
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assessing backbone solvation effects in the conformational propensities of amino acid residues in unfolded peptides†
Niranjan V.
Ilawe
a,
Alexandra E.
Raeber
a,
Reinhard
Schweitzer-Stenner
*b,
Siobhan E.
Toal‡
b and
Bryan M.
Wong
 *a
*a
aDepartment of Chemical & Environmental Engineering, and Materials Science & Engineering Program, University of California, Riverside, 900 University Avenue, Riverside, CA 92521, USA. E-mail: bryan.wong@ucr.edu; Web: http://www.bmwong-group.com Tel: +1-951-827-2163
bDepartment of Chemistry, Drexel University, 3141 Chestnut Street, Philadelphia, PA 19104, USA. E-mail: rschweitzer-stenner@drexel.edu; Web: http://www.schweitzer-stenner.com Tel: +1-215-895-2268
First published on 27th August 2015
Abstract
Conformational ensembles of individual amino acid residues within model GxG peptides (x representing different amino acid residues) are dominated by a mixture of polyproline II (pPII) and β-strand like conformations. We recently discovered rather substantial differences between the enthalpic and entropic contributions to this equilibrium for different amino acid residues. Isoleucine and valine exceed all other amino acid residues in terms of their rather large enthalpic stabilization and entropic destabilization of polyproline II. In order to shed light on these underlying physical mechanisms, we performed high-level DFT calculations to explore the energetics of four representative GxG peptides where x = alanine (A), leucine (L), valine (V), and isoleucine (I) in explicit water (10 H2O molecules with a polarizable continuum water model) and in vacuo. We found that the large energetic contributions to the stabilization of pPII result, to a major extent, from peptide–water, water–water interactions, and changes of the solvent self-energy. Differences between the peptide–solvent interaction energies of hydration in pPII and β-strand peptides are particularly important for the pPII ⇌ β equilibria of the more aliphatic peptides GIG and GLG. Furthermore, we performed a vibrational analysis of the four peptides in both conformations and discovered a rather substantial mixing between water motions and peptide vibrations below 700 cm−1. We found that the respective vibrational entropies are substantially different for the considered conformations, and their contributions to the Gibbs/Helmholtz energy stabilize β-strand conformations. Taken together, our results underscore the notion of the solvent being the predominant determinant of peptide (and protein) conformations in the unfolded state.
Introduction
Our current understanding of protein folding processes relies on a detailed knowledge of the conformations they can adopt in their folded and unfolded states. For some time it was commonly believed that the unfolded states of peptides and proteins were characterized by a random sampling of all possible backbone conformations.1–4 This predominant view is largely based on Flory's classical independent site model, which allows the various protein residues to sample the entire allowed region of the Ramachandran space.2 However, over the last 15 years, considerable evidence has accumulated suggesting that significantly more restricted conformational ensembles of individual amino acid residues are present in unfolded peptides5–12 and restricted coil libraries.13,14 Specifically, amino acid residues of short peptides in solution as well as in truncated coil libraries were found to exhibit much more restricted conformational sampling and to differ in terms of their conformational propensities. A recent conformational analysis of GxG peptides in water based on vibrational spectroscopic and NMR data revealed that most residues do predominantly sample the upper left quadrant of the Ramachandran plot, which can be subdivided into two sub-conformational ensembles associated with the β-strand and less extended polyproline II (pPII) conformations.12,15,16 Individual amino acid residues differ in terms of the equilibrium between these two conformations – while alanine shows a clear preference for pPII, β-strand like conformations are slightly more preferred for valine and isoleucine. The ΔGpPII-β values associated with these equilibria range between −3.5 and 2 kJ mol−1, with most values lying in the R·T energy range.17 However, these ΔGpPII-β values were found to be composed of much larger ΔH and ΔS values, which also exhibit much larger variations with changing residues than ΔG. For two subsets of data, the analysis resulted in isoequilibria, e.g. temperatures at which the ΔG values of the different members of the subset were practically identical. This was interpreted as an indication that peptide–water interactions are the key determinant of the residue's propensities. In this context it was particular striking that valine and isoleucine were found to exhibit rather large ΔH and TΔS values (in the 40–60 kJ mol−1 range at room temperature) that exceed those of other residues with aromatic or aliphatic side chains. The fact that the ΔH and TΔS values of isoleucine exceed the corresponding values for the isomeric amino acid leucine by a factor of ∼2.5 was particular astonishing and has thus far been rationalized, in general terms, as an intricate interplay between backbone and side chain solvation.17 However, these results cannot be understood in terms of solvent accessible surface areas that are normally considered to be the decisive parameters in side chain–solvent interactions. Specifically, since isoleucine (I) and leucine (L) have practically the same accessible surface area, which exceeds that of valine (V),18 this suggests a hierarchy with I, L > V, contrary to the observed I > V ≫ L hierarchy.Multiple lines of evidence suggest that interactions between unfolded peptides/proteins and the solvent are pivotal for conformational preferences exhibited by individual amino acid residues. This notion is particularly applicable to alanine which exhibits the highest propensity for pPII, a conformation generally found in proline-rich proteins and peptides.19–28 In non-aqueous solvents like DMSO and primary alcohols, the sampling of pPII is generally reduced or even absent.25,28 The role of water in stabilizing the pPII conformation of alanine residues was first proposed by Han et al. based on density functional theory (DFT) calculations of N-acetyl-L-alanine N′-methylamide (alanine dipeptide) in complexes with four water molecules.19 These authors attributed the stabilization of pPII to water bridges between CO and NH groups of the two peptide linkages of the molecule. Garcia, by means of molecular dynamics (MD) simulations with a modified AMBER force field, suggested that pPII conformations allowed for an optimal packing of water molecules in the hydration shell.21 Fleming et al. proposed a correlation between pPII propensity and changes in the solvent accessibility area of side chains associated with transitions between pPII and β-strand like conformations.18 However, this notion is at odds with results of MD studies reported by Mezei et al., who found instead that the pPII preference of alanine over β-strand results from stronger backbone–water interactions in the former.24 Computational studies by Drozdov et al.20 as well as Avbelj and Baldwin29,30 led these authors to suggest a more indirect role of solvation in the stabilization of pPII, namely the screening of electrostatic interactions between peptide groups, which would produce a preference for more extended β-strand conformations. Amino residues with side chains bulkier than alanine were predicted to perturb the hydration shell and thus destabilize pPII, a notion consistent with experimental results.31 A recent DFT-based study by Lanza and Chiacchio on N-acetyl-L-alanine amide complexes with up to 13 water molecules revealed the existence of water clusters that were hydrogen bonded to the functional groups of the peptide backbone (i.e. CO and NH). They concluded that pPII conformations exhibit an energetic preference that leads to a greater reduction of water mobility,32 further suggesting that pPII is energetically favored and entropically disfavored, in agreement with experimental data.31 A more recent study by these authors confirmed this view in which the number of water molecules in the hydration shell was substantially increased up to 37.33 A somewhat lower number of water molecules (22) was found to be necessary for obtaining stabilization energies of pPII relative to the β-strand that are comparable with experimentally obtained values.
The present study is aimed at shedding some light on the influence of side chains on the backbone hydration of GxG peptides. Toal et al. recently reported rather diverse enthalpy and entropy values for the pPII ⇌ β-strand equilibria of fifteen GxG peptides in aqueous solution.31 To this end, we performed several DFT calculations on a selected set of the earlier investigated GxG peptides surrounded by 10 explicit water molecules with a polarizable continuum water model. As representative peptides, we selected a set of aliphatic amino acid residues, namely, alanine (A), leucine (L), valine (V), and isoleucine (I) for our studies, as these residues showed the largest variance in thermodynamic values.17 Alanine was also selected owing to its exceptional high pPII propensity (0.72 in GAG) for which multiple molecular dynamics (MD) and some quantum chemical studies suggest hydration as the main reason.20–24,29,30,32,34–37 The other three aliphatic side residues were selected because the above thermodynamic analysis has yielded large and surprisingly different ΔH and TΔS values for their respective pPII ⇌ β equilibria. The experimental ΔH and ΔS values for these peptides as reported by Toal et al. are displayed in Fig. 1.31 In this study, we focus on determining the energetics of the pPII ⇌ β equilibria for the investigated peptides rather than on a thorough exploration of the energy landscapes of these peptides. In view of the established preponderance of pPII and β-strand conformations in their Ramachandran plots, this restriction is justified.12,15,16
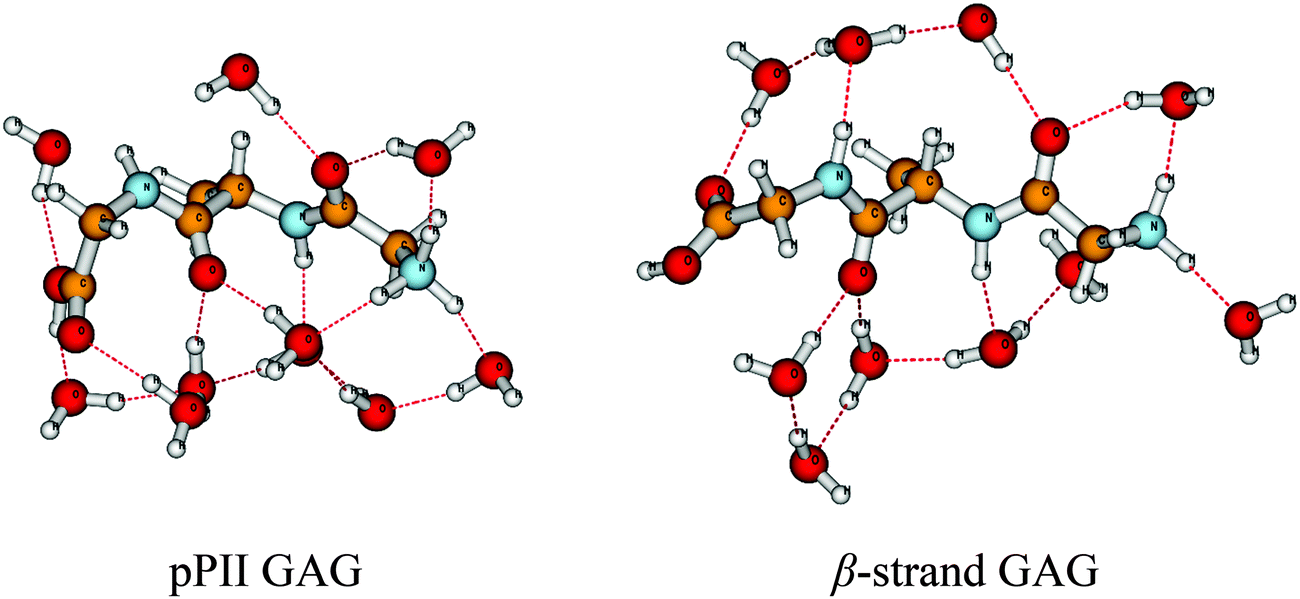 | ||
| Fig. 1 pPII and β-strand conformation of a cationic GAG + 10 H2O complex obtained from geometry optimizations as described in the text. | ||
Computational methods
All quantum chemical calculations in this study utilized density functional theory (DFT) calculations in conjunction with the dispersion-corrected, range-separated ωB97X-D functional as implemented in the Gaussian 09 package. Previous investigations by us38–40 and others41 have shown that these dispersion corrections in conjunction with nonlocal exchange are essential for accurately calculating both the hydrogen-bonding interactions and thermochemistry of water-based complexes. All geometries were optimized with a large cc-pVTZ basis set in the presence of an aqueous polarizable continuum model (PCM). The specific PCM model used in this work is the implementation devised by Tomasi and co-workers42–46 which creates a solute cavity via a set of overlapping spheres to calculate the solvent reaction field. The initial geometries of the water complexes were taken from Lanza and Chiacchio.32 Geometry optimizations and harmonic frequencies at the same level of theory were calculated to verify that these stationary points were local minima.Results and discussion
Energetics of GxG pPII ⇌ β equilibria in water
Quantum calculations were performed for the selected cationic GxG peptides in vacuo and in a bath of 10 explicit H2O molecules with the entire system embedded in a polarizable continuum solvent model, as described in the Computational methods section. The number of water molecules is substantially less than what has recently been employed by Lanza and Chiacchio.33 However, as shown in Fig. 1 our approach allowed us to provide a minimal hydration of all functional groups of the peptide backbone, which includes 2 water molecules as hydrogen-bonded donors for the two carbonyl groups and one water molecule as a hydrogen-bond acceptor for the two amide protons. The remaining six water molecules were used to hydrate the terminal groups and to connect the different water molecules hydrogen bonded to the peptide. It is important to note that there exists many conformational isomers in each tripeptide, and we have only focused on a single conformer in each of the four GxG tripeptides. However, the goal of this study is the comparison of pPII ⇌ β equilibria rather than an accurate calculation of absolute values of energy differences or a thorough sampling of the conformational space of water molecules in the hydration shell. The chosen setup of water molecules does not hydrate the side chains of the chosen x-residues, hence the contributions from side chain solvent interactions are neglected in our study (as they were in the studies of Lanza and Chiacchio32,33). Furthermore, the previous studies of Lanza and Chiacchio investigated both AAA and AdP-type peptides, while our work focuses on the different GxG peptide geometries. While a comparison between our work and Lanza and Chiacchio's study is not entirely straightforward, we were able to confirm that the backbone geometry for our computed GAG structure has a similar topology with their previously published structure for AdP. This comparison, as well as our results described further below, demonstrate that our approach is capable of elucidating the influence of the side chains on backbone hydration. We selected the fully protonated form to directly correspond to the experiments this study is referring to.12,16,31 Since we were solely interested in the pPII ⇌ β equilibria of the investigated peptides, we carried out two fully unconstrained geometry optimizations per peptide, one starting in the pPII conformation and the other in the β-strand region. After a full optimization, a normal mode analysis was carried out. These calculations served three purposes: (1) determining the internal energy difference between β and pPII, (2) analyzing, in particular, the manifold of peptide modes that are vibrationally mixed with the hydration shell of 10 water molecules, and (3) checking for the appearance of any imaginary frequencies which would indicate that the observed conformation does not represent a true minimum in the energy landscape of the system.Tables 1 and 2 list the dihedral angles of the obtained optimized structures, the energy differences between the pPII and β-strand geometries, and the permanent dipole moments for the various GxG systems in vacuo and in explicit water, respectively. Table 2 also lists the dihedral angles of earlier reported maxima of pPII and β-strand sub-distributions obtained from amide I′ profiles and various J-coupling constants.12,15,16,47 Most of the obtained values are in reasonable and some in excellent agreement with the reported experimentally-derived values. Exceptions are, to a minor extent, the ϕ-value of the β-strand conformation of GLG and, to a major extent, the ψ-value of the β-strand GVG. The experimentally-obtained values of pPII are generally very well reproduced by our calculations. Generally, our structure optimization with the larger cc-pVTZ basis set does a better job of reproducing experimentally obtained positions of energy minima (population maxima) than MD simulations, which generally yield rather extended structures for β-strand conformations.27,48–54
| ϕ [°] | ψ [°] | ΔUe (kJ mol−1) | |μ| (D) | |
|---|---|---|---|---|
| GAG-pPII | −65.86 | 148.09 | 7.60 | 18.67 |
| GAG-β | −159.49 | 161.17 | 14.69 | |
| GLG-pPII | −72.94 | 153.38 | 4.89 | 18.31 |
| GLG-β | −115.17 | 153.68 | 14.06 | |
| GVG-pPII | −83.33 | 161.85 | 6.83 | 17.71 |
| GVG-β | −134.33 | 138.28 | 13.00 | |
| GIG-pPII | −80.58 | 163.63 | 5.10 | 17.55 |
| GIG-β | −132.13 | 131.66 | 12.85 |
| ϕ [°] | ψ [°] | ΔUe (kJ mol−1) | |μ| (D) | |
|---|---|---|---|---|
| GAG-pPII | −71.93 (−74) | 163.59 (152) | −21.24 | 3.89 |
| GAG-β | −137.84 (−115) | 131.55 (120) | 18.93 | |
| GLG-pPII | −70.42 (−76) | 160.81 (145) | −16.42 | 3.89 |
| GLG-β | −121.24 (−98) | 132.98 (145) | 20.13 | |
| GVG-pPII | −77.81 (−80) | 169.07 (170) | −18.25 | 3.70 |
| GVG-β | −130.26 (−120) | 118.29 (170) | 18.69 | |
| GIG-pPII | −78.84 (−77) | 169.58 (170) | −20.34 | 3.96 |
| GIG-β | −129.81 (−118) | 119.37 (115) | 18.39 |
Fig. 1 depicts the conformations resulting from the geometry optimization of the investigated GAG + 10 H2O configurations. The optimization process, to a major extent, has maintained the water-mediated hydrogen bonding network and the hydration of the backbone groups, consistent with the initial setup. These conformations resemble what Ben Naim has termed the HϕI interaction, which are hydrophilic in character.55 A notable difference is observed for the arrangements of the water molecules above the peptide backbone. In the β-strand conformation, a water-mediated hydrogen bonding loop is maintained between the N- and the C-terminal which involves hydrogen bonding to and from the amide group of the C-terminal and the carbonyl group of the N-terminal peptide. In the pPII conformation, however, this loop is broken owing to the oxygen atom of the C-terminal's water molecule being now hydrogen bonded to the amide proton of the C-terminal peptide. On the contrary, the hydrogen bonding network below the peptide backbone is more extended in the pPII conformation. A distance analysis revealed that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–O–H, NH⋯O, and H–O–H⋯OH2 distances are generally shorter in the β-strand than in the corresponding pPII conformations, thus stabilizing the former. Corresponding bending angles are also closer to their ideal 180° value in the β-strand conformation than in pPII. Such a stabilization of the β-strand conformation by hydrogen bonding was earlier reported by Mezei et al. for a polyalanine peptide in explicit water.24 Lanza and Chiacchio reported a similar finding for their AcANH2·13H2O complex.32
O⋯H–O–H, NH⋯O, and H–O–H⋯OH2 distances are generally shorter in the β-strand than in the corresponding pPII conformations, thus stabilizing the former. Corresponding bending angles are also closer to their ideal 180° value in the β-strand conformation than in pPII. Such a stabilization of the β-strand conformation by hydrogen bonding was earlier reported by Mezei et al. for a polyalanine peptide in explicit water.24 Lanza and Chiacchio reported a similar finding for their AcANH2·13H2O complex.32
Based on the above analyses, one is tempted to suspect an overall energetic stabilization of β-strand over pPII. However, the corresponding electronic energy differences, ΔUe(pPII,β) = Ue(pPII) − Ue(β), as plotted in Fig. 2 do not meet this expectation. These ΔUe(pPII,β) values can be directly compared to the experimental ΔH(pPII,β) (= H(pPII) − H(β)) values reported by Toal et al.31 Since no pressure and significant volume changes are involved, one can expect that the enthalpy ΔH equals the internal energy ΔUint; i.e. the Gibbs free energy equals the Helmholtz energy. Therefore, in the following, we interpret the experimentally-obtained ΔH values reported by Toal et al. as internal energies that can generally be decomposed into electronic, vibrational, rotational, and translational contributions. For our system we can certainly ignore translation and rotation and we can further expect that the electronic part exceeds by far the vibrational contribution (vide infra). Hence, it is justified to compare the computational ΔUe with the experimental ΔUint values reported by Toal et al.27 The calculated and experimental ΔU values are practically identical for GLG, whereas calculated values for GVG and GIG are lower than the experimental values by factors of 2 and 3, respectively. Surprisingly, our calculation leads to an overestimation of ΔU for GAG; the value we obtained more resembles the experimentally-obtained AAA and alanine dipeptide structures.27 However, our calculated values reproduced the experimental hierarchy of I > V > L, which is one of the primary goals of this study. The corresponding ΔU-values for GxG in vacuo are also displayed in Fig. 2. They are all positive, indicating a stabilization of the β-strand conformation and confirming that water is indeed pivotal for pPII stabilization. Interestingly, GLG and GIG in vacuo now exhibit very similar energies, as one would expect from the fact that their side chains are chemical isomers. This suggests that the dissimilarity between pPII/β energy differences obtained (both experimentally and computationally) for hydrated leucine and isoleucine (Fig. 1) is in part a direct result of differences in side-chain and backbone solvation.31 Since the only difference between these two residues is the position of –CH3 branching on the side-chain (i.e., β-branching in isoleucine and γ-branching is leucine), it is likely that the hydration shell about this group is the pivotal determinant. In general, these results strongly corroborate the notion that the unfolded state of peptides in water would be dominated by pPII-like conformations if the favorable hydration energy (Fig. 1) was not compensated and sometimes overcompensated by entropic contributions. They further suggest that the pPII ⇌ β equilibrium enthalpy/energy of GxG peptides is predominantly determined by peptide–solvent interactions.28 Large differences between ΔU (and also between corresponding ΔS values) for different amino acid residues can give rise to large disparities between their respective pPII ⇌ β equilibria at conditions significantly above and below their compensation temperatures. This further suggests a sequence dependence of the conformational entropies of unfolded states of peptides and proteins at high (thermal denaturation) and low temperatures (cold denaturation).56
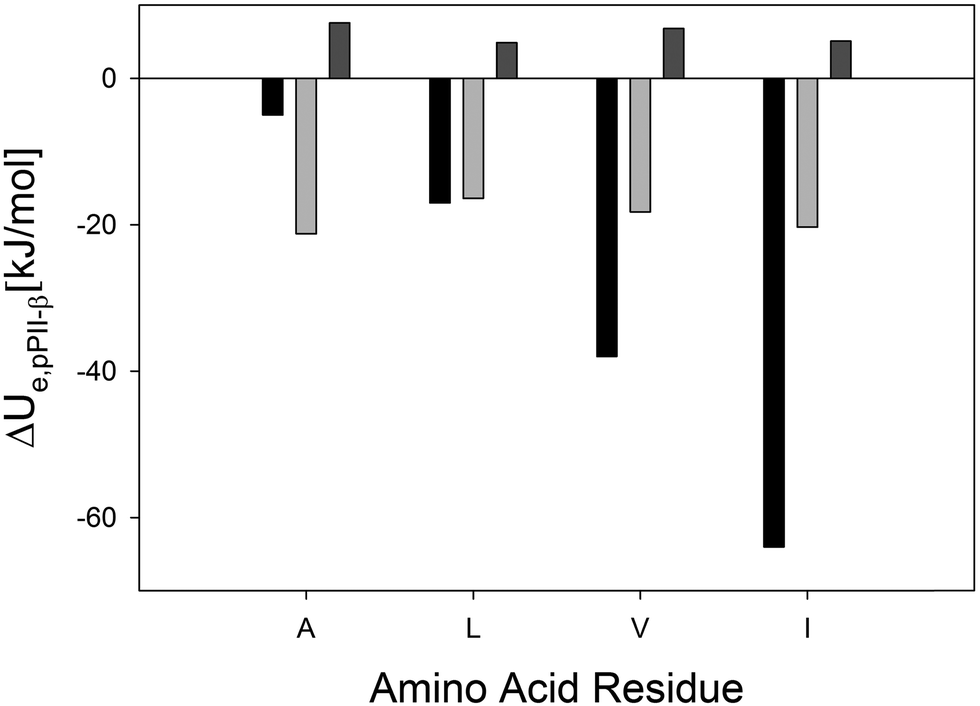 | ||
| Fig. 2 Comparison of experimental enthalpies (black bars)28 and calculated energies (light grey: explicit water; grey: in vacuo) for the pPII ⇌ β-strand equilibrium of the indicated amino acid residues in cationic GxG peptides in H2O. | ||
The discrepancy between the calculated energetics and the β-strand stabilization suggested by the hydrogen bond network analysis is surprising, but not unprecedented. Fleming et al., from an MD-based analysis of a 12-residue polyalanine in explicit water, found no evidence for water bridges in pPII at all.18 Nevertheless, they obtained a strong stabilization of pPII, attributable to peptide–water interactions, in agreement with our findings for all the peptides investigated. The DFT-based optimization of various AcANH2·nH2O complexes by Lanza and Chiacchio yielded water bridges for both conformations, but with more stable ones in β-strands. With regard to the total energy, however, hydration was found to stabilize pPII energetically.32 The underlying reason for this overall stabilization of pPII remains elusive in all these studies.
Taken together, our analysis suggests that it is difficult to pinpoint the reason for the enthalpic (energetic) stabilization of pPII in water. It is clear from our study and previous experimental results that the interaction between the peptide and the first hydration shell is pivotal in this regard. Furthermore, the present calculations confirm the notion of Toal et al.31 that the hydration energy difference between pPII and β-strand is heavily side chain dependent. However, the net electronic energies that emerged from our calculations are likely to be a superposition of competing contributions, which might involve through-space electrostatic in addition to through-hydrogen-bond interactions. To quantify these various interactions, we carried out a detailed analysis of the configurations for all four peptides using the energy-decomposition analysis of Mirkin and Krimm.57 Within this energy-decomposition scheme, one can rigorously define the total energy of a composite system as the sum of the energies of its individual components plus the interaction energy between them. For example, the energy Ue(jswn), of the conformation j = P, β in explicit water is given by
| Ue(jswn) = Ue(js) + Ue,j(wn) + Ue,i(j), | (1) |
| ΔUe(Psβswn) = ΔUe(Psβs) + ΔUe,Pβ(wn) + ΔUe,i(Pβ), | (2) |
| ΔUe(Psβswn)a (kJ mol−1) | ΔUe(Psβs)b (kJ mol−1) | ΔUe,Pβ(wn)c (kJ mol−1) | ΔUe,i(Pβ)d (kJ mol−1) | |
|---|---|---|---|---|
| a Electronic energy difference between pPII and β solvated systems consisting of the peptide and 10 water molecules. b Electronic energy difference between individual pPII and β structures in the geometry of the solvated system. c Electronic energy difference between 10 water molecules associated with the pPII and β structures. d Electronic interaction energy difference between pPII and β systems. | ||||
| GAG | −21.24 | 10.88 | −11.28 | −20.84 |
| GLG | −16.42 | 7.49 | −13.26 | −10.65 |
| GVG | −18.25 | 8.61 | −13.70 | −13.15 |
| GIG | −20.34 | 7.85 | −13.13 | −15.06 |
Estimation of vibrational entropies and energies
Generally, DFT energy calculations do not provide any direct information about entropies and, therefore, do not enable one to gauge relative stabilities of conformations. However, we wondered to what extent the hydration shell could contribute to entropic differences between the considered conformations solely through peptide–water and water–water vibrational mixing. Such vibrational mixing is well-established for amide I and the water bending modes.58,59 However, similar interactions between low-frequency modes of the peptide and collective water modes have not been fully investigated on a quantum chemical level. Our vibrational analysis reveals that nearly all modes below 700 cm−1 are involved in peptide–water mixing. As an example, Fig. 3 shows four mixed water–peptide modes of the GAG–water complex, two for pPII and two for β-strand. | ||
| Fig. 3 Representative mixed water-peptide modes of the GAG–water complex. For both the pPII and β-strand conformations, there is a strong mixing between water and peptide modes. | ||
The strong mixing between the water and peptide modes clearly indicates that peptide–water interactions constitute a dynamic entity with significant coherent vibrational dynamics. It is obvious that the degree of this mixing and, thus, the vibrational energy density should depend on the backbone conformation. To check this conjecture we calculated the difference between the (harmonic) entropies of pPII and β at room temperature by utilizing the well-known relationship:
 | (3) |
 the wavenumber of the ith peptide-H2O vibration, R the gas constant, kB the Boltzmann constant, and T the absolute temperature. To avoid any arbitrary cutoffs, we included the entire vibrational manifold of N modes as obtained from the DFT based normal mode calculations. We are aware that this approach neglects the anharmonicity to be expected for low frequency modes and possible inaccuracies of our calculated frequencies. However, it is reasonable to expect that our calculations should provide us with reliable trends in the magnitude and sign of hydration-induced differences between the vibrational entropies of pPII and β-strand conformations. This is corroborated by recent comparisons between experimentally-obtained and computed entropy differences between pPII and β-strand of AcANH2 in water.32,33
the wavenumber of the ith peptide-H2O vibration, R the gas constant, kB the Boltzmann constant, and T the absolute temperature. To avoid any arbitrary cutoffs, we included the entire vibrational manifold of N modes as obtained from the DFT based normal mode calculations. We are aware that this approach neglects the anharmonicity to be expected for low frequency modes and possible inaccuracies of our calculated frequencies. However, it is reasonable to expect that our calculations should provide us with reliable trends in the magnitude and sign of hydration-induced differences between the vibrational entropies of pPII and β-strand conformations. This is corroborated by recent comparisons between experimentally-obtained and computed entropy differences between pPII and β-strand of AcANH2 in water.32,33
The vibrational entropy contributions to the Helmholtz energies of the investigated GxG peptides as obtained by eqn (3) are visualized in Fig. 4. Apparently, all vibrational entropies stabilize the β-strand conformation in explicit water by contributing between 5 to 18 kJ mol−1 at room temperature. Interestingly, the obtained side chain dependence of the vibrational entropies correlate with the corresponding ΔU values. Even though they display the same I > V > L hierarchy, they underestimate the respective experimental values for these residues and overestimate the entropy of GAG. The underestimated entropy of GLG, GVG, and GIG can again be related to the inability of the considered 10 water molecules to fully hydrate these peptides' side chains. However, our results clearly suggest that the vibrational dynamics of the combined peptide–water system is a major source of the entropic contribution to the Gibbs or Helmholtz energy differences between pPII and the β-strand, in agreement with Lanza and Chiacchio.33
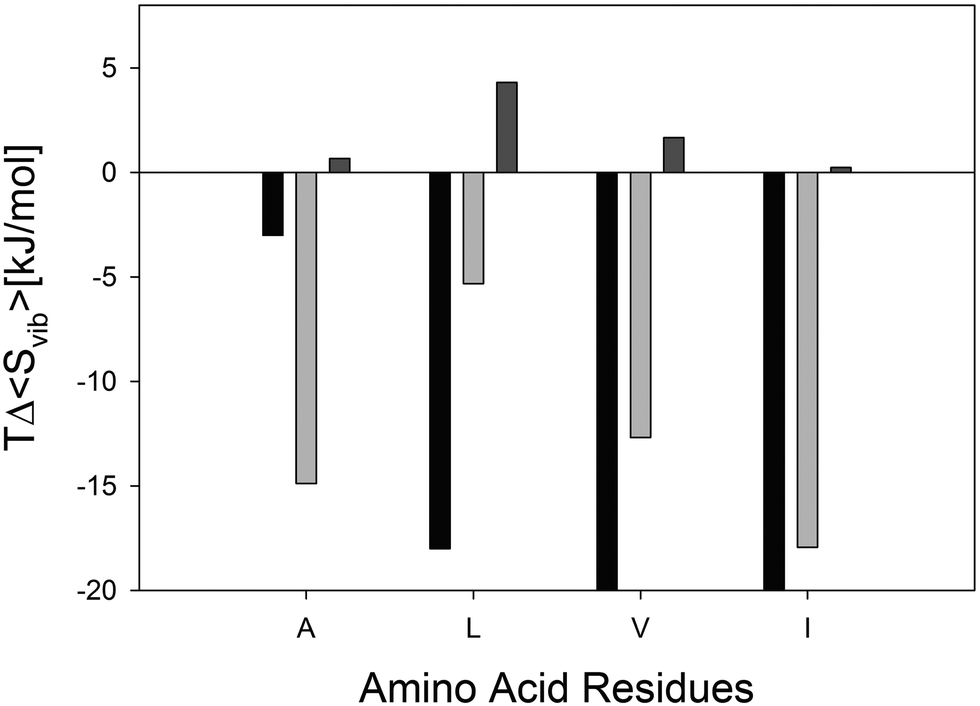 | ||
| Fig. 4 Difference between the Gibbs/Helmholtz energy contributions of the vibrational entropy of the indicated amino acid residue in cationic GxG peptides in explicit water (grey) and in vacuo (light grey) in the pPII and β-strand conformation. Experimentally obtained TΔS values of the corresponding the experimentally obtained pPII ⇌ β-strand equilibrium are plotted in black. In vacuo values for L and I are so small that they are barely visible on the chosen scale. | ||
In order to further corroborate the contributions of hydration to the vibrational entropy differences between pPII and β-strand conformations, we calculated the entropy differences between the respective vibrational manifolds of the investigated GxGs without solvent molecules. The respective contributions to the total free energy are also plotted in Fig. 4. For GAG and GIG, these (positive) contributions are negligible. For GLG and GVG, the calculations actually yielded a room temperature stabilization of the pPII conformation by 4.3 and 1.6 kJ mol−1, respectively. Together, all these calculations demonstrate that peptide solvation inverts the sign of the internal energy and entropy contribution to the Helmholtz energy governing the pPII ⇌ β equilibrium.
Finally, for the sake of completeness, we also estimated the vibrational contribution to the internal energy for the above considered vibrational manifold of GxG·10H2O. This can be done by using the textbook equation:
 | (4) |
Conclusion
Taken together with our earlier reported experimental results, the complementary DFT calculations reveal the dominant role of backbone hydration in stabilizing the pPII conformation of amino acid residues of short peptides in water. Specifically, these interactions stabilize the pPII conformation energetically but destabilizing it entropically. We were able to reproduce an earlier observed I > V > L hierarchy of the experimentally-obtained ΔH/ΔU and TΔS values associated with the pPII ⇌ β equilibrium of GxG peptides in water on a qualitative level. We decomposed the obtained energies of the investigated pPII ⇌ β equilibria into intramolecular (peptide and water) and intermolecular contributions (water–peptide). The results of this procedure revealed that peptide–water interactions and solvent self-energies promote the energetic stabilization of pPII over water. Our results show that the hydration of the peptide backbone critically depends on the backbone structure and on the nature of the side chain. This notion seems to be consistent with the conditional solvation model of Ben Naim, but at variance with a model constructed from context free analogues of side chains.55 It is further consistent with predictions from electrostatic theories.30 Moreover, we provide evidence for the notion that the rather large entropic differences between pPII and β-strand are assignable to the vibrational dynamics of the peptide–water entity. Our calculations with only ten water molecules actually underestimate these contributions for residues with large and bulky side chains. Mezei et al. performed a molecular-dynamics-based analysis of a polyalanine 12-mer in explicit water and reported that the number of water molecules in the first hydration shell of both pPII and β is approximately 120.24 Any further characterization of peptide hydration shells in terms of hydrogen bonding and orientational distribution based on quantum chemical calculations will be the subject of future studies.Acknowledgements
S. E. T. was in part supported by a research fellowship of the College of Arts and Sciences which is gratefully acknowledged. B.M.W. acknowledges the National Science Foundation for the use of supercomputing resources through the Extreme Science and Engineering Discovery Environment (XSEDE), Project No. TG-CHE150040.References
- D. A. Brant and P. J. J. Flory, J. Am. Chem. Soc., 1965, 87, 2791–2800 CrossRef CAS.
- P. J. Flory, Statistical Mechanics of Chain Molecules, Wiley & Sons, New York, 1969 Search PubMed.
- G. N. Ramachandran, C. Ramachandran and V. Sasisekharan, J. Mol. Biol., 1963, 7, 95–99 CrossRef CAS.
- C. Tanford, Adv. Protein Chem., 1968, 23, 121–282 CrossRef CAS.
- Z. Shi, K. Chen, Z. Liu, A. Ng, W. C. Bracken and N. R. Kallenbach, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17964–17968 CrossRef CAS.
- Z. Shi, C. A. Olson, G. D. Rose, R. L. Baldwin and N. R. Kallenbach, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9190–9195 CrossRef CAS PubMed.
- Z. Shi, K. Shen, Z. Liu and N. R. Kallenbach, Chem. Rev., 2006, 106, 1877–1897 CrossRef CAS PubMed.
- S. Woutersen and P. Hamm, J. Phys. Chem. B, 2000, 104, 11316–11320 CrossRef CAS.
- S. Woutersen and P. Hamm, J. Chem. Phys., 2001, 114, 2727–2737 CrossRef CAS.
- J. Graf, P. H. Nguyen, G. Stock and H. Schwalbe, J. Am. Chem. Soc., 2007, 129, 1179–1189 CrossRef CAS PubMed.
- F. Eker, X. Cao, L. Nafie and R. Schweitzer-Stenner, J. Am. Chem. Soc., 2002, 124, 14330–14341 CrossRef CAS PubMed.
- A. Hagarman, T. J. Measey, D. Mathieu, H. Schwalbe and R. Schweitzer-Stenner, J. Am. Chem. Soc., 2010, 132, 540–551 CrossRef CAS PubMed.
- A. K. Jha, A. Colubri, K. F. Freed and T. R. Sosnick, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13099–13104 CrossRef CAS PubMed.
- J. DeBartolo, A. Jha, K. F. Freed and T. R. Sosnick, in Protein and Peptide Folding, Misfolding, and Non-Folding, ed. R. Schweitzer-Stenner, John Wiley & Sons, Hoboken, 2012, pp. 79–98 Search PubMed.
- A. Hagarman, D. Mathieu, S. Toal, T. J. Measey, H. Schwalbe and R. Schweitzer-Stenner, Chemistry, 2011, 17, 6789–6797 CrossRef CAS PubMed.
- R. Schweitzer-Stenner, A. Hagarman, S. Toal, D. Mathieu and H. Schwalbe, Proteins, 2013, 81, 955–967 CrossRef CAS PubMed.
- S. E. Toal and R. Schweitzer-Stenner, Biomolecules, 2014, 4, 725–773 CrossRef CAS PubMed.
- P. J. Fleming, N. C. Fitzkee, M. Mezei, R. Srinivasan and G. D. Rose, Protein Sci., 2005, 14, 111–118 CrossRef CAS.
- W.-G. Han, K. J. Jakanen, M. Elstner and S. Suhai, J. Phys. Chem. B, 1998, 102, 2587–2602 CrossRef CAS.
- A. N. Drozdov, A. Grossfield and R. V. Pappu, J. Am. Chem. Soc., 2004, 126, 2574–2581 CrossRef CAS PubMed.
- A. E. Garcia, Polymer, 2004, 120, 885–890 Search PubMed.
- S. Gnanakaran and A. E. Garcia, J. Phys. Chem. B, 2003, 107, 12555–12557 CrossRef CAS.
- A. Kentsis, M. Mezei, T. Gindin and R. Osman, Proteins: Struct., Funct., Genet., 2004, 55, 493–501 CrossRef CAS PubMed.
- M. Mezei, P. J. Fleming, R. Srinivasan and G. D. Rose, Proteins: Struct., Funct., Genet., 2004, 55, 502–507 CrossRef CAS PubMed.
- F. Eker, X. Cao, L. Nafie, Q. Huang, K. Griebenow and R. Schweitzer-Stenner, J. Phys. Chem. B, 2003, 107, 358–365 CrossRef CAS.
- Z. Liu, K. Chen, A. Ng, Z. Shi, R. W. Woody and N. R. Kallenbach, J. Am. Chem. Soc., 2004, 126, 15141–15150 CrossRef CAS.
- S. Toal, D. Meral, D. Verbaro, B. Urbanc and R. Schweitzer-Stenner, J. Phys. Chem. B, 2013, 117, 3689–3706 CrossRef CAS PubMed.
- S. Toal, A. Omidi and R. Schweitzer-Stenner, J. Am. Chem. Soc., 2011, 133, 12728 CrossRef CAS.
- F. Avbelj, in Protein and Peptide Folding, Misfolding and Non-Folding, ed. R. Schweitzer-Stenner, John Wiley & Sons, Hoboken, 2012, pp. 131–158 Search PubMed.
- F. Avbelj and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5742–5747 CrossRef CAS PubMed.
- S. E. Toal, D. J. Verbaro and R. Schweitzer-Stenner, J. Phys. Chem. B, 2014, 118, 1309–1318 CrossRef CAS PubMed.
- G. Lanza and M. A. Chiacchio, ChemPhysChem, 2014, 15, 2785–2793 CrossRef CAS PubMed.
- G. Lanza and M. A. Chiacchio, Phys. Chem. Chem. Phys., 2015, 17, 17101–17111 RSC.
- F. Avbelj and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10967–10972 CrossRef CAS PubMed.
- F. Avbelj, S. G. Grdadolnik, J. Grdadolnik and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1272–1277 CrossRef CAS PubMed.
- W. Li, M. Qin, Z. Tie and W. Wang, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 84, 041933 CrossRef.
- C. D. Poon, E. T. Samulsi, C. F. Weise and J. C. Weisshaar, J. Am. Chem. Soc., 2000, 122, 5612–5613 CrossRef.
- M. A. Hossain, M. A. Saeed, A. Pramanik, B. M. Wong, S. A. Haque and D. R. Powell, J. Am. Chem. Soc., 2012, 134, 11892–11895 CrossRef CAS.
- M. A. Saeed, A. Pramanik, B. M. Wong, S. A. Haque, D. R. Powell, D. K. Chand and M. A. Hossain, Chem. Commun., 2012, 48, 8631–8633 RSC.
- M. A. Saeed, B. M. Wong, F. R. Fronczek, R. Venkatraman and M. A. Hossain, Cryst. Growth Des., 2010, 10, 1486–1488 CAS.
- E. E. Dahlke, R. M. Olson, H. R. Leverentz and D. G. Truhlar, J. Phys. Chem. A, 2008, 112, 3976–3984 CrossRef CAS PubMed.
- A. Fortunelli and J. Tomasi, Chem. Phys. Lett., 1994, 231, 34–39 CrossRef CAS.
- J. Tomasi, R. Bonaccorsi, R. Cammi and F. J. O. Delvalle, THEOCHEM, 1991, 80, 401–424 CrossRef CAS.
- F. J. O. Delvalle, R. Bonaccorsi, R. Cammi and J. Tomasi, THEOCHEM, 1991, 76, 295–312 CrossRef.
- F. J. O. Delvalle and J. Tomasi, Chem. Phys., 1991, 150, 139–150 CrossRef.
- M. A. Aguilar, F. J. O. Delvalle and J. Tomasi, Chem. Phys., 1991, 150, 151–161 CrossRef CAS.
- K. Rybka, S. E. Toal, D. J. Verbaro, D. Mathieu, H. Schwalbe and R. Schweitzer-Stenner, Proteins, 2013, 81, 968–983 CrossRef CAS PubMed.
- K. Kwac, K. K. Lee, J. B. Han, K. I. Oh and M. Cho, J. Chem. Phys., 2008, 128, 105106 CrossRef PubMed.
- B. Zagrovic, J. Lipfert, E. J. Sorin, I. S. Millett, W. F. van Gunsteren, S. Doniach and V. S. Pande, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11698–11703 CrossRef CAS PubMed.
- S. Gnanakaran and A. E. Garcia, Proteins, 2005, 59, 773–782 CrossRef CAS PubMed.
- M. Feig, J. Chem. Theory Comput., 2008, 4, 1555–1564 CrossRef CAS.
- S. Hovmoller, T. Zhou and T. Ohlson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 768–776 CrossRef PubMed.
- Y. Mu, D. S. Kosov and G. Stock, J. Phys. Chem. B, 2003, 107, 5064–5073 CrossRef CAS.
- Y. Mu and G. Stock, J. Phys. Chem. B, 2002, 106, 5294–5301 CrossRef CAS.
- A. Ben Naim, Molecular Theory of Water and Aquesous Solutions-Part I: Understanding Water, World Scientific, Singapore, 2009 Search PubMed.
- R. Schweitzer-Stenner and S. E. Toal, Phys. Chem. Chem. Phys., 2014, 16, 22527–22536 RSC.
- N. G. Mirkin and S. Krimm, Biopolymers, 2012, 97, 789–794 CrossRef CAS PubMed.
- X. G. Chen, R. Schweitzer-Stenner, S. A. Asher, N. G. Mirkin and S. Krimm, J. Phys. Chem., 1995, 99, 3074–3083 CrossRef CAS.
- G. Sieler and R. Schweitzer-Stenner, J. Am. Chem. Soc., 1997, 119, 1720 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Energy decomposition of the solvated peptides at the ωB97X-D/cc-pVDZ level of theory and reference Cartesian coordinates and energies for all 16 peptides. See DOI: 10.1039/c5cp03646a |
| ‡ Present address: Department of Biophysics and Biochemistry, Yale University, New Haven, CT 06250, USA. |
| This journal is © the Owner Societies 2015 |