Appearance of annular ring-like intermediates during amyloid fibril formation from human serum albumin†
Received
30th June 2015
, Accepted 30th July 2015
First published on 3rd August 2015
Abstract
The self-assembly of proteins triggered by a conformational switch into highly ordered β-sheet rich amyloid fibrils has captivated burgeoning interest in recent years due to the involvement of amyloids in a variety of human diseases and a diverse range of biological functions. Here, we have investigated the mechanism of fibrillogenesis of human serum albumin (HSA), an all-α-helical protein, using an array of biophysical tools that include steady-state as well as time-resolved fluorescence, circular dichroism and Raman spectroscopy in conjunction with atomic force microscopy (AFM). Investigations into the temporal evolution of nanoscale morphology using AFM revealed the presence of ring-like intermediates that subsequently transformed into worm-like fibrils presumably by a ring-opening mechanism. Additionally, a multitude of morphologically-diverse oligomers were observed on the pathway to amyloid formation. Kinetic analysis using multiple structural probes in-tandem indicated that HSA amyloid assembly is a concerted process encompassing a major structural change that is primarily mediated by hydrophobic interactions between thermally-induced disordered segments originating in various domains. A slower growth kinetics of aggregates suggested that the protein structural reorganization is a prerequisite for fibril formation. Moreover, time-dependent Raman spectroscopic studies of HSA aggregation provided key molecular insights into the conformational transitions occurring within the protein amide backbone and at the residue-specific level. Our data revealed the emergence of conformationally-diverse disulfides as a consequence of structural reorganization and sequestration of tyrosines into the hydrophobic amyloid core comprising antiparallel cross β-sheets.
1. Introduction
Amyloid fibrils are ordered protein aggregates and are characterized by a cross-β motif which is comprised of β-strands that run perpendicular to the fibril axis and are arranged in hydrogen-bonded ribbon-like β-sheets.1–7 The conversion of soluble monomeric protein into intractable amyloid aggregates is implicated in a multitude of human disorders that include Alzheimer's disease, Parkinson's disease, type II diabetes, and the spongiform encephalopathies.8–16 Owing to the extraordinary supramolecular architectural characteristics possessed by these amyloids, they are not just limited to diseases but have also been discovered in living organisms to perform an array of important biological functions.17–19 In addition, the robust mechanical strength possessed by amyloids makes them attractive candidates for nanotechnological applications as bionanomaterials.20–23 On the pathway of amyloid formation, various intermediates with different morphologies have been observed. An increasing body of evidence suggests that these oligomeric intermediates might be more cytotoxic than the matured fibrils.15,24 The morphology of these intermediates dictates the toxicity of the amyloid species and various morphologies have been observed for oligomeric as well as fibrillar species. The annular pore-like nanostructures are one of the most commonly observed morphologies that have been shown to exert cytotoxic effects in neurodegenerative diseases by virtue of disrupting the biological membranes.25–31 Thus, the morphologies of these oligomeric intermediates might play a very crucial role in the pathogenesis of amyloid diseases. Therefore, understanding the morphological as well as conformational characteristics of the oligomeric intermediates and the underlying molecular mechanism of amyloid formation is important not only for designing better therapeutics to target amyloid disorders but also for developing novel bionanomaterials. Most proteins, if not all, are capable of undergoing structural transition from their native state to amyloid fibrils under certain experimental conditions in vitro, suggesting that the amyloid formation might be a generic property of polypeptides.16 The mechanism of amyloid formation has been investigated in a variety of model proteins such as lysozyme, β-lactoglobulin, β2-microglobulin, insulin, islet amyloid polypeptide and serum albumins.31–40 Amongst these proteins, human serum albumin (HSA) is an all α-helical protein and has been considered a good model system for protein aggregation studies due to its propensity to easily aggregate under in vitro conditions.
HSA is a single polypeptide chain consisting of 585 amino acids and is composed of three homologous domains I–III and each domain has two sub-domains, A and B (Fig. 1A).41,42 It has physiological importance as a carrier protein in transporting fatty acids, drugs and other organic compounds in the circulatory system.41,43 Several studies on HSA have shown that varying certain conditions like pH, ionic strength, and temperature can have a drastic effect on the native structure of HSA, for example, low pH and high temperature lead to the destabilization of its native structure.44–47 The higher temperature mimics the heat stress condition that leads to heat shock or unfolded protein response which results in protein aggregation and imbalance of protein homeostasis.48 Also, the high temperature provides protein molecules with a sufficient amount of energy to overcome the high activation barrier thereby accelerating the aggregation reaction and making it more accessible to the laboratory timescale. In the case of a multidomain protein like HSA, the increase in temperature might lead to partial destabilization of its well-organized native structure. The resultant partially destabilized state might have resemblance with the disordered state of proteins having a higher propensity to self-assemble than the native form that otherwise lacks any structural attributes that suggest predilection towards amyloid formation.9,49–51 HSA aggregation has been investigated under different pH, temperature and solution conditions using a variety of tools.52–56 However, an in-depth molecular insight into the structural transition from an α-helical native to cross β sheet-rich amyloid state via oligomeric intermediates still remains elusive. In this work, we have utilized steady-state as well as time-resolved fluorescence spectroscopy, circular dichroism and atomic force microscopy to unravel the mechanism of HSA fibrillation. Efforts were directed towards elucidating the protein conformational – and size changes as a function of time with an emphasis on delineating the key structural transitions in a domain-specific manner. Additionally, Raman spectroscopic measurements at various aggregation stages provided a wealth of structural insights into alterations in the protein backbone amides and side chain residues as a function of amyloid assembly.
 |
| | Fig. 1 (A) The crystal structure of human serum albumin (PDB ID: 1UOR) generated using PyMol (DeLano Scientific, CA). The disulfides, tryptophan (in domain II) and the free cysteine (in domain I) are shown in cyan, green and yellow, respectively. The amyloid formation kinetics monitored by (B) ThT fluorescence and (C) CD. The continuous black lines through the data points represent the fits obtained using a single-exponential kinetic model (the inset in both the figures shows the spectra for zero time point (black) and final time point (red) of fibrillation). | |
2. Experimental methods
2.1 Materials
Albumin from human serum (HSA), 8-anilino-1-naphthalenesulfonic acid ammonium salt (ANS), thioflavin-T (ThT), and sodium hydrogen phosphate (monobasic) were obtained from Sigma (St. Louis, MO). 6-Acryloyl-2 (dimethylamino) naphthalene (acrylodan) and 5-((((2-iodoacetyl)amino)ethyl)amino) naphthalene-1-sulfonic acid (IAEDANS) were procured from Molecular Probes, Invitrogen Inc. Milli-Q water was used for the preparation of all solutions. The pH of the buffers was adjusted to ∼7.4 using a Metrohm pH meter at ∼25 °C.
2.2 Methods
Aggregation reaction.
The HSA stock solution was prepared in 10 mM phosphate buffer (pH 7.4). The accurate protein concentration was estimated by measuring the absorbance at 280 nm using a molar absorption coefficient (ε) of 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 219 M−1 cm−1.52 For the aggregation reaction, the stock solution was diluted using the same buffer containing 50 mM NaCl to a final protein concentration of 100 μM. The resultant solution was heated at 65 ± 2 °C in a heating block, preset at the required temperature under quiescent conditions. Aliquots were taken out at regular intervals for the kinetics measurements. For aggregation studies using labeled HSA, the labeled protein concentration was 5% of the total protein concentration (100 μM) in the aggregation reaction mixture.
219 M−1 cm−1.52 For the aggregation reaction, the stock solution was diluted using the same buffer containing 50 mM NaCl to a final protein concentration of 100 μM. The resultant solution was heated at 65 ± 2 °C in a heating block, preset at the required temperature under quiescent conditions. Aliquots were taken out at regular intervals for the kinetics measurements. For aggregation studies using labeled HSA, the labeled protein concentration was 5% of the total protein concentration (100 μM) in the aggregation reaction mixture.
Arrhenius plot.
In order to monitor the temperature dependence of the aggregation reaction using ThT fluorescence, the reaction was carried out at 55 °C, 65 °C, 70 °C and 75 °C. ThT fluorescence observed under different temperature conditions could be fitted to single exponential. The activation energy was obtained using the Arrhenius equation,| | | k = Aexp(−Ea/RT) | (1) |
where A is the pre-exponential factor, T is the absolute temperature, R is the universal gas constant and Ea is the activation energy. The slope of ln(k) versus 1/T plot yielded the activation energy (Ea) for the reaction.
Circular dichroism (CD) measurements.
The CD spectra were acquired using a Chirascan Spectrophotometer (Applied Photophysics, UK) in a 1 mm path length quartz cell with a scan range of 200–260 nm and 1 nm as step size. The spectra were corrected for the buffer signal and were plotted in Origin 8.5 software.
Atomic Force Microscopy (AFM) imaging.
AFM images of oligomers and amyloid fibrils were acquired on MultiView 2000™ instrument (Nanonics Imaging Ltd, Jerusalem, Israel) operating in intermittent contact mode based on the phase feedback technique. For AFM imaging, the aliquots were withdrawn after 10 min and 2 h. The sample withdrawn after 10 min was diluted 4000-fold and the one which was withdrawn after 2 h was diluted 2000-fold using an aggregation reaction buffer filtered through a 0.22 μm syringe filter. The sample (10 μL in volume) was deposited on freshly cleaved and buffer-washed muscovite mica (Grade V-4 mica from SPI, PA). The mica surface was incubated with the sample for 20 min before washing it with filtered Milli-Q water and dried under a gentle stream of nitrogen gas. AFM scanning was performed under ambient environmental conditions using a Cr-coated cantilevered glass probe of diameter ∼10 nm oscillating at a resonance frequency of ∼32 kHz. AFM height images of 400 × 400 pixels in resolution, covering areas of 10 μm × 10 μm were collected with a sample delay of 4 ms. Smaller areas (2.5 μm × 2.5 μm or 1.45 μm × 1.45 μm) were scanned to image finer morphological features. The AFM images were acquired using the Quartz software (provided with MultiView) and were processed using the WSxM software provided with our AFM instrument.
Fluorescence labeling of HSA with acrylodan and IAEDANS.
The labeling of the free cysteine in domain I with acrylodan and IAEDANS was carried out separately in 10 mM phosphate buffer (pH 7.4) according to the protocol described previously.57 Briefly, acrylodan/IAEDANS and HSA were mixed in a 10:1 molar ratio and allowed to stir in the dark for 24 h at room temperature. Stock solutions of acrylodan (in acetonitrile) and IAEDANS (in dimethyl sulfoxide (DMSO)) were prepared freshly. The labeled protein was then passed through a PD-10 column to remove any unreacted dye and was further concentrated using AMICON ultra (30 kDa cutoff; from Millipore). Finally, the reaction mixture was dialyzed against 10 mM phosphate buffer (pH 7.4) over a 4 day period at 4 °C; with buffer change after every 12 h. The concentration of the labeled protein was estimated using ε365 = 12800 M−1 cm−1, for acrylodan58 and ε337 = 6100 M−1 cm−1 for AEDANS.59
Steady-state fluorescence measurements.
All the steady-state fluorescence measurements were performed on a FluoroMax-4 spectrofluorometer (Horiba Jobin Yvon, NJ) at ∼25 °C. The aliquots of the protein sample were taken out at different time intervals and were diluted 5-fold in 10 mM phosphate buffer (pH 7.4) for all the steady-state fluorescence measurements, except for the AEDANS-labeled protein. The AEDANS samples were not diluted before the fluorescence measurements. The samples were excited at 295 nm (Trp), 350 nm (ANS), 360 nm (acrylodan), 337 nm (AEDANS) and 450 nm (ThT). The concentration of ANS and ThT used for ANS and ThT binding experiments, respectively, was 10 μM. The steady-state fluorescence anisotropies were measured at 350 nm (Trp), 470 nm (ANS), 500 nm (acrylodan) and 475 nm (AEDANS). The steady-state fluorescence anisotropy (rss) is given by the following relationship:60,61| | | rss = (I∥ − I⊥G)/(I∥ + 2I⊥G) | (2) |
where I∥ and I⊥ are fluorescence intensities collected using parallel and perpendicular geometry, respectively. The perpendicular components were always corrected using a G-factor.
Time resolved fluorescence measurements.
All the time-resolved fluorescence data were acquired using a time-correlated single photon counting (TCSPC) setup (Fluorocube, Horiba Jobin Yvon, NJ). For fluorescence lifetime measurements, the peak count was fixed to 10000 and the excitation and emission polarizers were oriented at the magic angle (54.7°). For time-resolved fluorescence anisotropy measurements, the peak difference was 10000 counts and the orientation of the emission polarizer was 0° and 90° with respect to the excitation polarizer for parallel fluorescence intensities (I∥) and perpendicular fluorescence intensities (I⊥), respectively. The emission monochromator for AEDANS was fixed at 500 nm with a bandpass of 4 nm. The 375 nm laser diode was used as a light source for the excitation of AEDANS. The instrument response function (IRF) was collected using Ludox (colloidal silica). The width (FWHM) of IRF was ∼250 ps. The anisotropy decays were analyzed by globally fitting I∥(t) and I⊥(t).60 Briefly, the fluorescence intensity decay curves were deconvoluted taking IRF into account using| |  | (3) |
where αi and τi represent the contributions and lifetime of the different lifetime components, respectively. The fitted parameters were then used as input for the anisotropy decay analysis and I∥(t) and I⊥(t) were fitted using:| |  | (4) |
| |  | (5) |
where I∥(t) and I⊥(t) represent the parallel and perpendicular fluorescence intensities, respectively. I(t) represents the fluorescence intensity at magic angle (54.7°) and r(t) is the anisotropy decay function. The perpendicular fluorescence intensities were corrected for G-factor which was obtained for AEDANS using ANS in 100% ethanol. The anisotropy decays were then analyzed using a biexponential anisotropy decay function that describes the fast (local) and slow (global) motion of a fluorophore.60,62| | | r(t) = r(0)[βfastexp(−t/ϕfast) + βslowexp(−t/ϕslow)] | (6) |
where r(0) represents the intrinsic anisotropy of a fluorophore; ϕfast and ϕslow represent the fast and slow rotational correlation times, respectively, with βfast and βslow as their respective amplitudes. For estimating r(0) for all the fluorophores, the fluorophores were taken in 100% glycerol and the decays were collected. The r(0) so obtained was used for analyzing the anisotropy data with a window of ±0.005. The slow correlation time represents the global motion of a fluorophore and hence can be used for estimating the size of a molecule using the Stokes–Einstein relationship given by| | 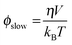 | (7) |
where η is the viscosity, kB is the Boltzmann constant, T is the temperature which was kept as 25 °C for all the experiments and V is the molecular volume given by V = (4/3)πRh3 where Rh is the hydrodynamic radius of the molecule.
Raman spectroscopy.
Raman spectra of the samples were collected on an inVia Raman microscope with 180° scattering geometry (Renishaw, UK). The samples were prepared freshly. The aliquots were withdrawn at regular time intervals and the samples were precipitated with acetone (100% v/v). The precipitates were then deposited on a glass slide covered with aluminium foil. A HPNIR laser with 785 nm wavelength was used as an excitation source and focused into the sample spot using a 50× objective lens (Nikon, Japan). The scattered light was collected through the same objective and was allowed to pass through an edge filter (785 nm) in order to remove the Rayleigh scattering. A 1200 l per mm grating was used for the dispersion of the scattered light and the signal was finally detected using an air-cooled CCD detector. The data acquisition was done using Wire 3.1 software provided with our Raman spectrometer. All the spectra were averaged over 50 scans with an exposure time of 10 s for the spectral range 400–2500 cm−1, 1600–1700 cm−1 and 1200–1300 cm−1. All the data were corrected for tilt in the baseline using the cubic spline interpolation method and smoothened in Wire 3.1 software. The baseline corrected and smoothened Raman spectra were plotted in Origin 8.5 software. The deconvolution of Raman spectra was done in Origin 8.5 software as described in our previous publication.63 The spectral range for the estimation of the secondary structure content was 1620–1700 cm−1 (amide I) and 1220–1300 cm−1 (amide III). The peaks were first analyzed by the peak analyzer option in Origin 8.5 software. Mainly three bands were observed at 1640–1700 cm−1 and 1220–1300 cm−1 that correspond to α-helix (1645–1654 cm−1, 1264–1272 cm−1), β-sheet (1665–1674 cm−1, 1230–1240 cm−1) and coils, turns or extended conformation (1680–1690 cm−1, 1245–1255 cm−1). These bands were then fitted using multiple peak-fitting with the Gaussian function.
3. Results and discussion
3.1 Kinetics of amyloid fibril formation and secondary structural changes
The progression of HSA fibrillation at high temperature was monitored by thioflavin-T (ThT) which is a well-known amyloid marker.64 A sharp increase in ThT fluorescence, devoid of any lag phase, was observed within 1 h of incubation indicating a rapid structural transition from the native α-helical state to the amyloidogenic state (Fig. 1B). Typically, amyloid formation via the nucleation-dependent polymerization mechanism exhibits a lag phase followed by an assembly phase.14,50 The absence of a lag phase in HSA fibrillation suggests that the nuclei are formed immediately upon incubation at high temperature which is in contrast to that observed during a nucleation-dependent amyloid assembly. This observation might indicate a “downhill polymerization” mechanism50 and is in agreement with the previous reports on HSA fibrillation.52 However, the reaction rate observed by us was faster. A single exponential fit of the time-course of ThT fluorescence was adequate to describe the fibril formation kinetics suggesting a quasi-two-state transition. Additionally the observed rate of fibrillation, monitored by ThT fluorescence, increased with an increase in temperature and a linear Arrhenius plot was obtained (Fig. S1, ESI†). The activation energy estimated (eqn (1) in Experimental methods) from the slope of the Arrhenius plot was found to be ∼157 kJ mol−1 (∼37.5 kcal mol−1) which is higher than that of bovine serum albumin.65
The CD spectrum of native HSA shows two minima at 208 and 222 nm indicating the characteristics of an α-helical conformation and is consistent with its crystal structure.41,42 As the aggregation reaction progressed, a significant decrease in the ellipticity at 222 nm (θ222) was observed (Fig. 1C). We also observed an increase in θ215/θ222 and θ205/θ222 as a function of time that signified the emergence of β-sheet and disordered structures, respectively, at the expense of the α-helices during amyloid formation. The CD signature of the amyloid fibrils remained unchanged even after prolonged incubation at room temperature. Next, AFM imaging was performed to gain insights into the nanoscale morphology of early oligomeric intermediates and amyloid fibrils.
3.2 Nanoscale morphological transitions
Atomic force microscopy (AFM) is a non-invasive and chemical-free imaging technique which has emerged as a promising tool for investigating nanoscale topographical features of protein amyloids.66–68 In our study, we utilized AFM in a time-dependent manner to visualize the morphologies of early oligomeric species and their subsequent transition to matured fibrillar species (Fig. 2). Therefore, prior to incubating the sample at elevated temperature, AFM imaging was performed. We observed small spherical particles of heights ranging from 2–4 nm and upon statistical analysis, a unimodal height distribution centered at ∼2.5 nm was obtained (Fig. S2A and C, ESI†) indicating that even at 100 μM concentration, HSA remained predominantly monomeric. Upon incubation at higher temperature, AFM imaging revealed a heterogeneous co-existence of nanoscopic ring-like aggregates and protofibrils (Fig. 2A and Fig. S2B, ESI†). The statistical analysis of the topography of ring-like species revealed a height range of 2–8 nm (Fig. S2D, ESI†), similar to that of protofibrils (Fig. S3, ESI†), and an inner diameter of 30–50 nm (Fig. S3, ESI†). The 3D image of a single ring has been shown for clarity (Fig. 2B). It is possible that the smaller rings anneal to form protofibrils (Fig. 2G inset) that subsequently anneal to form bigger rings. At later stages of aggregation (≥2 h), a mixed population of crescent-shaped intermediates, sliced rings and worm-like fibrils was observed (Fig. 2C, H and I and insets). Such morphological diversity among HSA aggregates in addition to a predominant population of worm-like fibrils suggested that probably, the early ring-like intermediates unravel to form curly/worm like fibrils (Fig. 2H and I and insets). The height of these fibrils ranged between 3 and 6 nm and the average diameter for the sliced rings was 40–50 nm. These fibrils do not undergo any further growth and sliced rings were evident even after prolonged incubation (Fig. S3A, ESI†). Therefore, a careful investigation of HSA aggregation using AFM indicated a complex process comprising a time-dependent, stepwise evolution of morphologically-diverse nanoscopic species on the pathway to amyloid fibrils. At first, the predominantly monomeric HSA molecules associate and transform into ring-like oligomeric intermediates that are apparently metastable. Subsequently, these ring-like species undergo further transition to worm-like fibrils presumably, by a ring-opening mechanism. The proposed mechanism might hold true for many disease-related proteins that form ring-like nanostructures in the intermediate stages of amyloid aggregation.27–31
 |
| | Fig. 2 AFM images and the height profiles of (A) oligomeric ring-like structures formed after 10 min (2.5 μm × 2.5 μm area) and (C) worm-like fibrils formed after 2 h incubation at 65 °C (1.45 μm × 1.45 μm area). The 3D images of (B) a single ring-like structure and (D) fibrils show the topography of the observed morphologies. (E and F) The AFM line-profiles of the images are shown in height plots. (G–I) The AFM images showing transition of oligomeric ring-like structures (with figure G insets showing the individual as well as annealed ring-like structures) to worm-like fibrils (with figure H and I insets showing the transition from ring-like intermediates to fibrils). | |
3.3 Conformational and dynamical changes during amyloid formation
After establishing the morphological characteristics of various nanoscopic species en-route to HSA amyloid fibrillation, next we embarked upon probing the protein structural changes using fluorescence spectroscopy with an emphasis on delineating the conformational changes in various domains. Fluorescence spectroscopy is a highly sensitive and multi-parametric technique for studying protein conformational − and dynamical changes in a site-specific manner whereby intrinsic and/or extrinsic fluorophores can be employed. The steady-state fluorescence spectrum provides insights into the local environment around the fluorophore whereas the steady state fluorescence anisotropy provides information about an overall size of the protein with a presumption that the overall size changes predominate over the local dynamics.60,61 HSA contains a free cysteine (Cys 34) in domain I and a single tryptophan (Trp 214) in domain II (Fig. 1A).41–43 In order to probe the conformational changes around domain I during HSA fibrillation, the free thiol group of Cys 34 was covalently modified with two distinct extrinsic fluorophores namely, acrylodan and IAEDANS whereas the conformational changes around domain II were probed by Trp which is an intrinsic fluorophore. Additionally, time-resolved fluorescence decay measurements provided quantitative estimates about the average sizes of the HSA aggregates.
3.3.1 Structural changes in domain I.
The local conformational changes in domain I were monitored by acrylodan (covalently attached to Cys 34) due to its high sensitivity towards the micro-environment.69 An increase in acrylodan fluorescence with a concomitant blue shift in emission maxima of ∼22 nm (from 499 nm to 477 nm; Fig. S4A and B in ESI†) suggested that the region near Cys 34 gets buried into the hydrophobic environment during aggregation. The emergence of more hydrophobic regions in HSA aggregates could be due to non-specific association between thermally-unfolded, disordered structures. The sequestration of Cys 34 into hydrophobic regions is also accompanied by an enhancement in the aggregate size since an increase in the acrylodan fluorescence anisotropy was observed during HSA fibrillation (Fig. S4C, ESI†). We also monitored the steady-state fluorescence anisotropy kinetics of AEDANS (covalently attached to Cys 34 in domain I) since it has a longer fluorescence lifetime (≥10 ns) and therefore, is more sensitive to the overall size-growth of aggregates.70 As aggregation progressed, the AEDANS fluorescence anisotropy showed a time-dependent increase indicating the formation of large-sized aggregates and hence, corroborated the acrylodan anisotropy data (Fig. 3A).
 |
| | Fig. 3 Kinetics of amyloid formation monitored by (A) AEDANS covalently attached to the free cysteine and (B) Trp steady-state fluorescence anisotropy. The continuous black lines through the data points represent the fits obtained using a single-exponential kinetic model. (C) Time-resolved fluorescence anisotropy decay for AEDANS at three different stages of aggregation. The red lines represent the bi-exponential anisotropy decay fits using eqn (6) (see Table S1 for the recovered parameters associated with depolarization kinetics, ESI†). | |
3.3.2 Structural changes in domain II.
Investigations into the structural changes in domain II utilizing Trp 214 fluorescence revealed a drop in the fluorescence intensity along with a blue shift of ∼10 nm (from 340 nm to 330 nm) in the emission maxima (Fig. S5A, ESI†). The blue shift indicates that during aggregation Trp 214 gets relocated into a more hydrophobic environment and the nearby residues such as histidines and cystines probably quench its fluorescence resulting in a drop in the intensity. Additionally, changes in Trp fluorescence indicate changes in the tertiary structure during aggregation. Next, we monitored the steady-state fluorescence anisotropy of Trp 214 which increased as a function of time, suggestive of the formation of larger aggregates and hence, reaffirmed the acrylodan and AEDANS anisotropy results (Fig. 3B). Next, in order to investigate the changes in hydrophobicity of HSA during amyloid aggregation, alterations in ANS (non-covalently bound, extrinsic probe) fluorescence intensity were monitored as a function of time since ANS is a well-known hydrophobic environment reporter.71,72
3.3.3 Changes in overall hydrophobicity.
Free ANS is scarcely fluorescent in water with an emission maximum at ∼515 nm that undergoes a significant blue shift to ∼475 nm with a simultaneous rise in intensity upon binding to hydrophobic pockets.73 Additionally, it has been demonstrated that the cytotoxicity of amyloidogenic entities could be correlated with the changes in ANS fluorescence intensity.74 Monomeric native HSA contains at least three hydrophobic, ANS binding sites (in domains II and III) that primarily differ by higher and lower binding affinities.75,76 In our study, initially we observed a high ANS fluorescence intensity (0 min sample; prior to heating) indicating that ANS was bound to the hydrophobic pockets of the predominantly monomeric protein at room temperature. As aggregation was triggered upon raising the temperature, we observed a progressive drop in ANS fluorescence at ∼475 nm which saturated to ∼50% of the initial intensity within one hour of amyloid fibrillation (Fig. S5B and C, ESI†). Since ANS fluorescence provides information about the overall hydrophobicity, we speculate that at elevated temperature, the hydrophobic sites specifically harboring the ANS get deformed due to the thermal unfolding of domains II and III77 and consequently, ANS molecules can no longer bind to the hydrophobic pockets. However, the fact that the ANS emission does not undergo any red-shift from ∼475 nm (Fig. S5B, ESI†) suggests that probably, ANS molecules still remain bound to the hydrophobic clusters of HSA aggregates. Additionally, a rise in the ANS fluorescence anisotropy (Fig. S5D, ESI†) during amyloid aggregation indicated the formation of larger aggregates which is in accordance with our tryptophan anisotropy data. Taken together, our results suggest that both domains I and II undergo conformational changes during HSA fibrillation.
3.3.4 Dynamical signature and average sizes of the aggregates.
In order to gather insights into the dynamical changes and the size-growth associated with HSA amyloid formation, time-resolved fluorescence measurements were performed on Cys 34 (domain I) covalently linked with AEDANS, which is considered a suitable fluorophore for studying the depolarization measurements due to the long fluorescence lifetime.70 The fluorescence anisotropy decay of AEDANS in the native form (0 min), ring-like intermediates (10 min) and worm-like fibrils (120 min) showed bi-exponential depolarization kinetics comprising a fast and a slow rotational correlation times that represent the local motion of the probe and the global motion (tumbling) of the protein molecules/aggregates, respectively (Fig. 3C and eqn (6) in Experimental methods). The results from the detailed analyses of the time-resolved fluorescence data are summarized in Table S1 (ESI†). The longer component (global) was used for estimating the average hydrodynamic sizes of the predominantly monomeric, oligomeric and fibrillar HSA (eqn (7) in Experimental methods) that were found to be 3.4 ± 0.1 nm, 3.9 ± 0.1 nm and 5.2 ± 0.7 nm, respectively. The observed increase in the longer rotational correlation time for oligomers and fibrils is suggestive of the dampening of the global motion resulting from the association of protein molecules to form oligomeric species that subsequently form large-sized amyloid fibrils. Therefore, taken together, all of the fluorescence measurements revealed that both domains I and II get progressively buried into hydrophobic regions which evolve due to non-specific association between temperature-induced disordered segments that are generated during aggregation. The emergence of hydrophobic clusters is also supported by ANS fluorescence at ∼475 nm although the hydrophobic pockets in native HSA probably get deformed upon increase in temperature. The progressive increase in the average aggregate sizes, as indicated by steady-state anisotropy, was further confirmed by the time-resolved fluorescence data. Next, we embarked upon delineating finer structural changes at the molecular level during various stages of the assembly process using Raman spectroscopy.
3.4 Structural insights into amyloid assembly using Raman spectroscopy
Raman spectroscopy being a very powerful technique allows us to pinpoint the fine differences among various protein conformations on the basis of mainly backbone amides (amide I: 1620–1700 cm−1 and amide III: 1220–1300 cm−1) and several characteristic side-chain vibrations at a residue-specific level such as Cys, Trp, and Tyr.63,78–80 In our study, we monitored the Raman spectra of HSA aggregates in a time dependent manner, especially the amide I and amide III bands (Fig. 4) since the deconvolution of the peaks in these regions gave an estimate of secondary structural elements such as α-helices, β-sheets and random coil/extended conformations at different aggregation stages (Tables S2 and S3, ESI†). Additionally, detailed analyses of amide III region provided information about the Ramachandran ψ dihedral angle which allowed us to predict the β-sheet packing in amyloid nanostructures.81 Moreover, the tyrosines and disulfides provided some key insights into the protein conformational transitions during amyloid aggregation.
 |
| | Fig. 4 (A) Raman spectra (400–2000 cm−1) at different time points of HSA aggregation with black arrows showing amide I and amide III regions. (B) The amide I (1620–1700 cm−1) and (C) amide III region (1220–1300 cm−1) at different time points of aggregation showing transition from a predominantly α-helical to β-sheet-rich structure. The deconvolution of (D) amide I region for zero (0 min) and final time point (120 min) along with (E) amide III region for the same time points is shown. (The individual peaks obtained upon fitting are shown in blue, red, green and magenta. The cumulative peaks are shown in cyan.) | |
3.4.1 Insights into the backbone amide I and III regions.
The Raman spectrum obtained for HSA prior to incubation at 65 °C (0 min) showed a major amide I peak at 1658 cm−1, indicative of an α-helical structure along with two minor peaks at 1679 cm−1 and 1690 cm−1 which correspond to coils/extended conformation (Fig. 4B). As aggregation progressed upon raising the temperature, the amide I band showed a transition from the initially α-helix rich state (1658 cm−1) to the β-sheet rich state (1670 cm−1) via oligomeric intermediates rich in extended conformation/random coil content (1660 cm−1) (Fig. 4B). For instance, the 10 min sample comprising oligomeric species and the ring-like structures retained a high content of random coil and extended conformation along with α-helices and β-sheets (Table S2, ESI†). For subsequent time points, the contents of α-helices, random coils and extended conformations dropped significantly with an appreciable increase in the β-sheet content as is evident from the shift in amide I peak at 1658 cm−1 to 1670 cm−1 (Fig. 4B) which is a characteristic of the amyloid architecture. The kinetics of the growth of 1670 cm−1 band at the expense of 1658 cm−1 band was similar to that of CD and ThT fluorescence (Fig. S6, ESI† and Fig. 4).
The amide III peaks of the native HSA (0 min sample) were centered at 1245 cm−1 and 1270 cm−1 (Fig. 4C), which correspond to coils/turns and α-helices, respectively. The amide III peak at 1270 cm−1 almost completely disappeared with time suggesting an attenuation in the α-helical content (Fig. 4C) whereas the peak at 1243 cm−1 gained prominence. This observation suggested that the substantial drop in α-helical content was concomitant with an increase in the β-sheet content, thus corroborating the results obtained from the amide I region. The peak observed at 1243 cm−1 is in agreement with the results obtained for insulin filaments and ovalbumin amyloid pores.63,82 Further analysis of the amide III peak (at 1243 cm−1), using the approach reported previously,81 enabled us to estimate the Ramachandran ψ dihedral angle which was found to be ≈+143°; suggestive of antiparallel β-sheet conformation. Interestingly, the analysis of amide III band for early ring-like nanoscopic species formed after 10 min also showed the presence of a higher amount of random coil and extended conformation (Table S3, ESI†) compared to the later times that showed more ordered β-sheet rich structures. Therefore, based on the results obtained from amide I and amide III regions, we suggest that HSA fibrillation proceeds through a loss of α-helical content in conjunction with an increase in the random coil content followed by reorganization into antiparallel β-sheet rich amyloid fibrils.
3.4.2 Insights into residue-specific changes during aggregation.
After gaining information about protein backbone conformational changes, we investigated the side chain vibrations at the residue-specific level. The peaks observed at 830 and 850 cm−1 represent the Fermi doublet for tyrosine. The intensity ratio of 850 to 830 is an indicator of the hydrogen bonding strength between the hydroxyl group of tyrosine and the surrounding solvent molecules.78,79 The I850/I830 ratio for the native conformation was ∼1.3 which decreased to ∼1.0 during aggregation. This indicates weakening of the H-bonding between the tyrosine moiety and the neighboring water molecules in β-sheet rich conformations which in turn implies that the tyrosines get progressively buried during the aggregation. However, the weakening of the hydrogen bonding is an average assessment since HSA contains 18 tyrosines. We next monitored the changes in the disulfide Raman intensities since HSA contains 17 disulfide bridges that contribute towards the overall structural rigidity but allow sufficient conformational flexibility to HSA upon changes in solution conditions.83 Typically, the disulfide S–S stretching frequency appears at ∼510 ± 5 cm−1 as observed during insulin82,84 and lysozyme aggregation.85 In our study, we observed multiple peaks in 500–600 cm−1 for the disulfide bonds wherein a major peak at ∼507 cm−1 remained conserved throughout the aggregation. Some new peaks (∼520 cm−1, ∼550 cm−1) emerged during aggregation and the peak at 507 cm−1 became broader (Fig. S6C, ESI†). Analysis of all the Raman disulfide peaks revealed a strong dependence of S–S wavenumber position on the torsional angle χ(C–S–S–C) which provides information about the internal rotation about C–S and C–C bonds in Cα–Cβ–S–S–Cβ′–Cα′ conformations.86,87 The peak at 510 ± 5 cm−1 (∼507 cm−1, in our case) is suggestive of the gauche–gauche–gauche conformation (χ: ±60°) that was confirmed by careful analysis of the disulfides in the crystal structure using PyMol (Fig. 1A). The additional peaks observed at 525 ± 5 cm−1 and at 540 ± 5 cm−1 are attributed to gauche–gauche–trans (χ: ±80–90°) and trans–gauche–trans conformations, respectively. Our results indicate the emergence of multiple, heterogeneous disulfide conformers during the transformation from the α-helical state to cross-β sheet-rich amyloid fibrils comprising antiparallel β-sheets. Additionally, the broadening of peak position during the transition from the native to the amyloid state is suggestive of structural rearrangement as a consequence of intermolecular non-covalent interactions and “perturbations in local environment”85 that emerge during HSA amyloid assembly.
3.5 Mechanism of HSA amyloid fibrillation using multiple structural probes
In this study, multiple structural probes were employed to discern the time-dependent evolution of molecular events underlying HSA amyloid formation. We followed the aggregation kinetics by CD, steady-state as well as time-resolved fluorescence, and Raman spectroscopy that revealed a lag phase independent assembly (Fig. 5). The time-dependence of various spectroscopic readouts could be satisfactorily described using a single exponential kinetics and an apparent first order rate constant of (5 ± 0.8) × 10−2 min−1 was obtained for all the probes (Fig. S5, S6A and B, ESI†), except for ANS anisotropy. The similarity in the rates suggested that the structural reorganization and amyloid formation occur simultaneously. However, the aggregate growth rate, monitored by ANS fluorescence anisotropy, was found to be somewhat slower compared to the structural changes (Fig. 5 and Fig. S5B, ESI†). These findings suggest that the amyloid formation involves a major structural change that triggers the self-assembly process followed by a slow growth phase.
 |
| | Fig. 5 The plot comparing the apparent first order rate constants recovered from the amyloid formation kinetics monitored using multiple structural probes viz. ThT and ANS fluorescence intensity; Trp, ANS and AEDANS fluorescence anisotropy; circular dichroism (ratio of ellipticity at 205 nm to 222 nm); amide I vibration from Raman spectroscopy (ratio of 1670 cm−1 to 1658 cm−1 bands). See also Fig. S5 and S6 (ESI†) for the kinetics data. | |
4. Conclusions
In summary, we have investigated the mechanism of amyloid fibril formation from human serum albumin using a host of biophysical tools. The kinetic analyses of multiple spectroscopic readouts indicated that the amyloid formation involves a major conformational change from a predominantly monomeric all α-helical to a cross-β sheet rich structure in a lag phase-independent manner. Raising the temperature triggers the formation and accumulation of flexible, non-native unfolded or disordered conformers that facilitate the protein self-assembly mediated by various non-covalent including hydrophobic interactions. Also, almost all the domains participate in antiparallel β sheet-rich amyloid assembly whereby tryptophan, tyrosines, and the lone cysteine get sequestered into the hydrophobic interior. Additionally, AFM imaging revealed the formation of a multitude of morphologically-diverse nanoscopic intermediates during amyloid aggregation. We speculate that the nanoscopic ring-like intermediates might be metastable structures that subsequently mature into worm-like fibrils through a unique mechanism that involves both annealing and unraveling of rings. The antiparallel β-sheet structures have been linked with amyloid-induced toxicity and have been observed for various toxic, annular pore-like oligomers of disease-related proteins or peptides. The ring-like intermediates observed by us might have resemblance with the annular pores that can interact with the membrane and disrupt the membrane by pore formation. The elucidation of the cascade of molecular events and morphological transition is important for designing small-molecule inhibitors for anti-amyloid therapeutics. We believe that such mechanistic studies will open new avenues for the controlled fabrication of exotic bionanomaterials having a wide array of nano-biotechnological applications.
Acknowledgements
We thank the members of the Mukhopadhyay laboratory for critically reading the manuscript. We thank IISER Mohali for the financial support, the Ministry of Human Resources Development, Govt. of India for the Centre of Excellence grant, the Council of Scientific and Industrial Research, New Delhi (research grant to S.M., research fellowships to S.A. and A.K.) and the Department of Science and Technology, New Delhi (for the SERB Young Scientist Scheme to M.B.). We thank Prof. N. Periasamy (TIFR Mumbai) for the time-resolved fluorescence data analysis software.
References
- I. K. Lednev, Biophys. J., 2014, 106, 1433–1435 CrossRef CAS PubMed.
- R. V. Pappu, Biophys. J., 2014, 107, 795–797 CrossRef CAS PubMed.
- R. Tycko and R. B. Wickner, Acc. Chem. Res., 2013, 46, 1487–1496 CrossRef CAS PubMed.
- C. J. Sarell, P. G. Stockley and S. E. Radford, Prion, 2013, 7, 359–368 CrossRef CAS PubMed.
- J. Greenwald and R. Riek, Structure, 2010, 18, 1244–1260 CrossRef CAS PubMed.
- O. S. Makin and L. C. Serpell, FEBS J., 2005, 272, 5950–5961 CrossRef CAS PubMed.
- R. Nelson, M. R. Sawaya, M. Balbirnie, A. O. Madsen, C. Riekel, R. Grothe and D. Eisenberg, Nature, 2005, 435, 773–778 CrossRef CAS PubMed.
- J. Brettschneider, K. Del Tredici, V. M. Lee and J. Q. Trojanowski, Nat. Rev. Neurosci., 2015, 16, 109–120 CrossRef CAS PubMed.
- V. N. Uversky, V. Davé, L. M. Iakoucheva, P. Malaney, S. J. Metallo, R. R. Pathak and A. C. Joerger, Chem. Rev., 2014, 114, 6844–6879 CrossRef CAS PubMed.
- T. P. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed.
- B. H. Meier and A. Bockmann, Curr. Opin. Struct. Biol., 2014, 30C, 43–49 Search PubMed.
- D. Eisenberg and M. Jucker, Cell, 2012, 148, 1188–1203 CrossRef CAS PubMed.
- I. Moreno-Gonzalez and C. Soto, Semin. Cell Dev. Biol., 2011, 22, 482–487 CrossRef CAS PubMed.
- J. Lee, E. K. Culyba, E. T. Powers and J. W. Kelly, Nat. Chem. Biol., 2011, 7, 602–609 CrossRef CAS PubMed.
- M. Stefani and C. Dobson, J. Mol. Med., 2003, 81, 678–699 CrossRef CAS PubMed.
- F. Chiti, P. Webster, N. Taddei, A. Clark, M. Stefani, G. Ramponi and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3590–3594 CrossRef CAS.
- D. M. Fowler, A. V. Koulov, W. E. Balch and J. W. Kelly, Trends Biochem. Sci., 2007, 32, 217–224 CrossRef CAS PubMed.
- M. M. Barnhart and M. R. Chapman, Annu. Rev. Microbiol., 2006, 60, 131–147 CrossRef CAS PubMed.
- H. Wu, Cell, 2013, 153, 287–292 CrossRef CAS PubMed.
- I. Cherny and E. Gazit, Angew. Chem., Int. Ed., 2008, 47, 4062–4069 CrossRef CAS PubMed.
- T. Scheibel, R. Parthasarathy, G. Sawicki, X.-M. Lin, H. Jaeger and S. L. Lindquist, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4527–4532 CrossRef CAS PubMed.
- C. Li and R. Mezzenga, Nanoscale, 2013, 5, 6207–6218 RSC.
- U. Shimanovich, I. Efimov, T. O. Mason, P. Flagmeier, A. K. Buell, A. Gedanken, S. Linse, K. S. Åkerfeldt, C. M. Dobson, D. A. Weitz and T. P. J. Knowles, ACS Nano, 2015, 9, 43–51 CrossRef CAS PubMed.
- M. Stefani, Prog. Neurobiol., 2012, 99, 226–245 CrossRef CAS PubMed.
- M. Bucciantini, E. Giannoni, F. Chiti, F. Baroni, L. Formigli, J. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson and M. Stefani, Nature, 2002, 416, 507–511 CrossRef CAS PubMed.
- S. M. Butterfield and H. A. Lashuel, Angew. Chem., Int. Ed., 2010, 49, 5628–5654 CrossRef CAS PubMed.
- K. N. Dahlgren, A. M. Manelli, W. B. Stine, L. K. Baker, G. A. Krafft and M. J. LaDu, J. Biol. Chem., 2002, 277, 32046–32053 CrossRef CAS PubMed.
- H. A. Lashuel and P. T. Lansbury, Q. Rev. Biophys., 2006, 39, 167–201 CrossRef CAS PubMed.
- M. Stöckl, N. Zijlstra and V. Subramaniam, Mol. Neurobiol., 2013, 47, 613–621 CrossRef PubMed.
- H. A. I. Lin, R. Bhatia and R. Lal, FASEB J., 2001, 15, 2433–2444 CrossRef CAS PubMed.
- A. Nath, A. D. Miranker and E. Rhoades, Angew. Chem., Int. Ed. Engl., 2011, 50, 10859–10862 CrossRef CAS PubMed.
- A. Cao, D. Hu and L. Lai, Protein Sci., 2004, 13, 319–324 CrossRef CAS PubMed.
- D. Hamada and C. M. Dobson, Protein Sci., 2002, 11, 2417–2426 CrossRef CAS PubMed.
- J. L. Jiménez, E. J. Nettleton, M. Bouchard, C. V. Robinson, C. M. Dobson and H. R. Saibil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9196–9201 CrossRef PubMed.
- P. Taboada, S. Barbosa, E. Castro and V. Mosquera, J. Phys. Chem. B, 2006, 110, 20733–20736 CrossRef CAS PubMed.
- M. Bhattacharya, N. Jain and S. Mukhopadhyay, J. Phys. Chem. B, 2011, 115, 4195–4205 CrossRef CAS PubMed.
- R. Mishra, K. Sörgjerd, S. Nyström, A. Nordigården, Y.-C. Yu and P. Hammarström, J. Mol. Biol., 2007, 366, 1029–1044 CrossRef CAS PubMed.
- T. Eichner, A. P. Kalverda, G. S. Thompson, S. W. Homans and S. E. Radford, Mol. Cell, 2011, 41, 161–172 CrossRef CAS PubMed.
- S. E. Hill, J. Robinson, G. Matthews and M. Muschol, Biophys. J., 2009, 96, 3781–3790 CrossRef CAS PubMed.
- D. E. Schlamadinger and A. D. Miranker, Biophys. J., 2014, 107, 2559–2566 CrossRef CAS PubMed.
- S. Curry, H. Mandelkow, P. Brick and N. Franks, Nat. Struct. Mol. Biol., 1998, 5, 827–835 CAS.
- S. Sugio, A. Kashima, S. Mochizuki, M. Noda and K. Kobayashi, Protein Eng., 1999, 12, 439–446 CrossRef CAS PubMed.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed.
- M. Dockal, J. Biol. Chem., 2000, 275, 3042–3050 CrossRef CAS PubMed.
- N. K. Das, N. Ghosh, A. P. Kale, R. Mondal, U. Anand, S. Ghosh, V. K. Tiwari, M. Kapur and S. Mukherjee, J. Phys. Chem. B, 2014, 118, 7267–7276 CrossRef CAS PubMed.
- A. K. Shaw and S. K. Pal, J. Photochem. Photobiol., B, 2008, 90, 69–77 CrossRef CAS PubMed.
- W. Qiu, L. Zhang, O. Okobiah, Y. Yang, L. Wang, D. Zhong and A. H. Zewail, J. Phys. Chem. B, 2006, 110, 10540–10549 CrossRef CAS PubMed.
- K. Richter, M. Haslbeck and J. Buchner, Mol. Cell, 2010, 40, 253–266 CrossRef CAS PubMed.
- V. N. Uversky and A. L. Fink, Biochim. Biophys. Acta, 2004, 1698, 131–153 CrossRef CAS PubMed.
- R. V. Pappu, X. Wang, A. Vitalis and S. L. Crick, Arch. Biochem. Biophys., 2008, 469, 132–141 CrossRef CAS PubMed.
- Q. Qiao, G. R. Bowman and X. Huang, J. Am. Chem. Soc., 2013, 135, 16092–16101 CrossRef CAS PubMed.
- J. Juárez, P. Taboada and V. Mosquera, Biophys. J., 2009, 96, 2353–2370 CrossRef PubMed.
- J. Juárez, P. Taboada, S. Goy-López, A. Cambón, M.-B. Madec, S. G. Yeates and V. Mosquera, J. Phys. Chem. B, 2009, 113, 12391–12399 CrossRef PubMed.
- A. Stirpe, M. Pantusa, B. Rizzuti, L. Sportelli, R. Bartucci and R. Guzzi, Int. J. Biol. Macromol., 2011, 49, 337–342 CrossRef CAS PubMed.
- J. Juárez, M. Alatorre-Meda, A. Cambón, A. Topete, S. Barbosa, P. Taboada and V. Mosquera, Soft Matter, 2012, 8, 3608 RSC.
- S. Movaghati, A. A. Moosavi-Movahedi, F. Khodagholi, H. Digaleh, E. Kachooei and N. Sheibani, Colloids Surf., B, 2014, 122, 341–349 CrossRef CAS PubMed.
- R. Wang, S. Sun, E. J. Bekos and F. V. Bright, Anal. Chem., 1995, 67, 149–159 CrossRef CAS.
- K. Flora, J. D. Brennan, G. A. Baker, M. A. Doody and F. V. Bright, Biophys. J., 1998, 75, 1084–1096 CrossRef CAS.
- E. N. Hudson and G. Weber, Biochemistry, 1973, 12, 4154–4161 CrossRef CAS.
-
J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, 2006 Search PubMed.
-
N. Jain and S. Mukhopadhyay, in Applied Spectroscopy and the Science of Nanomaterials, ed. P. Misra, Springer, Singapore, 2015, ch. 3, vol. 2, pp. 41–57 Search PubMed.
- S. Arya and S. Mukhopadhyay, J. Phys. Chem. B, 2014, 118, 9191–9198 CrossRef CAS PubMed.
- M. Bhattacharya, N. Jain, P. Dogra, S. Samai and S. Mukhopadhyay, J. Phys. Chem. Lett., 2013, 4, 480–485 CrossRef CAS.
- H. Levine, Protein Sci., 1993, 2, 404–410 CrossRef CAS PubMed.
- N. K. Holm, S. K. Jespersen, L. V. Thomassen, T. Y. Wolff, P. Sehgal, L. A. Thomsen, G. Christiansen, C. B. Andersen, A. D. Knudsen and D. E. Otzen, Biochim. Biophys. Acta, 2007, 1774, 1128–1138 CrossRef CAS PubMed.
- I. Usov, J. Adamcik and R. Mezzenga, Faraday Discuss., 2013, 166, 151 RSC.
- L. R. Volpatti, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, ACS Nano, 2013, 7, 10443–10448 CrossRef CAS PubMed.
- K. K. M. Sweers, K. O. van der Werf, M. L. Bennink and V. Subramaniam, ACS Nano, 2012, 6, 5952–5960 CrossRef CAS PubMed.
- R. E. Hibbs, T. T. Talley and P. Taylor, J. Biol. Chem., 2004, 279, 28483–28491 CrossRef CAS PubMed.
- S. Mukhopadhyay, P. K. Nayak, J. B. Udgaonkar and G. Krishnamoorthy, J. Mol. Biol., 2006, 358, 935–942 CrossRef CAS PubMed.
- A. Hawe, M. Sutter and W. Jiskoot, Pharm. Res., 2008, 25, 1487–1499 CrossRef CAS PubMed.
- K. E. Marshall, K. L. Morris, D. Charlton, N. O'Reilly, L. Lewis, H. Walden and L. C. Serpell, Biochemistry, 2011, 50, 2061–2071 CrossRef CAS PubMed.
- E. Daniel and G. Weber, Biochemistry, 1966, 5, 1893–1900 CrossRef CAS.
- B. Bolognesi, J. R. Kumita, T. P. Barros, E. K. Esbjorner, L. M. Luheshi, D. C. Crowther, M. R. Wilson, C. M. Dobson, G. Favrin and J. J. Yerbury, ACS Chem. Biol., 2010, 5, 735–740 CrossRef CAS PubMed.
- K. Takehara, K. Yuki, M. Shirasawa, S. Yamasaki and S. Yamada, Anal. Sci., 2009, 25, 115–120 CrossRef CAS.
- I. M. Kuznetsova, A. I. Sulatskaya, O. I. Povarova and K. K. Turoverov, PLoS One, 2012, 7, e40845 CAS.
- B. Farruggia, F. Rodriguez, R. Rigatuso, G. Fidelio and G. Picó, J. Protein Chem., 2001, 20, 81–89 CrossRef CAS.
-
D. Nemecek, J. Stepanek and G. J. Thomas, Current Protocols in Protein Science, John Wiley & Sons, Inc., 2001 DOI:10.1002/0471140864.ps1708s71.
- R. Tuma, J. Raman Spectrosc., 2005, 36, 307–319 CrossRef CAS PubMed.
- V. A. Shashilov, V. Sikirzhytski, L. A. Popova and I. K. Lednev, Methods, 2010, 52, 23–37 CrossRef CAS PubMed.
- A. V. Mikhonin, S. V. Bykov, N. S. Myshakina and S. A. Asher, J. Phys. Chem. B, 2006, 110, 1928–1943 CrossRef CAS PubMed.
- T. Zako, M. Sakono, N. Hashimoto, M. Ihara and M. Maeda, Biophys. J., 2009, 96, 3331–3340 CrossRef CAS PubMed.
- M. Bhattacharya, N. Jain, K. Bhasne, V. Kumari and S. Mukhopadhyay, J. Fluoresc., 2011, 21, 1083–1090 CrossRef CAS PubMed.
- K. Huang, N. C. Maiti, N. B. Phillips, P. R. Carey and M. A. Weiss, Biochemistry, 2006, 45, 10278–10293 CrossRef CAS PubMed.
- R. Remmele, Jr., P. McMillan and A. Bieber, J. Protein Chem., 1990, 9, 475–486 CrossRef.
- N. Biswas, A. J. Waring, F. J. Walther and R. A. Dluhy, Biochim. Biophys. Acta, 2007, 1768, 1070–1082 CrossRef CAS PubMed.
- H. E. Van Wart and H. A. Scheraga, J. Phys. Chem., 1976, 80, 1823–1832 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The Arrhenius plot, the steady-state fluorescence data for acrylodan and ANS, aggregation kinetics data from circular dichroism and Raman spectroscopy, and detailed analyses of AFM images and the data recovered from time-resolved fluorescence and Raman spectroscopy (Fig. S1–S6, Tables S1–S3). See DOI: 10.1039/c5cp03782d |
|
| This journal is © the Owner Societies 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence







