
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface effect on the electromelting behavior of nanoconfined water†
Arnab
Bose
,
Atanu K.
Metya
and
Jayant K.
Singh
*
Department of Chemical Engineering, Indian Institute of Technology Kanpur, Kanpur-208016, India. E-mail: jayantks@iitk.ac.in; Fax: 91-512-2590104; Tel: +91-512-2596141
First published on 31st July 2015
Abstract
Electric field induced phase transitions of confined water have an important role in cryopreservation and electrocrystallization. In this study, the structural and dynamical properties of nano-confined water in nano-slit pores under the influence of an electric field varying from 0 to 10 V nm−1 are investigated under ambient conditions using molecular dynamics simulations. In order to replicate the nature of different materials, a systematic approach is adopted, including pore-size and lattice constant variations in different lattice arrangements viz., triangular, square and hexagonal, with hydrophilic and hydrophobic surface–fluid interactions. The structural behavior of water is investigated using radial distribution functions, bond order parameters and hydrogen bond calculations; the dynamical properties are analyzed using lateral and rotational diffusivity calculations. The lateral diffusivity with increasing electric field E increases by order(s) of magnitude during electromelting. The pore-size, lattice constant, lattice arrangement and hydrophobic/hydrophilic nature of the pore surface strongly influence the electromelting behavior for E ≤ ∼7 V nm−1. Higher values of lattice constants and/or hydrophobic pores enhance the electromelting behavior of nanoconfined water.
1. Introduction
Electric fields play a vital role in many biological, environmental and industrial aspects, such as the cryopreservation of living cells and tissues, the protection of crops, power lines and pipe lines from freezing, electrocrystallization, snow making, various food processing operations, and in making various nano-materials like graphene.1–3 A recent quasielastic neutron scattering study on water confined in a silica nano-pore2 reported that after applying an electric field of 2.5 kV mm−1, the translational diffusivity of the confined water is enhanced due to the dissociation of intermolecular hydrogen bonds. On the other hand, an electric field of around 1 kV mm−1 is observed experimentally to promote the formation of ice in a gold nano-confinement of 0.7 nm pore-size under ambient conditions.4 Electric fields of comparable strength are found in various clay materials, amino acid crystals,5 electrified thunder clouds6 and charged surfaces.1,7 At weaker electric fields, the nature of the confinement and the geometry of the surface play an important role in ice formation. For example, mica, a clay material, possesses a pseudo-hexagonal symmetry which promotes cooperative hydrogen bonding with an adjacent layer of the water, and acts as an ice nucleating agent.8 On the other hand, aluminum phosphate, another clay material, prevents crystallization in its cylindrical nano-pores to a glassy state over a wide range of temperatures (173 K ≤ T ≤ 293 K) as the curvature of the cylindrical nano-pore prevents the formation of ice.9 Unlike hydrophilic clay surfaces,10 hydrophobic confinements11–15 like carbon nanotubes (CNTs), graphite- or graphene-slit pores do not form hydrogen bonds with water molecules. Thus, the adjacent layer possesses higher mobility and a weaker hydrogen bond network than the unconstrained bulk phase.16,17 According to molecular dynamics (MD) studies, various electric field induced ice structures are observed in 1 nm slit graphene pores (0 ≤ E ≤ 1.5 V nm−1) and in ∼1 nm CNTs (0 ≤ E ≤ 8 V nm−1) at 1 bar lateral/axial pressure.18,19 Another MD study suggested that hydrophobic pores of size ∼0.7 nm restricted the mobility of the adsorbed water layer at room temperature.20 The pore-size of confinements has an important effect on the freezing of confined water in the absence of an electric field, as shown recently by Zangi and Mark.21,22 These authors reported the freezing of monolayer and bilayer water under ambient conditions in different pore-sizes (0.57–1.14 nm) of quartz confinement. The freezing of water under ambient conditions in a gold nano-confinement of size 0.7 nm has been studied experimentally.4 Qiu and Guo23 reported the freezing of water in a 0.79 nm quartz confinement under ambient conditions using MD simulations, where an electric field of the order of 2.5 V nm−1 was used to melt (electromelting) the ice monolayer. Under such a strong electric field, the long range structural order of the ice layer breaks, increasing the lateral diffusivity by many orders of magnitude (10−8 → 10−5 cm2 s−1) during electromelting.21,23 According to a density functional theory study, electric fields beyond a threshold value of ∼3.5 V nm−1 are able to dissociate bulk water with a sustainable ionic current due to a series of proton jumps.24 However, nano-confined water is not seen to dissociate under a higher electric field of 5 V nm−1 if dense water layers do not allow enough space for the hydrated ions.23 In a recent MD study,23 electric fields (E) in the 0 ≤ E ≤ 10 V nm−1 range were able to melt the ice layer in a nano-slit made up of triangularly arranged atoms of a model quartz material under ambient conditions. However, it is not clear if such behavior is feasible for different materials. To address the above query, this work presents a systematic analysis of the structural and dynamical properties of confined water in slit pores over a range of electric fields: 0 ≤ E ≤ 10 V nm−1. In order to represent different types of material, various lattice arrangements, namely, triangular, square and hexagonal, have been adopted with the pertinent lattice constant range. Many elements, including copper (Cu), nickel (Ni), gold (Au), silver (Ag), yttrium (Y), rhodium (Rh), scandium (Sc), iridium (Ir), etc., possess 2D triangular lattices with 0.2–0.3 nm interatomic distances. Similarly, 2D hexagonal lattice arrangements are also abundant in nature within the 0.1–0.2 nm interatomic distance range, such as the basal (0001) planes of magnesium (Mg), cobalt (Co), zinc (Zn), carbon (C), titanium (Ti), zirconium (Zr), hafnium (Hf), etc. On the other hand, the (100) plane of the BCC lattice of alkali-metals (Li, Na, K, Rb, Cs) has a 2D square lattice and their various compounds fall in the 0.2–0.25 nm interatomic distance range.25,26 The focus of this molecular dynamics study is to investigate the generality of the behavior of electromelting of nano-confined water in variable slit pores with different lattice geometries under an electric field in the range 0–10 V nm−1.2. Models and methodology
A slit pore is designed by placing two layers of surfaces in parallel separated by a distance H. H, in this work, is in the range 0.77–0.83 nm. Surface particles are arranged according to the different lattice arrangements and lattice constants (see Fig. S1–S3, ESI†). To address the characteristics of the various materials available in nature, we have adopted different surface–fluid interactions. Two distinct interactions are considered to model surface particles. In one case, interactions are taken as those of graphene,27 representing hydrophobic interactions. In the other case, quartz (SiO2) surface interactions are considered, representing hydrophilic interactions.21 The TIP4P/200528 model is used for the water–water interaction. The surface–water interaction parameters are considered according to the scheme of Zangi and Mark,21 and are summarized in Table 1. A cutoff distance of 0.9 nm is set for the Lennard–Jones (LJ) interaction.23 The particle–particle particle–mesh (PPPM) method29 is used for calculating the long-range interactions with a cutoff distance of 1.4 nm. Zangi and Mark21,22 have shown that a bilayer of water forms within the two parallel walls in the range 0.72 ≤ H ≤ 0.85 nm with density variation 0.97–0.87 gm cm−3. Therefore, to investigate the phase transition of water from solid to liquid under an external electric field, we have fixed the number of water molecules at 778, corresponding to an initial density of 0.96 gm cm−3 for H = 0.79 nm, confined between two parallel plates with an area of ∼7.2 × 7.2 nm2 along the x–y plane. The electric field, when applied, is in the z-direction, perpendicular to the surface.23 The equation of motion of the surface particles is not integrated, i.e., surface particles are kept fixed. To imitate the confinement in between two infinite plates, periodic boundary conditions are applied in the x and y-directions, and in the +z direction, a 5 nm vacuum is applied on top of the upper plate and in the −z direction a 5 nm vacuum is used beneath the lower plate to avoid unrealistic interactions.| Surface-type | Oxygen-surface | Hydrogen-surface | ||
|---|---|---|---|---|
| ε (kcal mol−1) | σ (nm) | ε (kcal mol−1) | σ (nm) | |
| Hydrophilic | 0.1986 | 0.3160 | 0.0992 | 0.2840 |
| Hydrophobic | 0.1139 | 0.3352 | 0.0569 | 0.2647 |
All MD simulations are conducted using the LAMMPS package.30 An NPxyT ensemble is utilized. Time integration of the equations of motion is performed at a 300 K temperature and at 1 bar lateral pressure, using the Nosé-Hoover thermostat and a barostat with 1 ps and 2.5 ps relaxation times, respectively.31–33 All of the systems under study have been equilibrated for 5 ns, followed by an additional 10 ns to evaluate structural and dynamic properties, with a time step of 2 fs.
2.1 Analysis
 | (1) |
 | (2) |
| 〈Δxi2 + Δyi2〉 = 4Dxyt, | (3) |
| 〈Δϕi2〉 = 2Dϕt, | (4) |
3. Results and discussion
We first present the effects of pore-size and surface lattice arrangement on the structure of water in extremely narrow pores, in the absence of an electric field. Fig. 1 presents the 2D radial distribution function of oxygen–oxygen for different pore-sizes for different lattice arrangements.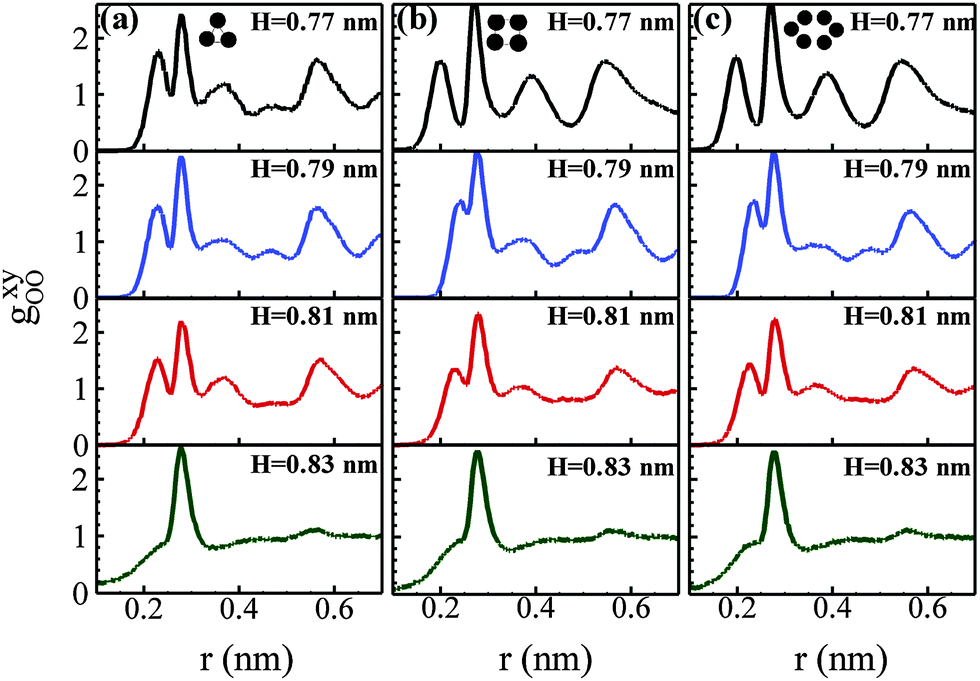 | ||
| Fig. 1 In-plane 2D oxygen–oxygen RDF as a function of pore-size in the absence of an electric field in the hydrophilic confinement of (a) a triangular lattice with a = 0.23 nm, (b) a square lattice with a = 0.17 nm and (c) a hexagonal lattice with a = 0.142 nm. | ||
In the case of a triangular lattice arrangement, multiple peaks in the RDF are observed for H = 0.77–0.81 nm. Similar behaviors are seen for square (Fig. 1b) and hexagonal arrangements of surface particles (Fig. 1c), though ordered behavior is slightly suppressed for H = 0.81 nm. The RDF curves, for pore-size less than 0.81 nm, consist of a split-peak, which is an indication of crystalline phase. As the pore- size increases, the split-peak vanishes, and a dominant single peak appears. The location of the prominent first peak is at 0.28 nm for all cases. We estimate the 2D oxygen–oxygen coordination number (CN) by integrating the RDF up to the minimum of the prominent first peak. For pore-size H = 0.77–0.81 nm, the average coordination number is within the range 4.1 to 4.4 for all surface arrangements. At H = 0.83 nm, the coordination number is high at 4.8, indicating liquid-like behavior. This nature is observed for all of the surface arrangements. The diffusivity plots, shown in Fig. 2a, further corroborate the above findings.
 | ||
| Fig. 2 (a) Variation of lateral diffusion coefficient with pore-size for three different hydrophilic slit pores in the absence of an electric field at 300 K and 1 bar lateral pressure. Error bars are calculated by the block average method. (b) Mean square displacement (MSD) as a function of time in triangular, a = 0.23 nm slit-pores at various pore heights. | ||
The lateral diffusivity in the hydrophilic pores increases from ∼10−8 to ∼10−4 cm2 s−1 with increasing pore-size. The lateral diffusivity (Dxy) of water is on the order of 10−8 for H = 0.77–0.79 nm. The diffusivity is enhanced by 2 to 3 orders with a slight increase in the pore-size, as evident for H = 0.81 and 0.83 nm. The low mobility of water molecules at lower pore-sizes, along with the RDF plots, clearly indicates that there is an ordered network structure among the confined water molecules for low values of H = 0.77–0.79 nm. At higher H values, the MSD starts to diverge, as can be seen for the pore-size H = 0.83 (see Fig. 2b), indicating liquid-like behavior. In contrast to the hydrophilic surface, the hydrophobic surface (graphitic pore) offers around one order higher diffusivity for confined water molecules due to its weaker affinity for water molecules (see Fig. S7, ESI†). Our present study is in line with previous MD simulations.21,23,34 Therefore, we have considered the pore-size H = 0.79 nm where the ordered structure is intact, and have extended our study to different arrangements of surface-particles, namely, square and hexagonal with variable lattice constants. In order to understand the effect of lattice constant on the structure and dynamics of confined water, we choose a fixed pore-size, H = 0.79 nm. In each pore, the lateral diffusivity is calculated when a Fickian regime is achieved, as shown in Fig. 2b for triangular, hydrophilic slit pores (a = 0.23 nm).
Fig. 3 presents the 2D-RDF of O–O in a hydrophilic pore in the absence of an electric field with the variation of lattice constant, a, of 0.211–0.300 nm, 0.160–0.230 nm and 0.142–0.200 nm for triangular, square and hexagonal surface-particle arrangements, respectively.
 | ||
| Fig. 3 Variation of in-plane 2D oxygen–oxygen radial distribution function, at various lattice constants at fixed pore-size (H = 0.79 nm), for (a) triangular, (b) square and (c) hexagonal lattice arrangements under hydrophilic surface–fluid interactions in the absence of an electric field. Snapshots of the corresponding lattice constants are given in insets. | ||
The RDF plots clearly indicate that for a fixed type of lattice arrangement and surface–fluid interaction, the split-peak starts to vanish, and the position of the prominent peak decreases slightly with increase in the lattice constant. For example, with triangular lattice surface particles, the dominant first peak of the in-plane RDF is at 0.28 nm for a = 0.211 and 0.23 nm, but at higher values, a = 0.25 and 0.30 nm, the peak position shifts to 0.27 nm. We observe that the coordination number decreases with increase in lattice constant for all lattice arrangements. In the case of triangular lattice surface particles, the calculated coordination number is around 4.1 for a lattice constant less than 0.25 nm, whereas a lower coordination number (3.8) is seen for 0.3 nm. For the square lattice arrangement, a = 0.16, 0.17 nm and a = 0.20, 0.23 nm CNs are 4.4 and 3.9, respectively. A similar trend is also observed for the hexagonal lattice arrangement. Clearly, at lower lattice constants, the fluid attains an ordered structure that gets destroyed at higher lattice constants. In the case of hydrophobic interactions, the effect of the lattice constant is more dramatic. For example, in the case of a triangular surface (see Fig. S5, ESI†) an increase in a (in nm) from 0.23 to 0.25 or 0.30 decreases the RDF peaks significantly, leading to a disordered structure, as indicated by the single peak in the RDF plot. Therefore, the lower values of lattice constant allow the confined water molecules to stay in an ordered network, whereas the higher values promote disorderedness.
While lattice constant plays an important role, the lattice arrangement can also affect the properties significantly. For example, in the case of a square arrangement with a = 0.23 nm (Fig. 3b), for hydrophilic interactions, the RDF has considerably fewer peaks compared to the number seen for the triangular arrangement (Fig. 3a), signifying a disordered water network. Similar behavior is seen for hydrophobic surfaces (see Fig. S5, ESI†), which is well supported by the lateral diffusivity behavior (see Fig. S7, ESI†). The low mobility of water molecules at the lower lattice constant clearly indicates that there is an ordered network structure among the confined water molecules for low values of the lattice constant. Furthermore, the ordered water network becomes more prominent at lower values of lattice constant when the lattice arrangement varies in the order: triangular → square → hexagonal. These observations are due to the number of interaction sites (i.e., the number of surface-particles), which is directly related to the lattice constant and arrangement of the surface atoms. Since, for a given lattice constant, the surface-atom-density decreases in the order: triangular → square → hexagonal, the water network becomes less prominent in the same order, which affects accordingly the configuration energy of water in the same order (see Table S1, ESI†). Furthermore, for a fixed lattice constant (a = 0.2 nm), in the case of square lattice surfaces, the ordered structure is conspicuous under hydrophilic confinement, whereas it is less prominent in the case of hydrophobic confinement. Therefore, it is important to note that the ordered network of water under confinement in the absence of an electric field depends on the lattice properties, as well as on the surface–fluid interaction. From the RDF and diffusivity results, we conclude that the ordered network of water exists for a narrow range of lattice constants. To this end, we have taken a = 0.23, 0.17 and 0.142 nm for triangular, square and hexagonal, respectively, as representative lattice constants for our electric field-induced phase transition studies.
Now, we turn our attention to explore the effects of the electric field on the structural and dynamic properties of water under nano-confinement. To explore the effect of electric field on the arrangement of the water molecules under various nano-confinements, we have calculated the structural properties such as the in-plane bond order parameters and the radial distribution function. Fig. 4 presents the bond order parameters with the variation of electric field (0–10 V nm−1) for different lattice properties and surface–fluid interactions.
 | ||
| Fig. 4 Variation of order parameters with electric field in pores of (a) triangular, (b) square and (c) hexagonal lattices with a = 0.23, 0.17 and 0.142 nm, respectively, under hydrophilic surface–fluid interaction. Error bars are smaller than the symbol size. The corresponding RDFs are shown in panel d–i. Snapshots at different electric fields are shown in the insets. The grids are connecting lines among atoms to show the existence of symmetries. | ||
In the case of the triangular arrangement (a = 0.23 nm), at E = 0 V nm−1, the values of Ψ4 and Ψ6 are 0.53 and 0.36, respectively, under hydrophilic confinement. The in-plane order parameter value clearly indicates that the water molecules have a propensity to remain in the square symmetry, though some water molecules remain in triangular symmetry in the confinement of the triangular lattice arrangement. The corresponding snapshots (Fig. 4d) also show the existence of symmetries within a water layer for the triangular lattice. On the other hand, for the square (a = 0.17 nm) and hexagonal (a = 0.142 nm) hydrophilic confinements, the higher values of Ψ4 (>0.5) compared to Ψ6 (<0.05) indicate that the water molecules prefer to remain in the square symmetry. The existence of such ordered structures is corroborated by the RDF plots as shown in Fig. 4d–f. Furthermore, analysis of the peak locations suggests that the lattice constant of the surface is not commensurate with the structure of the water layer. We observed both the square and hexatic symmetry for the triangular substrate at lower electric field. This result implies that the triangular confined system might have some kind of geometric preference for both symmetries. The snapshots shown in Fig. S6 (ESI†) clearly depict oxygen atoms in square and hexatic symmetries. To address this conjecture, we evaluate the average configuration energy for different confined systems. The average configurational energy of confined water is approximately −18.54 kcal mol−1 and −16.52 kcal mol−1 for the square and hexagonal arrangements, respectively (see Table S1, ESI†), in the absence of an electric field. Whereas for the triangular arrangement, the energy value is approximately −13.21 kcal mol−1, which is closer to the configurational energy values of the bulk water under ambient conditions.40 The higher configuration energy, due to the large oxygen–oxygen distance in triangular confinement, arises due to the low surface density of the surface particles. This allows defects in the system (see Fig. S6, ESI†), and thus promotes both forms of water symmetry. This is also evident from the rdf plots in Fig. 4d–f, which show the location of the first peak at 0.398 nm, 0.375 nm, 0.375 nm for triangular (a = 0.23 nm), square (a = 0.17 nm) and hexagonal (a = 0.142 nm), respectively. With increase in the electric field, both the Ψ4 and Ψ6 values for the triangular arrangement, and only the Ψ4 values for the square and hexagonal arrangements decrease sharply, indicative of electromelting behavior. At E = 10 V nm−1, the height of the dominant peak decreases, and the RDF approaches unity, as shown in Fig. 4g–i. It should be noted that there is a small variation in the lattice constant under the influence of the electric field (see Table S2, ESI†). In addition, to check the sensitivity of the LJ parameters, ε and σ of substrate–oxygen interactions, towards the order parameters, we have varied ε and σ by ±5%. Typically, the sensitivity of σ is greater than that of ε at lower electric field, whereas at higher electric field the order parameters are insensitive to both LJ parameters.
Fig. 5 presents the lateral diffusivity of water under different arrangements of surface particles and surface–fluid interactions with the electric field varying from 0 to 10 V nm−1. The figure clearly indicates a lower value of diffusivity of ∼10−8 cm2 s−1 for the hydrophilic surface and ∼10−6 cm2 s−1 for the hydrophobic surface, representing the order-like structures when the applied electric field is in the range 0 to ∼3.5 V nm−1. Further increase in the electric field increases the diffusivity to ∼10−5 cm2 s−1, indicative of the electromelting or disordered state. In the lower range of electric field, the lateral diffusivity is higher in hydrophobic pores in comparison to hydrophilic pores. During electromelting, the effect of electric field on the diffusivity of water in a hydrophilic nano-pore displays interesting behavior, where the electromelted state (Dxy ∼ 10−5 cm2 s−1) is achieved much earlier for the triangular (E = 3.5 V nm−1) and hexagonal (E = 5 V nm−1) lattice surface in comparison to that of the square (E = 7 V nm−1) lattice surface. It is noted that once the confined water is electromelted, the overall behavior of diffusivity with further increase in electric field is similar for both the hydrophilic and hydrophobic nano-pores. Furthermore, at low electric field, the variation in the lateral diffusivity (∼10−8 → ∼10−7 cm2 s−1) demonstrates that the surface particle arrangement can have a drastic influence on the diffusivity in the hydrophilic or hydrophobic confinements. Importantly, our results are also in line with a previous MD study23 in terms of the incremental jump in lateral diffusivity value (i.e., electromelting) in various confinements under atmospheric conditions. Next, we evaluate the rotational diffusivity to understand the effect of electric field under different nano-confinements. Fig. 6 presents the rotational diffusivity vs. electric field for a hydrophilic surface in various lattice arrangements. At E = 0 V nm−1, the rotational diffusivity is of the same order (∼10−5 rad2 ps−1) in all three cases. The values of the rotational diffusivity are of the order 10−5 rad2 ps−1 when the electric field is in the range 0–1 V nm−1. With further increase in the electric field, the rotational diffusivity values decrease from ∼10−5 rad2 ps−1 to ∼10−7 rad2 ps−1, which are independent of the surface–fluid interactions and the lattice properties (rotational diffusivity for the hydrophobic surface is not shown). The drop in the rotational diffusivity is due to the reorientation of water molecules in a fashion such that their dipoles are aligned in the direction of the applied electric field. It should be noted that the rotational diffusivity decreases monotonously with increasing electric field, which is in contrast to the behavior seen for the lateral diffusivity, where lateral diffusivity first increases and remains constant for a range of E. On the other hand, the rotational diffusivity continuously decreases even during the electromelting stage.
 | ||
| Fig. 5 Variation of lateral diffusion coefficient in confinement as a function of electric field at 0.79 nm pore-size in different lattice arrangements of the surface particles under hydrophilic and hydrophobic surface–fluid interactions. | ||
 | ||
| Fig. 6 Variation of rotational diffusivity with electric field in various hydrophilic nanopores. Error bars are calculated by the block average method. | ||
To explore the variation of ordering in the water-layers, we have evaluated the average number of hydrogen bonds (HBs) per water molecule (nHB) under different confinements with the electric field varying in the range 0–10 V nm−1. Fig. 7 presents the effect of electric field on the intralayer and interlayer hydrogen bond network within hydrophilic and hydrophobic nanopores of various lattice arrangements. The total number of hydrogen bonds (intralayer HB + interlayer HB) in all confinements is less than the reported value for bulk water under ambient conditions.40
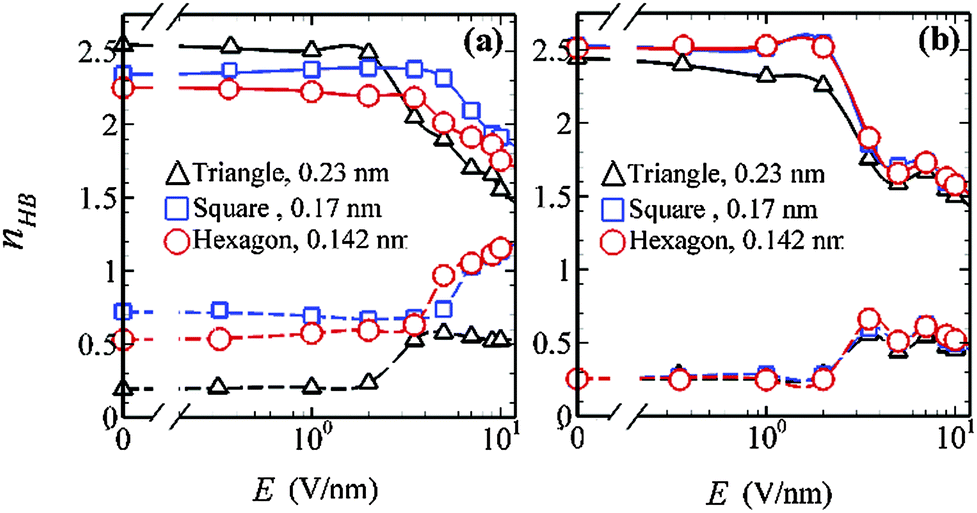 | ||
| Fig. 7 Variation of average number of intralayer and interlayer hydrogen bonds per water molecule as a function of electric field for triangular, square and hexagonal lattices with a = 0.23, 0.17 and 0.142 nm, respectively. Panels (a) and (b) are for hydrophilic and hydrophobic surface–fluid interactions, respectively. Solid lines are average intralayer HBs and dashed lines are average interlayer HBs. Error bars are smaller than the symbol size. | ||
In hydrophilic confinement, the average number of hydrogen bonds within a single layer (intralayer HBs) is ∼2.5, indicating the presence of an in-plane ordered network of water in the 0 to ∼3.5 V nm−1 electric field range. With further increase in the electric field, intralayer HBs decrease due to disruption of the HB network within the plane. However, the average number of interlayer HBs increases. This suggests that the intralayer HBs of the ordered water layer are largely interrupted with increasing electric field, leading to electromelting. At higher values of electric field (E = 10 V nm−1), the dipoles of the water molecules become oriented in the direction of the applied electric field. Thus, the average number of intralayer hydrogen bonds decreases, whereas the interlayer hydrogen bonds increase, signifying a stronger interlayer connectivity at high E values. Furthermore, to see the onset of the melting behavior with increasing electric field, we have estimated the average number of intralayer and interlayer HBs within a 0 ps to 50 ps time scale. Fig. 8 represents the variation of the average number of intralayer and interlayer HBs with respect to time at different electric fields (E = 1, 5 and 7 V nm−1) for triangular, hydrophilic (a = 0.23 nm and H = 0.79 nm) confinement.
 | ||
| Fig. 8 Instantaneous number of hydrogen bonds per water molecule as a function of time at different electric fields for (a) intralayer HBs and (b) interlayer HBs. | ||
The intralayer and interlayer HBs do not change much at low values of the electric field (E = 1 V nm−1), which indicates strong order in the system. On the other hand, at higher electric field values (E = 5 and 7 V nm−1), the number of intralayer HBs drops sharply within 10 ps, as evident from Fig. 8a. Thus, the breaking of the intra-layer HB network indicates that melting has started at the solid–liquid interface with increasing electric field. Interlayer hydrogen bonding, on the other hand, increases slightly at low electric field, as seen for E = 5 V nm−1 compared to E = 1 V nm−1. This is primarily because water molecules tend to bind with molecules with other layers upon loss in intra-layer hydrogen bonding to minimize the configuration energy. However, further increase in the electric field overcomes such a tendency, as seen for E = 7 V nm−1. Similar behavior is also observed in the case of square and hexagonal lattice arrangements.
Finally, we have explored the effect of pore-size and lattice constant on lateral diffusivity for a particular lattice arrangement. Fig. 9a presents the variation in the lateral diffusivity of water under square (a = 0.17 nm) hydrophilic confinement with the electric field for different pore-sizes.
 | ||
| Fig. 9 Lateral diffusivity as a function of applied electric field with square lattice arrangement of the surface particles (a) at various pore-sizes, with lattice constant a = 0.17 nm under hydrophilic confinement, and (b) at various lattice constants with pore-size H = 0.79 nm. | ||
At H = 0.77 nm, the lateral diffusivity jumps from 10−8 → 10−6 cm2 s−1 during electromelting within the electric field range: 3.5–10 V nm−1. The incremental jump is significant (10−8 → 10−5 cm2 s−1) and takes place in a shorter electric field range, 3.5–7 V nm−1 at 0.79 nm pore-size. The jump in lateral diffusivity during electromelting is smaller at H = 0.81 nm and ultimately vanishes at H = 0.83 nm. This is mainly due to the change in the structural property from a crystalline phase to a liquid phase with increasing pore-size (see Fig. 1), which reduces the difference between their diffusivity values. Similar behavior is also observed in triangular (a = 0.23 nm) and hexagonal (a = 0.142 nm) lattice arrangements under hydrophilic surface–fluid interactions (see Fig. S8a and c, ESI†). Fig. 9b presents the variation of the lateral diffusivity with electric field at different lattice constants in a square slit pore with 0.79 nm pore-size. Under hydrophilic surface–fluid interactions, lateral diffusivity has a three orders of magnitude incremental jump (10−8 → 10−5 cm2 s−1) in the 3.5–5 V nm−1 electric field range at a = 0.180 nm. At a = 0.215 nm, the incremental jump in lateral diffusivity is one order of magnitude smaller (10−7 → 10−5 cm2 s−1), indicating that larger lattice constants promote melting behavior with an earlier approach to the liquid-like structure with increasing electric field. All else being equal, electromelting is enhanced under hydrophobic interactions, with a smaller incremental jump in lateral diffusivity compared to that seen in hydrophilic slit pores. A similar dependence of lateral diffusivity on lattice constants is observed in triangular and hexagonal slit pores (see Fig. S8b and d, ESI†).
4. Conclusions
We have reported the electric field-induced phase transition of water in various confinements with different lattice properties and surface–fluid interactions. Electromelting is attributed to the success of electric field induced disordering over surface induced ordering.23 A strong influence of pore-size, lattice constants, lattice arrangements and interaction nature on the electromelting (E ≤ ∼7 V nm−1) behavior has been observed. The electromelted region (higher lateral diffusivity regions in the presence of an electric field) shrinks with decreasing pore-size. We note that the lattice constant from 0.1 to 0.3 nm with various surface-particle arrangements closely resembles many metals and non-metal surfaces. However, based on the current results the electromelting of nanoconfined fluids may vary in the case of polar or charged surfaces due to the simplified model considered in this work. Hence, the precise nature of electromelting behavior depends strongly on the lattice properties, pore-size and the strength and nature of the substrate–fluid interactions.Acknowledgements
This work was supported by the Department of Science and Technology, Govt of India. Computational resources were provided by the HPC cluster of the Computer Center (CC), Indian Institute of Technology Kanpur.Notes and references
- D. Ehre, E. Lavert, M. Lahav and I. Lubomirsky, Science, 2010, 327, 627 CrossRef PubMed.
- S. O. Diallo, E. Mamontov, W. Nobuo, S. Inagaki and Y. Fukushima, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 86, 021506 CrossRef CAS.
- W.-K. Lee, S. Tsoi, K. E. Whitener, R. Stine, J. T. Robinson, J. S. Tobin, A. Weerasinghe, P. E. Sheehan and S. F. Lyuksyutov, Nano Res., 2013, 6, 767–774 CrossRef CAS.
- E.-M. Choi, Y.-H. Yoon, S. Lee and H. Kang, Phys. Rev. Lett., 2005, 95, 085701 CrossRef.
- M. Gavish, J.-L. Wang, M. Eisenstein, M. Lahav and L. Leiserowitz, Science, 1992, 256, 815–818 CAS.
- P. V. Hobbs, Ice Physics, Clarendon Press, Oxford, 1974 Search PubMed.
- X. X. Zhang, Y. J. Lu and M. Chena, Mol. Phys., 2013, 111, 3808–3814 CrossRef CAS PubMed.
- W. Cantrell and G. E. Ewing, J. Phys. Chem. B, 2001, 105, 5434–5439 CrossRef CAS.
- F. G. Alabarse, J. Haines, O. Cambon, C. Levelut, D. Bourgogne, A. Haidoux, D. Granier and B. Coasne, Phys. Rev. Lett., 2012, 109, 035701 CrossRef CAS.
- D. Asay and S. H. Kim, J. Chem. Phys., 2006, 124, 174712 CrossRef PubMed.
- A. B. Farimani and N. R. Aluru, J. Phys. Chem. B, 2011, 115, 12145–12149 CrossRef PubMed.
- Y. Maniwa, H. Kataura, M. Abe, A. Udaka, S. Suzuki, Y. Achiba, H. Kira, K. Matsuda, H. Kadowaki and Y. Okabe, Chem. Phys. Lett., 2005, 401, 534–538 CrossRef CAS PubMed.
- A. L. Ferguson, N. Giovambattista, P. J. Rossky, A. Z. Panagiotopoulos and P. G. Debenedetti, J. Chem. Phys., 2012, 137, 144501 CrossRef PubMed.
- J. C. Rasaiah, S. Garde and G. Hummer, Annu. Rev. Phys. Chem., 2008, 59, 713–740 CrossRef CAS PubMed.
- N. Severin, P. Lange, I. M. Sokolov and J. P. Rabe, Nano Lett., 2012, 12, 774–779 CrossRef CAS PubMed.
- M. C. Gordillo and J. Marti, J. Phys.: Condens. Matter, 2010, 22, 284111 CrossRef CAS PubMed.
- M. C. Gordillo and J. Marti, J. Phys. Chem. B, 2010, 114, 4583–4589 CrossRef CAS PubMed.
- Z. Qian and G. Wei, J. Phys. Chem. A, 2014, 118, 8922–8928 CrossRef CAS PubMed.
- Y. He, G. Sun1, K. Koga and L. Xu, Sci. Rep., 2014, 4, 1–5 Search PubMed.
- H. Mosaddeghi, S. Alavi, M. H. Kowsari and B. Najafi, J. Chem. Phys., 2012, 137, 184703 CrossRef PubMed.
- R. Zangi and A. E. Mark, J. Chem. Phys., 2003, 119, 1694–1700 CrossRef CAS PubMed.
- R. Zangi and A. E. Mark, Phys. Rev. Lett., 2003, 91, 025502 CrossRef.
- H. Qiu and W. Guo, Phys. Rev. Lett., 2013, 110, 195701 CrossRef.
- A. M. Saitta, F. Saija and P. V. Giaquinta, Phys. Rev. Lett., 2012, 108, 207801 CrossRef.
- Chemistry of the Elements, ed. N. N. Greenwood and A. Earnshaw, Butterworth-Heinemann, Oxford, 2nd edn, 1997 Search PubMed.
- Structural Inorganic Chemistry, ed. A. F. Wells, Oxford University Press, 3rd edn, 1962 Search PubMed.
- N. Patra, B. Wang and P. Kral, Nano Lett., 2009, 9, 3766–3771 CrossRef CAS PubMed.
- J. L. F. Abascal and C. Vega, J. Chem. Phys., 2005, 123, 234505 CrossRef CAS PubMed.
- R. W. Hockney and J. W. Eastwood, Computer Simulation using Particles, IOP Publishing Ltd., Bristol, England, 1988 Search PubMed.
- S. J. Plmimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef PubMed.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef.
- J. M. Thijssen, Computational Physics, Cambridge University Press, Cambridge, 2nd edn, 2007 Search PubMed.
- G. Rosenthal and S. H. L. Klapp, Int. J. Mol. Sci., 2012, 13, 9431–9446 CrossRef CAS PubMed.
- C. K. Das and J. K. Singh, Theor. Chem. Acc., 2013, 132, 1351 CrossRef.
- C. Ghatak and K. G. Ayappa, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2001, 64, 051507 CrossRef CAS.
- S. Han, M. Y. Choi, P. Kumar and H. E. Stanley, Nat. Phys., 2010, 6, 685–689 CrossRef CAS PubMed.
- A. Kusumi, Y. Sako and M. Yamamoto, Biophys. J., 1993, 65, 2021–2040 CrossRef CAS.
- D. Rozmanov and P. G. Kusalik, J. Chem. Phys., 2012, 136, 044507 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp03778f |
| This journal is © the Owner Societies 2015 |