
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
SEI-component formation on sub 5 nm sized silicon nanoparticles in Li-ion batteries: the role of electrode preparation, FEC addition and binders†
Tony
Jaumann
*a,
Juan
Balach
 a,
Markus
Klose
a,
Steffen
Oswald
a,
Ulrike
Langklotz
b,
Alexander
Michaelis
b,
Jürgen
Eckert
ab and
Lars
Giebeler
ab
a,
Markus
Klose
a,
Steffen
Oswald
a,
Ulrike
Langklotz
b,
Alexander
Michaelis
b,
Jürgen
Eckert
ab and
Lars
Giebeler
ab
aLeibniz Institute for Solid State and Materials Research (IFW) Dresden e.V., Institute for Complex Materials, P.O. Box 27 01 16, D-01171 Dresden, Germany. E-mail: t.jaumann@ifw-dresden.de; Fax: +49 351 4659 307; Tel: +49 351 4659 307
bTechnische Universität (TU) Dresden, Institut für Werkstoffwissenschaft, Helmholtzstraße 7, D-01069 Dresden, Germany
First published on 27th August 2015
Abstract
Silicon is a promising negative electrode for secondary lithium-based batteries, but the electrochemical reversibility of particularly nanostructured silicon electrodes drastically depends on their interfacial characteristics, commonly known as the solid electrolyte interface (SEI). The beneficial origin of certain electrolyte additives or different binders is still discussed controversially owing to the challenging peculiarities of interfacial post-mortem investigations of electrodes. In this work, we address the common difficulties of SEI investigations of porous silicon/carbon nanostructures and study the addition of a fluoroethylene carbonate (FEC) as a stabilizing additive as well as the use of two different binders, carboxymethyl cellulose/styrene-butadiene rubber (CMC/SBR) and polyacrylic acid (PAA), for the SEI formation. The electrode is composed of silicon nanocrystallites below 5 nm diameter allowing a detailed investigation of interfacial characteristics of silicon owing to the high surface area. We first performed galvanostatic long-term cycling (400 times) and carried out comprehensive ex situ characterization of the cycled nanocrystalline silicon electrodes with XRD, EDXS, TEM and XPS. We modified the preparation of the electrode for post-mortem characterization to distinguish between electrolyte components and the actual SEI. The impact of the FEC additive and two different binders on the interfacial layer is studied and the occurrence of diverse compounds, in particular LiF, Li2O and phosphates, is discussed. These results help to understand general issues in SEI formation and to pave the way for the development of advanced electrolytes allowing for a long-term performance of nanostructured Si-based electrodes.
1. Introduction
Lithium-ion batteries (LIBs) are currently the first choice for portable electronic devices due to their high energy density and good reversibility. For a significant further enhancement of the energy density, the development of new electrode materials with higher capacity is inevitable.1,2 Silicon is a promising candidate to replace graphite as a commonly used anode in LIBs since it offers capacities up to 10 times higher than graphite and a low discharge potential. In addition, silicon showed high potential as a safe anode for future LIBs.3–6 A critical aspect concerning silicon anodes is the so called “solid electrolyte interface” (SEI) which is a passive surface film typically formed on anode materials by decomposition of the electrolyte components during the first cycle. If the SEI is stable, it prevents further decomposition of the electrolyte and allows good Li-ion conductivity. For instance, a graphite-based SEI formed during cycling in ethylene carbonate shows these positive properties which are a major reason for the extremely high reversibility of graphite as an anode.7 This passive layer is supposed to be insufficiently stable on silicon due to continuous volume expansion and contraction of up to 300% during cycling.8 The reiterating volume change causes a continuously growing passive interface which electrically isolates the active silicon species and results in fast capacity fading of silicon nanostructures whereas bulk silicon rather suffers from pulverization during lithiation. There are several strategies to stabilize the SEI on silicon. Liu et al. proposed a carbon encapsulation of silicon nanoparticles as an artificial SEI with void space between silicon and carbon to compensate the volume expansion.9 Silicon nanoparticles below 20 nm seem to stabilize the SEI10 due to highly reactive surface atoms which lead to the development of high-performance silicon nanostructures.11,12 Another promising and facile approach that aims to stabilize the SEI formation on silicon is the use of electrolyte additives and modified conductive salts.13–15 These modifications can improve the reversibility exceptionally owing to the formation of a stable SEI during the initial cycles. One of the most promising candidates is fluoroethylene carbonate (FEC).13,16,17 This compound drastically improves the cycle stability without complexly designed silicon structures. Intensive research has been carried out to determine the origin for the stabilization in order to design more efficient additives.18 In this regard silicon nanowires,19 thin silicon films13,15,20–22 and silicon nanoparticles23–25 with 50–100 nm in size have been examined. As the first group Choi et al. attributed the enhanced stability to a pronounced formation of LiF and Si–Fx surface groups originating from the decomposition of FEC,13 which was recently confirmed.26 Other studies indicate the deposition of a smooth polycarbonate or a polymer-like layer on silicon as a main factor for stabilization.18,20 A major reason for partially controversial results is found in the large number of parameters affecting SEI formation. Electrochemical preparation,27 current rates28 and potential windows for galvanostatic cycling significantly influence the SEI formation,25 but more importantly, the procedures made for enabling post-mortem analyses.27 The washing procedure of the electrode prior to analysis can affect the surface composition, but it is necessary to remove electrolyte components. The SEI on silicon typically consists of a complex structure with an upper organic layer (semi-organic lithium carbonates) and a lower inorganic matrix (Li2CO3, LiF, etc.).24,25,29 In this regard, Tasaki et al. examined the solubility of different SEI components and showed that the organic salts can dissolve.30 The dissolution of compounds may cause a huge variety of results in the literature. For example, the concentration of phosphorus, the central atom of the complex anion of the electrolyte’s conductive salt, determined by X-ray photoelectron spectroscopy (XPS) on thin silicon films after cycling varies from 8 at% to 0.1 at% even under similar test conditions.20,21 It is often ambiguous what is part of the SEI and what are just residues of electrolyte components like conductive salt LiPF6 or solid co-solvents (i.e. ethylene carbonate). In addition, the role of the binder in the surface chemistry of silicon-based electrodes is not well investigated. It is believed that the functional groups of the binder (such as RCOOH) undergo chemical reaction with the silicon surface which should affect the surface chemistry.31 To avoid these interactions binder-free electrodes are used for interfacial investigation, but their practical application is not realistic yet.26,32,33 Therefore, an investigation with different binders will help to unveil their role in silicon-based anodes.In this study, we highlight three current and delicate issues of SEI investigations for nano-silicon electrodes in Li-ion batteries: (I) the washing procedure for reliable post-mortem electrode characterization is addressed which is based on a short sonication treatment verified as an effective non-destructive procedure to remove only electrolyte components from the surface. (II) We focus on the superior electrochemical performance due to the addition of FEC and discuss the role of interfacial compounds in silicon electrodes, in particular Li2O, LiF and phosphates formed during electrochemical cycling. (III) The significant effect of two well-known binders for silicon-based electrodes, namely carboxymethyl cellulose/styrene-butadiene rubber (CMC/SBR)31 and polyacrylic acid (PAA),34 on SEI formation on nanoparticulate silicon is fundamentally investigated. As the Si anodes prepared with the help of the aforementioned binders show similar electrochemical performance and characteristics in the pristine state, post-mortem characterization identifies different organic surface species induced by just changing the binder. A model for the SEI formation is proposed and major challenges obtained in comparing the literature results under unique experimental conditions are discussed. We highlight the SEI properties and the SEI formation mechanism on silicon nanocrystallites with 2–5 nm in size embedded in a porous carbon scaffold after 400 galvanostatic cycles at high current rates for example for highly reversible and highly stable nanoparticulate Si-based negative electrode materials.
2. Experimental
2.1 Preparation of a silicon–carbon nanocomposite
A silicon–carbon nanocomposite was prepared according to a recently published synthesis procedure.12 Briefly, a hydrogen silsesquioxane (HSQ) precursor was produced by polycondensation of trichlorosilane with water in the presence of a surfactant under an inert gas atmosphere. The dried HSQ precursor was annealed at 1100 °C which causes a transformation into a silica matrix with embedded silicon nanocrystallites of 2–5 nm in size determined by TEM and XRD.12 After annealing, the composite was wrapped into carbon through carbonization of sucrose and finally etched with hydrofluoric acid to remove the silica matrix. The porous silicon–carbon nanocomposite (nc-Si@C) was dried under vacuum.2.2 Electrochemical testing
A water-based slurry (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 = composite:Super P (TIMCAL):CMC/SBR (1:1 m/m)) was prepared in 1 M HCl by using a swing mill and sonication. The temperature of the slurry should not exceed 25 °C in order to avoid oxidation. The electrodes were prepared by drop coating onto 12 mm copper discs (9 μm thickness, MTI Corporation) and subsequently dried under vacuum to minimize oxidation. Ethanol was used as solvent for the PAA binder (Sigma-Aldrich, Mw ∼ 450000) to prepare the slurry (8:1:1 = composite:Super P:PAA). The final electrode was prepared by blade coating on copper foil and 12 mm copper discs were punched out. The mass loading was 2 ± 0.4 mg per electrode with about 33 wt% of total silicon on the electrode and a dried electrode thickness of 100 ± 9 μm. The electrodes were transferred into a glove box filled with argon (H2O < 1 ppm, O2 < 0.1 ppm) and finally dried at 100 °C in a vacuum overnight before assembling Swagelok test cells. We used 250 μl of the electrolyte and a glass fiber (Whatman) as a separator for each cell. 1 M LiPF6 in ethylene carbonate (EC):dimethyl carbonate (DMC) with 1:1 v/v (LP30, Selectilyte BASF) was used as an additive-free electrolyte reference. The electrolytes containing the additive FEC (>99%, Solvay Chemicals) are composed of DMC:EC:additive (2:1:1 v/v/v) with 1 M LiPF6 (>99.9%, <20 ppm water, ABCR Chemicals). All chemicals were used as received without further purification. For the counter electrode metallic lithium foil (Chempur, 250 μm thickness) was applied. Galvanostatic cycling was carried out between 0.01–1.2 V vs. Li/Li+ with a BaSyTec cell test system. All measurements were climatized at 25 °C. The specific capacity and current rates were calculated based on the mass of silicon. Investigations of the electrolyte decomposition were carried out in a three-electrode Swagelok cell, using lithium as counter and reference electrode and silicon–carbon nanocomposites as working electrodes. The experiment consisted of repetitive potentiostatic polarizations to 0.84 V vs. Li/Li+ for a duration of one hour, and subsequent measurements of the open circuit potential over four hours.
:1:1 = composite:Super P (TIMCAL):CMC/SBR (1:1 m/m)) was prepared in 1 M HCl by using a swing mill and sonication. The temperature of the slurry should not exceed 25 °C in order to avoid oxidation. The electrodes were prepared by drop coating onto 12 mm copper discs (9 μm thickness, MTI Corporation) and subsequently dried under vacuum to minimize oxidation. Ethanol was used as solvent for the PAA binder (Sigma-Aldrich, Mw ∼ 450000) to prepare the slurry (8:1:1 = composite:Super P:PAA). The final electrode was prepared by blade coating on copper foil and 12 mm copper discs were punched out. The mass loading was 2 ± 0.4 mg per electrode with about 33 wt% of total silicon on the electrode and a dried electrode thickness of 100 ± 9 μm. The electrodes were transferred into a glove box filled with argon (H2O < 1 ppm, O2 < 0.1 ppm) and finally dried at 100 °C in a vacuum overnight before assembling Swagelok test cells. We used 250 μl of the electrolyte and a glass fiber (Whatman) as a separator for each cell. 1 M LiPF6 in ethylene carbonate (EC):dimethyl carbonate (DMC) with 1:1 v/v (LP30, Selectilyte BASF) was used as an additive-free electrolyte reference. The electrolytes containing the additive FEC (>99%, Solvay Chemicals) are composed of DMC:EC:additive (2:1:1 v/v/v) with 1 M LiPF6 (>99.9%, <20 ppm water, ABCR Chemicals). All chemicals were used as received without further purification. For the counter electrode metallic lithium foil (Chempur, 250 μm thickness) was applied. Galvanostatic cycling was carried out between 0.01–1.2 V vs. Li/Li+ with a BaSyTec cell test system. All measurements were climatized at 25 °C. The specific capacity and current rates were calculated based on the mass of silicon. Investigations of the electrolyte decomposition were carried out in a three-electrode Swagelok cell, using lithium as counter and reference electrode and silicon–carbon nanocomposites as working electrodes. The experiment consisted of repetitive potentiostatic polarizations to 0.84 V vs. Li/Li+ for a duration of one hour, and subsequent measurements of the open circuit potential over four hours.
2.3 Characterization of morphology
Prior to post-mortem characterization, the Swagelok cells were disassembled after 400 cycles in the glove box and the silicon electrodes were washed in 5 ml DMC (<20 ppm water, Selectilyte BASF) twice and once in 5 ml DMC in a sonication bath for 5 min, if not otherwise mentioned. For sonication, the samples were sealed in a container to avoid exposure to air/water and put into a continuous ultrasonic cleaner (VWR symphony). After sonication, the samples were kept overnight for sedimentation of the dispersed material, in order to remove the solvent. The electrodes were finally dried under vacuum for 1 h. The preparation for each characterization method was done in the glove box in order to avoid contact with air or moisture. X-ray powder diffraction (XRD) experiments were performed in transmission geometry with Cu Kα1 radiation on an STOE Stadi P diffractometer with a curved Ge(111) crystal monochromator and a 6°-position sensitive detector. The sample was pressed between Kapton tape to avoid exposure to air during scanning. The scan range was 10° < 2θ < 90° with a step size of Δ2θ = 0.01° with three repetitions. Rietveld analyses were carried out assuming isotropic crystallite size distribution. Scanning electron microscopy (SEM) was realized with a Gemini 1530 from LEO equipped with a Bruker EDXS detector. TEM was performed using a Tecnai F30 equipped with a field emission gun and operated at 300 kV. A dispersion of the electrode material was dropped onto a copper grid with a lacey carbon film. During sample transfer into the electron microscope the exposure to air was minimized, but could not be completely excluded. X-ray photoemission spectroscopy (XPS) was carried out using a Physical Electronics PHI 5600 CI system with Mg Kα radiation (350 W) at a pass energy of 29 eV and a step size of 0.1 eV. The used Mg source (1253.6 eV) causes lower charging effects than a commonly monochromatic Al source (1486.6 eV) allowing a precise investigation of insulating SEI components. A special transfer chamber was used to prevent air exposure. The binding energies were normalized to LiF F 1s (685.5 eV) to correct for surface charging effects.35 The pristine electrode was normalized to carbon C 1s (284.5 eV). Elemental concentrations from the XP spectra were calculated using standard single-element sensitivity factors. The core level signals were fitted with a Gaussian function (MagicPlot Software) using a basic linear background after normalization to 1. Ar+ sputtering was performed at 3.5 keV corresponding to 3.5 nm min−1 for SiO2. The silicon content in the nc-Si@C composite was determined by thermogravimetry (Netzsch Jupiter STA 449C) after combustion in synthetic air at 900 °C (10 °C min−1 heating rate). From the amount of residual silica the silicon-to-carbon ratio was calculated.3. Results and discussion
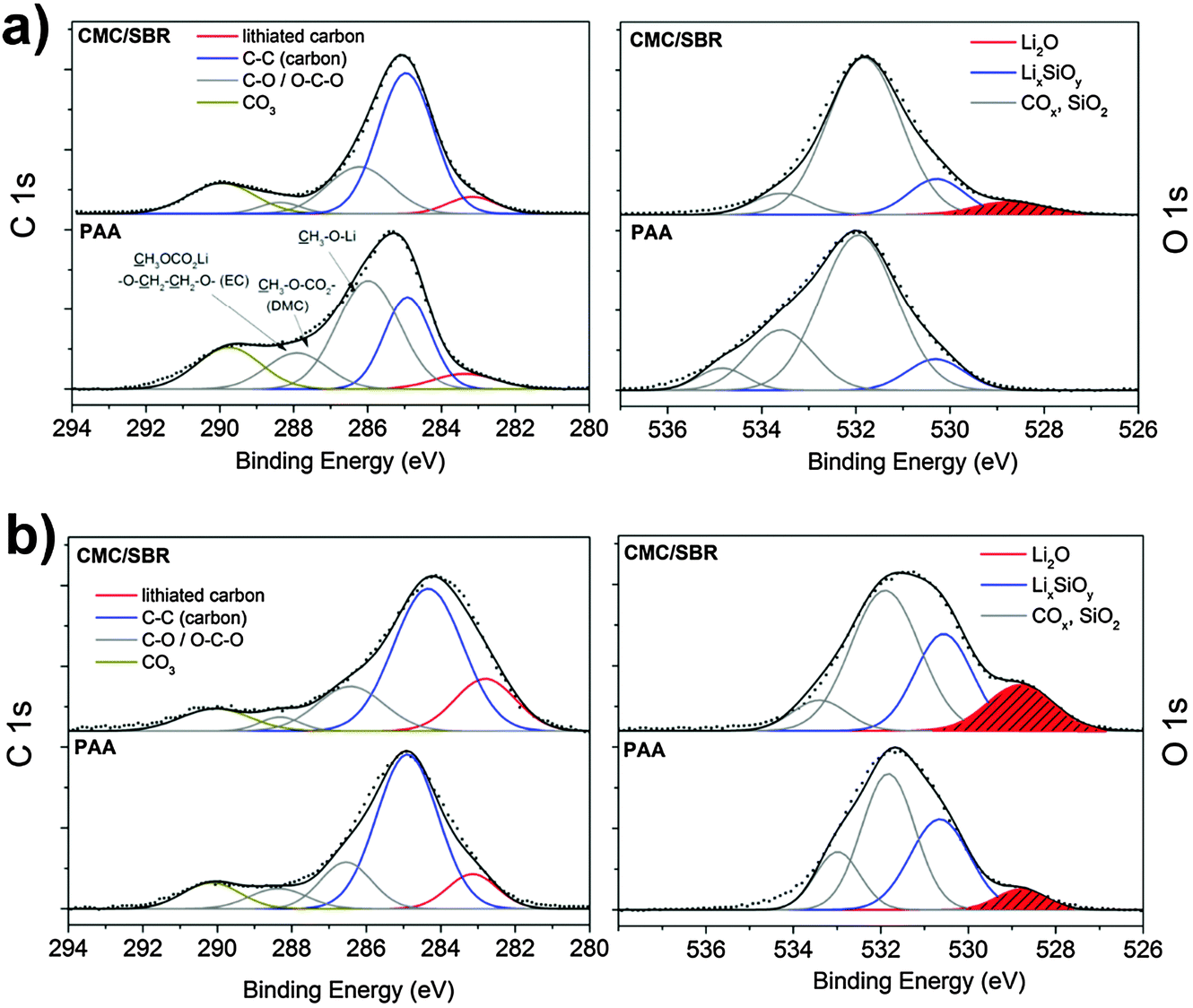
3.1 Properties of the pristine electrode
Structure and electrochemical performance of the porous silicon–carbon nanocomposite was extensively studied in our recently published report.12 Herein, we address the surface properties of silicon after slurry-based electrode preparation which is crucial to understand the SEI formation on electrode materials in Li-ion batteries. In general, the hydride-terminated silicon nanoparticles at hand are meta-stable and slowly oxidize in air due to omnipresent radicals.36 Thus, the surface properties of the silicon nanoparticles can change during the slurry-based electrode preparation in ambient atmosphere. To get an impression on the reactivity, surface species and structure of the nanoparticulate silicon are primarily characterized by XPS and XRD in order to evaluate structural changes during electrode preparation with the two different binders. The results are displayed in Fig. 1a which shows the typical X-ray diffraction pattern of cubic silicon. Data analysis according to the Rietveld method proves the presence of silicon nanocrystallites of less than 5 nm in size after preparation with water- (CMC/SBR) and ethanol-based (PAA) slurry. This observation is in agreement with the results for the as-synthesized nanocomposite.12 The surface species were identified by XPS (Fig. 1b). In both samples, similar surface composition is observed as expected (Table S1, ESI†). The Si 2p binding energy position shows two maxima at 99.9 eV and 104 eV corresponding to silicon and silicon dioxide, respectively.37 The binding energy position between these maxima is attributed to silicon suboxides. Both oxides are the result of silicon oxidation due to handling in air and the incomplete etching process of the silicon–carbon nanocomposite.12,38 A native oxide layer is present on any silicon-based material and the high concentration of oxides in our material originates from the high surface area. Similar amounts of silicon dioxides and suboxides independent of the electrode preparation are found. Deconvolution of the C 1s binding energy shows peak maxima at 284.5 eV and 286 eV in both binders corresponding to C–C and C–O bonds, respectively, as a result of the porous carbon scaffold. Additional functional groups at 288–290 eV (ether, carboxy and carbonate groups) stem from the binder.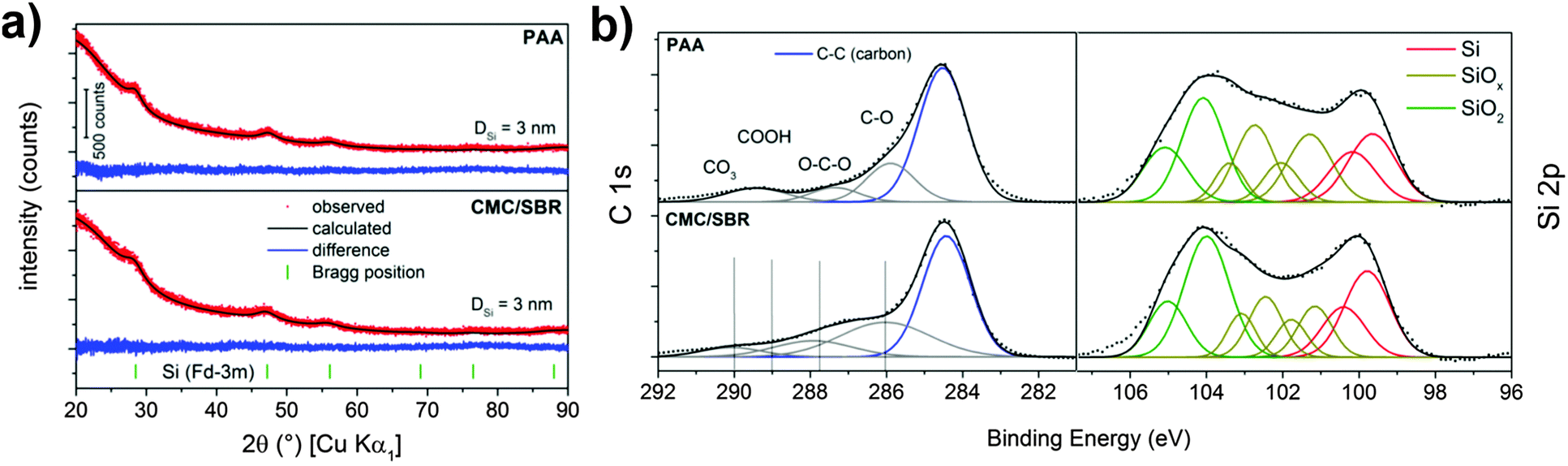 | ||
| Fig. 1 (a) XRD pattern and (b) XP spectra of the pristine silicon electrodes prepared with PAA and CMC/SBR as binders. | ||
3.2 Electrochemical characterization
Electrochemical experiments were conducted in a conventional carbonate-based electrolyte composed of 1 M LiPF6 in EC/DMC (1:1 v/v) (LP30) with and without partial substitution of EC with FEC (1 M LiPF6) in EC/DMC/FEC (1:2:1 v/v/v) (LP30 + FEC) according to a recent publication.16 The delithiation capacity vs. cycle number of the silicon–carbon nanocomposite depending on the two binders and the FEC addition is depicted in Fig. 2. The initial capacity is almost equal for all samples independent of binders and FEC addition. The initial drop and subsequent increase of capacity may be attributed to an activation process or to a delayed uptake of electrolyte into the porous structure.39 This special behavior is not observed during cycling of the electrode at low current rates (0.5 A g−1) confirming our assumption (Fig. S1a, ESI†). The largest difference in reversibility is observed depending on the FEC addition whereas the binder seems to affect the reversibility negligibly. However, it will be shown that the results of SEI investigation vary depending on binders.
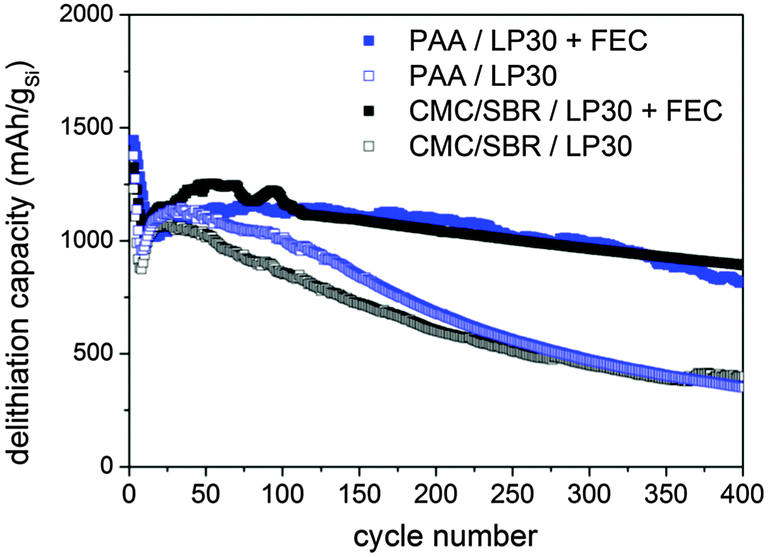 | ||
| Fig. 2 Galvanostatic cycling between 0.01–1.2 V vs. Li/Li+ at 2.5 A g−1 (two pre-cycles at 0.5 A g−1) of the silicon electrode depending on binders and FEC addition. | ||
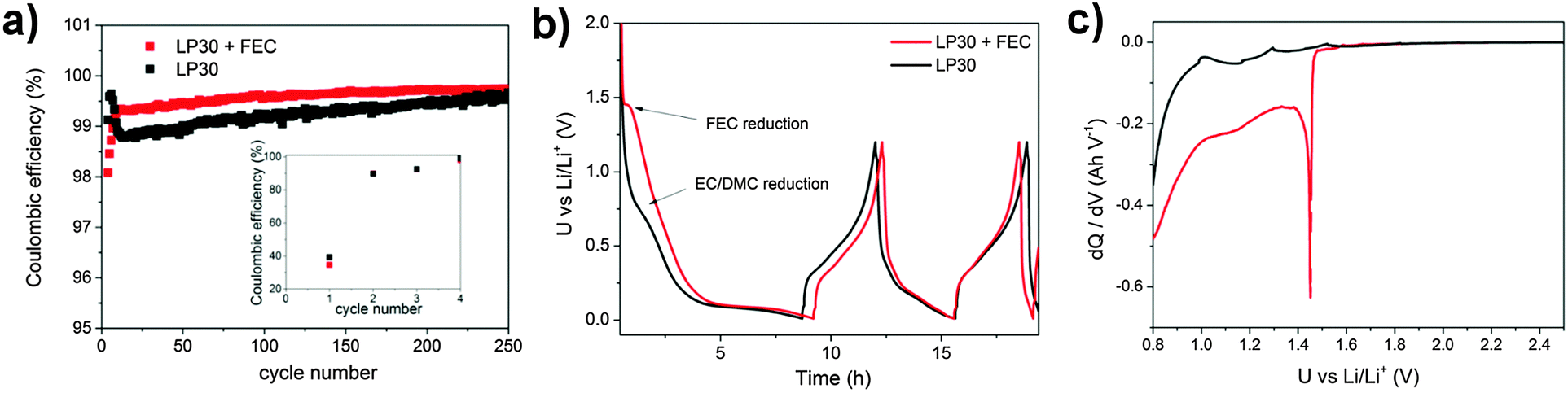 | ||
| Fig. 3 Electrochemical characterization of the silicon electrode with CMC/SBR as a binder depending on FEC addition. (a) Coulombic efficiency of the galvanostatic cycling between 0.01–1.2 V vs. Li/Li+ at 2.5 A g−1 (the first two cycles at 0.5 A g−1), (b) the first two discharge/charge curves and (c) the differential capacity of the first discharge. | ||
3.3 Post-mortem analysis of the SEI
All electrodes were galvanostatically cycled 400 times between 0.01–1.2 V vs. Li/Li+ at a current rate of 2.5 A gSi−1 (∼1 mA cm−2) (shown in Fig. 2) and disassembled in the delithiated state.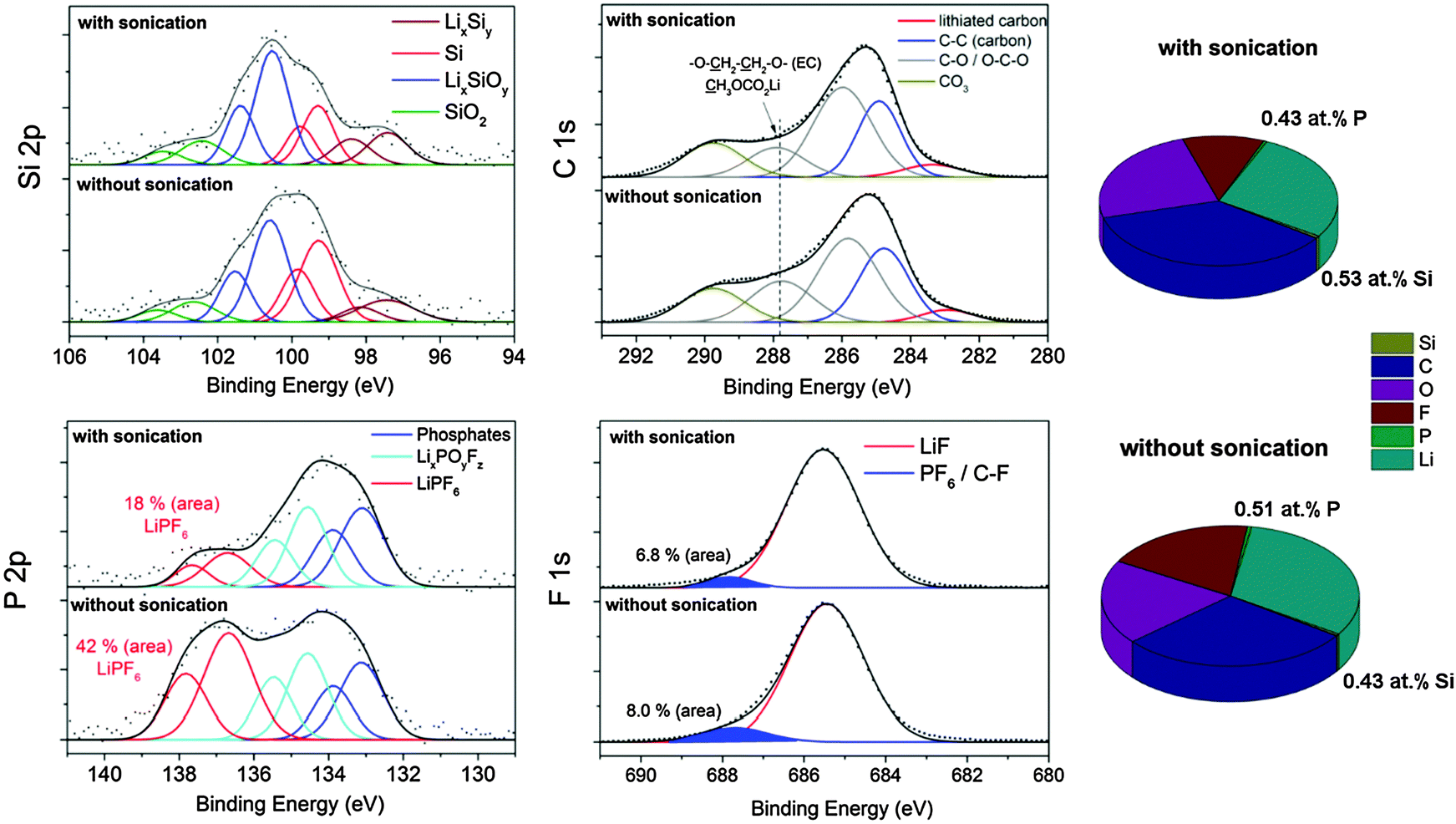 | ||
| Fig. 4 XP spectra (left) and concentration (right) of the silicon electrode with PAA as a binder after 400 times cycling in LP30 with FEC addition depending on the washing procedure (washed 3 times in DMC with a final sonication process or 3 times in DMC without sonication). | ||
(1) From deconvolution of the P 2p spectrum, we determined 0.08 mol% LiPF6 with sonication and almost three times more without sonication (0.24 mol% LiPF6). The occurrence of LiPF6 compounds is verified by the appearance of a binding energy position at ∼688 eV in the F 1s spectrum.
(2) The binding energy position at ∼288 eV in the C 1s spectrum decreases by approximately 20% after sonication which we majorly attribute to a lowered O–CH2–CH2–O/O–CHF–CH2–O concentration originating from EC/FEC and to the partial removal of easily soluble semi-organic carbonates such as (CH2OCO2Li)2, CH3OCO2Li as typical upper SEI components.30,41 The removal of these components is also be observed in the O 1s spectrum (Fig. S3, ESI†) because the intensity of the binding energy position at 534 eV corresponding to C–O species is lowered after sonication. Note that the concentration of the CO3 (290 eV) and C–O (286 eV) species is nearly equally independent of the washing procedure indicating retained Li2CO3 and CH3–OLi.41
All other elements show no change in their chemical characteristics. Importantly, the Si 2p spectrum is higher resolved after sonication due to the higher concentration of silicon and the binding energies remain constant indicating a leftover of reaction-sensitive Si/LixSiy nanoparticles during the washing procedure. Phosphates (POx) are a major part of the SEI whereas PF6 species seems to be mainly a residue of the electrolyte. Although it was proposed that LiPF6 may be part of the SEI,25 our results show that this species is easily soluble and likely dissolves during cycling whereas the vast majority of COx species and phosphates remain. Hence, PF6-containing compounds cannot be substantial part of the SEI on small-sized silicon nanoparticles. In summary, a short sonication procedure is a promising non-destructive way to remove softly-bond electrolyte components. However, the partial removal of the upper organic SEI components (in particular CH3OCO2Li) cannot be excluded, but it allows characterization of the interfacial characteristics of silicon and the upper organic SEI and is a suitable technique for non-destructive depth-profiling of the SEI.
| Sample | Si (at%) | C (at%) | O (at%) | F (at%) | P (at%) |
|---|---|---|---|---|---|
| w/o FEC | 6.8 | 42.1 | 48.8 | 2.2 | 0 |
| With FEC | 10 | 37.8 | 39.5 | 11.1 | 0.7 |
X-ray diffraction gives insights about structure and morphology of the entire post-mortem electrode and the results are depicted in Fig. 5. No reflections appear in the case of the additive-free sample, indicating a completely amorphous material. Note that the silicon reflections typically disappear after cycling due to an amorphization process during lithiation.42 In the case of FEC, we observe the reflections of LiF with a cubic crystal structure (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). From the XRD pattern a crystallite size of roughly 4 nm is determined by Rietveld analysis. The formation of crystalline LiF is also confirmed by TEM (Fig. 5b). The bright-field images show an agglomeration of the porous carbon scaffold attached to silicon nanoparticles after cycling. A high-resolution analysis is impossible because the high-energy electron beam decomposes the SEI components. However, selected area electron diffraction (SAED) proves the formation of nanocrystalline LiF only for the addition of FEC. LiF is often detected by XPS measurements, but has not yet been reported in diffraction experiments after cycling. It is likely formed in the initial cycles where a two-phase transition is observed which suggests a precipitation from the liquid phase. The role of fluorides in Li-ion batteries is controversially discussed.43 LiF is recognized as an insulator for both lithium ions and electrons and its formation should be avoided.44 In contrast to this, LiF is often associated, among other compounds, as a SEI-stabilizing component for silicon-based electrodes resulting in an enhanced cycling stability.13,26,45,46 The rather controversial literature reports suggest that LiF may be neither beneficial nor disadvantageous for the reversibility. From the aspect of diffraction experiments, we can detect LiF only in the presence of FEC. From this point of view LiF indeed supports a better cycling stability. However, since most of the material is amorphous, further information for evaluation is provided by XPS measurements.
m). From the XRD pattern a crystallite size of roughly 4 nm is determined by Rietveld analysis. The formation of crystalline LiF is also confirmed by TEM (Fig. 5b). The bright-field images show an agglomeration of the porous carbon scaffold attached to silicon nanoparticles after cycling. A high-resolution analysis is impossible because the high-energy electron beam decomposes the SEI components. However, selected area electron diffraction (SAED) proves the formation of nanocrystalline LiF only for the addition of FEC. LiF is often detected by XPS measurements, but has not yet been reported in diffraction experiments after cycling. It is likely formed in the initial cycles where a two-phase transition is observed which suggests a precipitation from the liquid phase. The role of fluorides in Li-ion batteries is controversially discussed.43 LiF is recognized as an insulator for both lithium ions and electrons and its formation should be avoided.44 In contrast to this, LiF is often associated, among other compounds, as a SEI-stabilizing component for silicon-based electrodes resulting in an enhanced cycling stability.13,26,45,46 The rather controversial literature reports suggest that LiF may be neither beneficial nor disadvantageous for the reversibility. From the aspect of diffraction experiments, we can detect LiF only in the presence of FEC. From this point of view LiF indeed supports a better cycling stability. However, since most of the material is amorphous, further information for evaluation is provided by XPS measurements.
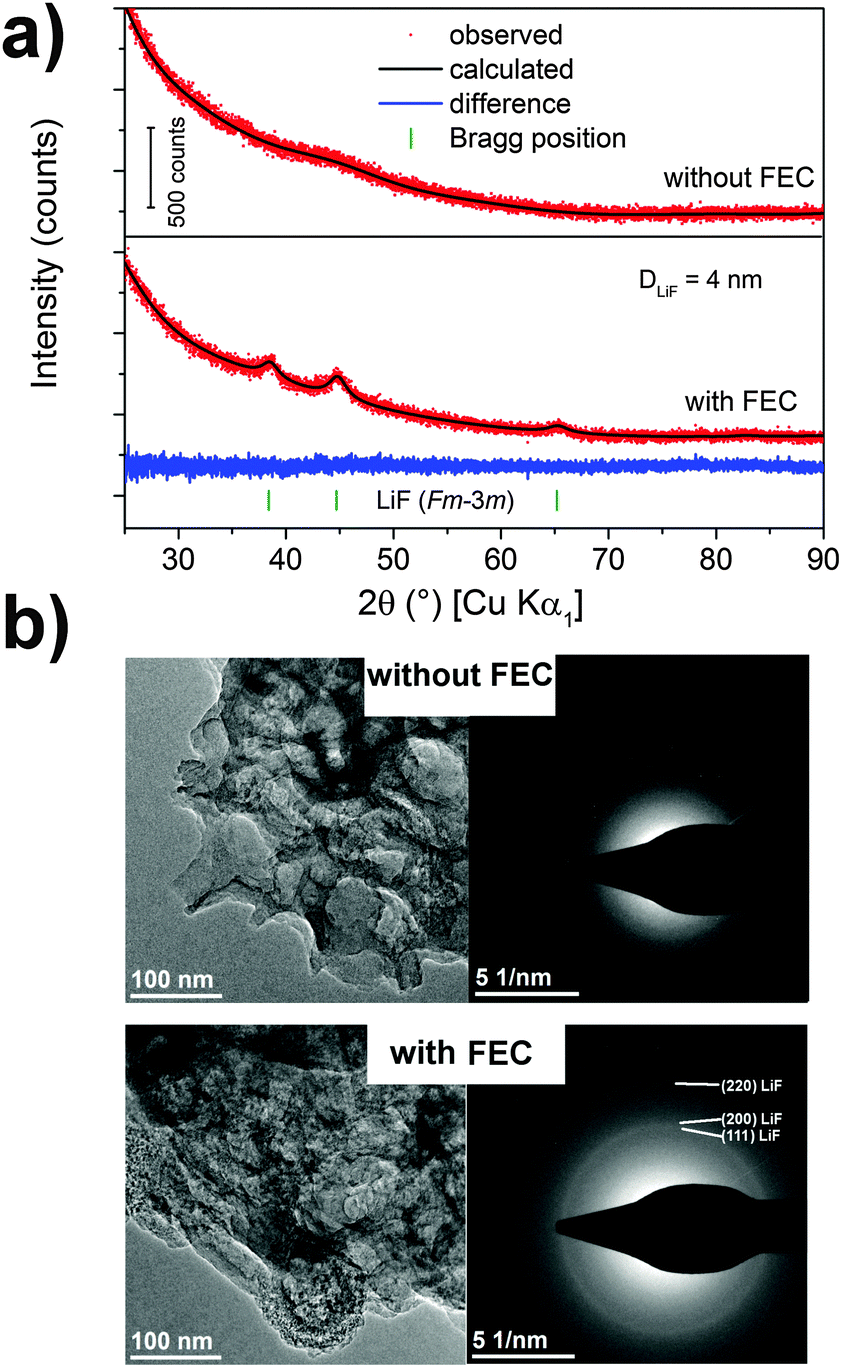 | ||
| Fig. 5 (a) XRD pattern and (b) TEM bright-field images (left) with selected area electron diffraction (SAED) (right) of the cycled silicon electrodes with CMC/SBR depending on FEC addition. | ||
Fig. 6 displays the XPS results and the chemical composition. We detect a concentration of ∼1 at% silicon for both samples. Higher amounts of lithium in the FEC sample indicate lithium salts as major components of the interfacial layer on silicon. Further differences are found in the carbon/oxygen ratio and the fluorine concentration. With FEC the carbon/oxygen ratio is higher (1.31) than without additive (0.8). The fluorine and phosphorus concentration is up to five times higher with FEC suggesting high decomposition of the additive and an integration of hydrolysis products of LiPF6 into the SEI. Higher amounts of phosphorus with FEC addition were reported by Elazari et al. and Chen et al. on, respectively, thin silicon films and silicon nanoparticles, suggesting pronounced decomposition of the conductive salt as well as the integration of the products into the SEI.17,22 However, almost no phosphorus is detected without the additive.
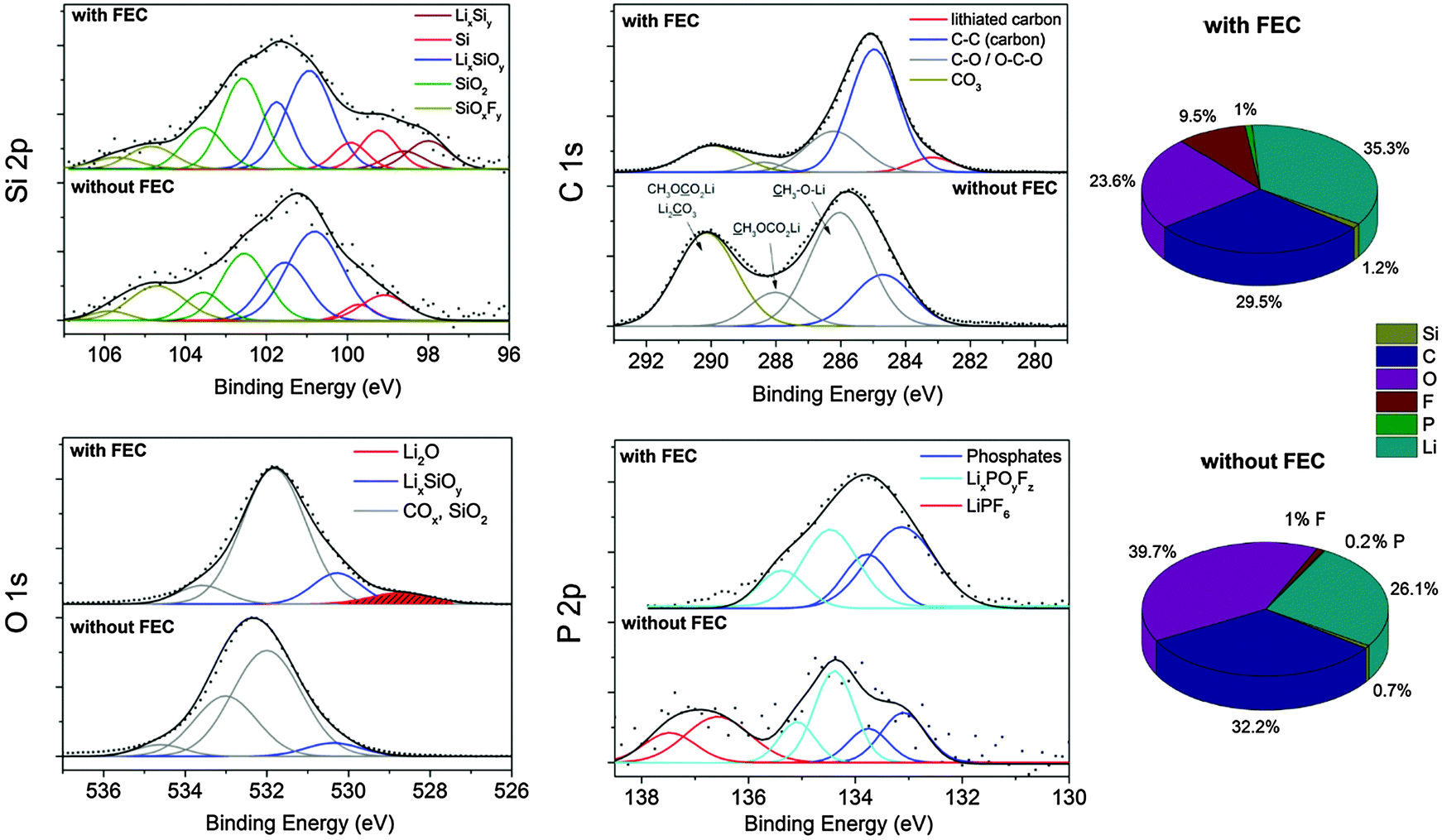 | ||
| Fig. 6 XP spectra and concentrations of the silicon electrodes with CMC/SBR as a binder cycled 400 times depending on FEC addition. | ||
Both Si 2p spectra in Fig. 6 show signals with a maximum at approximately 101 eV which correspond to LixSiOy.24 This compound is an initial reaction product of lithium with SiO2 which is present in large concentrations on the pristine electrode (Fig. 1). From literature24 it is known that SiO2 undergoes different reactions during cycling which are summarized here:
| SiO2 + 4Li → Si + 2Li2O | (I) |
| 2SiO2 + 4Li → Si + Li4SiO4 | (II) |
| SiO2 + HF → SiOxFy + H2O | (III) |
The total amount of carbon–oxygen (COx) species on the pristine electrode was estimated to be 34%. After cycling in the additive-free electrolyte the C 1s binding energy position features a maximum at 285.7 eV majorly corresponding to C–O species and to conductive carbon (C–C species at 284.5 eV).47 A second local maximum at 290 eV is observed caused by lithium carbonates and diverse semi organic lithium carbonates. These results evidence the presence of (CH2OCO2Li)2, CH3OCO2Li, CH3OLi and Li2CO3 as typical SEI components in alkyl carbonates.41 The total amount of COx species increased to 83% indicating a strong electrolyte decomposition. With FEC as an additive the dominant signal is shifted to 284.9 eV which majorly corresponds to the conductive carbon scaffold as a result of the thin SEI. The low energy signal at 283 eV corresponds to lithiated carbon.48 Only low amounts of CO3 and C–O species with a total concentration of 39% are present which is only slightly higher than for the pristine electrode (34%). This observation provides the proof of a negligible decomposition of the electrolyte solvents DMC and EC. The low concentration of CO3/C–O species and the high intensity of C–C/C–H bonds point to the formation of a thin layer-like polymer, presumably a vinyl polymer with functional C–O/CO3 groups, on the silicon–carbon composite. It was proposed that FEC can decompose to HF and vinylene carbonate which oligomerizes to polycarbonates as the stable SEI on silicon.19 Vinylene carbonate has been proven to be an effective stabilizer in LIBs owing to the formation of stable polycarbonates.49 Nakai et al.20 proposed HF and Li2CO3 formation by a ring opening reaction of FEC and its subsequent polymerization to a vinyl polymer. A recent report17 suggests the formation of poly(-vinyl carbonates).
The binding energy position of the Li 1s core level spectra (Fig. S4, ESI†) is located at about 56 eV for both samples and corresponds to various lithium salts in the SEI.48 The characteristics in the O 1s spectrum are similar. For the additive-free a peak maximum at 532.5 eV is observed. The addition of FEC shifts this peak maximum to lower energies (531.5 eV). Higher energies indicate the presence of more COx species whereas the lower O 1s binding energy position is attributed to the presence of LixSiOy which is supposed to be a major component of the SEI on silicon.24 Taking into account the integrated peak area and the silicon/oxygen concentration, we determined a stoichiometric oxygen number of about 3 for the additive-free sample and 4 with FEC according to reaction (II). Interestingly, we observe a peak maximum at 528.5 eV only in the FEC sample which corresponds to Li2O. Li2O is typically formed in the first cycle due to the reaction of SiO2 with lithium, as explained earlier (reaction (I)). Philippe et al.24 reported that Li2O was reversibly detected in nano-silicon anodes even after many cycles but seems to disappear owing to a dissolution by HF from the decomposition of FEC and LiPF6:
| Li2O + 2HF → 2LiF + 2H2O | (IV) |
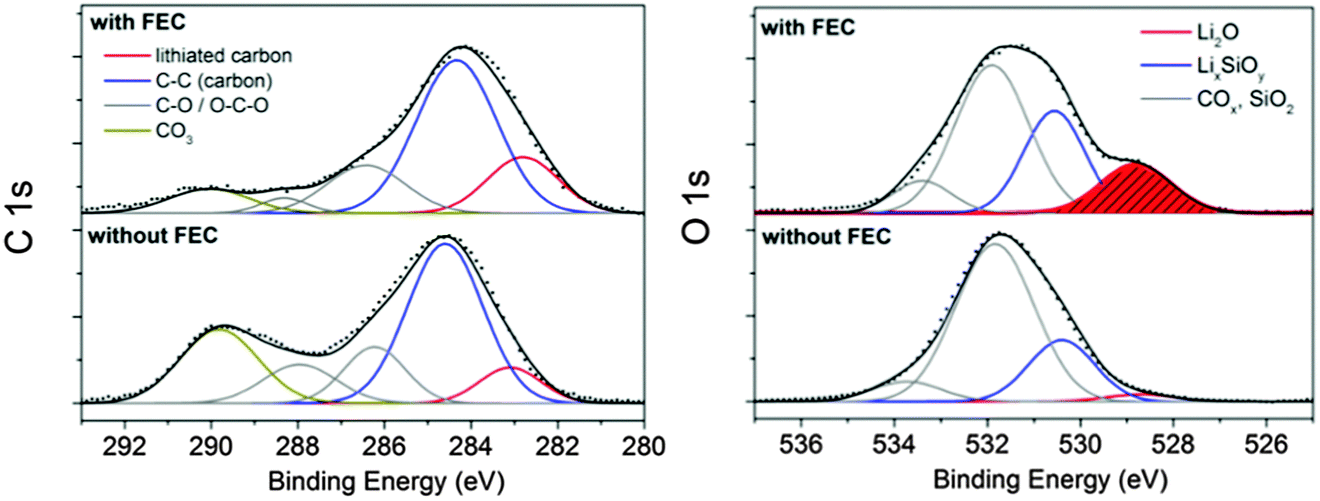 | ||
| Fig. 7 XP spectra of the silicon electrodes with CMC/SBR as a binder cycled 400 times depending on FEC addition after Ar+ ion sputtering (∼15 nm surface removal). | ||
The F 1s spectrum (Fig. S5, ESI†) of the additive-free sample in the non-sputtered state consists of a signal at 687.7 eV with low intensity and a distinct signal at 685.5 eV corresponding to traces of LiPF6, LixPOyFz and LiF, respectively. The phosphorus species are confirmed by the P 2p spectrum (Fig. 6). The signal at 137.5 eV points to traces of the conductive salt and the one at 134.5 eV to partially fluorine-substituted phosphates (reaction (V)). The presence of fluorine-substituted phosphates as part of the SEI after cycling in a conventional carbonate-based electrolyte has been reported previously.19,22,25 The concentrations of these phosphates seem to vary noticeably. Our results suggest that negligible concentrations of partially fluorinated phosphates participate in the SEI formation on silicon in the additive-free electrolyte. With addition of FEC, the F 1s spectrum exhibits solely one signal at 685.5 eV corresponding to (nanocrystalline) LiF as already confirmed with XRD. The LiF cannot be present as a closed layer since a crystallite size of 4 nm (determined by diffraction experiments) in a layer-like structure would cause the absence of any other signal in the XP spectrum as a result of the low attenuation length of these photons (2–3 nm) at this energy. Thus, the overall morphology of LiF is present as nanoparticles or as a fragmentary scaffold. This finding contradicts a recent work suggesting that a layer-like structure of LiF stabilizes the SEI.26 Our findings support other reports attributing the enhanced reversibility to an organic polymeric film structure.40 The P 2p spectrum (Fig. 6) reveals a binding energy position at 134.0 eV proving the presence of majorly phosphates and fluorine-substituted phosphates. The presence of FEC causes the hydrolysis of considerable amounts of LiPF6 to (fluorinated) phosphates which are integrated into the inorganic part of the SEI. The general reaction path of the phosphate formation was described in literature.25 (reaction (V)).
| PF6− + H2O → POF3− + 2HF | (V) |
| Sample | Li (at%) | Si (at%) | C (at%) | O (at%) | F (at%) | P (at%) |
|---|---|---|---|---|---|---|
| CMC/SBR | 35.23 | 1.17 | 29.44 | 23.57 | 9.48 | 0.95 |
| PAA | 27.55 | 0.53 | 36.08 | 24.32 | 11.09 | 0.43 |
In addition, the carbon and fluorine concentration is higher and the C 1s energy (Fig. 8a) reveals significantly more COx compounds on the surface with PAA as binder. In particular, the concentration of DMC/EC and their related compounds increases considerably whereas the lithium concentration is considerably lower with PAA suggesting lower proportions of lithium salts as major constituents of the SEI. The Li2O identified as retained interfacial compound with FEC addition is solely observed after sputtering (Fig. 8b) indicating a thicker passive layer with the PAA binder. The P 2p spectrum reveals higher concentration of LiPF6 with the PAA binder (Fig. 4 and 6). From these results, we conclude that the overall chemical species in the SEI are similar, but the amount of electrolyte-related components such as EC/DMC and LiPF6 is higher when PAA is used. This observation may be the result of a stronger interaction of electrolyte components with PAA, for instance with a pronounced swelling in the electrolyte since PAA is a well-known superadsorber. Magasinski et al. analyzed the swelling of PAA and CMC and found no significant difference.34 However, they used diethyl carbonate (DEC) whereas the smaller molecule DMC as solvent in LP30 may behave differently. Furthermore, the analyzed molecular weight was considerably lower. Our assumption is supported by a study of Bordes et al. dealing with nano-Si/graphene in PAA as a binder cycled in LP30 with FEC.18 The test conditions and electrode composition were comparable to the conditions chosen for our work. They observed no silicon on the electrode surface by XPS after 50 cycles18 which is conform to our study since only little concentration of silicon was observed with PAA. In conclusion, a different binder does not change the overall structure of the SEI, but it can considerably change the amount of surface species on the electrode which originates from the swelling of the binder with electrolyte as well as stronger or weaker interactions of electrolyte-related components with the individual binder. It can impede the detection of crucial SEI components due to a thicker layer of electrolyte(-related) species on the electrode surface, as shown for Li2O as a typical interfacial SEI component on silicon.
 | ||
| Fig. 8 XP spectra of the silicon electrodes cycled 400 times in LP30 with FEC depending on the binder (a) no sputtering and (b) after sputtering with an Ar+ beam (∼15 nm surface removal). | ||
4. Conclusion
The electrochemical analysis of the nanocrystalline silicon/carbon electrode with FEC addition revealed an enhanced reversibility, but initially a higher electrolyte consumption compared to the additive-free electrolyte. After a few cycles the decomposition with FEC addition is negligible suggesting the formation of a protective layer. The CMC/SBR or the PAA binder does not influence the overall electrochemical performance. Galvanostatic long-term cycled electrodes (400 times) were characterized depending on FEC addition and binders. A modified washing procedure of the electrodes including a sonication step was investigated which allows the evaluation of the actual SEI and residues of electrolyte components appearing very similar to that specified by common surface sensitive characterization techniques. The results suggest that retained LiPF6 salt plays a negligible role in the SEI of silicon/carbon electrodes. The addition of FEC causes an extremely low electrolyte decomposition (EC/DMC) even after 400 cycles. The formation of considerable amounts of nanocrystalline LiF was observed by XRD, TEM and XPS. The crystallite size of 4 nm suggests a particle-like rather than a closed film-like morphology. From this result we propose that fluorides have neither a beneficial nor a disadvantageous effect on the reversibility and are just decomposition by-products at most. We attribute the enhanced reversibility with FEC to a very thin (<4 nm) polymer layer (presumably a vinyl polymer) on the silicon/carbon electrode. A high concentration of Li2O in the FEC-containing electrolyte suggests that the diffusion of HF (formed from fluorinated compounds) through the SEI is prevented. Note that Li2O is a reaction product of the successive decomposition of the native SiO2 layer on silicon with the electrolyte in the first cycle. The additive-free electrolyte causes high electrolyte decomposition and shows no ability to prevent dissolution of Li2O. Phosphorus species seem to play a negligible role in the additive-free electrolyte. In contrast, high concentrations of phosphate are observed with FEC addition which may result from an increased amount of water traces. The water traces are attributed to a higher HF release from the fluorinated additive and its subsequent reaction with oxides and carbonates (SiO2, Li2O, Li2CO3). Our results suggest that both the FEC and the SiO2 play a critical role in the reversibility of silicon-based anodes. The application of another binder does not completely change the overall structure of the SEI, but it can complicate the analysis due to the occurrence of more electrolyte-related components on the surface caused by swelling effects or a stronger interaction with the PAA binder. Fig. 9 illustrates a simplified schematic view of the silicon–carbon nanostructure before and after cycling depending on FEC addition.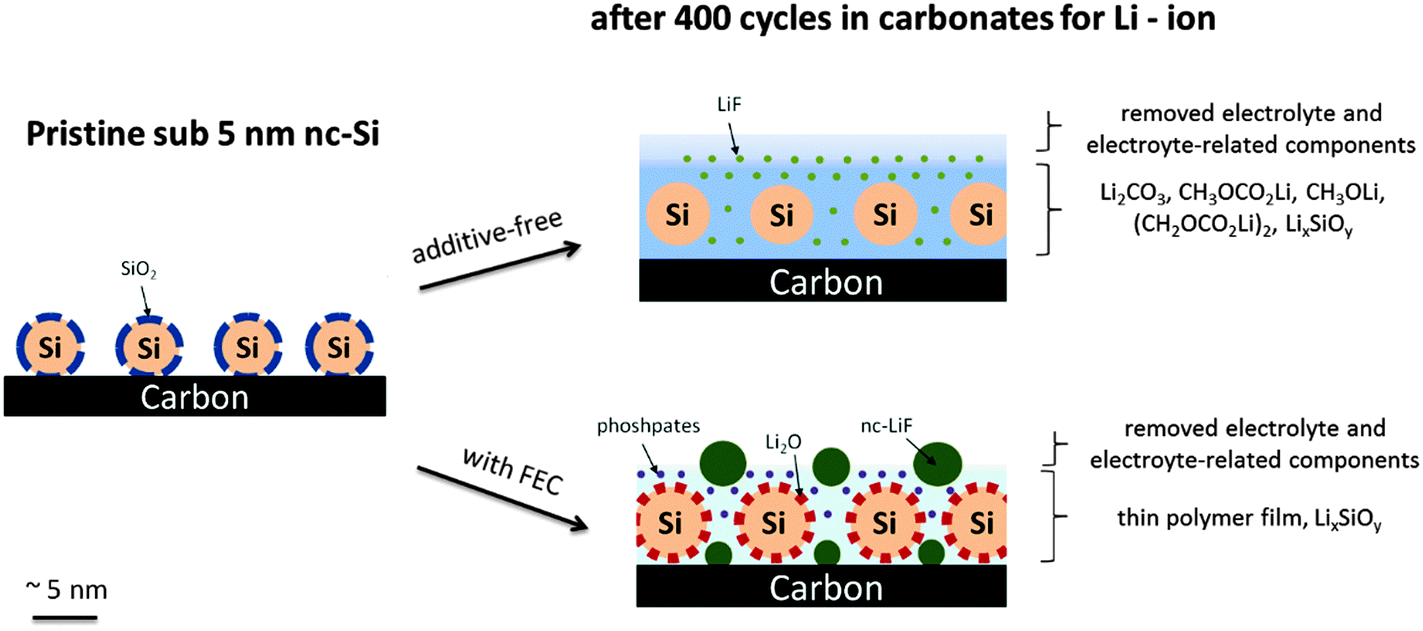 | ||
| Fig. 9 Simplified schematic illustration of the silicon–carbon nanostructure after long-term cycling depending on FEC addition. | ||
Acknowledgements
The authors thank Silke Hampel and Alexander Schubert for their valuable technical support. We gratefully acknowledge Solvay Chemicals for providing the fluoroethylene carbonate and acknowledge the financial support from the German Federal Ministry of Education and Research (BMBF) through the Excellent Battery – WING center “Batteries – Mobility in Saxony” (Grant No. 03X4637B and 03X4637C).References
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–30 CrossRef CAS.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS.
- C.-M. Park, J.-H. Kim, H. Kim and H.-J. Sohn, Chem. Soc. Rev., 2010, 39, 3115–3141 RSC.
- J. Brückner, S. Thieme, F. Böttger-Hiller, I. Bauer, H. T. Grossmann, P. Strubel, H. Althues, S. Spange and S. Kaskel, Adv. Funct. Mater., 2014, 24(9), 1284 CrossRef.
- J. Balach, T. Jaumann, M. Klose, S. Oswald, J. Eckert and L. Giebeler, J. Phys. Chem. C, 2015, 119(9), 4580 CAS.
- Y. Yang, M. T. McDowell, A. Jackson, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2010, 10, 1486–1491 CrossRef CAS.
- D. Aurbach, J. Power Sources, 2000, 89, 206–218 CrossRef CAS.
- H. Wu, G. Chan, J. W. Choi, I. Ryu, Y. Yao, M. T. McDowell, S. W. Lee, A. Jackson, Y. Yang, L. Hu and Y. Cui, Nat. Nanotechnol., 2012, 7, 310–315 CrossRef CAS.
- N. Liu, H. Wu, M. T. McDowell, Y. Yao, C. Wang and Y. Cui, Nano Lett., 2012, 12, 3315–3321 CrossRef CAS.
- H. Kim, M. Seo, M.-H. Park and J. Cho, Angew. Chem., Int. Ed., 2010, 49, 2146–2149 CrossRef CAS.
- J. Song, S. Chen, M. Zhou, T. Xu, D. Lv, M. L. Gordin, T. Long, M. Melnyk and D. Wang, J. Mater. Chem. A, 2014, 2, 1257–1262 CAS.
- T. Jaumann, M. Herklotz, M. Klose, K. Pinkert, S. Oswald, J. Eckert and L. Giebeler, Chem. Mater., 2015, 27, 37–43 CrossRef CAS.
- N.-S. Choi, K. H. Yew, K. Y. Lee, M. Sung, H. Kim and S.-S. Kim, J. Power Sources, 2006, 161, 1254–1259 CrossRef CAS.
- B. Philippe, R. Dedryvère, M. Gorgoi, H. Rensmo, D. Gonbeau and K. Edström, J. Am. Chem. Soc., 2013, 135, 9829–9842 CrossRef CAS.
- S. Dalavi, P. Guduru and B. L. Lucht, J. Electrochem. Soc., 2012, 159, A642–A646 CrossRef CAS.
- Y.-M. Lin, K. C. Klavetter, P. R. Abel, N. C. Davy, J. L. Snider, A. Heller and C. B. Mullins, Chem. Commun., 2012, 48, 7268–7270 RSC.
- X. Chen, X. Li, D. Mei, J. Feng, M. Y. Hu, J. Hu, M. Engelhard, J. Zheng, W. Xu, J. Xiao, J. Liu and J.-G. Zhang, ChemSusChem, 2014, 7, 549–554 CrossRef CAS.
- A. Bordes, K. Eom and T. F. Fuller, J. Power Sources, 2014, 257, 163–169 CrossRef CAS.
- V. Etacheri, O. Haik, Y. Goffer, G. A. Roberts, I. C. Stefan, R. Fasching and D. Aurbach, Langmuir, 2012, 28, 965–976 CrossRef CAS.
- H. Nakai, T. Kubota, A. Kita and A. Kawashima, J. Electrochem. Soc., 2011, 158, A798–A801 CrossRef CAS.
- J. S. Kim, D. Byun and J. K. Lee, Curr. Appl. Phys., 2014, 14, 596–602 CrossRef.
- R. Elazari, G. Salitra, G. Gershinsky, A. Garsuch, A. Panchenko and D. Aurbach, J. Electrochem. Soc., 2012, 159, A1440–A1445 CrossRef CAS.
- I. A. Profatilova, C. Stock, A. Schmitz, S. Passerini and M. Winter, J. Power Sources, 2013, 222, 140–149 CrossRef CAS.
- B. Philippe, R. Dedryvère, J. Allouche, F. Lindgren, M. Gorgoi, H. Rensmo, D. Gonbeau and K. Edström, Chem. Mater., 2012, 24, 1107 CrossRef CAS.
- B. Philippe, R. Dedryvère, M. Gorgoi, H. Rensmo, D. Gonbeau and K. Edström, Chem. Mater., 2013, 25, 394–404 CrossRef CAS.
- K. Schroder, J. Alvarado, T. A. Yersak, J. Li, N. Dudney, L. J. Webb, Y. S. Meng and K. J. Stevenson, Chem. Mater., 2015, 27, 5531–5542 CrossRef CAS.
- K. W. Schroder, H. Celio, L. J. Webb and K. J. Stevenson, J. Phys. Chem. C, 2012, 116, 19737–19747 CAS.
- C. Pereira-Nabais, J. Światowska, A. Chagnes, A. Gohier, S. Zanna, A. Seyeux, P. Tran-Van, C.-S. Cojocaru, M. Cassir and P. Marcus, J. Phys. Chem. C, 2014, 118, 2919–2928 CAS.
- R. Dedryvère, L. Gireaud, S. Grugeon, S. Laruelle, J.-M. Tarascon and D. Gonbeau, J. Phys. Chem. B, 2005, 109, 15868–15875 CrossRef.
- K. Tasaki, A. Goldberg, J.-J. Lian, M. Walker, A. Timmons and S. J. Harris, J. Electrochem. Soc., 2009, 156, A1019–A1027 CrossRef CAS.
- J.-S. Bridel, T. Azaıs, M. Morcrette, J.-M. Tarascon and D. Larcher, Chem. Mater., 2010, 22, 1229–1241 CrossRef CAS.
- A. Xiao, L. Yang, B. L. Lucht, S.-H. Kang and D. P. Abraham, J. Electrochem. Soc., 2009, 156(4), A318–A327 CrossRef CAS.
- S.-H. Kang, D. P. Abraham, A. Xiao and B. L. Lucht, J. Power Sources, 2008, 175, 526–532 CrossRef CAS.
- A. Magasinski, B. Zdyrko, I. Kovalenko, B. Hertzberg, R. Burtovyy, C. F. Huebner, T. F. Fuller, I. Luzinov and G. Yushin, ACS Appl. Mater. Interfaces, 2010, 2, 3004–3010 CAS.
- S. Oswald, Appl. Surf. Sci., 2015, 351, 492–503 CrossRef CAS.
- W. J. I. DeBenedetti and Y. J. Chabal, J. Vac. Sci. Technol., A, 2013, 31, 050826 Search PubMed.
- M. L. Mastronardi, E. J. Henderson, D. P. Puzzo, Y. Chang, Z. B. Wang, M. G. Helander, J. Jeong, N. P. Kherani, Z. Lu and G. A. Ozin, Small, 2012, 8, 36–47 CrossRef.
- Y. Yu, C. M. Hessel, T. D. Bogart, M. G. Panthani, M. R. Rasch and B. A. Korgel, Langmuir, 2013, 29, 1533–1540 CrossRef CAS.
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 Search PubMed.
- C. Xu, F. Lindgren, B. Philippe, M. Gorgoi, F. Björefors, K. Edström and T. Gustafsson, Chem. Mater., 2015, 27, 2591–2599 CrossRef CAS.
- D. Aurbach, B. Markovsky, I. Weissman, E. Levi and Y. Ein-Eli, Electrochim. Acta, 1999, 45, 67–86 CrossRef CAS.
- J. Li and J. R. Dahn, J. Electrochem. Soc., 2007, 154, A156–A161 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS.
- Z. Chen and K. Amine, J. Electrochem. Soc., 2006, 153, A1221–A1225 CrossRef CAS.
- N. Azimi, W. Weng, C. Takoudis and Z. Zhang, Electrochem. Commun., 2013, 37, 96–99 CrossRef CAS.
- H. Kim, F. Wu, J. T. Lee, N. Nitta, H.-T. Lin, M. Oschatz, W. I. Cho, S. Kaskel, O. Borodin and G. Yushin, Adv. Energy Mater., 2014, 1401792 Search PubMed.
- K. László, K. Josepovits and E. Tombácz, Anal. Sci., 2001, 17, i1741–i1746 Search PubMed.
- K. Ciosek Högström, S. Malmgren, M. Hahlin, H. Rensmo, F. Thebault, P. Johansson, K. Edström, M. Armand and J.-M. Tarascon, J. Phys. Chem. C, 2013, 117, 23476–23486 Search PubMed.
- L. Martin, H. Martinez, M. Ulldemolins, B. Pecquenard and F. Le Cras, Solid State Ionics, 2012, 215, 36–44 CrossRef CAS.
- M. Hoffmann, M. Zier, S. Oswald and J. Eckert, J. Power Sources, 2015, 288, 434–440 CrossRef CAS.
- S. Sim, P. Oh, S. Park and J. Cho, Adv. Mater., 2013, 25, 4498–4503 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp03672k |
| This journal is © the Owner Societies 2015 |