
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A DFT study of adsorption of imidazole, triazole, and tetrazole on oxidized copper surfaces: Cu2O(111) and Cu2O(111)-w/o-CuCUS†
Dunja
Gustinčič
ab and
Anton
Kokalj
*a
aDepartment of Physical and Organic Chemistry, Jožef Stefan Institute, Jamova 39, SI-1000 Ljubljana, Slovenia. E-mail: tone.kokalj@ijs.si; Web: http://www.ijs.si/ijsw/K3-en/Kokalj Fax: +386-1-251-93-85; Tel: +386-1-477-35-23
bFaculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, SI-1000 Ljubljana, Slovenia
First published on 28th September 2015
Abstract
Azoles and their derivatives are known for their corrosion inhibition ability for copper. For this reason the bonding of imidazole, triazole, and tetrazole—used as archetypal models of azole corrosion inhibitors—to Cu2O(111) and Cu2O(111)-w/o-CuCUS was characterized using density functional theory (DFT) calculations. The former surface contains coordinatively-saturated (CSA) and coordinatively-unsaturated (CUS) Cu sites, whereas the latter lacks the CUS sites. We find that the molecules preferentially bond with a single unsaturated N atom to a surface Cu ion and concomitantly form a hydrogen bond with the surface O ion. They adsorb rather strongly at CUS sites with an adsorption energy of about −1.6 eV (as calculated with the PBE functional), whereas the bonding at CSA sites is about three times weaker thus being similar as on metallic Cu(111). The impact of van der Waals dispersion interactions on molecular adsorption bonding is also addressed. Depending on specifics of the adsorption structure, they strengthen the adsorption bonding by about 0.2–0.5 eV. Due to this specific bonding enhancement, dispersion interactions alter the relative stability of adsorption modes for tetrazole. An atomistic thermodynamics approach was used to construct two-dimensional phase diagrams for all the three molecules. In the viable range of oxygen chemical potential only three phases appear in the phase-diagrams, two of which are the high coverage (1 × 1) molecular phases (one on Cu2O(111) and the other on Cu2O(111)-w/o-CuCUS) and the third is clean Cu2O(111)-w/o-CuCUS. The current results indicate that molecular adsorption at CUS sites is strong enough to compensate the thermodynamic deficiency of stoichiometric Cu2O(111) thus making it more stable than Cu2O(111)-w/o-CuCUS, unless the conditions are too oxygen rich and/or for azole lean. This finding may tentatively suggest that the corrosion inhibition capability of azoles stems from their ability to passivate reactive surface sites.
1 Introduction
Azole molecules and their derivatives are well known for their ability to slow down the corrosion of metals,1,2i.e., many of them are efficient corrosion inhibitors. Although the atomic scale mechanism of how organic corrosion inhibitors work is usually not known, it is widely accepted that the adsorption of inhibitors onto surfaces represents an important step in achieving the inhibitory effect.3 From this point of view, it is therefore important to characterize the molecule–surface bonding, although it represents only one aspect towards the atomic-scale understanding of corrosion protection mechanisms (for a more thorough approach, which involves a deconstruction of various relevant elements and their integration into a multiscale model, see the recent paper of Taylor4). Moreover, the interaction of organic molecules with surfaces has also been receiving considerable attention due to its importance in many other technological applications as well as for reasons of scientific curiosity.In previous publications, the adsorption of imidazole, triazole, and tetrazole—used as archetypal models of azole inhibitors—has been characterized on Cu(111) by means of DFT calculations5,6 to provide an atomic-scale insight into the chemistry of azole–copper bonding. It has been shown that neutral molecules bind weakly with Cu(111) via unsaturated N heteroatoms through the σ-type bonding and the magnitude of adsorption energy decreases from imidazole to tetrazole. However, oxide-free copper surfaces are more relevant at acidic pH, but under other conditions copper surfaces are often oxidized.7 In order to explain how the adsorption bonding of azole molecules depends on the oxidation state of copper, we extend the previous studies5,6 by investigating the adsorption of these molecules on oxidized copper surfaces.
In general, the interaction of corrosion inhibitors has been explicitly modeled by DFT methods mainly on bare metallic surfaces; the reason is likely related to the fact that metallic surfaces are structurally and electronically simpler than oxidized surfaces. Computational DFT studies concerning the adsorption of azole inhibitors on oxidized copper surfaces are very scarce, i.e., Jiang8 and Peljhan and Kokalj9,10 considered the adsorption of benzotriazole on Cu2O, and Blajiev and Hubin11 considered two thiadiazole derivatives. But several other studies modeled adsorption of probe molecules on Cu2O, such as CO, NO, H2O and CO2,12–19 and also cylic organic molecules, such as 2-chlorophenol,20 bromobenzene, aniline21 and cyclohexanol.22 Soon23 emphasized the importance of surface defects on copper oxide surfaces and showed that several non-stoichiometric surfaces are more stable than stoichiometric Cu2O(111). Due to this several studies of molecular adsorption were performed on non-stoichiometric surfaces.9,10,20,21
In the current paper, we investigate the bonding of imidazole, triazole, and tetrazole (their molecular structures are shown in Scheme 1) on Cu2O(111) and Cu2O(111)-w/o-CuCUS surfaces; the latter surface is considered, because it is thermodynamically more stable than the former in ambient oxygen atmosphere.23 The difference between the two surfaces is that the latter lacks the coordinatively unsaturated (CUS) Cu sites; the notation Cu2O(111)-w/o-CuCUS thus stands for “Cu2O(111) without the Cu CUS sites”. Molecular adsorption is currently considered at a solid/vacuum interface, although in the context of corrosion inhibition it would be more appropriate to consider adsorption at a solid/water interface. This choice is due to obvious modeling reasons, and moreover because the adsorption from an aqueous phase is a rather involved phenomenon with a number of competitive effects, such as molecule–surface, molecule–water, and surface–water interactions. Hence, it is appropriate to start with a simpler system that allows a more direct chemical characterization of the molecule–Cu2O bonding, which is the issue that the current paper is targeted at.
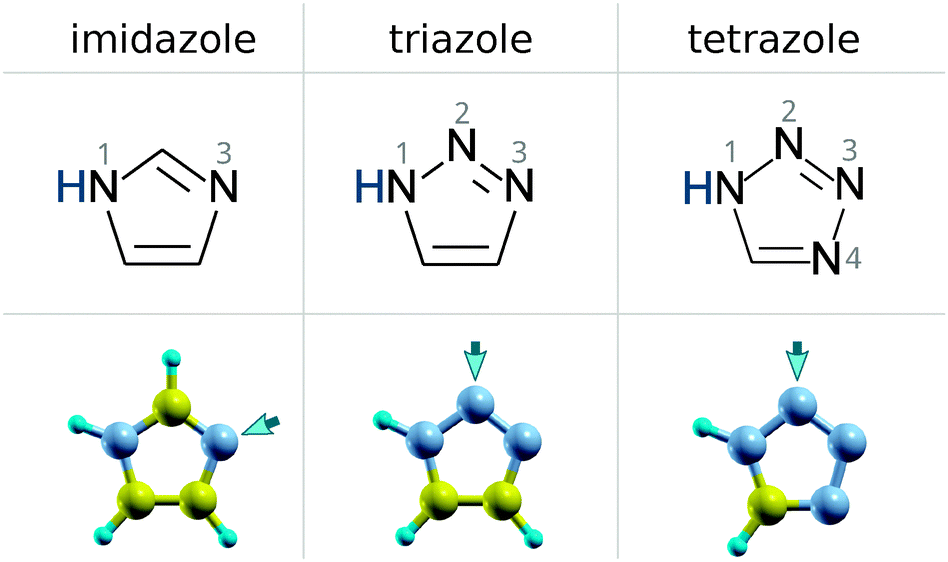 | ||
| Scheme 1 Skeletal formulae of imidazole, triazole, and tetrazole with the numbering of N atoms (top) and ball-and-stick models of optimized molecular structures (bottom). The arrows indicate the N atoms that preferentially bond with the considered Cu2O surfaces (see Section 3). | ||
2 Technical details
2.1 Computational
The calculations were performed in the framework of DFT using the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE).24 In addition to the plain PBE functional, adsorption calculations were also performed using a PBE-D′′ functional, which includes a reparametrized empirical dispersion correction of Grimme25,26 that consists of a damped C6R−6 like energy term on top of the PBE. The double prime in the PBE-D′′ label is used to indicate the reparametrization of the original method. This reparametrization is slightly different from our previous PBE-D′ reparametrization of ref. 27,‡ which was also used in ref. 9, 10 and 31–34. By default, all the presented results refer to the PBE functional, unless explicitly stated otherwise.A pseudopotential method with ultrasoft pseudopotentials was used.35,36 All calculations were done using the PWscf code from the Quantum ESPRESSO distribution,37 whereas visualization and molecular graphics were produced by the XCRYSDEN graphical package.38 The Kohn–Sham orbitals were expanded in a plane-wave basis set up to a kinetic energy cutoff of 30 Ry (240 Ry for the charge density cutoff). Brillouin zone (BZ) integrations were performed employing the special-point technique39 using a Marzari–Vanderbilt cold smearing40 of 0.01 Ry.
2.2 Model of oxidized copper surfaces
Oxidized copper surfaces were modeled by Cu2O slabs without a metal support underneath. This model is appropriate for cases where the oxide layer on top of metal is not ultrathin, because the reactivity of few Å thick oxide films supported on metals can be very different from the reactivity of surfaces of bulk oxides.41 In a recent study,42 the average thicknesses of Cu2O oxide layers formed on Cu immersed in 3 wt% NaCl solution were estimated to be about 2.2 ± 0.3 nm for the non-inhibited sample and 1.3 ± 0.2 nm for the sample inhibited by benzotriazole. These thicknesses seem therefore sufficient to make the current model adequate.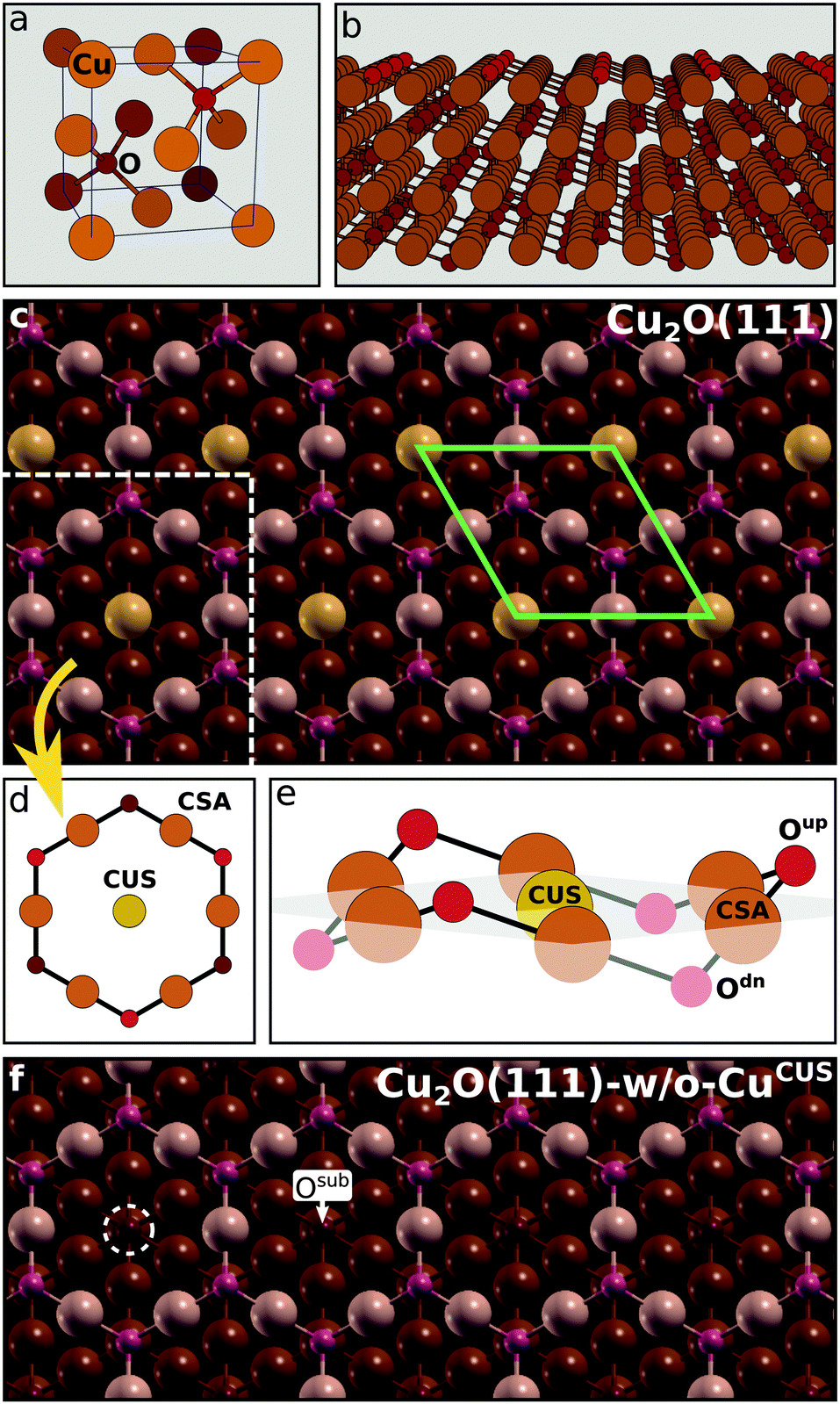 | ||
| Scheme 2 (a) Unit cell of Cu2O bulk, chosen such that Cu is at the origin. Smaller red and bigger brown balls represent O and Cu ions, respectively. (b) The side view of a four O–Cu–O trilayer thick Cu2O(111) slab. (c) The top view of Cu2O(111) with the surface Cu ions colored brighter; it can be seen that the surface trilayer is constructed of centered hexagons. The (1 × 1) surface cell is indicated by a greenish parallelogram. (d) Top and (e) side views of a single centered hexagon with the designation of pertinent Cu and O ions (for brevity reasons copper sites are labeled as CUS and CSA with the Cu being omitted). In (c)–(e), the CUS ions are colored a bit more yellowish than the CSA ions. (f) The top view of Cu2O(111)-w/o-CuCUS, which lacks the CUS sites. One CUS vacancy at the left is indicated by a white dashed circle and towards the right an Osub ion is explicitly marked. | ||
The structure of Cu2O(111)-w/o-CuCUS, which lacks the CUS sites, is shown in Scheme 2f. The O ions below the CUS vacancies are labeled as Osub (“sub” stands for subsurface; both the CUS vacancies and Osub are indicated in Scheme 2f). Although Cu2O(111)-w/o-CuCUS displays a sizable magnetic moment (e.g., about 1 μB per CUS vacancy44), several test calculations revealed that the effect of magnetism on the total energy is marginal,9 hence all the presented results refer to spin unpolarized slab calculations.
2.3 Surface free energy calculations
Surface free energies were calculated using symmetric (1 × 1)-slabs in which both surfaces are equivalent. The slabs consisting of 6, 7, and 9 O–Cu–O trilayers were used. The in-plane lattice spacing was fixed to the calculated equilibrium bulk lattice parameter, while other degrees of freedom were relaxed. The BZ integrations were performed using a 3 × 3 × 1 uniformly shifted k-mesh.It has been shown that to a first approximation total energies can be used to represent Gibbs free energies of solids,23,45 hence the surface free energies (γsurf) were calculated by fitting the equation:9
| Eslab(Ntl) = 2Aγsurf + NtlEtl + ΔNstoichOμO, | (1) |
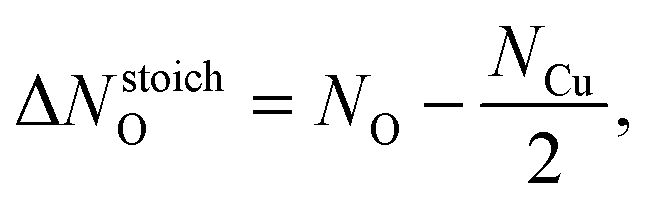 | (2) |
While plotting the γsurf as a function of μO, a viable range of μO should be considered. The oxygen poor (Olean) and rich (Orich) limits§ are chosen according to eqn (3a) and (3b), respectively:23,45
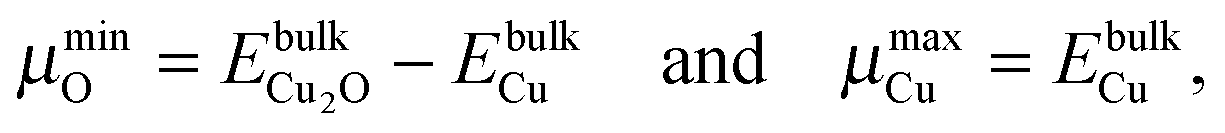 | (3a) |
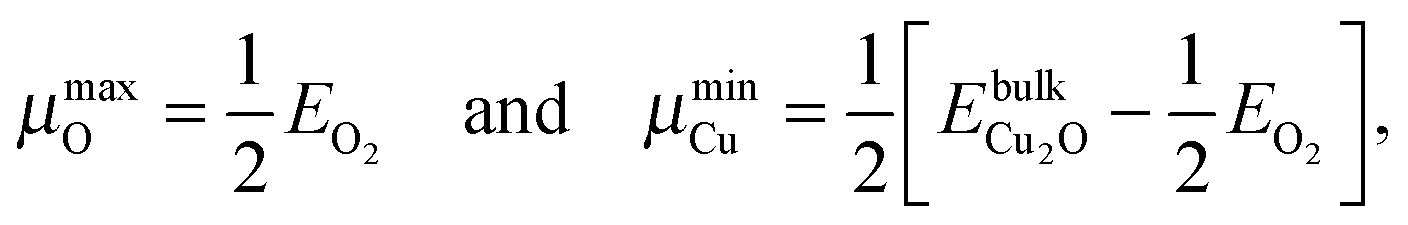 | (3b) |
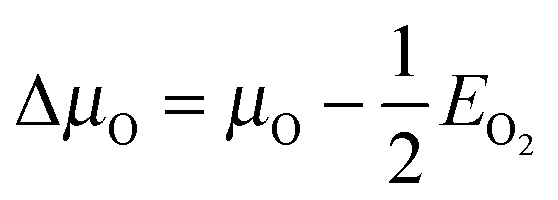 . Hence at the Olean limit ΔμO = −1.27 eV and at the Orich limit ΔμO = 0 eV. If the surface is in ambient gas phase, the μO depends on the temperature, T, and oxygen partial pressure, p, which within the ideal-gas approximation can be written as
. Hence at the Olean limit ΔμO = −1.27 eV and at the Orich limit ΔμO = 0 eV. If the surface is in ambient gas phase, the μO depends on the temperature, T, and oxygen partial pressure, p, which within the ideal-gas approximation can be written as 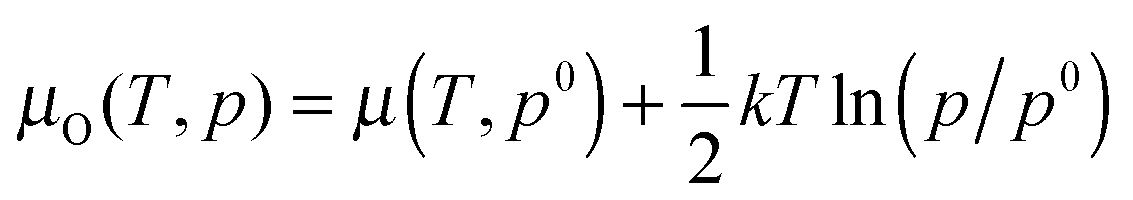 , where p0 stands for a standard pressure and k is the Boltzmann constant. This implies that low values of μO correspond to high T and low p (vice versa for the high values of μO).
, where p0 stands for a standard pressure and k is the Boltzmann constant. This implies that low values of μO correspond to high T and low p (vice versa for the high values of μO).
2.4 Adsorption calculations
The adsorption calculations were performed using slabs consisting of four O–Cu–O trilayers. The molecules were adsorbed on the top side of the slab and a dipole correction of Bengtsson46 was applied to cancel an artificial electric field that develops along the direction normal to the slab due to periodic boundary conditions imposed on the electrostatic potential. The thickness of the vacuum region—the distance between the top of ad-molecules and the adjacent slab—was set to about 20 Å. Adsorption properties were calculated with (1 × 1), (2 × 2), and (3 × 3) supercells, using the 3 × 3 × 1, 2 × 2 × 1, and 1 × 1 × 1 uniformly shifted k-meshes for the BZ integrations, respectively.The adsorption energy was calculated as:
| Eads = Emol/slab − (Eslab + Emol), | (4) |
Thermodynamic stability of adsorption structures that differ in surface coverage is evaluated by means of the adsorption surface free energy, γads, as a function of the molecular chemical potential, μmol. To a first approximation γads can be related to adsorption energy and μmolvia the relation:
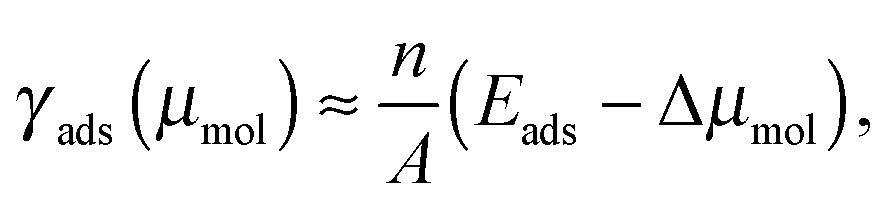 | (5) |
The stabilization of surface free energy due to a molecular adsorption can be estimated as:
![[small gamma, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0dd.gif) surf(μO) ≈ γsurf(μO) + εads, surf(μO) ≈ γsurf(μO) + εads, | (6) |
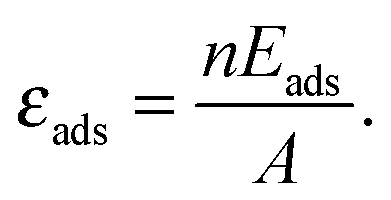 | (7) |
The one-dimensional treatment of eqn (5) can be extended by treating the adsorption surface free energy as a two-dimensional function of μmol and μO. We utilize the following approximate relation:
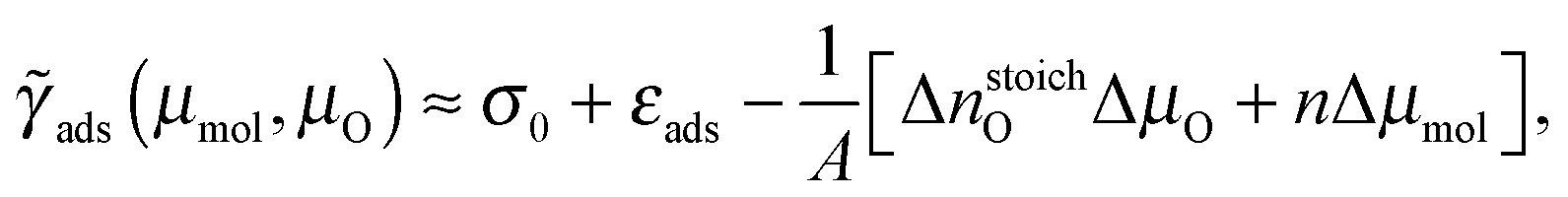 | (8) |
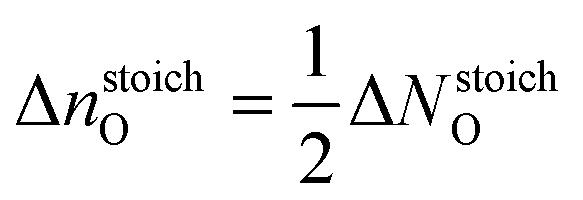 and σ0 is the surface energy at the oxygen rich limit (ΔμO = 0), i.e.,
and σ0 is the surface energy at the oxygen rich limit (ΔμO = 0), i.e.,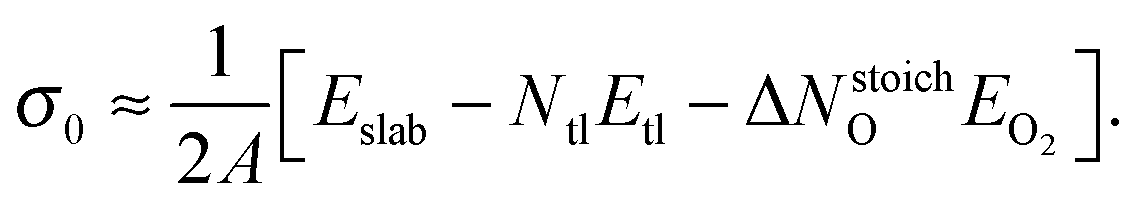 | (9) |
| Δρ(r) = ρmol/slab(r) − ρslab(r) − ρmol(r), | (10) |
The local geometry of adsorption structures are specified as Nlist + Hbond, where Nlist specifies with which N atoms a molecule bonds to the surface. If the adsorbed molecule also forms a hydrogen bond with the surface then the Hbond specifier characterizes it. Here are two examples: (1) the label N2 + N1H⋯Oup indicates that a molecule bonds with its N2 atom to the surface and also forms the N1–H⋯Oup hydrogen bond; (2) the label N2 + N3 indicates that a molecule bonds to the surface via its N2 and N3 atoms without any hydrogen bond.
The global geometry of adsorption phases are designated either as “(N × N)-mol@site” or as “(N × N)-mol@surface”, where (N × N) represents a periodic pattern that molecules form with respect to the surface unit-cell, site designates a specific site the molecules bond to (CSA or CUS), and surface specifies the surface (Cu2O(111)-w/o-CuCUS or Cu2O(111)). For example, (3 × 3)-mol@Cu2O(111)-w/o-CuCUS stands for one molecule adsorbed per (3 × 3) supercell on Cu2O(111)-w/o-CuCUS.
Near an adsorbed molecule at the CSA site not all Oup ions are equivalent, hence a more precise naming convention of O ions is utilized for unambiguous specification of adsorption structures (see Scheme 3). The molecule adsorbed at the CSA site can form a hydrogen bond with either the first or the second nearest neighbor Oup ions, which are named as Oupnear and Oupfar, respectively. For brevity reasons, Oupnear will be occasionally referred implicitly as Oup (but Oupfar will be always referred explicitly). In contrast, for a molecule adsorbed at the CUS site there are three equivalent nearby Oup ions.
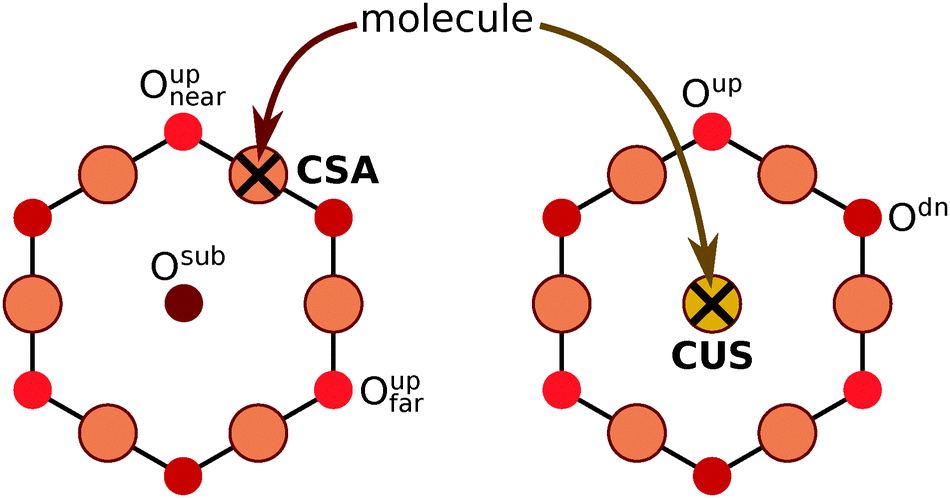 | ||
| Scheme 3 Definition of various O sites near an adsorbed molecule at the CSA site on Cu2O(111)-w/o-CuCUS (left) and CUS site (right). The molecule adsorbed at the CSA site can form a hydrogen bond with either Oupnear, Oupfar, or Osub. In contrast, the molecule adsorbed at the CUS site can only form a hydrogen bond with one among the three equivalent nearby Oup ions. | ||
3 Results and discussion
The calculated surface free energies of Cu2O(111) and Cu2O(111)-w/o-CuCUS as a function of the oxygen chemical potential are shown in Fig. S1 in the ESI;† they are in good agreement with those reported previously by Soon.23 Given that Cu2O(111)-w/o-CuCUS is considerably more stable than stoichiometric Cu2O(111), it is taken as a reference and a starting point for the current investigation of the adsorption of azoles on oxidized copper surfaces. This model is used to ascertain the molecular bonding at CSA sites. In addition, the adsorption is also modeled at individual CUS sites, which can be regarded as extraneous or defect sites on Cu2O(111)-w/o-CuCUS; the corresponding model therefore contains only as many CUS ions as adsorbed molecules. This model is used to ascertain the molecular bonding at CUS sites.3.1 Adsorption structures and energies
Several adsorption geometries were investigated for each molecule adsorbed at CSA and CUS sites and the corresponding results are presented in Fig. 1 and 2, respectively. These figures plot the dependence of adsorption energies on the nearest neighbor intermolecular distance, Rnn.¶ Beneath these plots the structures of pertinent adsorption modes—optimized at the lowest considered coverage using the (3 × 3) supercell—are shown; dipole-moment vectors of isolated molecules, oriented as in the respective adsorption state, are drawn superimposed with the adsorbed molecules and the N–Cu and H⋯O bond distances are also stated. It is worth noting that currently only the adsorption of intact molecules is considered, because the dissociation of the molecular N1–H bond, to form OH with the nearby surface O ion (note that on the Cu2O surface an H binds more strongly to an O than a Cu ion), was found to be endothermic by more than 1 eV for imidazole and over 0.1 eV for triazole and tetrazole; this issue will be considered in more detail in the forthcoming publication.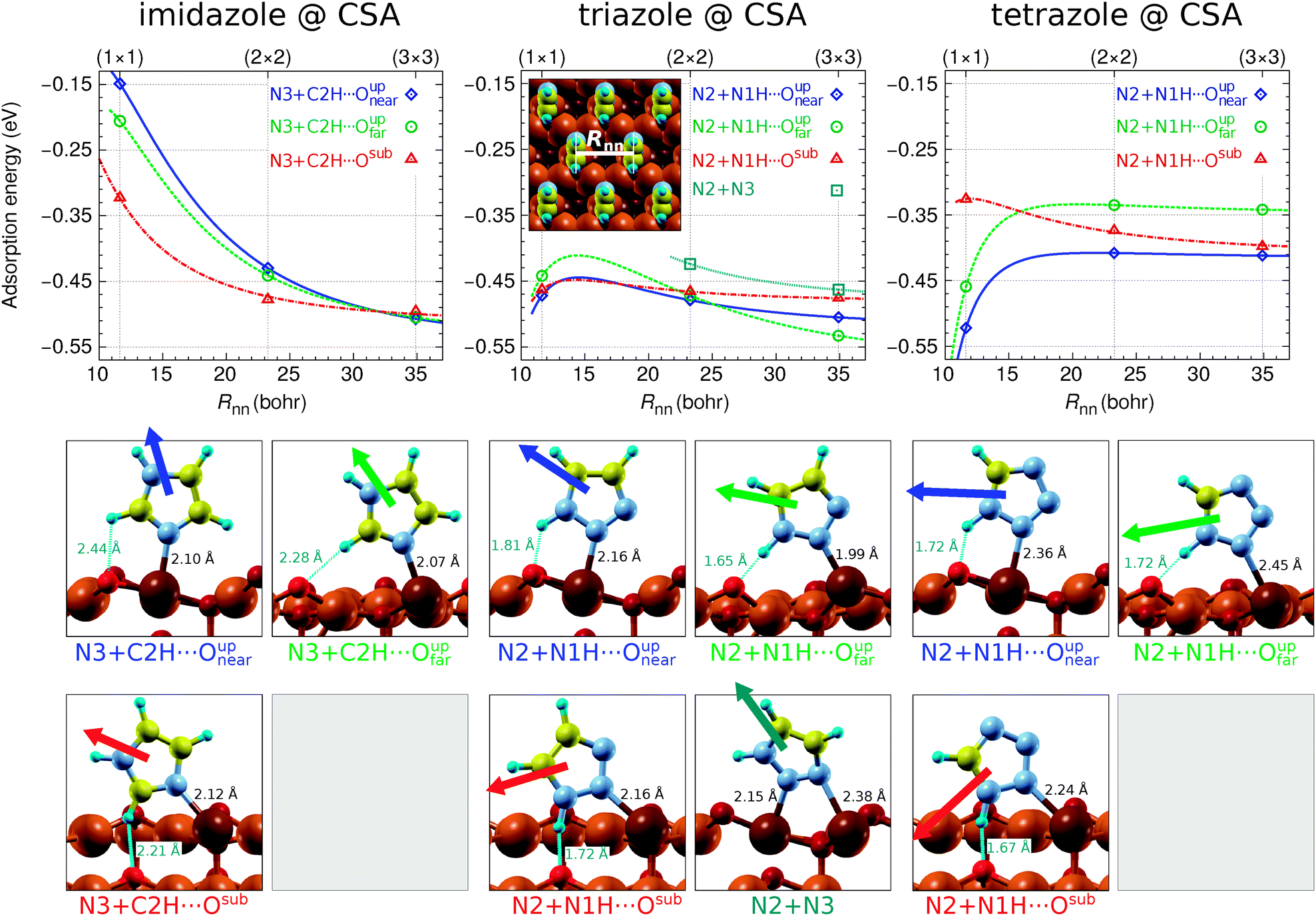 | ||
| Fig. 1 Top: Adsorption energy as a function of the nearest-neighbor intermolecular distance (Rnn, defined graphically in the inset at the center) for imidazole, triazole, and tetrazole bonded at a CSA site of Cu2O(111)-w/o-CuCUS. Curves are mainly drawn to guide the eye to make the plots more readable; they were calculated using the polarizable point-dipole model of ref. 47. The calculations were performed using the (1 × 1), (2 × 2), and (3 × 3) supercells. Bottom: Snapshots of pertinent adsorption modes, as obtained using the (3 × 3) supercell, superimposed with dipole vectors of isolated molecules. The corresponding N–Cu bond lengths and H⋯O distances are also stated. | ||
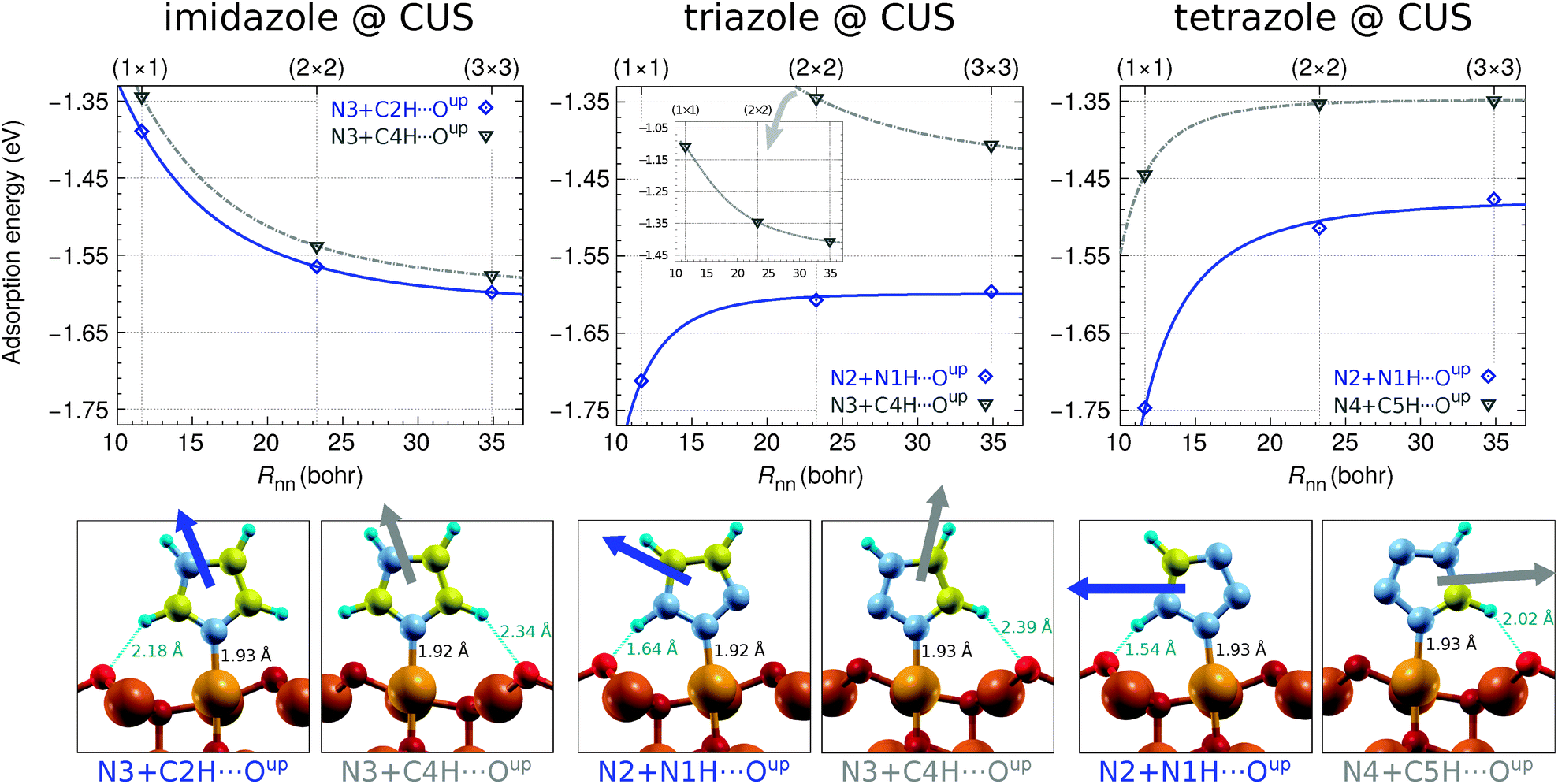 | ||
| Fig. 2 As in Fig. 1 but for the molecular bonding at a CUS site. Adsorption energies are calculated with respect to the high-symmetry position of CuCUS ions in the bare substrate to better represent the molecule–surface bond strengths. Note that the CuCUS ions of the bare substrate relax laterally to a more stable asymmetric position if the symmetry is broken (see Fig. 1 in ref. 9) with an energy stabilization of 0.13 eV at full (Θ = 1) and 0.04 eV at lower surface concentration (Θ ≤ 1/4) of extraneous CuCUS ions at Cu2O(111)-w/o-CuCUS. | ||
Imidazole binds with the N3 atom to the CSA and CUS sites and forms a weak C–H⋯O hydrogen bond with either Oupnear, Oupfar, or Osub ion when bonded at the CSA sites (Fig. 1, left) and with the Oup ion when adsorbed at the CUS sites (Fig. 2, left). Triazole and tetrazole bind preferably with the N2 atom to the CSA and CUS sites and form a N–H⋯O hydrogen bond (Fig. 1 and 2, middle and right) which is shorter and stronger than the C–H⋯O bond of imidazole (for the strength of these hydrogen bonds see Fig. S2 in the ESI†). That the N–H⋯O hydrogen bond is stronger than the C–H⋯O bond can be also inferred by comparing the adsorption energies of triazole N2 + N1H⋯Oupvs. N3 + C4H⋯Oup as well as tetrazole N2 + N1H⋯Oupvs. N4 + C5H⋯Oup structures at the CUS sites (Fig. 2, middle and right). Triazole can also bind with the N2 and N3 atoms to two neighboring CSA sites (N2 + N3 adsorption mode), but this mode is stable only at low coverage|| and even then it is inferior to the N2 bonding modes, presumably due to the lack of N–H⋯O bonding.
While on plain metallic Cu surfaces the dependence of adsorption energy on the Rnn or on the coverage (Θ, note that Θ ∝ R−2nn) can be straightforwardly understood in terms of the orientation of molecular dipoles—i.e., dipoles oriented normally (parallelly) to the surface result in lateral repulsive (attractive) interactions5,32,47—the situation is bit more complicated on the Cu2O(111)-w/o-CuCUS surface, because each CuCUS vacancy displays an inward pointing dipole of about 0.5 D and it is the cumulative (molecule + vacancies) dipole that matters. Nevertheless, the orientation of molecular dipoles seems still useful to roughly understand the lateral dependence. For example, imidazole displays the most repulsive and tetrazole the most attractive lateral interactions and, indeed, the dipoles of the former point largely upright and that of the latter almost parallel to the surface, e.g., see the left and right panels of Fig. 2 and also the dipole orientation of imidazole N3 + C2H⋯Oupnear and tetrazole N2 + N1H⋯Oupnear adsorption modes in Fig. 1.
The current results reveal that molecular bonding to the CSA sites is considerably weaker than that to the CUS sites. The strongest adsorption energy for the three molecules at the CSA sites is about −0.5 eV (Fig. 1), whereas at the CUS sites it is more than three times stronger, being about −1.6 eV for imidazole, −1.7 eV for triazole, and −1.75 eV for tetrazole (Fig. 2). This stronger bonding at CUS sites is also reflected in the N–Cu bond distances, which are about 1.9 Å at the CUS site and in the range between about 2.0 and 2.3 Å at the CSA sites. A similar strong bonding at the CUS sites was reported for benzotriazole.8–10 Several other molecules were also found to bind significantly stronger at CUS sites than at coordinatively saturated sites.16,18,21
A comparison with the previous results, obtained on metallic Cu(111),5 reveals that the bonding of imidazole, triazole, and tetrazole to CSA sites is similar in strength as their bonding to Cu(111). To facilitate the comprehension of trends, Fig. 3 compares adsorption energy magnitudes obtained on currently considered CSA and CUS sites to that on metallic Cu(111); the shown magnitudes refer to the most exothermic adsorption energies, regardless of the coverage. This figure clearly reveals that the bonding at CUS sites is considerably stronger than to Cu(111) and to CSA sites of Cu2O(111)-w/o-CuCUS. Indeed, the bonding at the CUS sites is even stronger than, for example, the bonding of benzotriazole at very low coordinated surface defects on metallic Cu surfaces, which was calculated to be about −1.3 eV.32
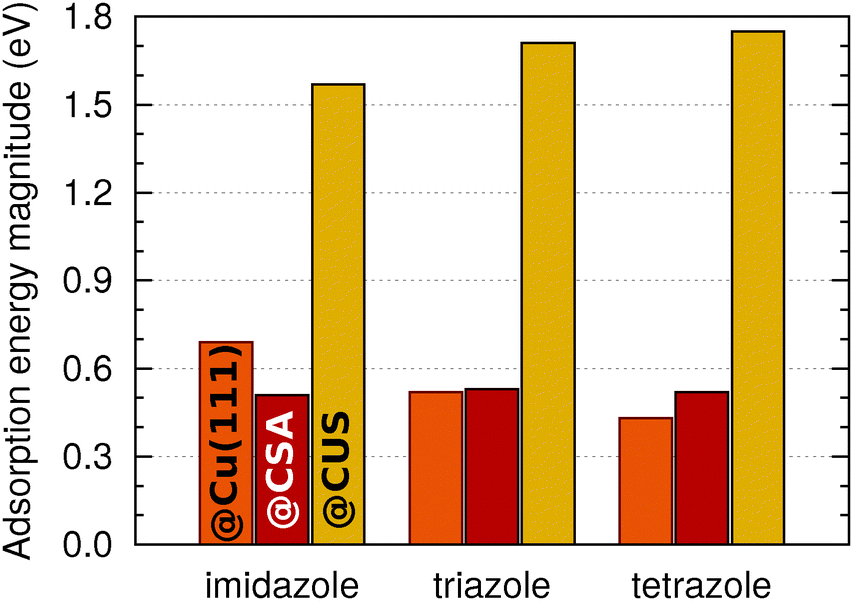 | ||
| Fig. 3 Magnitudes of adsorption energies of imidazole, triazole, and tetrazole on Cu(111) and at the CSA and CUS sites of Cu2O. The values for Cu(111) are taken from ref. 6. Only the largest magnitudes, irrespective of the coverage, are considered for a given molecule on a given site (or surface). | ||
A few more comments should be made with respect to the adsorption energy trends (cf.Fig. 1–3). On Cu(111) the magnitude of adsorption energy, |Eads|, decreases from imidazole to tetrazole at any coverage (for more details see ref. 5), whereas on Cu2O(111)-w/o-CuCUS the trends are bit more intricate due to the coverage dependence. At the lowest considered coverage, where the lateral dipolar intermolecular interactions are the smallest, the adsorption bonding strength follows the imidazole ≈ triazole > tetrazole trend on both the CSA and CUS sites,** whereas at a larger coverage the trend is affected by lateral dipolar interactions. Considering the largest adsorption energy magnitudes (cf.Fig. 3), irrespective of the coverage, the |Eads| trend at the CSA sites is imidazole ≈ triazole ≈ tetrazole, whereas at the CUS sites it is imidazole < triazole ≲ tetrazole.
3.2 Phase diagrams
The coverage dependence of molecular adsorption energies, presented in Fig. 1 and 2, allows us to construct phase diagrams to ascertain which adsorbate structures are thermodynamically the most stable at a given molecular chemical potential, μmol. It should be noted that only three discrete coverages are considered using one molecule per (N × N) supercell, where N ∈ [1,3]. To provide better comprehension of how densely the molecules cover the surface at these coverages, Fig. 4 plots the top view snapshots of adsorbed triazole. | ||
| Fig. 4 Topview of triazole adsorbed at extraneous CUS sites on Cu2O(111)-w/o-CuCUS at various coverages. From left to right: (1 × 1), (2 × 2), and (3 × 3) molecular phases. The molecules are drawn with the respective van der Waals radii, to give an impression of the representative molecular size, but the Cu2O surfaces are drawn with smaller radii to better show surface details. Note that for the (1 × 1) molecular phase the number of adsorbed molecules equals to the number of CUS sites on stoichiometric Cu2O(111), hence this phase corresponds to molecular adsorption on stoichiometric Cu2O(111). | ||
We first consider the adsorption surface free energy as a function of the molecular chemical potential (cf.eqn (5)). The corresponding plots of γads for the current molecules are shown in the top row panels of Fig. 5, where the upper plots correspond to the adsorption at CSA and lower plots at CUS sites. Thermodynamically the most stable structure at a given μmol is the one that displays the lowest adsorption surface free energy. In these plots the black horizontal line at γads = 0.0 meV Å−2 corresponds to a clean surface, which is the most stable at a low μmol, before any of the molecular line intersects it. It should be noted that for a given molecule at a given coverage only the structure with the most exothermic adsorption energy is considered here. It can be seen that for tetrazole and in part triazole, which display attractive lateral interactions, the high-coverage phases are always the most stable, unless the μmol is so low that the bare surfaces become the most stable. In addition, for triazole at the CSA sites, the lower coverage phases are competitive with the high-coverage phase around Δμmol ≈ −0.5 eV, where their γads lines intersect the clean surface line. In contrast, the situation is different for imidazole, because it displays repulsive lateral interactions. At the CSA sites, the high-coverage (1 × 1) phase is the most stable at Δμmol > −0.27 eV. Below this value the lower coverage phases are the most stable, first the (2 × 2) and then the (3 × 3), but only down to Δμmol = −0.51 eV, where the clean surface becomes the most stable. Note that only a few discrete coverages are considered in Fig. 5, hence there can be intermediate situations, but the point is that one passes from low- to high-coverage imidazole structures as the μmol increases from −0.51 eV to −0.27 eV. A similar relationship between high and lower coverage phases of imidazole also exists at the CUS sites with the exception that the stability intercepts are shifted to the left by about 1 eV due to much stronger molecular bonding at CUS compared to CSA sites.††
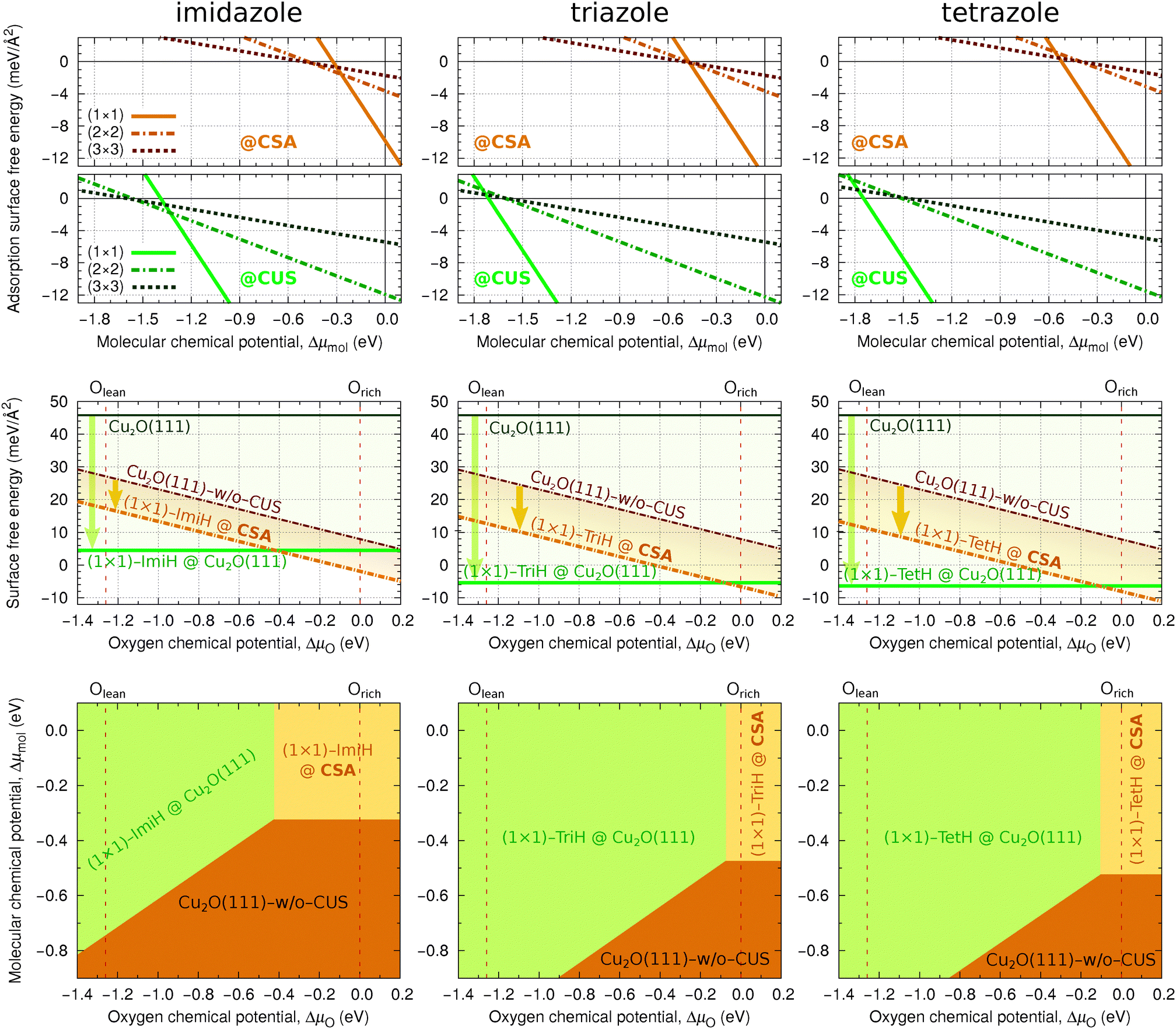 | ||
| Fig. 5 Adsorption phase diagrams of imidazole (left), triazole (middle), and tetrazole (right) on considered Cu2O surfaces. Top row: Adsorption surface free energy, γads, as a function of the molecular chemical potential (CSA sites are considered in the upper panels and CUS sites in the bottom panels). Middle row: Surface free energies, γsurf, of Cu2O(111) and Cu2O(111)-w/o-CuCUS and their stabilization due to high coverage molecular adsorption (for a given surface the upper γsurf line corresponds to a bare surface and the lower line to molecularly covered surface, while the vertical arrow indicates the stabilization due to high-coverage molecular adsorption). Bottom row: Two-dimensional phase diagrams as a function of molecular and oxygen chemical potentials. ImiH, TriH, and TetH stand for imidazole, triazole, and tetrazole, respectively, whereas “(1 × 1)-mol@CSA” is a shorthand label for (1 × 1)-mol@Cu2O(111)-w/o-CuCUS, where mol is either ImiH, TriH, or TetH. | ||
It should be noted that for the high-coverage (1 × 1) adsorption phases the number of adsorbed molecules equals to the number of CUS sites on stoichiometric Cu2O(111), which implies that (1 × 1) molecular phases at CUS sites correspond to molecular adsorption on stoichiometric Cu2O(111). Given that the high-coverage (1 × 1) molecular phases dominate in the γads(μmol) phase diagrams and the molecules bind much strongly to CUS than to CSA sites, opens a question whether this bonding enhancement is sufficient to stabilize the stoichiometric Cu2O(111) surface with respect to Cu2O(111)-w/o-CuCUS. This issue is addressed in the middle row panels of Fig. 5, which show how the high-coverage (1 × 1) molecular phases stabilize the surface free energies of Cu2O(111)-w/o-CuCUS and stoichiometric Cu2O(111) (cf.eqn (6)). It is apparent that the stabilization of stoichiometric Cu2O(111) due to adsorption of triazole and tetrazole is so large that the (1 × 1) molecularly covered Cu2O(111) dominates in the respective phase diagrams (horizontal thick green line). Only near the oxygen-rich limit, ΔμO ≳ −0.1 eV, the triazole and tetrazole covered Cu2O(111)-w/o-CuCUS become competitive. For imidazole the stabilization of stoichiometric Cu2O(111) is less pronounced and imidazole covered Cu2O(111)-w/o-CuCUS remains the most stable for ΔμO ≳ −0.4 eV, which represents about 30% span of the range between oxygen-lean and oxygen-rich limits of μO.
The above one-dimensional treatments can be extended by considering the adsorption surface free energy as a two-dimensional function of μmol and μOviaeqn (8). The bottom row panels of Fig. 5 show the resulting two-dimensional phase diagrams. These phase diagrams show only the most stable structures at agiven (μmol,μO). Regardless of the molecule, only three structures appear in the phase diagrams: (i) high coverage (1 × 1) molecular phase on stoichiometric Cu2O(111) in the top left region, (ii) high coverage (1 × 1) molecular phase on Cu2O(111)-w/o-CuCUS in the top right region, and (iii) bare Cu2O(111)-w/o-CuCUS in the bottom right region. For triazole and tetrazole, the phase diagrams are dominated by the (1 × 1)-mol@Cu2O(111) structure, whereas (1 × 1)-mol@Cu2O(111)-w/o-CuCUS exists only around the oxygen-rich limit when Δμmol ≳ −0.5 eV. Even for imidazole, which displays repulsive lateral interactions, only the high-coverage (1 × 1) molecular phases appear in the phase diagram, but their stability region is significantly reduced in favor of bare Cu2O(111)-w/o-CuCUS, i.e., the stability borders are shifted up-left compared to that of triazole or tetrazole.
Finally let us make a rather crude estimate, using the ideal-gas approximation, to which the ΔμO and Δμmol values would correspond at room temperature and partial pressure of p = 1 atm; the corresponding calculated values are ΔμO ≈ −0.3 eV and Δμmol ≈ −0.7 eV.‡‡ At these ΔμO and Δμmol values the Cu2O(111)-w/o-CuCUS phase is the most stable for imidazole, but for triazole and tetrazole this point lies close to the border with the (1 × 1)-mol@Cu2O(111) phase, which would prevail under more oxygen lean conditions. It is worth remarking that when van der Waals dispersion correction is taken into account, which enhances the molecular adsorption bonding (see Section 3.4), then the (ΔμO,Δμmol) = (−0.3 eV,−0.7 eV) point lies deep inside the region of high coverage molecular phases [either (1 × 1)-mol@Cu2O(111)-w/o-CuCUS or (1 × 1)-mol@Cu2O(111)].
3.3 Electronic structure analysis
To gain more insight into the chemistry of the molecule–surface bonding, Fig. 6 displays the charge density difference, Δρ(r), for imidazole, triazole, and tetrazole bound to the CSA (top row) and CUS (bottom row) sites. Only the most stable adsorption structures at a low coverage are considered, i.e., N3 + C2H⋯Oup for imidazole and N2 + N1H⋯Oup for triazole§§ and tetrazole. In the Δρ(r) plots, the red color represents electron charge accumulation and the blue color represents electron deficit regions. The formation of direct N–Cu bonds is clearly seen by the red colored charge accumulation lobes in the midst of these bonds. These electron charge accumulation lobes are weak at the CSA sites and much stronger at the CUS sites, which readily explains the much stronger molecular bonding at the latter. In addition to the N–Cu bonds, the molecules also interact at the surface with the X–H⋯Oup hydrogen bonds (X = C2 for imidazole or N1 for triazole and tetrazole). These H-bonds are characterized by the substantial charge accumulation located above the pertinent Oup ion and the charge deficit region of the nearby H atom. The intensities of these charge redistributions clearly reveal that the N–H⋯O bonds of triazole and tetrazole are considerably stronger than the C–H⋯O bonds of imidazole. For further characterization of the strength of N–H⋯O and C–H⋯O bonds see Fig. S2 in the ESI.† The Δρ(r) plots reveal that the strength of N–Cu bonds increases from tetrazole to imidazole (at the CSA sites), but the strength of the X–H⋯O hydrogen bonds follows the opposite direction, i.e., imidazole < triazole ≲ tetrazole (at both the CSA and CUS sites).¶¶ The latter is the reason that at a low coverage, where the lateral dipolar interactions are sufficiently small, the bonding strengths of imidazole and triazole are almost degenerate, but the bonding strength of tetrazole is about 0.1 eV less. The stronger N–H⋯O bond compared to the C–H⋯O bond is therefore able to compensate for the weaker N–Cu bond of triazole compared to that of imidazole, but falls somewhat short for compensating the even weaker N–Cu bond of tetrazole.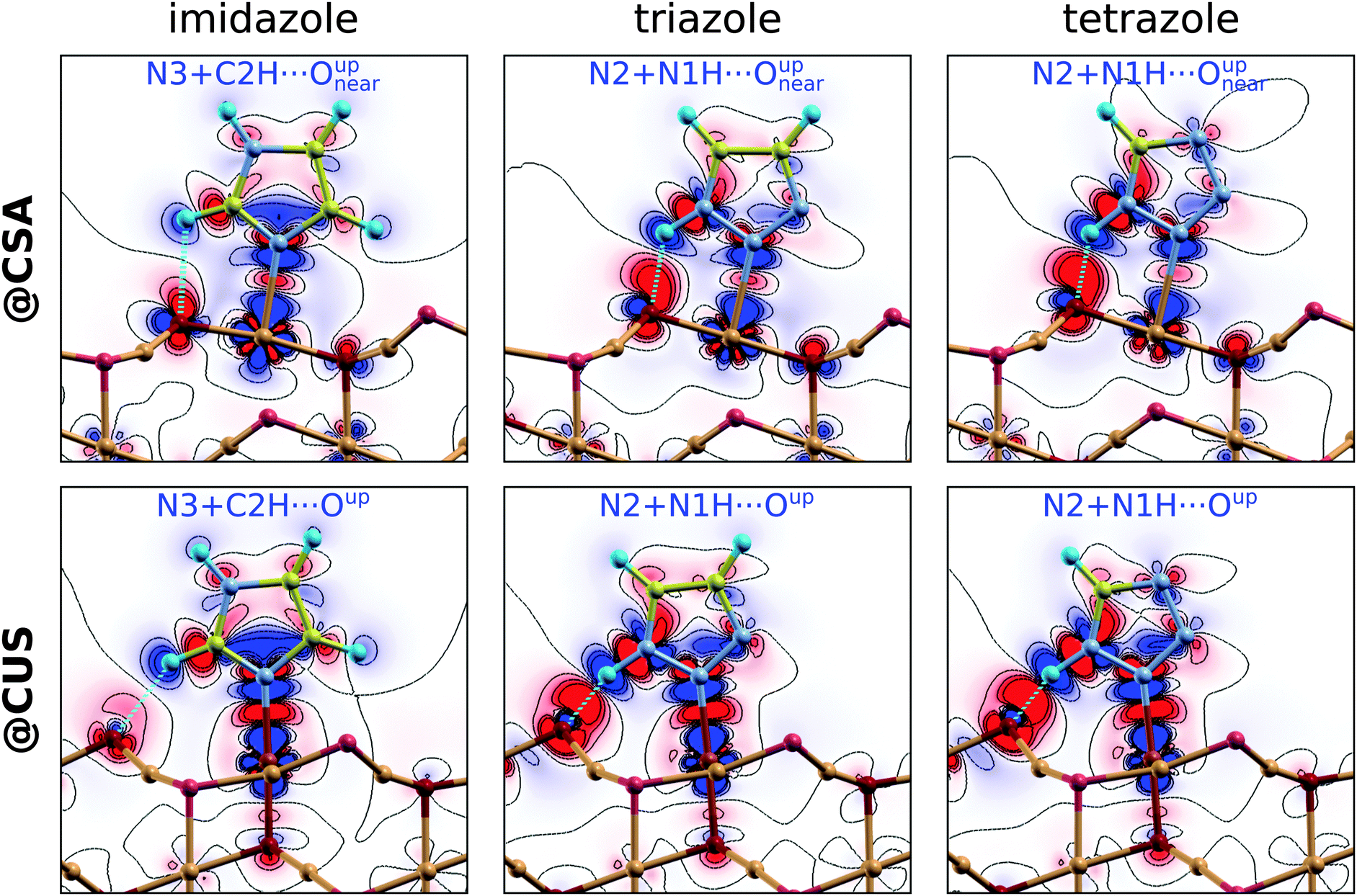 | ||
| Fig. 6 Electron charge density difference, Δρ(r), for imidazole, triazole, and tetrazole bonded at CSA (top) and CUS (bottom) sites. The plots are drawn with seven contours in a linear scale from −0.006 to +0.006 e a−30. The blue (red) color represents the electron deficit (excess) regions, i.e., electron charge flows from blue to red regions. | ||
A further analysis of the three-dimensional shape of Δρ(r) (not shown) reveals that the molecules interact with the surface through σ-type bonding, which is expected on the basis of the local symmetry of the N–Cu bonds. Even finer details of the molecule–surface interaction are provided for triazole in Fig. 7, where the same adsorption structure as in the Δρ(r) plot is considered, i.e., the N2 + N1H⋯Oup at the CSA and CUS sites. This figure displays the density of states projected (PDOS) to the molecule and the Cu atom beneath it, before and after the molecule–surface interaction sets in. These two cases will be termed before-interaction and after-interaction; for the former case, the molecule is up-shifted such that the N–Cu distance is 6 Å. In addition, the figure also shows the integrated local density of states (ILDOS) analysis as well as the density of states projected to individual molecular orbitals (MO-PDOS)49 of triazole, because these two techniques allow for unambiguous assignment of molecular PDOS peaks to individual molecular orbitals (MOs).
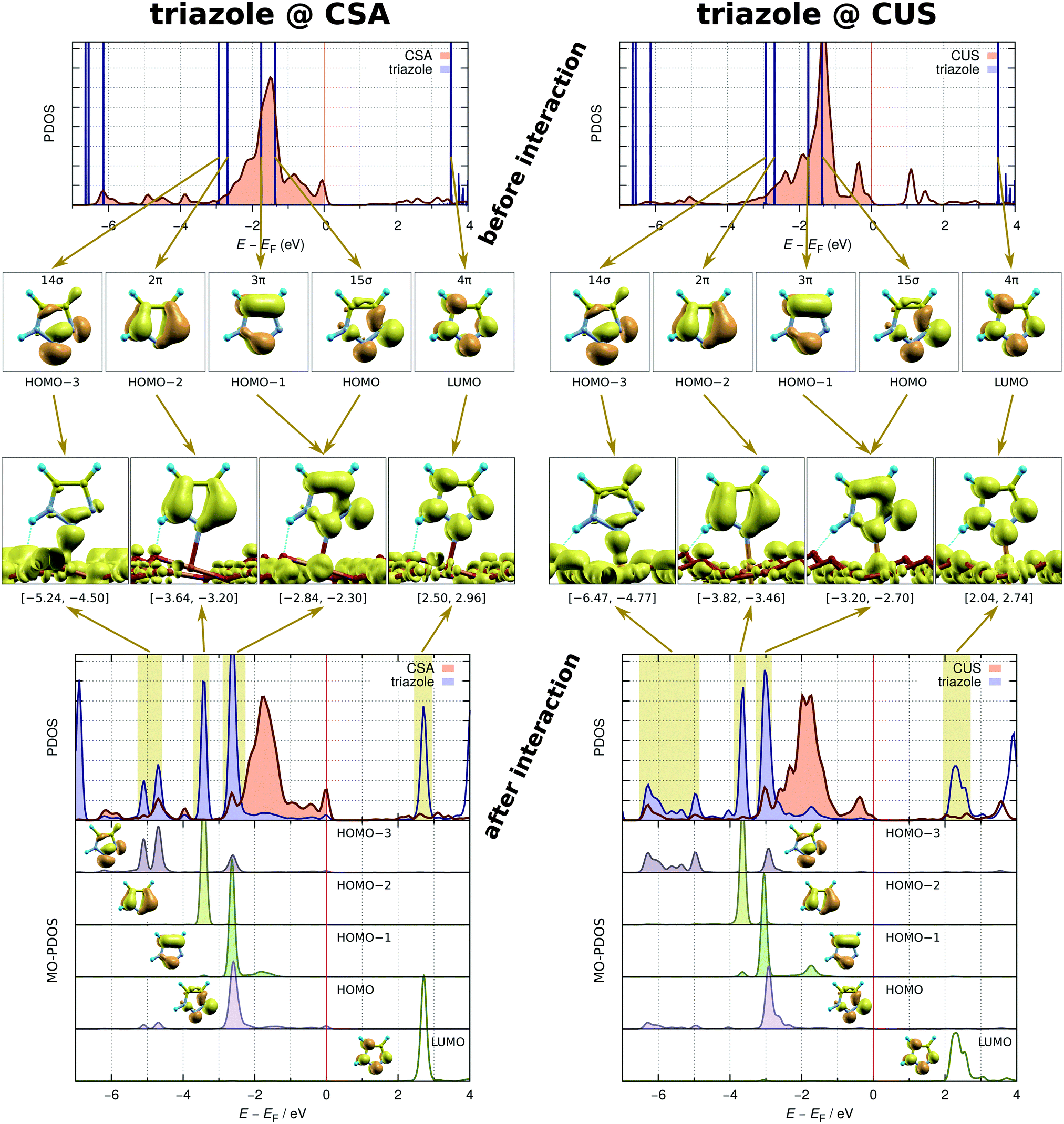 | ||
| Fig. 7 Bonding analysis of triazole at the CSA (left) and CUS (right) sites. From top to bottom: (1) density of states projected (PDOS) to the molecule (blue) and the Cu atom beneath it (brown) before the interaction sets in (i.e., the molecule is ∼6 Å above the surface). (2) Signed square modulus of molecular orbitals, i.e., sgn[ϕ(r)] × |ϕ(r)|2. (3) Integrated local density of states (ILDOS) of the interacting molecule/surface system; the integrated energy ranges are stated below the ILDOS plots and are also indicated by pale vertical bands beneath. (4) PDOS plots of the interacting molecule/surface system; below the PDOS plots the density of states is projected to individual molecular orbitals (MO-PDOS). | ||
The before-interaction PDOS plots reveal that four molecular orbitals (from HOMO−3 to HOMO (highest-occupied MO)) lie at the position of the metal d-band and are therefore considered in the analysis of the molecule–surface bonding; the LUMO (lowest-unoccupied MO) state, which lies more than 3 eV above the valence band edge, is also considered. Two among these MOs are σ-type orbitals (HOMO−3 and HOMO) and three are π-type orbitals (HOMO−2, HOMO−1, and LUMO). Upon interaction these molecular states downshift in energy. The downshift is larger and the molecular PDOS is more broadened for the CUS sites in accordance with the stronger molecule–surface interaction at the CUS compared to that at the CSA site. The downshift is the largest for the HOMO−3 σ-orbital, being about 2 eV at the CSA sites and about 3 eV at the CUS sites, and its PDOS peaks are the most broadened. These PDOS peaks are located at around −5 eV for the CSA sites and at around −6 eV for the CUS sites. The ILDOS analysis clearly reveals that only these low lying molecular peaks are involved in the interaction with the copper states,|||| whereas all the other molecular peaks are non-bonding with respect to the molecule–surface interaction. The MO-PDOS plots show that, in addition to HOMO−3, also the HOMO orbital marginally participates in the bonding low-lying peaks, but its predominant contribution is in the uppermost valence (occupied) molecular PDOS peak located at about −2.5 eV at the CSA sites and at about −3 eV at the CUS sites. The MO-PDOS plots further reveal that there is no back-donation of charge from the surface into the molecular LUMO, because the LUMO remains completely empty upon interaction, that is, the MO-PDOS shows no participation of the LUMO in the molecule–surface valence states.
3.4 Dispersion corrections
The results presented in the preceding Sections 3.1–3.3 were obtained using the PBE energy functional, which cannot describe van der Waals dispersion interactions. To shed some light onto how dispersion interactions affect the adsorption characteristics, they were estimated for the high coverage (1 × 1) adsorption phases and are presented in Table 1, which compares the corresponding PBE and dispersion corrected PBE-D′′ adsorption energies. In the following text the adsorbed molecules that form the hydrogen bond with Oup or Oupnear ions are referred as Oup modes and the molecules that form the hydrogen bond with Osub ions as Osub adsorption modes. Dispersion correction enhances the molecular adsorption bonding by about 0.3 eV for the Oup and about 0.5 eV for the Osub adsorption modes for all the three molecules; the N–Cu bonds correspondingly shorten by about 0.05 and 0.01 Å at the CSA and CUS sites, respectively. The reason that the Osub adsorption modes are stabilized stronger by dispersion interactions than the Oup modes is that for the former molecules are located closer to the surface than for the latter, because they are tilted into the CuCUS vacancies (see the respective snapshots in Fig. 1). Due to this stronger stabilization of the Osub adsorption modes, the relative stability of modes is altered for tetrazole at the CSA sites, i.e., without dispersion correction the Oup adsorption mode is by 0.2 eV more stable than the Osub adsorption mode (note that for triazole the two modes are almost degenerate, while for imidazole the Osub mode is more stable). But when dispersion correction is taken into account, the Osub adsorption modes are the most stable at the CSA sites for all the three molecules. The net stabilization due to dispersion interactions at the CSA sites—calculated as the difference between the most stable PBE and PBE-D′′ adsorption modes—is therefore stronger for imidazole and triazole (about 0.5 eV) than for tetrazole (0.3 eV); see the Δbest column in Table 1. In contrast, at the CUS sites, which lacks the Osub adsorption modes, the stabilization is only about 0.3 eV for all the three molecules.| Adsorption system | Site | Molecule | Adsorption structure | E ads (eV) | PBE-D′′–PBE | ||
|---|---|---|---|---|---|---|---|
| PBE | PBE-D′′ | Δ (eV) | Δbest (eV) | ||||
| a Respective adsorption energies are calculated with respect to high-symmetry position of CuCUS ions in the bare substrate. | |||||||
| (1 × 1)-mol@Cu2O(111)-w/o-CuCUS | CSA | ||||||
| Imidazole | N3 + C2H⋯Oupnear | −0.15 | −0.43 | −0.28 | −0.53 | ||
| N3 + C2H⋯Osub | −0.32 | −0.85 | −0.53 | ||||
| Triazole | N2 + N1H⋯Oupnear | −0.47 | −0.71 | −0.24 | −0.51 | ||
| N2 + N1H⋯Osub | −0.46 | −0.98 | −0.52 | ||||
| Tetrazole | N2 + N1H⋯Oupnear | −0.52 | −0.75 | −0.23 | −0.30 | ||
| N2 + N1H⋯Osub | −0.33 | −0.82 | −0.49 | ||||
| (1 × 1)-mol@Cu2O(111) | CUSa | ||||||
| Imidazole | N3 + C2H⋯Oup | −1.39 | −1.74 | −0.35 | |||
| Triazole | N2 + N1H⋯Oup | −1.71 | −2.02 | −0.31 | |||
| Tetrazole | N2 + N1H⋯Oup | −1.75 | −2.04 | −0.29 | |||
Dispersion interactions stabilize the adsorption structures mainly through the enhanced molecule–surface bonding. However, for the high-coverage phases the dispersion forces also affect the lateral molecule–molecule interactions due to proximity of neighboring molecules. The lateral molecule–molecule dispersion interactions were estimated*** only for imidazole at the CSA and CUS sites and amount to −0.04 eV per molecule for both sites. Given the empirical nature of the utilized dispersion correction, it can be straightforwardly inferred that it gives slightly weaker lateral molecule–molecule dispersion interactions for triazole and tetrazole compared to imidazole, because an N atom has a smaller C6 coefficient than a C atom. The current results therefore imply that the lateral molecule–molecule dispersion interactions contribute about 10% to the overall dispersion enhancement of the adsorption energy at the high coverage.
Dispersion corrections affect the adsorption phase-diagrams of Fig. 5 in two ways: (i) the enhancement of the adsorption energy downshifts the borders between the (1 × 1) adsorption phases and bare Cu2O(111)-w/o-CuCUS, i.e., the brown region in Fig. 5 is displaced to lower values of μmol. (ii) For imidazole and triazole the border between (1 × 1)-mol@Cu2O(111) [green region] and (1 × 1)-mol@CSA [yellow region] shifts to the left, i.e., to lower values of μO, because the dispersion bonding enhancement is larger at the CSA sites (≈0.5 eV) than at the CUS (≈0.3 eV) sites. In contrast, for tetrazole the dispersion bonding enhancement is almost the same at the two sites and the pertinent border remains unaffected. The resulting, dispersion corrected phase diagrams are presented in Fig. S3 in the ESI.†
Finally, some caution is in place concerning the currently used semi-empirical dispersion correction. Namely, PBE-D was currently reparametrized (PBE-D′′) to match the experimental adsorption energy of flat lying benzene on Cu(111), because the original PBE-D often overestimates the molecule–surface bonding.27,29,30,50 However, the current adsorption systems consist of Cu2O oxide surfaces and molecules that bond perpendicularly or tilted to the surface. For this reason the current dispersion corrected results may be taken more qualitatively than quantitatively.
4 Conclusions
Adsorption bonding of imidazole, triazole, and tetrazole to Cu2O(111)-w/o-CuCUS and Cu2O(111) was characterized by means of DFT calculations. The difference between these two surfaces is that the Cu2O(111)-w/o-CuCUS lacks the reactive CUS sites and is consequently thermodynamically more stable than Cu2O(111). We showed that the bonding of current azole molecules at CUS sites on Cu2O(111) is by about 1 eV stronger than at CSA sites on Cu2O(111)-w/o-CuCUS. The bonding at CUS sites is so strong that it compensates the thermodynamic deficiency of Cu2O(111), making it more stable than Cu2O(111)-w/o-CuCUS, provided the conditions are not too oxygen rich and/or azole lean. This finding, together with the previously observed trend that azoles bond significantly stronger to low coordinated defects on metallic Cu surfaces than to high coordinated flat facets,32,33 indicates that azoles have a strong affinity to preferentially adsorb at reactive undercoordinated or unsaturated surface sites, which in turn tentatively suggests that their corrosion inhibition capability may, at least in part, stem from their ability to passivate reactive surface sites.As for the chemical nature of the molecule–surface interaction, we showed that the current azole molecules preferentially bind to Cu2O surfaces via a single σ-type N–Cu bond and concomitantly form a hydrogen bond with a nearby surface O ion. Adsorption bonding is further enhanced by van der Waals dispersion interactions, in the range 0.23–0.53 eV, depending on specifics of the adsorption structure. This is a sizable enhancement, because it represents about 50–60% of the total adsorption bonding at the CSA and about 15–20% at CUS sites. At large intermolecular separations (or at a low coverage), where the lateral dipole–dipole effects are sufficiently small, the magnitude of adsorption energy follows the imidazole ≈ triazole > tetrazole order, but at a high coverage the trend is altered, because imidazole displays repulsive and tetrazole attractive lateral dipole–dipole interactions. Despite these differences in lateral interactions, atomistic thermodynamics analysis reveals that for all the three molecules only three among the considered phases appear in the phase diagrams: high coverage (1 × 1)-mol@Cu2O(111) and (1 × 1)-mol@Cu2O(111)-w/o-CuCUS molecular phases, and clean Cu2O(111)-w/o-CuCUS.
Acknowledgements
This work has been supported by the Slovenian Research Agency (Grant No. P2-0148 and P2-0393).References
- Y. I. Kuznetsov and L. P. Kazansky, Russ. Chem. Rev., 2008, 77, 219–232 CrossRef CAS PubMed.
- M. M. Antonijević, S. M. Milić and M. B. Petrović, Corros. Sci., 2009, 51, 1228–1237 CrossRef PubMed.
- J. O. Bockris and A. K. N. Reddy, Modern Electrochemistry, Kluwer Academic/Plenum Publishers, New York, Boston, Dordrecht, London, Moscow, 2nd edn, 2000, vol. 2B Search PubMed.
- C. D. Taylor, A. Chandra, J. Vera and N. Sridhar, Faraday Discuss., 2015, 180, 459–477 RSC.
- N. Kovačević and A. Kokalj, J. Phys. Chem. C, 2011, 115, 24189–24197 Search PubMed.
- N. Kovačević and A. Kokalj, Corros. Sci., 2013, 73, 7–17 CrossRef PubMed.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, NACE, Cebelcor, Houston, Texas, 2nd edn, 1974 Search PubMed.
- Y. Jiang, J. B. Adams and D. Sun, J. Phys. Chem. B, 2004, 108, 12851–12857 CrossRef CAS.
- A. Kokalj and S. Peljhan, J. Phys. Chem. C, 2015, 119, 11625–11635 CAS.
- A. Kokalj, Faraday Discuss., 2015, 180, 415–438 RSC.
- O. Blajiev and A. Hubin, Electrochim. Acta, 2004, 49, 2761–2770 CrossRef CAS PubMed.
- T. Bredow and G. Pacchioni, Surf. Sci., 1997, 373, 21–32 CrossRef CAS.
- M. Casarin and A. Vittadini, Surf. Sci., 1997, 387, L1079–L1084 CrossRef CAS.
- M. Casarin, C. Maccato, N. Vigato and A. Vittadini, Appl. Surf. Sci., 1999, 142, 164–168 CrossRef CAS.
- A. Soon, T. Söhnel and H. Idriss, Surf. Sci., 2005, 579, 131–140 CrossRef CAS PubMed.
- M. Li, J.-y. Zhang, Y. Zhang, G.-f. Zhang and T.-m. Wang, Appl. Surf. Sci., 2011, 257, 10710–10714 CrossRef CAS PubMed.
- H. Wu, N. Zhang, H. Wang and S. Hong, Chem. Phys. Lett., 2013, 568–569, 84–89 CrossRef CAS PubMed.
- L. I. Bendavid and E. A. Carter, J. Phys. Chem. C, 2013, 117, 26048–26059 CAS.
- Y. Shen, F. H. Tian, S. Chen, Z. Ma, L. Zhao and X. Jia, Appl. Surf. Sci., 2014, 288, 452–457 CrossRef CAS PubMed.
- M. Altarawneh, M. W. Radny, P. V. Smith, J. C. Mackie, E. M. Kennedy, B. Z. Dlugogorski, A. Soon and C. Stampfl, Appl. Surf. Sci., 2010, 256, 4764–4770 CrossRef CAS PubMed.
- I. C. Man, S. G. Soriga and V. Pârvulescu, Chem. Phys. Lett., 2014, 604, 38–45 CrossRef CAS PubMed.
- Z. Wang, X. Liu, D. Rooney and P. Hu, Surf. Sci., 2015, 640, 181–189 CrossRef CAS PubMed.
- A. Soon, M. Todorova, B. Delley and C. Stampfl, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 125420 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- V. Barone, M. Casarin, D. Forrer, M. Pavone, M. Sambi and A. Vittadini, J. Comput. Chem., 2009, 30, 934–939 CrossRef CAS PubMed.
- A. Kokalj and S. Peljhan, Langmuir, 2010, 26, 14582–14593 CrossRef CAS PubMed.
- E. R. McNellis, J. Meyer and K. Reuter, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 205414 CrossRef.
- K. Tonigold and A. Groß, J. Chem. Phys., 2010, 132, 224701 CrossRef PubMed.
- J. Klimeš and A. Michaelides, J. Chem. Phys., 2012, 137, 120901 CrossRef PubMed.
- A. Kokalj, S. Peljhan, M. Finšgar and I. Milošev, J. Am. Chem. Soc., 2010, 132, 16657–16668 CrossRef CAS PubMed.
- S. Peljhan and A. Kokalj, Phys. Chem. Chem. Phys., 2011, 13, 20408–20417 RSC.
- S. Peljhan, J. Koller and A. Kokalj, J. Phys. Chem. C, 2014, 118, 933–943 CAS.
- A. Kokalj, S. Peljhan and J. Koller, J. Phys. Chem. C, 2014, 118, 944–954 CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- Ultrasoft pseudopotentials for H, C, N, O, and Cu were taken from the Quantum Espresso PseudoPotential Download Page: http://www.quantum-espresso.org/pseudopotentials (files: H.pbe-rrkjus.UPF, C.pbe-rrkjus.UPF, N.pbe-rrkjus.UPF, O.pbe-rrkjus.UPF and Cu.pbe-d-rrkjus.UPF), 2015.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- A. Kokalj, J. Mol. Graphics Modell., 1999, 17, 176–179 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- N. Marzari, D. Vanderbilt, A. De Vita and M. C. Payne, Phys. Rev. Lett., 1999, 82, 3296–3299 CrossRef CAS.
- D. Costa, T. Ribeiro, F. Mercuri, G. Pacchioni and P. Marcus, Adv. Mater. Interfaces, 2014, 1, 1300072 Search PubMed.
- M. Finšgar, S. Peljhan, A. Kokalj, J. Kovač and I. Milošev, J. Electrochem. Soc., 2010, 157, C295–C301 CrossRef PubMed.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, Florida, USA, 74th edn, 1993 Search PubMed.
- C. Li, F. Wang, S. Li, Q. Sun and Y. Jia, Phys. Lett. A, 2010, 374, 2994–2998 CrossRef CAS PubMed.
- K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 65, 035406 CrossRef.
- L. Bengtsson, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 12301–12304 CrossRef CAS.
- A. Kokalj, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 045418 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision A.1, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- A. Baby, G. Fratesi, S. R. Vaidya, L. L. Patera, C. Africh, L. Floreano and G. P. Brivio, J. Phys. Chem. C, 2015, 119, 3624–3633 CAS.
- G. Mercurio, E. R. McNellis, I. Martin, S. Hagen, F. Leyssner, S. Soubatch, J. Meyer, M. Wolf, P. Tegeder, F. S. Tautz and K. Reuter, Phys. Rev. Lett., 2010, 104, 036102 CrossRef CAS.
Footnotes | ||||||
| † Electronic supplementary information (ESI) available: Calculated surface free energies (Fig. S1), charge density difference plots of hydrogen bonds (Fig. S2), and dispersion corrected adsorption phase diagrams (Fig. S3). See DOI: 10.1039/c5cp03647j | ||||||
| ‡ The reason for the reparametrization is that the original PBE-D overestimates a molecular bonding to copper surfaces,27–30 which can be attributed to a too large C6 value of a Cu atom.29 Both the PBE-D′ and the current PBE-D′′ are re-parametrized so as to match the experimental adsorption energy of a flat lying benzene on Cu(111). For PBE-D′ this was achieved in a dirty way by adjusting the s6 scaling parameter to the value of 0.47 (the original value is 0.75), because the Quantum ESPRESSO code did not allow to set the C6 values in the input. Consequently, PBE-D′ cannot handle well the lateral molecule–molecule dispersion interactions and is therefore mainly applicable for the molecule copper bonding at low coverage. In contrast, for PBE-D′′ the Quantum ESPRESSO code was modified to allow the specification of C6 parameters in the input and the C6 parameter of Cu was set to the value of 140 Ry Bohr−6 (the original value is 375 Ry Bohr−6), while the s6 was kept at its original value of 0.75. | ||||||
| § The oxygen poor limit can be defined as a point where the bulk Cu2O decomposes into bulk Cu and O2 gas, whereas at the oxygen rich limit oxygen gas condenses on the surface. | ||||||
| ¶ The issue of dependence of Eads on the coverage is relevant, because azole molecules have large permanent dipole moments that can result in long-range lateral dipole–dipole interactions between adsorbed molecules.5,32,47 | ||||||
| || In contrast with the CSA site, at the CUS site the N2 + N3 adsorption mode is not stable, i.e., it transforms to N2 bonding during the geometry optimization. | ||||||
| ** The reason that at a low coverage triazole bonds as strongly as imidazole to Cu2O(111)-w/o-CuCUS is due to the hydrogen bonding effects (this issue will be further discussed in Section 3.3). | ||||||
| †† For imidazole at the CUS site the high coverage (1 × 1) phase is the most stable at Δμmol > −1.33 eV. Below this value the (2 × 2) phase is the most stable and near its intercept with the clean surface line at Δμmol ≈ −1.6 eV also the low coverage (3 × 3) phase becomes competitive. | ||||||
| ‡‡ These values were calculated with the thermochemistry utility of the Gaussian 09 program package.48 | ||||||
| §§ It should be noted that for triazole at the CSA site, N2 + N1H⋯Oupfar is marginally more stable at a low coverage than the considered N2 + N1H⋯Oupnear (see Fig. 1), but the difference is insignificant. | ||||||
| ¶¶ The Δρ(r) plots clearly reveal that at the CSA site the strength of the N–Cu bond decreases from imidazole to tetrazole, which is consistent with the increasing N–Cu bond length in the same direction. In contrast, at the CUS site the N–Cu electron charge accumulation lobes as well as the N–Cu bond lengths appear to be very similar for all the three molecules. This suggests that at a low coverage the molecule–surface interaction should be the weakest for imidazole, because it lacks the strong N–H⋯O hydrogen bond. But this is not the case. The reason can be attributed to molecular chemical hardness, which increases from imidazole to tetrazole.5,6 Namely, at the CUS site the molecular electronic structure is sufficiently perturbed due to a strong molecule–surface interaction and the hybridization between molecular and copper states is the easiest for imidazole, which is chemically the softest among the three molecules. | ||||||
| |||| It should be noted that the ILDOS analysis shows only four plots per site, although five MOs are considered. The reason is that the HOMO−1 and HOMO states are located in the same energy region and cannot be separated, i.e., their PDOS peaks overlap, which is also evident from the respective MO-PDOS plots. | ||||||
*** Lateral molecule–molecule dispersion interactions (Elateraldisp) were estimated, by performing a set of single-point calculations, as follows:
|
| This journal is © the Owner Societies 2015 |