][TFSA] ([EMIM]+ = 1-ethyl-3-methylimidazolium) and [OMIM][TFSA] ([OMIM]+ = 1-methyl-3-octylimidazolium), with two rather similar noble metal surfaces, Ag(111) and Au(111). The chemical structure of the two ILs is displayed in Fig. 1. We focus on the adlayer structure formation in the submono- to monolayer coverage regime, where the resulting surface structures are unambiguously related to the first layer, which is in contact with the substrate. [TFSA]− based ILs, especially in combination with imidazolium or pyrrolidinium based cations, have emerged as promising candidates for application in Li ion batteries,10,14,15 underlining the interest in these compounds also from an application point of view. We are particularly interested in mapping out similarities and differences in the adsorption and structure formation behavior caused by the change of the cation and of the substrate, while the anion was kept the same. This will gain information on the influence of the cation and of the substrate structure on these properties, especially when also comparing with our previous results on the adsorption of another IL with identical anion, but rather different cation, [BMP][TFSA] (BMP = n-butyl-n-methylpyrrolidinium) on Au(111) and Ag(111).31
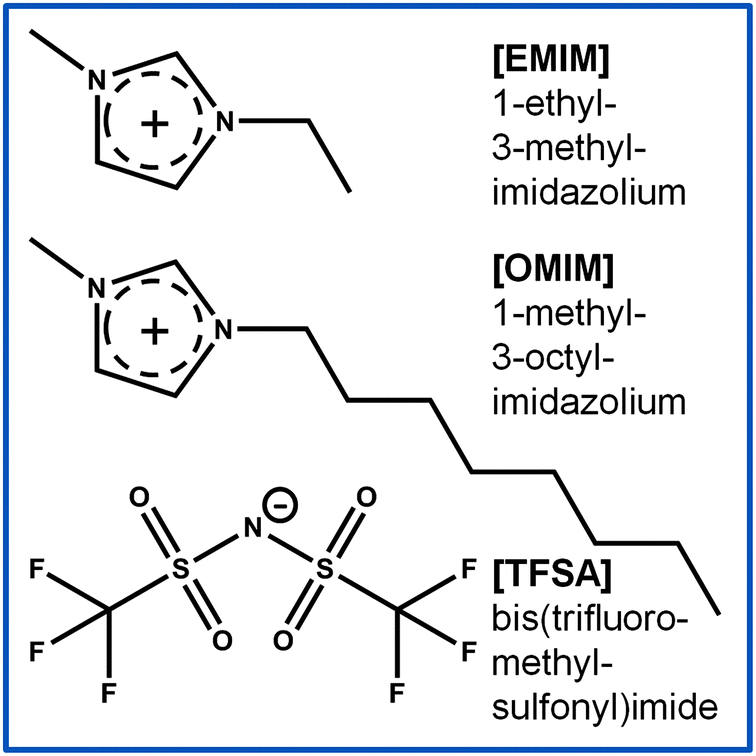 | ||
| Fig. 1 Structural formulas of the investigated ionic liquids. | ||
In the following we will present results on the structural characteristics of the adlayers on the different substrates at room temperature, followed by the results on the formation, structural characteristics and stability of the adlayer structure formed at low temperatures for each substrate–adsorbate system separately. We will start with adsorption on the structurally simpler Ag(111) surface, followed by adsorption on the reconstructed Au(111) surface.
Results
For all four adsorption systems, [EMIM][TFSA] and [OMIM][TFSA] adsorption on Au(111) and on Ag(111), respectively, STM imaging at room temperature did not resolve any specific adsorbate related structures, except for a significantly higher noise level than detected on the adsorbate free Au(111) or Ag(111) surfaces. On the other hand, the steps of the metal substrate surface as well as the herringbone surface reconstruction of the Au(111) sample did not show any significant modification. Hence, adsorption of the ILs does not lead to an IL induced modification of the surface itself. The latter had been reported by Atkin et al. for adsorption of [EMIM][TFSA] on Au(111) in an electrochemical environment, where they detected wormlike patterns on the Au surface upon adsorption at the open circuit potential (OCP), with holes of 1.5–2.0 nm depth in the Au surface.33 The pronounced noise during STM imaging is attributed to adsorbed IL species which move quickly on the substrate surface and pass through the tunnel junction. Hence, depending on the density of the adlayer this can be considered as a 2D gas or 2D liquid, where the adsorbed species are too mobile to be resolved on the time scale of the STM measurements.Similar results, pointing to the presence of a 2D liquid at room temperature, have been already reported for adsorption of other ILs on noble metal surfaces, such as [EMIM][TFSA] adsorption on Au(110),34 [BMP][FAP] on Au(111),28 and finally [BMP][TFSA] adsorption on Au(111)29 and Ag(111).30 It is not clear, however, whether the adsorbed IL species diffuse pairwise, similar to the existence of cation–anion pairs in the gas phase upon evaporation, as it was observed for various ILs (inter alia [EMIM][TFSA] and [OMIM][TFSA]),35,36 or whether they diffuse individually. Considering that for [BMP][TFSA] adsorption on Ag(111) DFT calculations and XPS measurements had indicated that the charge remained almost completely on the adsorbed species,30 we favor a correlated, pairwise motion on the surface. Comparing to results of in situ measurements in an electrochemical environment, our data are most closely related to measurements at OCP conditions, with no additional electrostatic charge on the substrate. Recently Elbourne et al.37 studied the interaction of [EMIM][TFSA] with HOPG by in situ AFM. They resolved row-like features at the OCP and concluded that the rows result from an ordered layer of the IL, with both anion and cation adsorbed directly to the surface. They proposed that cations and anions each form parallel rows with a sequence of anion–cation–cation–anion rows. The structures are first changing and afterwards at ±0.3 V no structures can be resolved any more. The formation of structures even at room temperature was argued to originate from a π–π interaction of the imidazolium ring and the HOPG surface. In that case, [EMIM][TFSA] seems to interact much more strongly with HOPG than with noble metal surfaces and the formation of these structures is very much depending on the substrate. In another in situ study, Wen et al.21 investigated the interaction of [BMP][TFSA] with Au(111) by Video-STM. While at the OCP no distinct features could be resolved, individual structures can be resolved at potentials <−1.0 V vs. Pt. These appear first as fluctuating structures in front of the substrate steps, and at <−1.4 V a close packed 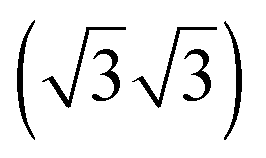 superstructure forms, which changes into a
superstructure forms, which changes into a 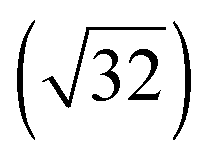 structure <−1.6 V. These structures were interpreted as layers of solely [BMP]+ cations which are electrostatically attracted to the negatively charged surface, with the butyl group lying flat on the surface in the first structure and the butyl group standing upright in the second case. Comparable results, with a mobile adsorption of both ion types around the PZC (point of zero charge; in this study at −0.3 V), and the formation of ordered and disordered structures at potentials >−0.1 V and at <−0.7 V, which were attributed to adsorption of solely the anions and solely the cations, respectively, were also reported for [BMIM][BF4] adsorption on Au(100).38,39 Similar results were also found for various alkylimidazolium based ILs in contact to Au(111) regarding cation adsorption at negative potentials40 and additionally for [BMIM][TFSA] on Au(111).41 In contrast, the present study performed under UHV conditions reveals the stable adlayer structures formed in the absence of an applied potential and of interactions with a bulk electrolyte. For [BMP][TFSA] on Ag(111)30 and Au(111)29 this results in co-adsorption of cation and anion with both ions directly interacting with the substrate.
structure <−1.6 V. These structures were interpreted as layers of solely [BMP]+ cations which are electrostatically attracted to the negatively charged surface, with the butyl group lying flat on the surface in the first structure and the butyl group standing upright in the second case. Comparable results, with a mobile adsorption of both ion types around the PZC (point of zero charge; in this study at −0.3 V), and the formation of ordered and disordered structures at potentials >−0.1 V and at <−0.7 V, which were attributed to adsorption of solely the anions and solely the cations, respectively, were also reported for [BMIM][BF4] adsorption on Au(100).38,39 Similar results were also found for various alkylimidazolium based ILs in contact to Au(111) regarding cation adsorption at negative potentials40 and additionally for [BMIM][TFSA] on Au(111).41 In contrast, the present study performed under UHV conditions reveals the stable adlayer structures formed in the absence of an applied potential and of interactions with a bulk electrolyte. For [BMP][TFSA] on Ag(111)30 and Au(111)29 this results in co-adsorption of cation and anion with both ions directly interacting with the substrate.
To obtain molecular resolution under UHV conditions and thus gain information on the adsorption behavior and on the molecule–surface interactions as well as on the interactions between the adsorbed IL species also for [EMIM][TFSA] and [OMIM][TFSA] on Ag(111) and Au(111), similar to [BMP][TFSA] the samples need to be cooled to reduce the mobility of the adsorbates. This was done by slowly (ca. 100 K h−1) cooling the entire STM set-up together with the adsorbate covered sample to liquid nitrogen (LN2) temperature. The results will be presented in the following for each adsorption system separately.
3.1 [OMIM][TFSA] adsorption on Ag(111)
At very low coverages, adsorbed [OMIM][TFSA] species are predominantly found in front of the descending steps of the Ag(111) surface. At higher coverages, islands form on the terraces, which, however, are always connected to the steps of the Ag(111) surface. Hence, upon cooling the sample to nearly LN2 temperature, condensation of the adsorbed IL species starts at the upper side of the steps, which act as preferred adsorption sites for the adsorbed IL molecules and hence as nucleation sites for 2D island formation. With higher coverages, the adsorbate islands grow across the terraces by condensation of further mobile adspecies at the perimeter of the adlayer islands. The formation of these islands indicates the existence of attractive interactions between the adsorbed species, similar to the adsorption of [BMP][TFSA] on Ag(111).30 DFT-calculations for the latter adsorption system had shown that the adsorbed ions keep most of their charge. We therefore relate the attractive intermolecular forces mainly to electrostatic interactions. Similar characteristics we assume also for [OMIM][TFSA] adsorption on Ag(111).[OMIM][TFSA] forms two types of adlayer structures on Ag(111) which are resolved in the STM image in Fig. 2a. This image, which was recorded on a surface covered by a submonolayer of [OMIM][TFSA], shows two terraces of a Ag(111) surface where one is largely overgrown with a long-range ordered structure (structure #1). This phase can also be denoted as a 2D crystalline structure. A high resolution image of this structure, recorded on the area marked by a white rectangle, is presented in Fig. 2b. The adlayer forms a regular pattern of round shaped protrusions (marked by black dots in the image) and elliptic protrusions (marked with black ovals in the image), where the latter ones are not as high as the round protrusions. The protrusions are aligned in rows parallel to the full black line in Fig. 2b; between each row of round shaped protrusions there is a row of elliptic protrusions (details see below). The rows of round shaped protrusions are arranged pairwise, with a shorter row-to-row distance within each pair than between the pairs of rows. A similar pairwise arrangement is also found along the rows. In this case, however, the differences between the distances within and between the pairs are less pronounced. Within each row of elliptic protrusions (along the full black line in Fig. 2b), the latter are oriented approximately in parallel, with their long axis rotated by 60 ± 10° with respect to the row direction. Between neighbored rows, the orientation of these protrusions changes by 120 ± 10°. The unit cell of the adlayer lattice is marked by a black parallelogram in Fig. 2b, its dimensions are 2.1 ± 0.1 nm × 2.3 ± 0.1 nm, with an angle of 82 ± 4° in between the lattice directions. The unit cell (cell area 4.9 ± 0.4 nm2) contains 4 round shaped protrusions and 8 elliptic protrusions, giving a density of 0.8 ± 0.1 round protrusions per nm2. The round shaped protrusions are always surrounded by 4 pairs of long shaped protrusions, and each of the pairs of longish protrusions by 4 round shaped protrusions.
 | ||
Fig. 2 STM images of a Ag(111) surface covered with [OMIM][TFSA] adsorbates: (a) the IL adsorbates arrange in domains with 2D crystalline (1) and 2D glass (2) structure; between the adsorbate covered areas bare Ag(111) surface (3) is visible (T = 110 K, UT = −1.16 V, IT = 130 pA). (b) High resolution image of the 2D crystalline structure; the size of the image is equal to the size of the white rectangle in (a); (cations: round shaped protrusions, anions: pairs of longish protrusions). The unit cell of the 2D crystalline structure is marked by a black parallelogram (dimension: 2.0 ± 0.1 nm × 2.2 ± 0.1 nm, directions: black line × dashed black line) (T = 118 K, UT = −1.10 V, IT = 90 pA). (c) 2D crystalline structure aligned parallel to well ordered Ag(111) steps along the 〈1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0〉 direction of the surface lattice. For better contrast the image was cut at the steps and the contrast was enhanced for each terrace on its own; terrace α is the lowest, γ the highest (T = 118 K, UT = −1.06 V, IT = 50 pA). (d) Domain of the 2D crystalline structure which is chiral to the ones shown in (a) and (b) (T = 148 K, UT = −1.10 V, IT = 80 pA). 0〉 direction of the surface lattice. For better contrast the image was cut at the steps and the contrast was enhanced for each terrace on its own; terrace α is the lowest, γ the highest (T = 118 K, UT = −1.06 V, IT = 50 pA). (d) Domain of the 2D crystalline structure which is chiral to the ones shown in (a) and (b) (T = 148 K, UT = −1.10 V, IT = 80 pA). | ||
The adlayer structure closely resembles that observed for [BMP][TFSA] on Ag(111), both with respect to the characteristic structural elements and their arrangement.30 The only significant difference is that in the latter case the distances between the round protrusions along the row direction marked by the black full line are, within the limits of the resolution, identical, leading to a unit cell with half the size as obtained here ([BMP][TFSA] – Ag(111): 2.3 ± 0.1 nm × 1.1 ± 0.1 nm, unit cell area 2.5 ± 0.3 nm2). This results in exactly the same density of 0.8 ± 0.1 round protrusions per nm2 for the latter adsorption system. Since [BMP][TFSA] and [OMIM][TFSA] appear nearly similar in the STM images, it is likely that both ILs have a comparable adsorption geometry on Ag(111), where the latter is known from combined STM results and DFT-D calculations.30 In that case, the [TFSA]− anion, which corresponds to a pair of elliptic protrusions,30 is adsorbed in a cis conformation, with the SO2 groups pointing to the surface, while the electron densities leading to the elliptic protrusions in the STM image are mainly generated by the CF3 groups pointing away from the surface. The round shaped protrusions then have to be associated with the [OMIM] cation. The DFT calculations for [BMP][TFSA] adsorption mentioned above had indicated that the cation adsorbs with the ring lying flat on the surface and the alkyl chain pointing away from the surface, where the latter is responsible for the round protrusion appearing in the STM images.30 Therefore we assume a similar adsorption geometry for the [OMIM] cation. Comparing this structure with the results of Cremer et al.25 and Krischok et al.,42 who concluded from their XPS25,42 and MIES (metastable induced electron spectroscopy)42 results that for room temperature [OMIM][TFSA] adsorption on Au(111) the alkyl chain is horizontal to the substrate surface, there seems to be a reorientation in the adsorption layer during cool down from room temperature to LN2 temperature, with the alkyl chains bending up from the surface when going to LN2 temperature. This may be rationalized by a tendency to order in an energetically more favorable adsorption geometry, with smaller distances between the differently charged adsorbed ions, and by the formation of islands. At temperatures above the 2D crystallization temperature, the thermal motion/energy in the system hinders the formation of ion pairs in their most favorable adsorption geometry, and therefore they move across the surface with little interactions to neighboring molecules. We speculate that in that case the adsorption of the cations may change such that the longer alkyl chain is oriented parallel to the surface because of the expected higher van der Waals interactions to the substrate.
The similarity of the 2D crystalline structures formed by [BMP][TFSA] and [OMIM][TFSA] on Ag(111) leads also to a very interesting conclusion: obviously the nature of the cation has little influence on the structure formation. [OMIM]+ is an imidazole with a 5-membered planar aromatic ring containing 2 nitrogen atoms, while [BMP]+ is a pyrrolidinium derivative with a 5-membered non-aromatic non-planar ring containing one nitrogen. Although the geometric and electronic structures of the cations differ considerably, which should result also in different interactions with the substrate and between neighbored cation and anion cations, we find no significant differences in the overall adsorption structures. This indicates that the resulting ordered adlayer structures are dominated by the [TFSA]−–substrate interactions, specifically by the variation of these interactions with adsorption site and adsorption geometry. Optimizing the anion adsorption geometry and thus the anion–substrate interactions seems to be the determining parameter for the adsorption structures found on Ag(111) for both [BMP][TFSA] and [OMIM][TFSA], overcompensating correlated variations in the cation–substrate and adsorbate–adsorbate interactions. It should be noted that such kind of structure determining role of the anions is specific for the present case. In particular for adsorption at negative potentials in an electrochemical environment, where one would expect the cations to be in direct contact with the electrode surface, these are likely to be responsible for the resulting adlayer structure. Accordingly, Borisenko et al. reported pronounced differences in the adsorption behavior of three different ILs, all containing the same [FAP]+ anion, but different cations ([BMP]+, [EMIM]+ and [HMIM]+) on Au(111) at negative potentials in a combined in situ STM and AFM study.43 Although molecular resolution could not be obtained, the change in cations was found to result in very different structures of the solid–liquid interface.43
Coming back to our STM measurements, in addition to structure #1, there are also larger disordered adlayer areas, as can be seen in Fig. 2a on the upper terrace (marked with ‘2’ in the image). Closer inspection reveals that the local structures are often similar to those in the long-range ordered phase, with short rows of round protrusions and the characteristic arrangement of anions. Hence, they exhibit a short-range order, but no long-range order. This structure will in the following be termed a 2D glass phase. The islands consisting of 2D glass domains also enclose adsorbate-free Ag(111) areas (marked with ‘3’ in Fig. 2a). Similar to [BMP][TFSA] adsorption on Ag(111),30 the 2D glass phase mainly appears on narrow terraces and along steps in areas where large ordered areas connect to steps. In addition, for [OMIM][TFSA] adsorption the 2D glass phase can form islands/domains also on wide terraces, which was not observed for [BMP][TFSA] adsorption on Ag(111).30 Finally, areas of structure #1 often include small disordered ‘defective’ areas with dimensions of a few nanometers. This contrasts the structure formation of [BMP][TFSA] on Ag(111),30 where the 2D crystalline domains typically extend with almost no defects over several 100 nm. Overall, the relative amount of 2D glass structure is clearly higher for [OMIM][TFSA] adsorption on Ag(111) than for [BMP][TFSA] adsorption. Most likely, the higher tendency to form the short-range ordered structure is related to slightly different space requirements of the ILs in the 2D crystalline structures, which allows a better fit of the adlayer to the substrate lattice for [BMP][TFSA] adsorption than for [OMIM][TFSA] adsorption.
The influence of the substrate lattice is on the other hand indicated by the fact that the 2D crystalline adlayer structure is not oriented randomly relative to the surface, but exhibits fixed rotational orientations with respect to the substrate lattice. In agreement with the threefold symmetry of the Ag(111) surface we found three rotational orientations of that phase, indicating that one lattice direction of the adlayer is oriented along the close-packed 〈10〉 direction of the substrate. Considering that the steps of a well prepared Ag(111) surface are largely oriented along the 〈10〉 directions and that the paired rows of the adsorbate structure are mostly either parallel (see Fig. 2c) or at angles of 60° or 120° (see Fig. 2a) to the steps, the rows of the adlayer structure must be oriented along the 〈10〉 directions as well. The long axes of the elliptic protrusions, which represent the anions, are rotated by 60° relative to the paired rows and are therefore also oriented in 〈10〉 direction. It was not possible to obtain molecular resolution images of the IL adlayers together with an atomically resolved Ag(111) surface in the same STM image to confirm this conclusion. This is due to the very different tunneling conditions that are necessary for atomic/molecular resolution imaging: for atomically resolved Ag(111) images tunneling currents higher than 300 pA are necessary in our setup, while at tunneling currents above ∼150 pA the tip starts to push away the adsorbed IL molecules and destroys the 2D crystalline domains. Furthermore, stable high resolution imaging is hindered by the moving admolecules and their ‘collisions’ with the tip.
In addition to the 3 rotational orientations the unit cell can be arranged in two different configurations by mirroring it at a vertical plane along the 〈10〉 direction, indicating that the structures are chiral. Two such structures are resolved in Fig. 2a, b and d. The images are rotated such that the molecule rows in the structure (marked with black lines in Fig. 2b and d) are oriented in the same direction. The second directions of the unit cells (marked with dashed black lines) in Fig. 2b and d are mirrored at a vertical plane along the 〈10〉 direction. In total, this yields 6 possibilities to align the ordered structure on the substrate surface (2 chiral structures in 3 directions each). Still, in all possible orientations found for the 2D crystalline adlayer structure, the longish protrusions align along the 〈10〉 direction. Since a pair of longish protrusions resembles one anion, this means that the anions always align in a special and well defined adsorption geometry relative to the substrate lattice. This observation supports our above assumption that the structure formation is dominated by the anion–substrate interactions and the tendency to optimize the [TFSA]− anion adsorption geometry.
The thermal stability of the adlayer structures was tested by slowly (ca. 70 K h−1) heating the STM set-up from 90 K to room temperature, while continuously recording STM images. Already at 90 K (see Fig. 2a and c) we find stripy features at the island boundaries to the surrounding 2D gas phase, both for the ordered and for the short-range ordered phase islands. These features arise from mobile molecules moving along the island boundaries or attaching to/detaching from the islands. The boundaries of the islands are therefore constantly changing, while the adsorbates within the 2D islands are immobile. With increasing temperature, the frizziness at the island boundaries increases and spreads across the 2D glass phase areas, while the central parts of the islands with 2D crystalline structure remain stable. For the 2D glass phase we cannot give a definite ‘melting’ temperature, since its mobility increases over a wide temperature range, but it was always completely dissolved when the 2D crystalline structure started to melt. An STM image taken close to the melting temperature of the 2D crystalline structure is given in the ESI† (Fig. S1a), where the coexistence of the 2D liquid (appearing as noisy features) and remaining 2D crystalline domains is visible.
The ordered structure melts at 165 ± 10 K in islands at submonolayer coverages and at 185 ± 10 K in a monolayer covered sample. The higher melting temperature in the monolayer coverage range reflects the mutual stabilization of the adsorbed species by (additional) repulsive interactions, while at lower coverages formation of ordered structures is dominated by attractive interactions. The stability of the adlayer structure closely resembles that of the [BMP][TFSA] adlayer structure on Ag(111), where a melting temperature of 180 ± 10 K was observed for both the monolayer and for the submonolayer coverage regime.30
Overall, the interaction of [OMIM][TFSA] with Ag(111) is dominated by molecular adsorption and the formation of a mobile 2D liquid at room temperature. This and the formation of 2D solid adlayer phases only at low temperatures, with melting temperatures below 200 K, indicate the presence of weak adsorbate–adsorbate interactions. The formation of very similar ordered and short-range ordered adlayer structures as observed previously for [BMP][TFSA] on Ag(111),30 as well as the similar orientation of the adsorbed [TFSA]− anions in both adsorption systems relative to the substrate lattice indicate that the structure formation is dominated by the anion–substrate interactions and the tendency to optimize the [TFSA]− anion adsorption geometry.
3.2 [EMIM][TFSA] adsorption on Ag(111)
[EMIM][TFSA] differs from [OMIM][TFSA] only by its shorter alkyl chain in the cation (octyl → ethyl). Therefore, any differences in the adsorption behavior and structure formation must be directly connected with this, or, reversely, if there are no significant differences, the length of the alkyl chain has no influence on the adsorption characteristics.The adlayer structures formed upon [EMIM][TFSA] adsorption indeed closely resemble those observed for the adsorption of [OMIM][TFSA] adsorption on Ag(111). As illustrated, e.g., in Fig. 3a, the same 2D crystalline structure is found on terraces wider than ∼20 nm, and also the short-range ordered 2D glass phase is formed. Similar to the findings for [BMP][TFSA] adsorption, the 2D glass phase is solely found on narrow terraces (<20 nm width) or as a narrow stripe directly along steps (see Fig. 3a and b). In addition, it sometimes appears at the perimeter of 2D crystalline domains. The relative amount of the 2D glass phase is lower than for [OMIM][TFSA]. Furthermore, the 2D crystalline areas show fewer defects than in the latter adlayer. Overall, the tendency for ordering in a 2D crystalline structure is more pronounced than for [OMIM][TFSA] adlayers, resembling more the case of [BMP][TFSA] adsorption.
 | ||
| Fig. 3 Ag(111) surface covered with islands/domains of [EMIM][TFSA]: (a and b) terraces with a width <20 nm are covered with adsorbates arranged in the 2D glass structure, on larger terraces mainly 2D crystalline domains are found. In different domains, the directions of the molecule rows in the 2D crystalline structures (marked with white lines) are rotated by 120° to each other. For better contrast, image (a) was cut at the steps and the contrast was enhanced for each terrace ((a) T = 108 K, UT = −1.13 V, IT = 60 pA; (b) T = 157 K, UT = −1.43 V, IT = 60 pA). (c) High resolution STM image of the 2D crystalline structure; the unit cell is marked by red parallelograms (size: 1.8 ± 0.2 × 2.1 ± 0.1 nm, lattice directions: white arrow × dashed arrow); the positions of the cations and anions are marked by black circles and pairs of black ovals in one of the parallelograms (T = 117 K, UT = −0.97 V, IT = 70 pA). (d) 2D crystalline domain of [EMIM][TFSA], showing some defects and small 2D glass domains in the long-range ordered structure (T = 130 K, UT = −1.20 V, IT = 60 pA). | ||
The 2D crystalline structure has the same type of unit cell as formed in the [OMIM][TFSA] adlayer, but with slightly different dimensions, as can be seen in the high resolution STM image in Fig. 3c, where the unit cell of the adlayer structure is marked again by a red parallelogram. For one unit cell also the round and elliptical protrusions are indicated by black circles and ovals, respectively. The unit cell has a size of 1.9 ± 0.2 nm × 2.3 ± 0.1 nm, with the longer side along the molecule rows, and an angle of 75 ± 8° in between the lattice vectors. On an area of 4.3 nm2 it includes 4 round and 8 elliptical protrusions (0.9 ± 0.1 round protrusions nm−2). Evaluating numerous images, we find that the exact dimensions of the unit cell vary slightly between different surface locations. It could be that the structure has slightly different dimensions, e.g., in the monolayer and the submonolayer regime, as it was also reported for [BMP][TFSA] on Au(111),29 but the differences are in the range of the standard deviation. Overall, the adlayer structures formed by [EMIM]−, [OMIM]− and [BMP][TFSA] on Ag(111) are very similar, therefore we conclude that (i) [EMIM][TFSA] adsorbs in a similar adsorption geometry as the other two ILs and that (ii) the round protrusions resemble the upstanding alkyl chains of the cation and a pair of longish protrusions represents one anion. Also the density of ion pairs per nm2 differs only slightly between [OMIM][TFSA] and [EMIM][TFSA], with 0.9 ± 0.1 nm−2 and 1.0 ± 0.1 nm−2. With the longer alkyl chain lying flat on the surface at coverages <0.8 ML, as it was postulated by Cremer et al. for [OMIM][TFSA] on Au(111) at room temperature25 the difference in the space requirement of both molecules should be much larger and, compared to [BMP][TFSA], the structure formation itself should also be different. A rough estimate of the difference in the structure sizes for flat lying alkyl chains can be given by using data on the ionic volumes for different IL ions calculated by Slattery et al.44 For [EMIM]+ they reported a volume of 0.156 nm3, for [OMIM]+ a value of 0.242 nm3. Assuming roughly spherical shapes, the spheres have a diameter of 0.35 nm ([EMIM]+) and 0.53 nm ([OMIM]+). This is a difference of 34%. Hence, when the longer alkyl chains would lie flat on the surface, the difference in the density of ion pairs should be larger than 34%, since the difference in volume is caused only by the longer alkyl chain and should be more pronounced in the surface direction when the alkyl chain is lying horizontally on the surface. In contrast, for an alkyl chain standing upright on the surface the difference should be smaller than this value. Since the measured difference in the density is only around 10%, which is close to the range of the error bar, this points to the latter adsorption geometry, in agreement with the results of DFT-D calculations for [BMP][TFSA] adsorption on Ag(111).30
The 2D crystalline structure of [EMIM][TFSA] shows the same orientation along the 〈10〉 lattice vectors of the Ag(111) substrate as it was found and discussed before for [OMIM][TFSA]. Different rotational domains, which are observed on different terraces, always include angles of 120 ± 5° between the directions of the close packed rows of round protrusions. This is illustrated in Fig. 3a, where the directions of the close packed rows in both structures are marked by white lines. The structure also forms parallel to well ordered Ag(111) steps (not shown). In addition, when comparing the unit cells of the adlayer in Fig. 3a and c, these appear as image and mirror image. Hence, also this adlayer structure is chiral. The 6 possibilities to align the structure on the substrate surface (3 sets of 2 chiral structures with the molecule rows of the structure parallel to each 〈10〉 direction of the surface lattice) are therefore identical to those for [OMIM][TFSA].
The thermal stability of the adlayer structures formed by [EMIM][TFSA] was tested in the same way as it was done for [OMIM][TFSA] adsorption. Similar to our findings for [OMIM][TFSA], the perimeters of the adsorbate islands become mobile already at 90 K, which is manifested by stripy features in the STM images. In contrast, the core of the adsorbate islands is stable under these conditions (compare e.g. in Fig. 3d). For the 2D crystalline structure, melting temperatures of 180 ± 10 K in the submonolayer and 212 ± 10 K in the monolayer regime were determined, while for the 2D glass structure the melting temperature could not be determined reliably because of the relatively low amount of this structure on the surface. The higher melting temperatures of the crystalline phase compared to the [OMIM][TFSA] adlayer phases, by 15 K for the submonolayer and by 27 K for the monolayer coverage regime, clearly reflect an influence of the longer alkyl chain of the cation on the stability of the adlayer structure. One possibility to explain this observation is that the stronger +I-effect of the octyl chain (electron donation via the covalent σ-bonds) compared to the ethyl chain leads to a slight weakening of the positive charge in the ring of the imidazolium cations and thus to weaker electrostatic interactions between the cations and anions in the structure. [BMP][TFSA], which lies with the length of its butyl-chain between ethyl and octyl, has nearly the same melting temperature as [OMIM][TFSA]. It could be that the effect of the compared to [BMP][TFSA] longer alkyl chain, which should lead to a lower melting temperature, is roughly compensated by the different electronic structure of the imidazolium ring compared to the pyrrolidinium ring.
The 2D melting temperatures determined here can be compared to the bulk melting temperatures of the respective ILs. Bulk [EMIM][TFSA] melts at 258 K (value is taken from the data sheet of the supplier), other studies reported values between 255 K45 and 265 K.46 For [BMP][TFSA], values between 252 K47 and 267 K46 were reported. For [OMIM][TFSA], no literature value is available. In general, the 2D melting temperatures for the adsorbed structures are slightly lower than the melting temperatures of the bulk ILs. The lower stability of the 2D crystalline structures is mainly attributed to weaker molecule–molecule interactions compared to the bulk due to the interaction with the surface.
The interaction of [EMIM][TFSA] with Ag(111) closely resembles that of [OMIM][TFSA], where the latter differs only by its longer alkyl chain in the cation. Based on the similar characteristics in structure formation and especially the similar densities of the adsorbate structures, we conclude that in the 2D crystalline phases the alkyl chains are standing upright and that also in this adsorption system the structure formation is dominated by the tendency to optimize the adsorption geometry of the anion on the surface.
3.3 [OMIM][TFSA] adsorption on Au(111)
On Au(111), [OMIM][TFSA] condenses into islands with monomolecular height like on Ag(111). Typical large-area STM images of a surface with submonolayer coverage are shown in Fig. 4a and 5a. Here it should be noted that on the adsorbate free areas the zig-zag double lines of the herringbone reconstruction48 are clearly visible; their orientation is indicated by the white zig-zag pattern in Fig. 4a. The images reveal a number of characteristic features: similar to observations for adsorption on Ag(111), the step edges are decorated by small islands (nuclei) of adsorbed [OMIM][TFSA] (Fig. 5a–c). Similar nuclei and also somewhat larger islands are observed at the bending points (‘elbows’) of the herringbone reconstruction. These islands exhibit the disordered structure discussed before (2D glass phase, see islands marked by ‘2’ in Fig. 4a). Furthermore, large islands are formed on the terraces, which largely consist of a 2D crystalline phase, similar to that observed for adsorption on Ag(111). These islands can extend across several domain boundaries of the underlying Au(111) reconstruction pattern formed by the bending points of the zig-zag lines (marked by dashed lines in Fig. 4a). On the other hand, these domain boundaries of the Au(111) reconstruction often coincide with the perimeters of the adsorbate islands (see Fig. 4a). Additionally, the islands generally have a lengthy form along these domains of the Au(111) reconstruction. It seems that an ordered domain preferably forms on top of one such Au(111) reconstruction domain. Obviously, the bending points of the Au(111) surface reconstruction seem to act as a barrier for further island growth, which is most likely attributed to the sudden misorientation between substrate and adlayer lattice. | ||
| Fig. 4 (a) STM image of a submonolayer of [OMIM][TFSA] adsorbed on Au(111): large islands consisting mainly of molecules arranged in a 2D crystalline structure extend across the surface (1); small islands with 2D glass structure are formed at the elbows of the Au(111) reconstruction (2), which is visible on the bare Au(111) surface areas (3) and is also visible through the 2D crystalline adlayer structures. For better visibility, one double line of the Au(111) corrugation lines is marked with white lines; the connections between bending points of the Au(111) surface reconstruction are marked with white dotted lines (T = 102 K, UT = −0.76 V, IT = 50 pA). (b) High resolution image of the 2D crystalline phase. The size of the image corresponds to the black rectangular in (a). The unit cell of the structure is indicated by a red parallelogram (size: 2.5 ± 0.1 nm × 3.5 ± 0.1 nm, arrow 1 × arrow 2) (T = 151 K, UT = −1.16 V, IT = 70 pA). (c) High resolution image of the 2D glass phase (T = 157 K, UT = −0.33 V, IT = 50 pA). (d) Two 2D crystalline domains next to each other; the domains are rotated by 120° to each other. Additionally, the two adlayer domains are mirror images to each other (T = 145 K, UT = −1.16 V, IT = 80 pA). (e) High resolution image of the boundary area between two long-range ordered domains (T = 149 K, UT = −1.16 V, IT = 90 pA). | ||
 | ||
| Fig. 5 (a) STM image of a Au(111) surface with low coverage of [OMIM][TFSA]. Preferred adsorption of the molecules takes place in front of the steps (T = 125 K, UT = −0.45 V, IT = 100 pA). (b) STM image of the area marked with dashed black lines in (a) with 2 min delay, showing that the bulk islands are stable at this temperature, only minor movement can be detected at the island boundaries (T = 125 K, UT = −0.45 V, IT = 110 pA). (c) High resolution image of the molecules adsorbed in front of descending steps. Directly at the step cations are adsorbed, followed by the anions (T = 127 K, UT = −0.45 V, IT = 70 pA). (d) High resolution images of an island boundary of a 2D crystalline island with a time delay of 2 min in between. In the white rectangle at the island boundary we find an attachment/detachment of an ion pair (T = 124 K, UT = −0.71 V, IT = 80 pA). | ||
Finally, we also observed adlayer islands containing different rotational domains of the adlayer structure. These different rotational domains are separated from each other by areas with 2D glass phase. Most likely, these islands developed by coalescence of separate islands, each consisting of one domain of ordered adlayer structure.
Next we will concentrate on the 2D crystalline structure. A high resolution STM image of that structure, recorded in the area imaged in Fig. 4a, is shown in Fig. 4b. The size and location of the latter image is indicated by the black rectangle in Fig. 4a. The long-range order in this structure differs somewhat from the 2D crystalline structure formed on Ag(111), but the appearance of the adsorbed ions is the same. Also in this case the structure is built up by round protrusions (marked as white dots in Fig. 4b) and pairs of elliptical protrusions (white ovals in Fig. 4b) with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. From the similarity in characteristic structural features we conclude that the general adsorption geometry of the IL ions is essentially identical to that of [OMIM][TFSA] and [EMIM][TFSA] on Ag(111). The anions (=elliptical protrusions) are aligned in rows (the direction of the rows is marked in Fig. 4b by arrow 1), and along these lines they are oriented parallel to each other. Comparing the orientation of the anions in different rows we find that this is identical always in two neighboring rows, while in the third one it is rotated by 120° relative to the preceding two rows. In the next two rows they are again oriented in the same way as in the two rows before, resulting in an ‘a–a–b–a–a–b’ ordering sequence. It is important to note that the “b” rows exhibit only 2/3 of the protrusions present in the “a” rows. The cations (round protrusions) are aligned in between the anion a-type rows, forming zig-zag lines with 4 cations per 3 anions in one neighboring a-type row. In total, this results in a different arrangement of the ions in the unit cell (see marked parallelogram in Fig. 4b) compared to the adlayer on Ag(111). In the latter case, cations and anions have the same number of neighbored adsorbed ions, the cations are surrounded by 4 anions and the anions by 4 cations. In the ordered structure of [OMIM][TFSA] on Au(111), the cations next to the “b” rows of the anions (see Fig. 4b) have 4 anions as neighbor. The cations between the “a” rows of anions have either 3 or 4 anions as neighbors (marked with a “3” or “4” in Fig. 4b), (note that since from the STM images we cannot unambiguously decide which two of the longish protrusions in the “a” rows form a pair and thus result from one anion, the assignment of 4 or 3 nearest neighbor anions to the cations in between the “a” rows in Fig. 4b could also be other way around). The anions in the “b” rows have always 4 neighbored cations; in the “a” rows 2/3 of the anions have also 4 nearest neighbors, but 1/3 has only 3 nearest neighbors. This gives a mean coordination of 3.75 neighbored counter ions per anion or cation, which is 0.25 less than in the 2D crystalline structure of [OMIM][TFSA] on Ag(111). The dimensions of the unit cell are 2.8 ± 0.1 nm along the direction marked by arrow 1 and 3.8 ± 0.1 nm along the direction of arrow 2, with an angle of 76 ± 5° in between the lattice vectors. The total area of the unit cell is 10.3 ± 0.5 nm2 and the density of round protrusions is 0.8 ± 0.1 nm−2, which on the other hand is identical to that on Ag(111). Compared to the adsorption of [BMP][TFSA] on Au(111),29 the arrangement of the ions in the structure relative to each other is exactly the same; also the density of [BMP][TFSA] on Au(111) was found to be 0.75 ± 0.05 nm−2 in a monolayer, which is in the limits of accuracy the same value as for [OMIM][TFSA]. In contrast, for submonolayers of [BMP][TFSA] on Au(111) a slightly smaller density of 0.61 nm−2 was found; while the ordered structure of [OMIM][TFSA] did not depend on the coverage. [OMIM][TFSA] adsorption on Au(111) results in an adlayer structure very similar to that formed for [BMP][TFSA] in the monolayer region. Overall, the similarity of adlayer structures for these ILs on Au(111) and Ag(111) further supports our above conclusion given for this type of ILs that the ordered adlayer structures are dominated by the substrate–anion interactions and that cation–substrate interactions seem to play a minor role.
:1. From the similarity in characteristic structural features we conclude that the general adsorption geometry of the IL ions is essentially identical to that of [OMIM][TFSA] and [EMIM][TFSA] on Ag(111). The anions (=elliptical protrusions) are aligned in rows (the direction of the rows is marked in Fig. 4b by arrow 1), and along these lines they are oriented parallel to each other. Comparing the orientation of the anions in different rows we find that this is identical always in two neighboring rows, while in the third one it is rotated by 120° relative to the preceding two rows. In the next two rows they are again oriented in the same way as in the two rows before, resulting in an ‘a–a–b–a–a–b’ ordering sequence. It is important to note that the “b” rows exhibit only 2/3 of the protrusions present in the “a” rows. The cations (round protrusions) are aligned in between the anion a-type rows, forming zig-zag lines with 4 cations per 3 anions in one neighboring a-type row. In total, this results in a different arrangement of the ions in the unit cell (see marked parallelogram in Fig. 4b) compared to the adlayer on Ag(111). In the latter case, cations and anions have the same number of neighbored adsorbed ions, the cations are surrounded by 4 anions and the anions by 4 cations. In the ordered structure of [OMIM][TFSA] on Au(111), the cations next to the “b” rows of the anions (see Fig. 4b) have 4 anions as neighbor. The cations between the “a” rows of anions have either 3 or 4 anions as neighbors (marked with a “3” or “4” in Fig. 4b), (note that since from the STM images we cannot unambiguously decide which two of the longish protrusions in the “a” rows form a pair and thus result from one anion, the assignment of 4 or 3 nearest neighbor anions to the cations in between the “a” rows in Fig. 4b could also be other way around). The anions in the “b” rows have always 4 neighbored cations; in the “a” rows 2/3 of the anions have also 4 nearest neighbors, but 1/3 has only 3 nearest neighbors. This gives a mean coordination of 3.75 neighbored counter ions per anion or cation, which is 0.25 less than in the 2D crystalline structure of [OMIM][TFSA] on Ag(111). The dimensions of the unit cell are 2.8 ± 0.1 nm along the direction marked by arrow 1 and 3.8 ± 0.1 nm along the direction of arrow 2, with an angle of 76 ± 5° in between the lattice vectors. The total area of the unit cell is 10.3 ± 0.5 nm2 and the density of round protrusions is 0.8 ± 0.1 nm−2, which on the other hand is identical to that on Ag(111). Compared to the adsorption of [BMP][TFSA] on Au(111),29 the arrangement of the ions in the structure relative to each other is exactly the same; also the density of [BMP][TFSA] on Au(111) was found to be 0.75 ± 0.05 nm−2 in a monolayer, which is in the limits of accuracy the same value as for [OMIM][TFSA]. In contrast, for submonolayers of [BMP][TFSA] on Au(111) a slightly smaller density of 0.61 nm−2 was found; while the ordered structure of [OMIM][TFSA] did not depend on the coverage. [OMIM][TFSA] adsorption on Au(111) results in an adlayer structure very similar to that formed for [BMP][TFSA] in the monolayer region. Overall, the similarity of adlayer structures for these ILs on Au(111) and Ag(111) further supports our above conclusion given for this type of ILs that the ordered adlayer structures are dominated by the substrate–anion interactions and that cation–substrate interactions seem to play a minor role.
The orientation of the adlayer structure relative to the substrate lattice can be evaluated by comparing with the orientation of the Au(111) reconstruction pattern. The double rows of the Au(111) reconstruction are known to run in 〈11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 〉 direction.48 In Fig. 4a, we find one large domain of the 2D crystalline structure in the middle and upper part of the image and two smaller domains at the bottom of the image. The direction in the respective structures, which corresponds to that of arrow 2 in Fig. 4b, is marked in Fig. 4a by a dashed black line. These directions are always parallel to the double rows in one domain of the Au(111) surface reconstruction and hence to a 〈11〉 direction of the Au(111) surface, with an angle of 120° between the different directions. This is also true for the domains shown in Fig. 4a, d and e, where these directions of the adlayer structures are also marked with dashed lines.
〉 direction.48 In Fig. 4a, we find one large domain of the 2D crystalline structure in the middle and upper part of the image and two smaller domains at the bottom of the image. The direction in the respective structures, which corresponds to that of arrow 2 in Fig. 4b, is marked in Fig. 4a by a dashed black line. These directions are always parallel to the double rows in one domain of the Au(111) surface reconstruction and hence to a 〈11〉 direction of the Au(111) surface, with an angle of 120° between the different directions. This is also true for the domains shown in Fig. 4a, d and e, where these directions of the adlayer structures are also marked with dashed lines.
Considering also the direction of the rows of round protrusions (arrow 1 in Fig. 4b), again two different structures can be formed for each 〈11〉 direction, which behave as image and mirror image. This is illustrated in Fig. 4d and e, where the directions in the adlayers corresponding to arrow 2 in Fig. 4b are marked with dashed black lines and the ones corresponding to arrow 1 are marked with full black lines. In each image the two marked domains are mirror images to each other. Hence, also in this case the resulting surface structure is chiral.
At comparable adlayer coverage, the ordered domains formed by the [OMIM][TFSA] adsorbates on Au(111) are much smaller than the ones on Ag(111). We attribute this to the Au(111) surface reconstruction, where both the steps and elbows act as nucleation centers for island formation, whereas on Ag(111) island formation always starts at the steps. Hence, the density of nucleation sites is much higher on Au(111) than on Ag(111).
Fig. 4c shows a high resolution image of the short-range ordered 2D glass phase (see above). The appearance of the molecules with round and elliptical protrusions is the same as observed in the 2D crystalline structure, pointing to a similar conformation of the adsorbed IL species in both phases. Also the short-range order in the 2D glass phase is resolved in that image, with cations and anions often lining up in short rows of up to around 10 adsorbed ions, which exhibit similar structural characteristics as the rows in the 2D crystalline phase. This holds true also for the orientation of the anions, where many of the elliptical protrusions are arranged in parallel to each other. Obviously, the anions prefer the same adsorption geometry relative to the surface as in the ordered structure. This is evident also from Fig. 4e, which shows a high resolution image of a small area of 2D glass structure between two 2D crystalline domains. Most of the elliptical protrusions are aligned in the same symmetric directions as observed in the long-range ordered adlayer structure.
The thermal stability of the adlayer phases was evaluated in the same way as described in the previous sections. Already at the lowest temperatures, the edges of the islands appear frizzy (see Fig. 4 and 5, except in Fig. 4a, where the scale is too large). In analogy to [OMIM][TFSA] and [EMIM][TFSA] adsorption on Ag(111) and to [BMP][TFSA] adsorption on Au(111) and Ag(111),31 we interpret this as either due to a high mobility of adsorbed IL species along the island edge, or, more likely, as rapid exchange of IL adsorbates between a surrounding 2D gas/liquid phase of mobile IL adsorbates and the respective islands (2D solid). On the other hand, the island cores are stable under these conditions. This is illustrated in the STM images in Fig. 5a, b and d, which were recorded at T = 125 K. Fig. 5a and b are images of the same IL island recorded with a time period of ca. 2 min in between, where Fig. 5b has a smaller scale than Fig. 5a (the area imaged in Fig. 5b is marked by a dashed black rectangle in Fig. 5a). On this large scale we find no significant changes of the island perimeter, and the core of the island is completely stable. On a smaller scale, however, changes can be detected, which is illustrated in Fig. 5d. This shows two images of the same area (but different from that in the images in Fig. 5a and b), which were recorded with a time delay of 2 min in between. Here we find clear differences in the perimeter structure, e.g., in the area marked by a white rectangle. In this case the position of one anion is changed and another one is missing completely. Overall, the island perimeters are mobile at 125 K on a molecular scale, but show only little mobility on a larger scale.
When warming the sample to higher temperatures, the mobility at the island edges gets more pronounced, but the molecules in the inner parts of the islands stay stable. The 2D glass islands ‘melt’ over a wide temperature range, but always at lower temperatures than the 2D crystalline ones. At 165 ± 10 K in the submonolayer regime and at 197 ± 10 K in the monolayer regime, the 2D crystalline structure melts, leaving a 2D liquid of the molecules on the surface, where the mobile IL adsorbates cannot be resolved by STM. Therefore, we cannot comment from these data on the proposal by Cremer et al.,25 who (based on ARXPS measurements performed at room temperature) proposed different adsorption geometries for the submono- and monolayer regime. For monolayer coverage they suggested that the cations are adsorbed in a similar geometry as favored in this work for low temperatures, with the alkyl chain pointing up, while at submonolayer coverage (<0.8 ML) they proposed that the alkyl chains are adsorbed flat on the surface. The larger space requirement of the latter adsorption geometry would be compatible with the lower density of the 2D liquid phase formed at room temperature in the submonolayer coverage regime, but not with that of the ordered structure present at low temperatures.
Comparison of adsorption of [OMIM][TFSA] on Au(111) with that on Ag(111) indicates that the influence of the substrate on the interaction of [OMIM][TFSA] with noble metal surfaces is small, both with respect to structure formation and to adsorbate–adsorbate interactions. The general appearance of the adsorbed species and also the orientation of the anions relative to the substrate are identical compared to those on Ag(111), leaving the adsorption geometry largely unchanged, though their arrangement relative to each other is slightly different. Different from the non-reconstructed Ag(111) surface, the herringbone reconstruction of Au(111) affects the shape of the adsorbate islands by limiting their size, and on the other hand providing additional nucleation centers at the elbows of the reconstruction.
3.4 [EMIM][TFSA] adsorption on Au(111)
Large-scale images of islands and domains of adsorbed [EMIM][TFSA] on Au(111) at submonolayer coverage are shown in Fig. 6. In the image in Fig. 6a we find two different 2D crystalline structures. The structure resolved on the right hand side of the image is very similar to that known from [OMIM][TFSA] and [BMP][TFSA] on Au(111) (structure #2). In contrast, the 2D crystalline structure appearing on the left of the image, which is denoted as structure #3, has not been observed for the other adsorption systems. | ||
| Fig. 6 [EMIM][TFSA] adsorbed on Au(111) at submonolayer coverage: (a) in addition to the 2D crystalline structure #2, another 2D crystalline structure (#3) is found. The image was cut at the steps of the Au(111) surface and the contrast was enhanced for the structures on the terraces, the appearance of different heights left from the screw dislocation at the left side of the image is an artifact of this treatment (T = 107 K, UT = −1.28 V, IT = 40 pA). (b) Various domains of structure #2 are visible in this STM image. The two large domains in the middle of the picture are aligned parallel to each other along the direction of arrow 2, which indicates also the direction of the corrugation lines of the Au(111) reconstruction domains on which they are largely located. The directions of the molecule rows in the domains (arrows 1) run at angles of 72° and −72° relative to the direction of arrow 2: these two domains are chiral. The upper part of the image shows a modified structure #2 (T = 113 K, UT = −1.28 V, IT = 80 pA). | ||
More detailed information on the structure of phase #2 is obtained from the high resolution image shown in Fig. 7a. The unit cell of the adlayer is marked by a red parallelogram, its size is 3.0 ± 0.1 nm × 3.4 ± 0.1 nm (arrow 1 × arrow 2) with an angle of 72° ± 5° in between, an area of 9.9 ± 0.5 nm2 and a density of 0.8 ± 0.1 ion pairs nm−2. The alignment and the general appearance of this structure are essentially identical to those in the 2D crystalline structures of [OMIM][TFSA] and [BMP][TFSA] on Au(111), with the same density as observed for [OMIM][TFSA] adsorption (0.8 ± 0.1 ion pairs nm−2). We therefore suggest that also in the present case the longer alkyl chain of the cation points upwards. The adlayer lattice is aligned along the 〈11〉 directions of the Au(111) surface (arrow 2 in Fig. 7a is oriented in 〈11〉 direction), as it was observed also for [OMIM][TFSA] on Au(111). Similar to the latter adsorption system, this adlayer lattice direction is oriented along the corrugation lines of the Au(111) surface reconstruction pattern. Also in this case the domain and island boundaries of the adsorbed 2D crystalline phases lie on top of lines along the bending points of the Au(111) reconstruction (see, e.g., in Fig. 6b). In the latter image, two pairs of the corrugation lines of the Au(111) herringbone reconstruction are marked by white lines, the bending points are located on the dotted lines. In the middle of the image, two large domains of the 2D crystalline adlayer structure are visible, whose boundaries partly are located on the bending lines of the herringbone reconstruction (dotted lines in Fig. 6b). These two domains are aligned parallel to each other based on the direction of arrow 2 (see also the white dashed line in this image). In contrast, the close packed rows of round protrusions, whose directions are indicated by arrows 1, are not parallel. Hence, these two domains are again examples for the mirror symmetric orientations expected in a chiral adlayer structure. Together with the three possible rotational orientations, we thus obtain 6 possible arrangements of the ordered adlayer, similar to our findings for the other two ILs ([OMIM][TFSA] and [BMP][TFSA]) on Au(111).
 | ||
| Fig. 7 High resolution STM images of the 2D crystalline structures formed by [EMIM][TFSA] on Au(111): (a) structure #2 (unit cell: 2.7 ± 0.1 nm × 3.1 ± 0.1 nm, arrow 1 × arrow 2). The lower part of the image shows the modified structure #2 described in the text (T = 153 K, UT = −1.54 V, IT = 70 pA). (b and c) Structure #3 with a unit cell of 3.2 ± 0.1 nm × 4.8 ± 0.1 nm (arrow 3 × arrow 4). (c) The positions of the anions are marked with pairs of black ovals in the unit cell (red parallelogram) ((b and c) T = 150 K, UT = −1.40 V, IT = 60 pA). (d) Domain boundary between both structures; the unit cell of #3 is rotated by 6° ± 2° to the axis of the unit cell of #2, which is parallel to the 〈11〉 substrate directions (T = 150 K, UT = −1.40 V, IT = 80 pA). | ||
For [EMIM][TFSA] adsorption we also observed a specific modification of structure #2 which is visible in the upper part of Fig. 6b. In this structure pairs of round protrusions along the direction of arrow 2 are rotated by 90°, which leads to a bending of the close packed rows of round protrusions. This is illustrated in the lower part of Fig. 7a, where one of such rotated pairs is marked by black dots, together with its ‘normal’ neighbor pairs (black circle). This structural modification is neither observed for [OMIM][TFSA] nor for [BMP][TFSA] on Au(111).
The second ordered phase formed by [EMIM][TFSA] on Au(111), structure #3, is resolved in Fig. 6a (large scale presentation) and in Fig. 7b and c (high resolution images). The unit cell of this structure is indicated by a red parallelogram in either of the images. Furthermore, the positions of the elliptical protrusions are marked with black ovals for better visibility in Fig. 7c. The unit cell has a size of 3.5 ± 0.1 nm × 5.3 ± 0.1 nm (arrow 3 × arrow 4, Fig. 7b), with an angle of 83° ± 4° in between, and an area of 19.0 ± 0.8 nm2. It contains 12 ion pairs. This leads to a density of 0.65 ± 0.08 ion pairs nm−2, which is lower than that of structure #2. Most obvious difference to structure #2 is that in structure #3 the adsorbed ions are not aligned in alternating rows of cations and anions, but in a more complex arrangement. In this structure the cations (round protrusions) are arranged in zig-zag lines, which are oriented along a direction illustrated in Fig. 6a by a full white line and by arrow 3, or directions rotated by ±120°. Compared to the dashed white line in Fig. 6a, which indicates the direction of arrow 2 in structure #2 and thus the 〈11〉 direction of Au(111), the white line is always rotated by 6° ± 2°. The presence of similar structural elements in structure #3 as in the other structure(s), with pairs of elliptical protrusions and round protrusions, indicates that these are also related to adsorbed anions and cations, respectively. Also similar to the other structures, the pairs of elliptical protrusions exist in different orientations, with the long axes oriented in one of the 〈10〉 directions. This is indicated in the high resolution image in Fig. 7d, where a domain of structure #2 appears next to a domain of structure #3. In structure #2 the axis marked by a dashed white line in Fig. 7d is parallel to the 〈11〉 direction of the Au(111) surface. The long axes of the longish protrusions are all with an accuracy of ±10° rotated by 30°/90° to the 〈11〉 direction and therefore oriented along the 〈10〉 direction. The orientation of the anions relative to the substrate in the new structure is therefore similar to that in structure #2, and also similar to the orientation of the anions of [BMP][TFSA]29 and [OMIM][TFSA] on Au(111). The adsorption geometry of the anion relative to the substrate is therefore the same (or very similar) as in the other 2D crystalline adlayer structures (and also in the 2D glass phase) of [OMIM][TFSA], [BMP][TFSA] and [EMIM][TFSA] on Au(111) and also on Ag(111). This further supports our above conclusion of a structure determining anion–substrate interactions in these adsorption systems.
In contrast to the similar adsorption geometry, the arrangements as well as the number of neighbored adsorbed anions in structure #2 and #3 differ, as described in the following. In the upper unit cell marked in Fig. 7c the number of neighbored anions is indicated on the positions of the respective cations. In the lower unit cell, the number of neighbored cations to each anion is indicated in the same way. The numbers vary between 2 and 5 neighbors for the anions and between 2 and 4 for the cations. Compared to structure #2, which had for both anion and cation only 3 or 4 neighbors, structure #3 shows a larger variation. Furthermore, the mean number of neighbors per anion or cation is lower in structure #3 (3.25 neighbors) than in structure #2 (3.75 neighbors). On the other hand, the density of the adlayer in structure #3 (0.65 ± 0.08 cations nm−2) is lower than in structure #2 (0.8 ± 0.1 cations nm−2). The reason for the different arrangement of the adsorbed ions relative to each other in these structures must be subtle differences in the adsorbate–adsorbate interactions in structure #3 and in structure #2. The overall energies of formation of these two structures, however, must be rather similar, since both adlayer structures coexist on the surface.
It is not yet clear why structure #3 (or an analogous structure) could not be observed for [OMIM][TFSA] and [BMP][TFSA] on Au(111), but its absence in these systems must be related to very subtle differences in the energetics, as concluded from the existence of similar #2 structures in all three adsorption systems.
Finally, also for this adsorption system we observed a 2D glass phase. High resolution images resolving its structure are presented in Fig. 8. The image in Fig. 8a resolves a domain of that phase between two domains of structure #2, Fig. 8b shows a part of a larger domain of the 2D glass structure. The arrangement of the adsorbed ions is very similar to that observed for [OMIM][TFSA] on Au(111) (see previous section and Fig. 4c). The anions as well as the cations are often arranged in short rows of cations and anions similar to the arrangement in the ordered structure #2. Furthermore, the adsorbed anions are oriented with their main axis along the 〈11〉 directions of the Au(111) surface, i.e., the long axes of the elliptical protrusions are orthogonal to that direction, identical to their orientation in structures #2 and #3.
 | ||
| Fig. 8 High resolution STM images of the 2D glass structure of [EMIM][TFSA] on Au(111) (a) between two domains of 2D crystalline structure #2 and (b) in a larger domain ((a) T = 152 K, UT = −1.40 V, IT = 70 pA; (b) T = 149 K, UT = −1.40 V, IT = 70 pA). | ||
Upon heating the samples slowly to room temperature, we determined melting temperatures of 240 ± 10 K for both 2D crystalline structures, of 205 ± 10 K for the 2D glass structure in the monolayer and of 120 ± 10 K for the 2D glass structure in the submonolayer regime. Compared to [OMIM][TFSA] on Au(111), which has 85/40 K (submonolayer/monolayer regime) lower melting points, this trend is the same as for the two ILs on Ag(111). As already discussed this can be attributed to the different alkyl chain length of the cation and its influence on the electrostatic interactions between the adsorbed ions. Comparing the adlayer melting temperatures on the different substrates Au(111) and Ag(111), we find an ∼30 K higher melting temperature for [EMIM][TFSA] adsorption on Au(111), while for [OMIM][TFSA] the melting temperatures are approximately identical on both substrates (submonolayer regime) or slightly higher on Au(111) (monolayer coverage). Hence, there is no clear trend for the influence of the two substrates Ag(111) and Au(111) on the stability of the 2D crystalline structures. They mainly differ in the local geometries on the surface due to the slightly different interatomic distances and of course by the reconstruction of the Au(111) surface layer, which is accompanied by also anisotropic atomic distances.
Overall, the interaction of [EMIM][TFSA] with Au(111) results in a larger structural variety than observed for its interaction with Ag(111), but also for interaction of the other ILs [OMIM][TFSA] and [BMP][TFSA] with Au(111). This difference must originate from subtle differences in the adsorbate–adsorbate interactions, caused by the different alkyl chain length. The main structural characteristics, in particular the anion adsorption configuration and also the effective adsorbate–adsorbate interactions, are, however, maintained and similar to those in the other adsorption systems considered here for comparison, [OMIM][TFSA] and [BMP][TFSA] on Au(111) and Ag(111). In all cases the optimization of the anion adsorption configuration seems to be the decisive parameter for the nature of the ordered adlayer structures.
3.5 Commensurability of the structures to the substrate lattice
The sizes of the unit cells given in the text above are those which were measured in the STM images. Since the 2D crystalline adlayer structures are strictly oriented along certain lattice directions it is likely that the molecules in each structure are also adsorbed on similar adsorption sites. In that case, the 2D crystalline structures must be commensurate to the substrate lattice. This was not the case in the structures discussed so far. In Fig. 9 and 10 we show drift corrected high resolution STM images together with commensurate structure models for the different molecule–substrate combinations investigated (Fig. 9a: [OMIM][TFSA] on Ag(111); Fig. 9b: [EMIM][TFSA] on Ag(111); Fig. 10a: [OMIM][TFSA] on Au(111); Fig. 10b and c: [EMIM][TFSA] on Au(111)). In all images we included a hexagonal network of black circles, which illustrate the Ag(111) or Au(111) substrate lattice. For Au(111) the network is contracted in the vertical 〈10〉 direction by 4.3% to account for the contracted outermost layer in the Au(111) herringbone reconstruction.48 The scale is identical for all images. The measured dimensions of the unit cells are drawn with red lines, the dashed red lines indicate the maximum area available for the unit cell considering the error range in the measured distances and orientations. The blue trapezoids indicate the limits for the unit cell considering the accuracy of the measured angles and side lengths.
 | ||
| Fig. 9 Models of the 2D crystalline structures of (a) [OMIM][TFSA] and (b) [EMIM][TFSA] on Ag(111) relative to the Ag(111) surface lattice (see underlaid lattice). Red lines show the “as measured” unit cell, dashed red lines and blue rectangles/trapezes show the error area, black lines show the best fit commensurate unit cells. The underlying STM pictures were drift corrected on the best fit commensurate unit cell. Grey spheres mark the positions of the cations, pairs of red ovals those of the anions. | ||
 | ||
| Fig. 10 Models of the 2D crystalline structures of (a) [OMIM][TFSA] and (b, c) [EMIM][TFSA] on Au(111), analogously to those in figure. | ||
It is not possible to decide from the STM images whether the anions or cations adsorb on on-top, bridge or on threefold hollow (hcp or fcc) sites, since atomic resolution of the substrate lattice could not be obtained in high resolution measurements of the adsorbed IL islands. Therefore in Fig. 9 and 10 we placed the round protrusions, which mark in these images the upper right corner of the unit cell, on a threefold hollow site. For a commensurate adlayer structure this means that all round protrusions (cations) at the corners of the unit cells are located on the same sites. The measured values for the unit cells do not fit exactly to a commensurate structure, but considering the error range in the STM measurements, commensurate unit cells are definitely possible, and also likely from the reasons given above. The commensurate unit cells fitting best to the measured sizes/orientations are marked in the images with black lines. The sizes of the best fit commensurate unit cells are summarized in Table 1. The positions of the round shaped protrusions (=cation positions) and of the pairs of longish protrusions (=anion positions) in the commensurate structure, averaged over several STM images, are marked with grey round spheres and red ovals. The positions of the cations vary slightly in different STM images. For the anions a determination of their exact positions is not possible because of their low apparent height. This is especially true for structure #3 of [EMIM][TFSA] on Au(111). Nevertheless, the adsorption sites of the anions on the substrate seem to be very similar in all structures. Finally we would like to mention that it cannot be ruled out that the cations exist in slightly different conformations. This is especially possible for the longer alkyl chains, which are mainly responsible for the roundish protrusions in the STM images30 This would explain slight variations in the positions of the round protrusions, while the imidazolium rings of the cations may still be adsorbed on identical adsorption sites (e.g., with N-atoms on top of substrate atoms) on the surface. Overall, determination of the exact adsorption geometry especially of the cations, but also of the anions requires additional support from other experimental methods and in particular from theory, from electronic structure calculations.
| Structure | Measured unit cell dimensions [nm] ± 0.1 nm | Measured density [ion pair nm−2] | Commensurate unit cell dimensions [nm] | Density [ion pair nm−2] | Matrix notation | Matrix notation (mirror structure) |
|---|---|---|---|---|---|---|
| [OMIM][TFSA] Ag(111) |
2.1 × 2.3
82° ± 4° |
0.8 ± 0.1 |
2.023 × 2.294
78.84° |
0.8778 |
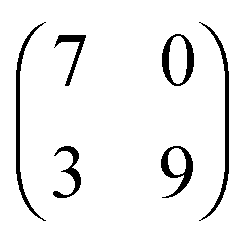
|

|
| [EMIM][TFSA] Ag(111) |
1.9 × 2.3
75° ± 8° |
0.8 ± 0.1 |
2.023 × 2.366
70.97° |
0.8778 |
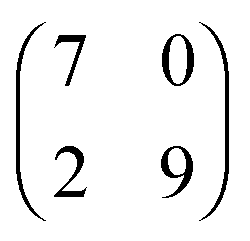
|
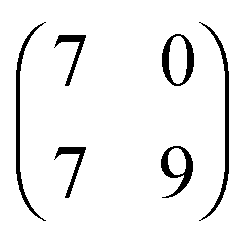
|
| [OMIM][TFSA] Au(111) |
2.8 × 3.8
76° ± 5° |
0.8 ± 0.1 |
2.530 × 3.744
76.21° |
0.8620 |
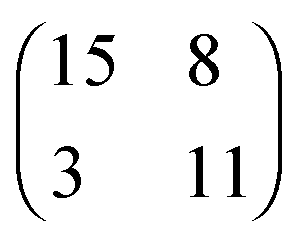
|
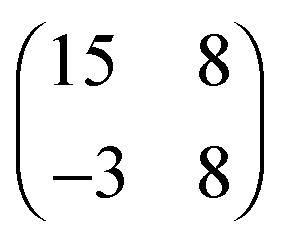
|
| [EMIM][TFSA]#2 Au(111) |
3.0 × 3.4
72° ± 5° |
0.8 ± 0.1 |
2.931 × 3.492
67.18° |
0.8313 |
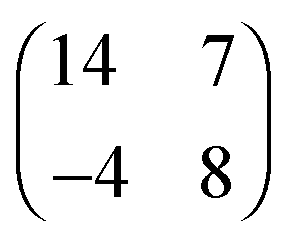
|
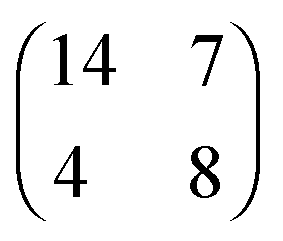
|
| [EMIM][TFSA]#3 Au(111) |
3.5 × 5.3
83 ± 4° |
0.65 ± 0.08 |
3.503 × 5.237
85.47° |
0.6563 |
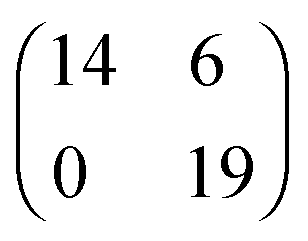
|
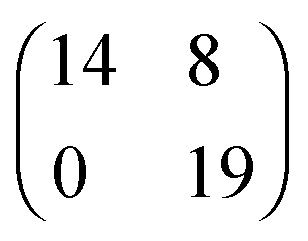
|
Conclusions
Based on the results of a comparative STM study on the structure, structure formation and thermal stability of (sub-)monolayers of two closely related ILs, [EMIM][TFSA] and [OMIM][TFSA], on two noble metal surfaces with similar local symmetry, Au(111) and Ag(111), which were performed under UHV conditions and at temperatures between 100 K and room temperature, and making use of previous findings on the structure and structure formation of an IL with similar anion, [BMP][TFSA] on these surfaces,29–31 we arrive at the following conclusions:(1) For all three ILs the ions remain intact upon evaporation. At room temperature, the adsorbed ILs form a 2D gas or a 2D liquid on each of the two substrates, depending on the coverage. Upon cooling to 100 K, the adsorbed species condense into monolayer islands on the surface. Island formation starts at descending steps and, in the case of Au(111), at the elbows of the surface reconstruction, followed by 2D growth over the terraces.
(2) In the condensed phase the adsorbed species either form a 2D crystalline phase or a short-range ordered 2D glass phase.
(3) From the close similarity of all adlayer structures with that of [BMP][TFSA] on Ag(111), with round shaped protrusions and pairs of elliptical protrusions, a similar density of these features and comparable lattice characteristics, we conclude on a similar origin of these features, with the round protrusions corresponding to the adsorbed cations with the alkyl chain pointing upwards, and the pairs of elliptical protrusions corresponding to the adsorbed anions.
(4) The rather similar adlayer structures and essentially identical adsorption geometry of the anions, despite the rather different nature of the cations in the three ILs, indicates that the adlayer structures are dominated by the optimization of the anion adsorption geometry, while the cation adsorption has little effect. Optimizing the [TFSA] anion adsorption geometry seems to be essential for the formation of the identified 2D crystalline structures. This is supported also by the observation of similar local adsorption configurations in the 2D glass phases. The little differences between adlayer structures formed for [EMIM][TFSA] and [OMIM][TFSA], which differ significantly in the length of the alkyl chain at the cation, further indicates that in the ordered phases the alkyl chains are standing upright rather than lying flat along the surface, in agreement with previous results of recent DFT-D calculations for [BMP][TFSA] on Ag(111).
(5) Substrate effects on the structure and structure formation are present, but weak. They are evident from differences between the 2D crystalline structures formed on Au(111) and Ag(111), both for the ILs investigated in this structure and for [BMP][TFSA]. Furthermore, the Au(111) herringbone reconstruction causes a higher amount of lattice defects and short-range ordered 2D glass phase, also the ordered domains are smaller on Au(111) than on Ag(111).
(6) The attractive interactions between the adsorbed ILs are weak, regardless of the IL and the nature of the noble metal substrate, even at 100 K the 2D solid phases of the ILs are in equilibrium with a 2D gas phase of adsorbed mobile species. The high mobility of the individual adsorbed species also points to a low diffusion barrier for the individual adsorbed species.
Overall, this comparative study gained detailed insight into the interaction of ILs containing [TFSA]− anions with noble metal surfaces, and the general features are expected to be characteristic also for the interaction of other types of ILs with noble metal surfaces.
Experimental
The measurements were performed in a UHV system with a base pressure <1 × 10−10 mbar, which is equipped with a variable temperature Aarhus-type STM (SPECS; Aarhus STM 150) and standard facilities for surface preparation and characterization. STM measurements can be carried out between 90–400 K, utilizing liquid N2 cooling and resistive sample heating. All images were acquired in constant current mode with tunnelling currents between 40 and 100 nA and tunnelling voltages of 1–2 V.The Au(111) and Ag(111) crystals were purchased from MaTeck GmbH with an orientation accuracy of <0.1° and a surface roughness <0.03 μm. The ILs were purchased from Merck ([EMIM][TFSA]) and from Io-Li-Tec (Germany) ([OMIM][TFSA]), both in the highest available quality (ultrapure). Cleaning of the Au(111) and Ag(111) surfaces was performed by repeated cycles of Ar+ sputtering (30 min, 4 μA, 0.5 keV) and heating (770 K, 30 min). The cleanness was checked with STM; for both substrates atomically clean surfaces were obtained with terraces of >200 nm width separated by monatomic steps. In the case of Au(111), the terraces exhibit the typical herringbone surface reconstruction pattern.48 The IL adlayers were prepared by physical vapor deposition using a triple Knudsen effusion evaporator (ventiotec, OVD-3). Prior to use, the glass crucibles were baked out at 870 K in UHV to remove vaporizable impurities from the glass. After cooling down and dismounting of the evaporator from the chamber, the ILs were filled into the crucibles, the evaporator is afterwards again installed at the UHV chamber and evacuated. The ILs are then degassed for 24 hours under UHV conditions at temperatures slightly below the evaporation temperature ([EMIM][TFSA] at 350 K; [OMIM][TFSA] at 300 K). This procedure ensures contamination-free IL vapor (checked by QMS). Residues such as H2O, Li+ or Na+ ions, or Al2O3 particles, which often appear even in “ultrapure” ILs,20,49 are either not vaporizable and thus remain in the crucible during evaporation, or have been removed by the degassing process. The evaporation rate was calibrated against the evaporator temperature by a quartz micro balance (intellemetrics IL150) and by mass spectrometric measurements, using a quadrupole mass spectrometer (Pfeiffer HiQuadQMG700) which was mounted with its ionization area into the IL beam. Defined IL adsorbate coverages were obtained by adjusting the evaporation time and the evaporation temperature, with evaporation temperatures between 350 K and 390 K for [EMIM][TFSA] and between 300 K and 330 K for [OMIM][TFSA].
A monolayer is defined as a closed layer of ions in direct contact to the surface, following the definition in our previous studies.29–31 This differs from the definition used by Cremer et al.,25 who defined a monolayer as a layer of ion pairs on top of each other. To avoid misunderstandings, all coverage data from Cremer et al. were converted by multiplying them with a factor of two.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft via the Collaborative Research Centre SFB 569 (Ulm) and by the German Federal Ministry of Education and Research via the project “Li-Eco-Safe” under contract number 03X4636C embedded in the BMBF-program “Werkstoffinnovationen für Industrie und Gesellschaft”. B.U. is grateful for a fellowship by the Fonds der Chemischen Industrie. We would like to thank Oliver Höfft for supplying us with a sample of the IL [OMIM][TFSA].References
- F. Endres, D. MacFarlane and A. Abbott, Electrodeposition from Ionic liquids, VCH Verlagsgesellschaft mbH, 2008 Search PubMed.
- F. Endres, ChemPhysChem, 2002, 3(2), 144–154 CrossRef CAS.
- S. Z. El Abedin, Z. Phys. Chem., 2006, 220(10), 1293–1308 CrossRef.
- F. H. Hurley and T. P. Wier Jr, J. Electrochem. Soc., 1951, 98, 207–212 CrossRef CAS.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39(21), 3772–3789 CrossRef CAS.
- T. Welton, Chem. Rev., 1999, 99(8), 2071–2084 CrossRef CAS PubMed.
- R. Sheldon, Chem. Commun., 2001, 2399–2407 RSC.
- Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13(10), 2619–2637 RSC.
- J. S. Wilkes, J. Mol. Catal. A: Chem., 2004, 214(1), 11–17 CrossRef CAS.
- B. Garcia, S. Lavallee, G. Perron, C. Michot and M. Armand, Electrochim. Acta, 2004, 49(26), 4583–4588 CrossRef CAS.
- J. H. Shin, W. A. Henderson and S. Passerini, J. Electrochem. Soc., 2005, 152(5), A978–A983 CrossRef CAS.
- H. Matsumoto, H. Sakaebe, K. Tatsumi, M. Kikuta, E. Ishiko and M. Kono, J. Power Sources, 2006, 160(2), 1308–1313 CrossRef CAS.
- S. Seki, Y. Kobayashi, H. Miyashiro, Y. Ohno, A. Usami, Y. Mita, N. Kihira, M. Watanabe and N. Terada, J. Phys. Chem. B, 2006, 110(21), 10228–10230 CrossRef CAS PubMed.
- A. Fernicola, F. Croce, B. Scrosati, T. Watanabe and H. Ohno, J. Power Sources, 2007, 174(1), 342–348 CrossRef CAS.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8(8), 621–629 CrossRef CAS PubMed.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195(9), 2419–2430 CrossRef CAS.
- N. Yabuuchi, K. Shimomura, Y. Shimbe, T. Ozeki, J. Y. Son, H. Oji, Y. Katayama, T. Miura and S. Komaba, Adv. Energy Mater., 2011, 1(5), 759–765 CrossRef CAS.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115(13), 6357–6426 CrossRef CAS PubMed.
- N. Borisenko, S. Z. El Abedin and F. Endres, J. Phys. Chem. B, 2006, 110(12), 6250–6256 CrossRef CAS PubMed.
- F. Endres and S. Zein El Abedin, Z. Phys. Chem., 2007, 221(9–10), 1407–1427 CrossRef CAS.
- R. Wen, B. Rahn and O. M. Magnussen, Angew. Chem., 2015, 127(20), 5889 CrossRef.
- T. Cremer, M. Killian, J. M. Gottfried, N. Paape, P. Wasserscheid, F. Maier and H. P. Steinrück, ChemPhysChem, 2008, 9(15), 2185–2190 CrossRef CAS PubMed.
- K. R. Lovelock, I. J. Villar-Garcia, F. Maier, H. P. Steinrück and P. Licence, Chem. Rev., 2010, 110(9), 5158–5190 CrossRef CAS PubMed.
- M. Sobota, I. Nikiforidis, W. Hieringer, N. Paape, M. Happel, H. P. Steinrück, A. Görling, P. Wasserscheid, M. Laurin and J. Libuda, Langmuir, 2010, 26(10), 7199–7207 CrossRef CAS PubMed.
- T. Cremer, M. Stark, A. Deyko, H. P. Steinrück and F. Maier, Langmuir, 2011, 27, 3662–3671 CrossRef CAS PubMed.
- T. Cremer, L. Wibmer, S. K. Calderon, A. Deyko, F. Maier and H. P. Steinrück, Phys. Chem. Chem. Phys., 2012, 14(15), 5153–5163 RSC.
- H. P. Steinrück, Phys. Chem. Chem. Phys., 2012, 14(15), 5010–5029 RSC.
- T. Waldmann, H. H. Huang, H. E. Hoster, O. Höfft, F. Endres and R. J. Behm, ChemPhysChem, 2011, 12, 2565–2567 CrossRef CAS PubMed.
- B. Uhl, T. Cremer, M. Roos, F. Maier, H. P. Steinrück and R. J. Behm, Phys. Chem. Chem. Phys., 2013, 15, 17295–17308 RSC.
- F. Buchner, K. Forster-Tonigold, B. Uhl, D. Alwast, N. Wagner, H. Farkhondeh, A. Groß and R. J. Behm, ACS Nano, 2013, 7(9), 7773–7784 CrossRef CAS PubMed.
- B. Uhl, F. Buchner, D. Alwast, N. Wagner and R. J. Behm, Beilstein J. Nanotechnol., 2013, 4(1), 903–918 CrossRef CAS PubMed.
- B. Uhl, F. Buchner, S. Gabler, M. Bozorgchenani and R. J. Behm, Chem. Commun., 2014, 50(62), 8601–8604 RSC.
- R. Atkin, S. Z. E. Abedin, R. Hayes, L. H. S. Gasparotto, N. Borisenko and F. Endres, J. Phys. Chem. C, 2009, 113(30), 13266–13272 CAS.
- R. Foulston, S. Gangopadhyay, C. Chiutu, P. Moriarty and R. G. Jones, Phys. Chem. Chem. Phys., 2012, 14, 6054–6066 RSC.
- J. P. Leal, J. M. S. S. Esperança, M. E. M. da Piedade, J. N. C. Lopes, L. P. N. Rebelo and K. R. Seddon, J. Phys. Chem. A, 2007, 111(28), 6176–6182 CrossRef CAS PubMed.
- D. Strasser, F. Goulay, M. S. Kelkar, E. J. Maginn and S. R. Leone, J. Phys. Chem. A, 2007, 111(17), 3191–3195 CrossRef CAS PubMed.
- A. Elbourne, S. McDonald, K. Voitchovsky, F. Endres, G. G. Warr and R. Atkin, ACS Nano, 2015, 9(7), 7608–7620 CrossRef CAS PubMed.
- Y. Z. Su, Y. C. Fu, J. W. Yan, Z. B. Chen and B. W. Mao, Angew. Chem., 2009, 121(28), 5250–5253 CrossRef.
- Y.-Z. Su, Y.-C. Fu, Y.-M. Wei, J.-W. Yan and B.-W. Mao, ChemPhysChem, 2010, 11(13), 2764–2778 CrossRef CAS PubMed.
- Y. Z. Su, J. W. Yan, M. G. Li, Z. X. Xie, B. W. Mao and Z. Q. Tian, Z. Phys. Chem., 2012, 226(9–10), 979–994 CrossRef CAS.
- C. Müller, S. Vesztergom, T. Pajkossy and T. Jacob, J. Electroanal. Chem., 2015, 737, 218–225 CrossRef.
- S. Krischok, A. Ulbrich, T. Ikari, V. Kempter, M. Marschewski and O. Höfft, Nucl. Instrum. Methods Phys. Res., Sect. B, 2014, 340, 51–57 CrossRef CAS.
- N. Borisenko, R. Atkin and F. Endres, Electrochem. Soc. Interface, 2014, 59–63 CAS.
- J. M. Slattery, C. Daguenet, P. J. Dyson, T. J. S. Schubert and I. Krossing, Angew. Chem., 2007, 119(28), 5480–5484 CrossRef.
- I. Krossing, J. M. Slattery, C. Daguenet, P. J. Dyson, A. Oleinikova and H. Weingärtner, J. Am. Chem. Soc., 2006, 128(41), 13427–13434 CrossRef CAS PubMed.
- G. B. Appetecchi, M. Montanino, M. Carewska, M. Moreno, F. Alessandrini and S. Passerini, Electrochim. Acta, 2011, 56(3), 1300–1307 CrossRef CAS.
- C. S. Stefan, D. Lemordant, P. Biensan, C. Siret and B. Claude-Montigny, J. Therm. Anal. Calorim., 2010, 102(2), 685–693 CrossRef CAS.
- J. V. Barth, H. Brune, G. Ertl and R. J. Behm, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42(15), 9307–9318 CrossRef CAS.
- F. Endres, S. Z. El Abedin and N. Borissenko, Z. Phys. Chem., 2006, 220(10), 1377–1394 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp03787e |
| This journal is © the Owner Societies 2015 |