
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chirally-modified metal surfaces: energetics of interaction with chiral molecules
Petr
Dementyev
a,
Matthias
Peter
a,
Sergey
Adamovsky
a and
Swetlana
Schauermann
*ab
aFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: schauermann@fhi-berlin.mpg.de
bInstitut of Physical Chemistry, Christian-Albrechts-Universität zu Kiel, Max-Eyth-Str. 1, 24118 Kiel, Germany
First published on 10th August 2015
Abstract
Imparting chirality to non-chiral metal surfaces by adsorption of chiral modifiers is a highly promising route to create effective heterogeneously catalyzed processes for the production of enantiopure pharmaceuticals. One of the major current challenges in heterogeneous chiral catalysis is the fundamental-level understanding of how such chirally-modified surfaces interact with chiral and prochiral molecules to induce their enantioselective transformations. Herein we report the first direct calorimetric measurement of the adsorption energy of chiral molecules onto well-defined chirally-modified surfaces. Two model modifiers 1-(1-naphthyl)ethylamine and 2-methylbutanoic acid were used to impart chirality to Pt(111) and their interaction with propylene oxide was investigated by means of single-crystal adsorption calorimetry. Differential adsorption energies and absolute surface uptakes were obtained for the R- and S-enantiomers of propylene oxide under clean ultrahigh vacuum conditions. Two types of adsorption behavior were observed for different chiral modifiers, pointing to different mechanisms of imparting chirality to metal surfaces. The results are analyzed and discussed in view of previously reported stereoselectivity of adsorption processes.
Introduction
Production and purification of enantiopure chemicals is a topic of vast practical importance arising from the fact that most biologically important molecules are chiral. Heterogeneous catalysts for enantioselective processes combine the chiral media to induce enantioselectivity into an underlying non-chiral substrate (e.g. metal) with the highly catalytically active metal nanoparticles and with this hold great potential to achieve tremendous synergetic effects.1 There is a rapidly emerging research field, which focuses on designing chirally-modified catalytic surfaces that have been successfully tested under laboratory conditions and batch reactors.2–4 Despite the impressive development in this field in the past years, the mechanistic details of the underlying microscopic processes are still under debate.5 In particular, it is not clear how the chiral surface binds the reactant molecules in a specific prochiral configuration that yields only one preferential enantiomer as a product. On the way to obtaining microscopic insights into the enantioselective surface chemistry, in situ spectroscopic characterization of the catalytic reactions has significantly progressed in powdered materials.6–8 Thus, Baiker and coworkers recently showed that hydrogen bonding between the pro-chiral reactant and chiral modifier molecules takes place.9 Molecular chirality has also attracted much interest in surface science research,10,11 where adsorption of chiral molecules on well-defined single-crystal surfaces and their interaction with other adsorbates have been extensively studied using a variety of surface sensitive techniques.12–27 A recent review by Gellman, Tysoe and Zaera summarizes the latest report on chiral surface chemistry performed on model single crystalline surfaces.28 As one of the major results of these studies, the authors put forward the idea that enantioselectivity arises from the energetic differences in the interaction of two opposite enantiomers with a chiral surface.Chiral differentiation can be generally accomplished on three types of metal surfaces: (i) intrinsically chiral terminations of higher Miller index surfaces; (ii) surfaces templated by ordered structures of chiral modifiers (this type of chirality is frequently referred to as “supramolecular chirality”) and (iii) the surfaces carrying individual chiral modifiers which form 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with pro-chiral reactant molecules. As shown by Gellman et al., natural chiral surfaces Cu(3,1,17)R&S can efficiently separate racemic mixture of aspartic acid.21 In the case of asymmetric surfaces, on which chirality is imposed by supramolecular assemblies of chiral adsorbates, the stereoselective adsorption sites are produced by so called “chiral pockets” in the ordered chiral overlayers. The interaction of chiral molecules with the chiral pockets in such supramolecular chiral surfaces has been extensively investigated by Tysoe's group.29–33 In particular, R- and S-propylene oxides (PO) were adsorbed on Pd(111) chirally modified with R- or S-2-butanol. At specific modifier coverage, significant enhancement (up to a factor of two) of the adsorbed amount of a homochiral enantiomer as compared to the enantiomer of the opposite chirality was detected in temperature-programmed desorption (TPD).29,30 A similar approach has been successfully applied by Zaera et al. to chirally modified Pt(111) surfaces.25,26,34 Pt(111) modified with 2-methylbutanoic acid was titrated by PO enantiomers and was found to exhibit a 25% higher TPD yield for the homochiral adsorbate, i.e. S-PO desorbing from (S)-2-methylbutanoic acid modified Pt(111). The energetic difference as measured by TPD was estimated to be ∼2 kJ mol−1.25 Another successful example of a specific enantioselective interaction is Pt(111) modified with 1-(1-naphthyl)ethylamine (NEA), which was found to split the PO desorption trace into two peaks.26 For the high temperature desorption trace, the TPD yield of PO in a homochiral adsorbate–modifier pair was found to be higher than for the adsorbate–modifier pair of the opposite chirality by a factor of four. The difference in the PO desorption peak positions was measured to be about 5 K, corresponding to the energy difference of 1–2 kJ mol−1. Two desorption states of PO were interpreted to be indicative of the formation of 1:1 complexes between the modifier and the probe molecules.
:1 complexes with pro-chiral reactant molecules. As shown by Gellman et al., natural chiral surfaces Cu(3,1,17)R&S can efficiently separate racemic mixture of aspartic acid.21 In the case of asymmetric surfaces, on which chirality is imposed by supramolecular assemblies of chiral adsorbates, the stereoselective adsorption sites are produced by so called “chiral pockets” in the ordered chiral overlayers. The interaction of chiral molecules with the chiral pockets in such supramolecular chiral surfaces has been extensively investigated by Tysoe's group.29–33 In particular, R- and S-propylene oxides (PO) were adsorbed on Pd(111) chirally modified with R- or S-2-butanol. At specific modifier coverage, significant enhancement (up to a factor of two) of the adsorbed amount of a homochiral enantiomer as compared to the enantiomer of the opposite chirality was detected in temperature-programmed desorption (TPD).29,30 A similar approach has been successfully applied by Zaera et al. to chirally modified Pt(111) surfaces.25,26,34 Pt(111) modified with 2-methylbutanoic acid was titrated by PO enantiomers and was found to exhibit a 25% higher TPD yield for the homochiral adsorbate, i.e. S-PO desorbing from (S)-2-methylbutanoic acid modified Pt(111). The energetic difference as measured by TPD was estimated to be ∼2 kJ mol−1.25 Another successful example of a specific enantioselective interaction is Pt(111) modified with 1-(1-naphthyl)ethylamine (NEA), which was found to split the PO desorption trace into two peaks.26 For the high temperature desorption trace, the TPD yield of PO in a homochiral adsorbate–modifier pair was found to be higher than for the adsorbate–modifier pair of the opposite chirality by a factor of four. The difference in the PO desorption peak positions was measured to be about 5 K, corresponding to the energy difference of 1–2 kJ mol−1. Two desorption states of PO were interpreted to be indicative of the formation of 1:1 complexes between the modifier and the probe molecules.
Despite these advances in the understanding of the enantiospecific interaction on chirally-modified surfaces, there is still a lack of detailed quantitative information on the interaction strength within the reactant-modifier pair. In previous studies, only indirect desorption-based methods, such as TPD, were used in order to estimate the interaction energies. However, these methods have a number of serious drawbacks, mainly arising from the fact that they can be correctly applied only for fully reversible processes and that the estimate of the desorption energies relies on some critical assumptions on the details of the desorption process. Additionally, the adsorbates and/or modifier-overlayers can undergo restructuring, be destroyed or decompose due to the increasing temperature.
As a strategy to overcome these problems, we applied for the first time single-crystal adsorption calorimetry (SCAC) based on molecular beam techniques as a direct method allowing the measurement of adsorption energies of chiral molecules onto chirally-modified metal surfaces. This technique enables the direct and very precise measurements of the adsorption energy of gaseous molecules on well-defined surfaces under isothermal conditions35,36 and does not rely on any assumptions on the desorption process. By carrying out the adsorption measurements under low temperature conditions, we can probe the adsorption energetics of very sensitive adsorption systems, such as e.g. chirally-modified surfaces interacting with light hydrocarbons. Additionally, SCAC allows for measuring adsorption energies as a very detailed function of the surface coverage of the adsorbates. The sticking coefficient measurement, which is an integral part of a SCAC experiment, provides an experimental possibility of studying the evolution of the surface coverage, for example, to monitor how adsorbates populate specific surface areas depending on the temperature or the coverage of the chiral modifier.
We investigate two previously characterized model chiral modifiers 1-(1-naphthyl)ethylamine (NEA) and 2-methylbutanoic acid (MBA) supported on Pt(111). The structural formulae of these molecules are shown in Fig. 1. As mentioned above, both chirally-modified surfaces were previously reported to exhibit appreciable enantioselectivity in the adsorption of PO enantiomers, both in terms of the adsorption energies and of the amount of the adsorbed PO.25,26 To verify these findings and to obtain accurate adsorption energies using a direct calorimetric measurement, we measured the PO adsorption energies as a detailed function of the PO coverage at a constant modifier coverage. Complementarily, we varied the surface coverages of the chiral modifiers to tune the properties of the chiral surface overlayers and determined the PO adsorption energies on these surfaces.
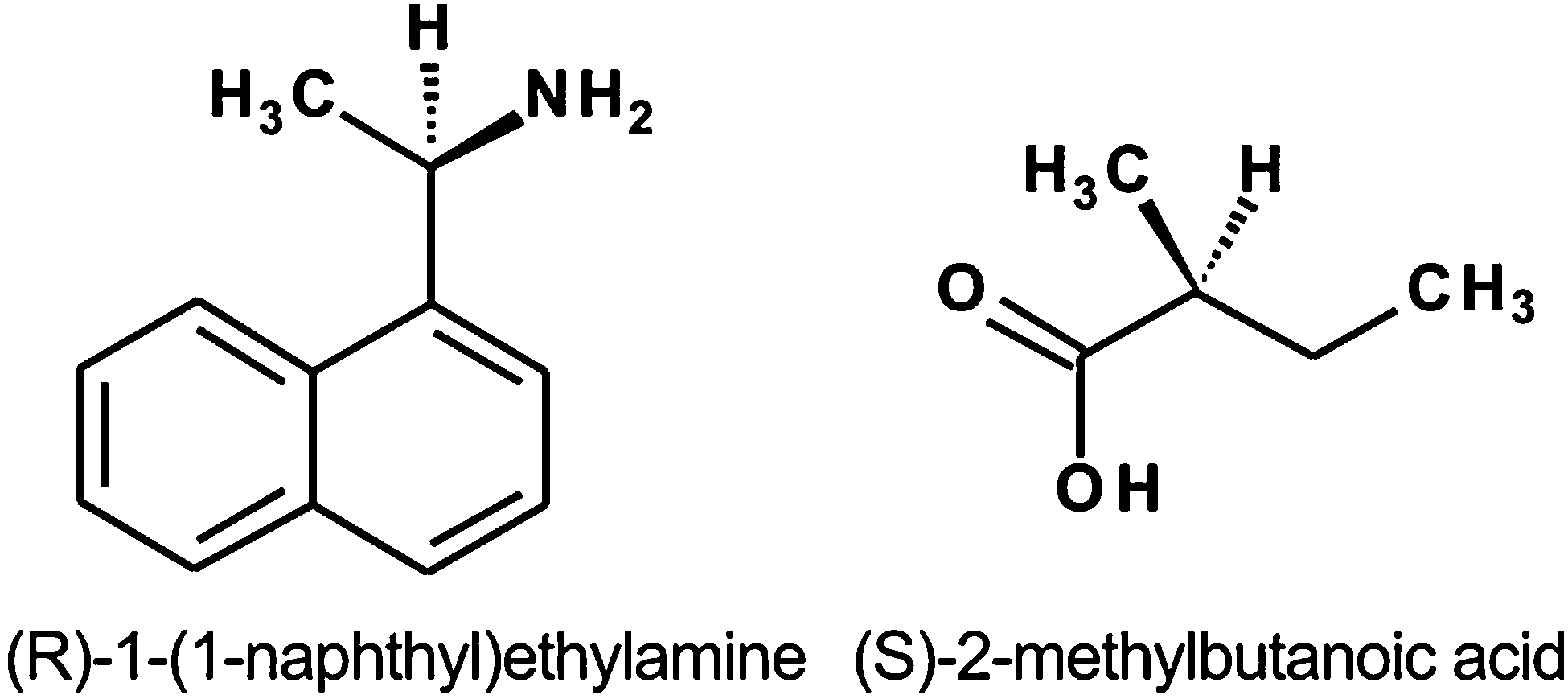 | ||
| Fig. 1 Structural formulae of the chiral modifiers studied. | ||
Two types of adsorption behavior were observed for different chiral modifiers, pointing to different mechanisms of imparting chirality to metal surface. The results will be analyzed and discussed in view of previously reported stereoselectivity of the same adsorption processes.
Experimental section
All experiments were carried out at the Fritz-Haber-Institut (Berlin, Germany) in an ultrahigh vacuum SCAC apparatus described in detail elsewhere.37 An ultrathin Pt(111) single crystal was cleaned by Ar sputtering followed by oxidation and annealing. Deposition of a modifier onto a clean metal surface was performed through a differentially pumped gas-doser. The vapor of R-NEA (≥99% (R)-(+)-1-(1-naphthyl)ethylamine, Sigma-Aldrich) at the saturation pressure was directly introduced into the gas-doser while the NEA flask was kept in an ice-water bath. For dosing S-MBA (98% (S)-(+)-2-methylbutanoic acid, Sigma-Aldrich) a backing pressure of 0.100 mbar was used. The amounts of deposited surface modifiers were controlled by the exposure time. The sample preparation procedure closely resembled the previously described preparation by Zaera and coworkers for better comparison.25,26 NEA was deposited onto a metal surface kept at a temperature below 100 K. MBA was deposited at approximately 100 K followed by annealing at 230 K. The obtained modifier coverage values were calibrated as discussed in the next section.A freshly prepared sample was transferred from the preparation chamber to the pre-cooled calorimeter. A molecular beam of either R- or S-enantiomers of propylene oxide (99% (R)-(+)-propylene oxide and 99% (S)-(−)-propylene oxide, Alfa Aesar, Sigma-Aldrich) was formed and cut into 266 ms pulses that were allowed to impinge on the sample surface. The energies were measured by a pyroelectric detector (6-μm thick β-PVDF ribbon from Piezotech), whose response was calibrated by a He–Ne laser of known intensity. For a detailed description of the calorimetric setup, see ref. 37. Propylene oxide was degassed by freeze–pump–thaw cycles prior to experiments. Sticking probabilities were determined via the King–Wells method38 with a mass spectrometer (Hiden Analytical, HAL 301/3F PIC) following the PO fragment at m/z = 58.
Further in the text we use the term adsorption energy, which we define as the following:
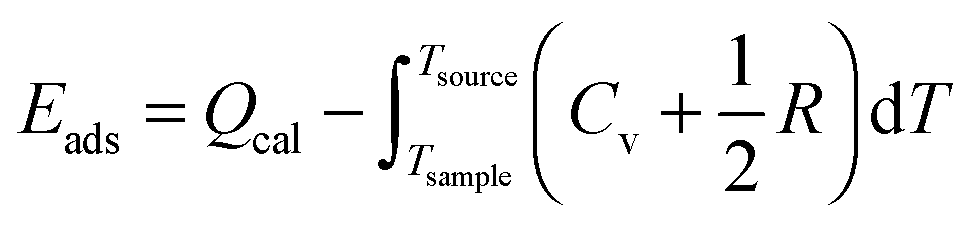
Results
a. PO adsorption on R-NEA-modified Pt(111)
NEA is a model chiral modifier, which has an aromatic moiety for binding to a metal surface and an amino group close to the chiral center capable of hydrogen-bonding.40 To control the modifier coverage we first calibrated the gas-dosing system. For this, carbon monoxide was used as a probe molecule for free metal sites. CO was dosed onto the surface pre-covered by NEA and sticking coefficient measurements were performed via the King–Wells method38 at room temperature. The measured sticking coefficients of carbon monoxide on Pt precovered with various amounts of NEA and the corresponding calibration curve are shown in Fig. 2. On bare Pt(111), the initial sticking coefficient was measured to be ∼0.7 and then strongly decay with increasing CO surface coverage. This behavior corresponds well to previously reported values of CO sticking coefficients on Pt(111).41 Saturation of a monolayer takes place at the CO coverage of ∼0.55 ML as referred to the absolute amount of Pt atoms 1.5 × 1015 cm−2. When NEA is deposited onto the surface, the initial sticking coefficient becomes smaller and fewer CO molecules are required to saturate the monolayer, which implies that NEA molecules do block the CO adsorption sites. When the NEA deposition time exceeds 20 s, the CO sticking coefficient becomes vanishingly small, indicating that NEA adsorbates completely cover the Pt surface and prevent CO adsorption. Note that on the clean Pt(111) substrate the sticking coefficient levels off at app. 0.1 and does not decrease to 0. This effect is related to the measurement of the sticking coefficient by applying short molecular beam pulses in such adsorption systems, where the saturation coverage strongly depends on the pressure of the adsorbing molecule. For CO adsorption on Pt(111), it was established in previous studies that the CO saturation coverage during the molecular beam is slightly higher than under the UHV conditions.42 Therefore, close to saturation the systems runs into a quasi steady state regime, in which adsorption during the pulse is compensated by desorption between the pulses and the final sticking coefficient remains non-zero. In contrast, on the NEA covered surfaces the final sticking coefficient was found to be zero, indicating that no additional CO can be added to this surface by applying higher CO pressures. The saturation coverage of CO is plotted as a function of the NEA deposition time in Fig. 2b. In the following, we define a relative coverage of NEA on Pt(111) ΘNEA as equal to 0 on the pristine surface and equal to 1 when CO adsorption is fully hindered.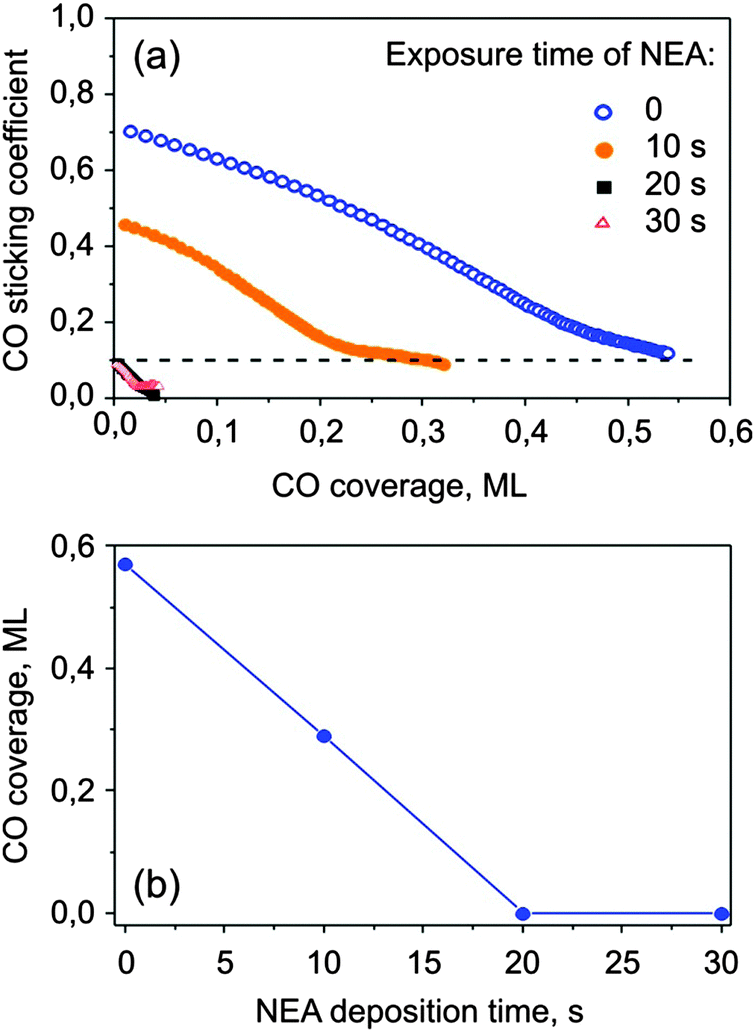 | ||
| Fig. 2 (a) Sticking coefficients of CO at 300 K on NEA covered Pt(111) for different NEA deposition times. The CO coverage is given as a ratio of the number of adsorbed CO molecules to the number of Pt surface atoms (1.5 × 1015 cm−2). (b) The CO saturation coverage (in ML) reached on the NEA-covered Pt(111) plotted as a function of the NEA deposition time. | ||
Adsorption of S- and R-PO was studied on samples with three different R-NEA coverage values as well as on pristine Pt at 110 K. The sticking probability of PO was determined to be unity at the experimental temperature (110 K) on all investigated surfaces. Fig. 3 shows adsorption energy of both the R- and S-enantiomers of PO as a function of the number of adsorbed PO molecules.
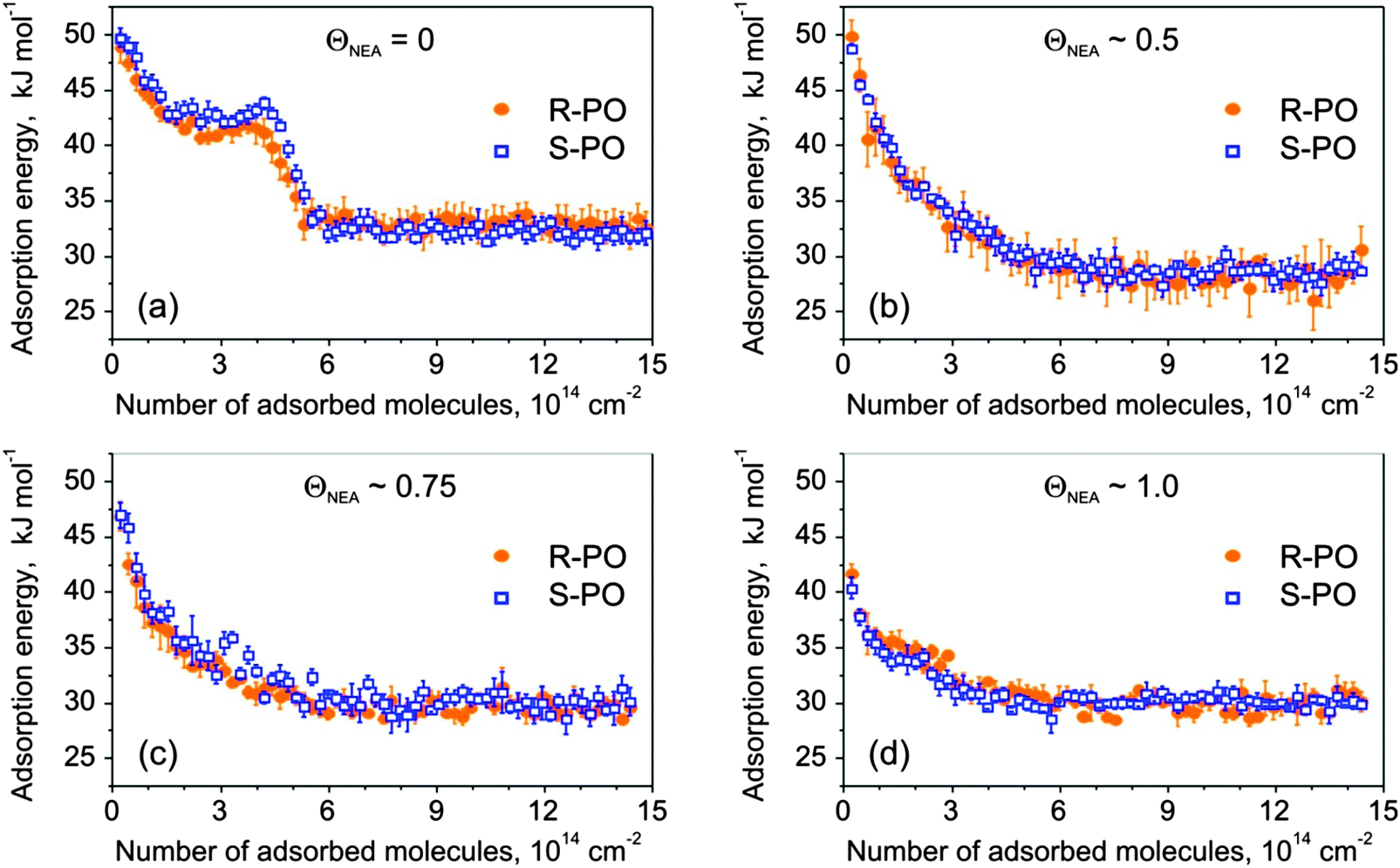 | ||
| Fig. 3 Adsorption energy of S- and R-PO as a function of the number of adsorbed molecules on: (a) bare Pt(111); as well as R-NEA-covered Pt(111) with the R-NEA relative coverage of (b) 0.5; (c) 0.75; (d) 1. The data points are averages of 3–5 independent measurements; the error bars show the error of the mean. | ||
On bare Pt(111) (Fig. 3a), PO shows non-monotonic changes in the adsorption energy with increasing coverage. The PO adsorption energy starts at 50 ± 1 kJ mol−1 and decreases to the level of 42–43 kJ mol−1 at the coverage of ∼1.5 × 1014 molecules cm−2, which corresponds to the adsorption of ∼0.1 PO molecule per one surface Pt atom. Above this coverage, the adsorption energy either goes through a small local maximum (S-PO) or rather remains constant (R-PO) up to the coverage of 4.1 × 1014 molecules cm−2, followed by a rapid decrease corresponding to the completion of the monolayer at the value of about 5.5 × 1014 molecules cm−2. This number agrees well with the previous IRAS and TPD data indicating that the PO monolayer on Pt(111) saturates after 1.6–2 L exposure corresponding to 4.2–5.3 × 1014 molecules cm−2.34 Starting from the coverage of ∼5.5 × 1014 molecules cm−2, a constant adsorption energy of 34 ± 1 kJ mol−1 was observed, which is related to PO adsorption in the second and subsequent layers. Almost no differences in PO adsorption on pristine Pt(111) surfaces were observed for both stereoisomers.
For NEA-covered surfaces, the adsorption behavior of both PO enantiomers strongly changes exhibiting adsorption energy monotonically decreasing (with the decay greater than linear) to constant energy values varying between 28 and 30 kJ mol−1. Note that even for the smallest NEA coverage the plateau-like character of the adsorption curves, as observed on pristine Pt(111), completely vanishes. One can also clearly see that for all modifier coverage, PO multilayer condensation in the second layer starts at approximately the same PO uptake of ∼6 × 1014 molecules cm−2, which is also true for bare Pt. This observation indicates that the complete PO monolayer on the NEA-modified surfaces contains not only PO molecules adsorbed on the metal but also those adsorbed on-top of NEA modifier and their total amount remains similar for all modifier coverages. Note that the adsorption energy of PO in the second layer is not very sensitive to the presence of NEA on the underlying surface.
The initial adsorption energies corresponding to the first PO pulses and the final energy values are plotted in Fig. 4 as a function of the NEA coverage. For both stereoisomers, the initial adsorption energy decreases upon going from bare Pt(111) to 1 ML NEA/Pt(111). Note that the adsorption energy of PO on 1 ML NEA is still higher than that on PO ice. This observation suggests a stronger interaction between PO and NEA than PO and PO ice. Within experimental error no difference between adsorption energies of different stereoisomers of PO on R-NEA was observed. The final energy on the surfaces partially covered with the modifier was found to be somewhat lower than those on bare and fully covered surfaces. These differences might be related either to our experimental accuracy or to the fact the PO adsorbed in the second and a few next layers might be still sensitive to the particular structure of the underlying substrate.
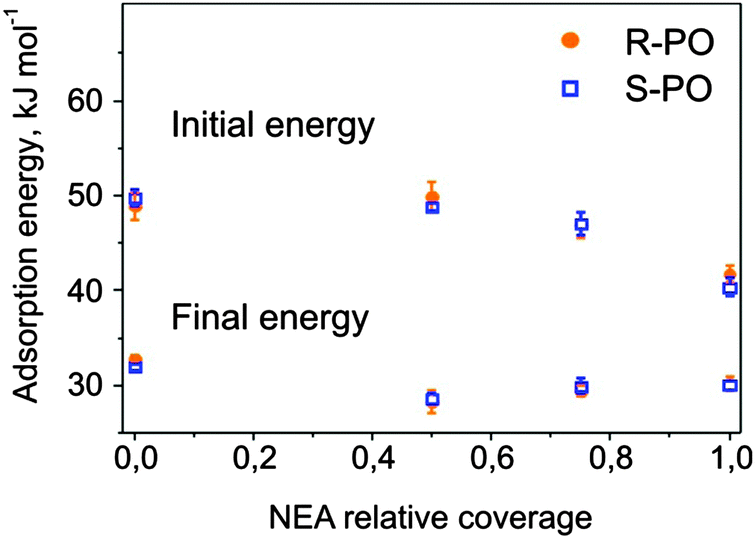 | ||
| Fig. 4 Initial and final adsorption energies of S- and R-PO plotted as a function of the NEA relative coverage. The initial adsorption energy is shown in the upper part, while the final adsorption energy is plotted in the lower part of the graph. | ||
b. PO adsorption on MBA-modified Pt(111)
The second model system investigated in this study is Pt(111) modified by S-MBA. This chiral modifier was previously shown to produce the carboxylate species upon thermal treatment.25,31 In contrast to NEA, the chiral center of MBA does not possess the ability to form hydrogen bonds. Instead, it is hypothesized that the adsorbed MBA molecules form supramolecular structures with a long-range order.25 This overlayer can be considered as a proxy for a templating-type of chirality induction.It was well-established in previous IRAS studies25 that MBA partly dehydrogenates on Pt(111) at 230 K to form 2-methylbutanoate species and further decomposes and partly desorbs above this temperature. These surface processes prevent the precise calibration of the deposited MBA amounts by measuring the CO sticking coefficient as described above for the case of NEA. In the following, we will report the amount of deposited MBA in terms of the deposition time and give some consideration to the absolute MBA coverage achieved in our deposition procedure. The bare Pt(111) was exposed to MBA at gradually increased exposure times and heated to 230 K to produce 2-methylbutanoate species. Adsorption of S- and R-PO was measured at 120 K, the corresponding adsorption energies are plotted in Fig. 5 as a function of the number of adsorbed PO molecules for different amounts of the surface modifier. Up to the deposition time of about 2.5 minutes, the energy curves were found to be very similar in shape to the ones obtained in the case of PO adsorption experiments on pristine Pt(111), exhibiting a plateau or a small maximum followed by a sharp decrease of the adsorption energy to ∼32 kJ mol−1. This behavior is dramatically different for the deposition times above 2.5 minutes: the adsorption energy decreases continuously with increasing PO coverage and the finally reached energy level lies well below 32 kJ mol−1. These observations can be rationalized if one assumes that the saturation of one monolayer of MBA occurs at the deposition time close to appr. 2.5 minutes. In the scope of this assumption, the similar form of the adsorption curves below the modifier exposure of 2.5 minutes corresponds to the situation where PO adsorbs on the free metal surface between the modifier molecules and exhibits the same adsorption behavior as on the pristine Pt surface. The change of the PO adsorption behavior above 2.5 minutes MBA exposure can be most likely attributed to the onset of formation of a second and further MBA layers so that almost no free metal surface is exposed and PO adsorbs on top of that layer very weakly.
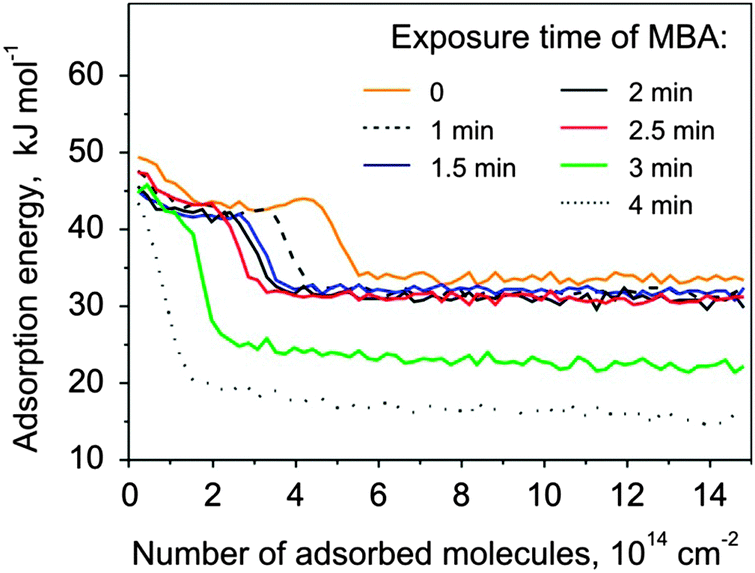 | ||
| Fig. 5 Adsorption energy of S-PO at 120 K on MBA covered Pt(111) for different MBA deposition times. | ||
Following this argument, we assume that a monolayer of MBA is reached after about 2.5 minutes exposure and use a term of relative modifier coverage with ΘMBA = 0 on pristine metal and ΘMBA = 1 after 2.5 minutes exposure.
Fig. 6 shows adsorption energies of both PO enantiomers as a function of PO coverage for four different modifier amounts. The adsorption energy on bare Pt was found to be almost the same as that displayed in Fig. 3a.
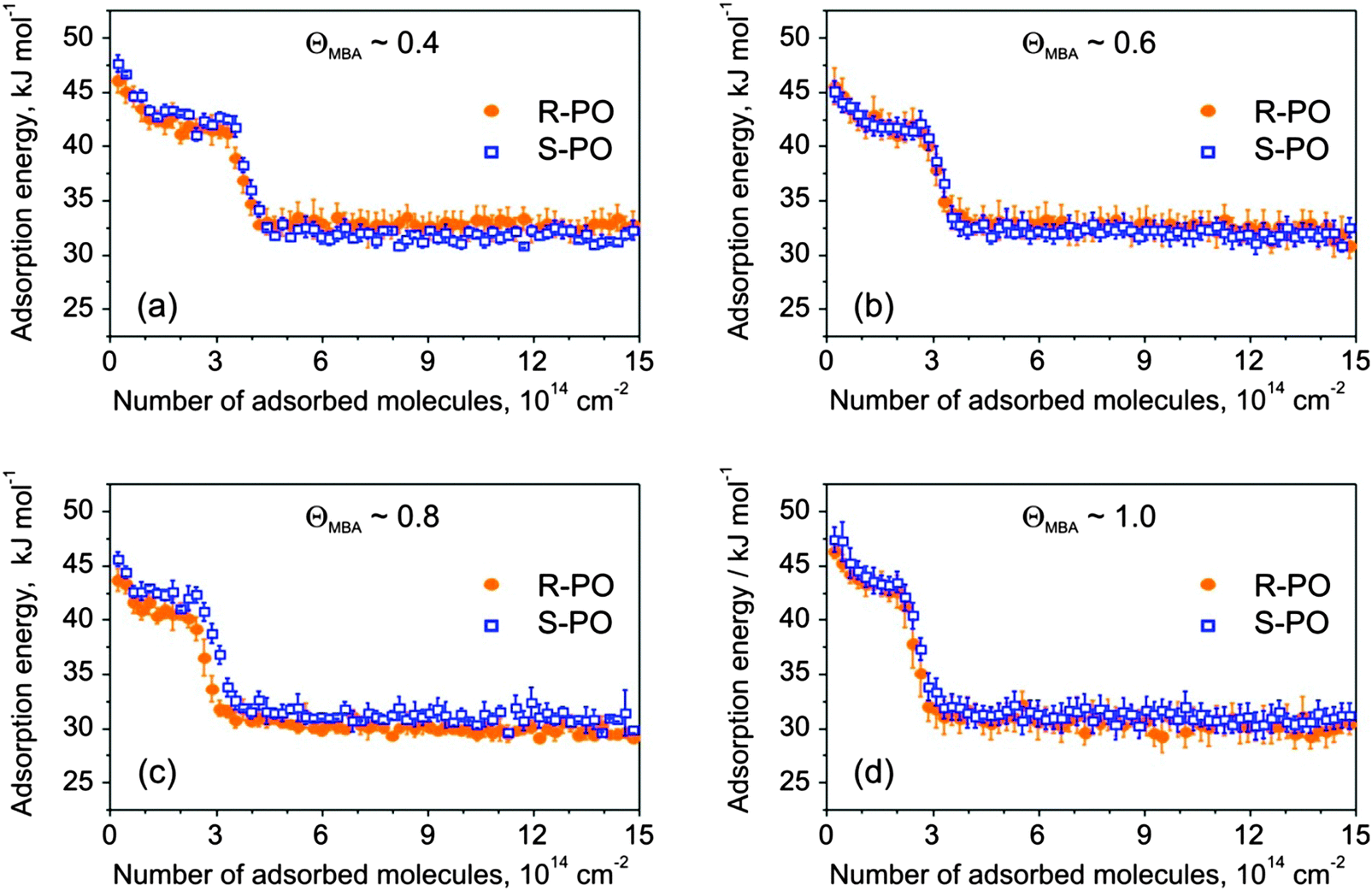 | ||
| Fig. 6 Adsorption energy of S- and R-PO as a function of the number of adsorbed molecules at 120 K on MBA-covered Pt(111) with the MBA relative coverage of: (a) 0.4; (b) 0.6; (c) 0.8; (d) 1. The data points are averages of 3–4 independent measurements. The error bars show the error of the mean. | ||
For all investigated MBA coverage, the initial adsorption energy of both S- and R-PO lies between 45 and 47 kJ mol−1 and decays to 30–32 kJ mol−1 in the multilayer regime. The PO adsorption behavior on MBA-covered Pt(111) drastically differs from that one on the NEA modified Pt surface. For all MBA modifier coverage, the adsorption energy is not strictly monotonically decaying (see Fig. 3b–d) but follows the pattern observed on the bare Pt(111) surface, i.e. it exhibits a clear plateau-like region followed by a fast decay to a constant level of 31 ± 1 kJ mol−1. As discussed above, we attribute the earlier stages of PO adsorption to adsorption on the metal, while the drop to the constant final value is related to PO adsorption in the second and the following layers. It is interesting to note that the presence of the MBA surface modifier almost does not change the shape of the measured curves and only the transition to the multilayer adsorption occurs at lower PO exposures in the presence of higher MBA amounts. This behavior suggests a very weak interaction between the PO molecules and the chiral surface modifier, which is unable to noticeably change the energetic situation on the Pt surface if present in sub-monolayer amounts. Most likely, PO adsorbs on the free metal patches available between the MBA molecules without showing any strong intermolecular interaction with the chiral surface modifier. This situation is in very sharp contrast to the case of NEA that was found to change the PO adsorption behavior even if present in the smallest surface concentrations. For comparison, we refer the reader to Fig. 3b, showing a strong change of the adsorption curve from a plateau-like on the bare Pt(111) to a strictly monotonically decaying when the smallest amount of NEA was deposited.
The initial and final adsorption energies were found to be approximately the same for all investigated surfaces. Only subtle differences in energy between the two enantiomers were observed for the intermediate PO coverage on 0.8 ML S-MBA/Pt(111) that might be indicative of a specific stereoselective interaction. Also the amounts of the adsorbed PO before the onset of the multilayer regime appear to be nearly identical for the two different enantiomers. In order to make a strict quantitative comparison of the amounts of PO adsorbed in one monolayer on different chirally modified surfaces, we treated the experimental data as following: the intermediate regions of the adsorption curves were best fitted by the Boltzmann function, and as a measure of the transition from sub-monolayer to multilayer PO adsorption the inflection points were taken. The results of this evaluation are presented in Fig. 7 showing the S- and R-PO uptake onto the metal surface (the PO coverage at the inflection point) as a function of the deposited MBA amount. One can see the PO uptake onto the metal decreases almost linearly with the increasing MBA coverage. When the modifier coverage is close to a monolayer, PO uptake is still almost half of that on bare platinum. This observation suggests that a monolayer of MBA forms an extended supramolecular structure, which leaves enough free metal space to accommodate large quantities of PO. Most likely, this metal pockets are too small for the modifier molecules themselves but large enough for the small probe molecules such as PO.
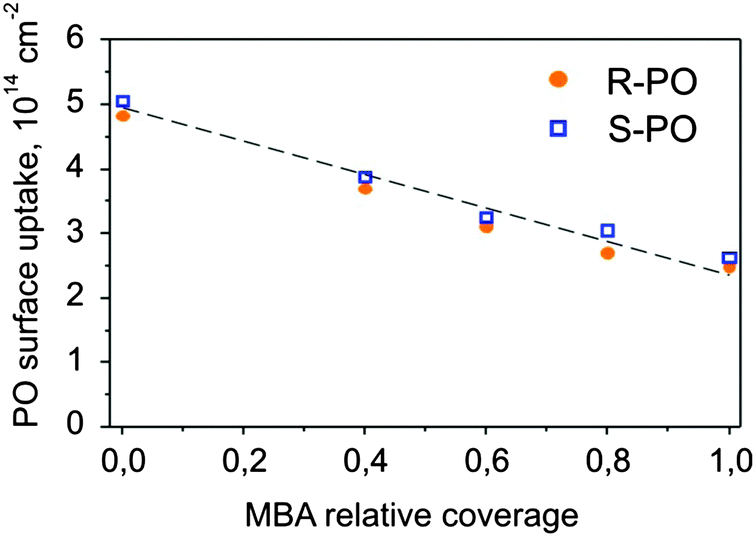 | ||
| Fig. 7 The number of PO molecules (S- and R-) adsorbed on one PO monolayer plotted as a function of the MBA relative coverage. | ||
Discussion
Based on the results obtained for both investigated chiral surface modifiers – NEA and MBA – it can be concluded that they show very different surface-modifying effects. Already the smallest sub-monolayer amounts of NEA very strongly change the adsorption behavior of both S- and R-PO on Pt(111) from a plateau-type adsorption discussed above to a strictly monotonic decrease of PO binding energy with increasing PO coverage. In contrast, on the surfaces modified with MBA, PO exhibits the same plateau-type of adsorption behavior as on the clean Pt(111) surface. The increasing amount of MBA results solely in a decreasing adsorption capacity of the surface toward PO adsorption, i.e. in smaller absolute amounts of PO accumulated in a monolayer, but does not affect the shape of the adsorption curve.Strong changes of the adsorption energy by NEA suggest a significant influence of the modifier molecules onto the incoming PO either directly, e.g. through dipole coupling or ensemble effects, or indirectly, e.g. through changing the electronic properties of the Pt surface. It is important to note that the PO binding energy on the pristine Pt(111) (∼50 ± 1 kJ mol−1) at the lowest PO coverage is comparable to that on the NEA-modified Pt(111) with the smallest investigated NEA coverage of 0.5 ML (50 ± 1.5 kJ mol−1, see Fig. 4). However, despite these vanishingly small differences in the binding energies, the presence of NEA decisively affects the adsorption behavior of PO. These observations can be rationalized as follows. When PO adsorbs on the pristine Pt(111) surface, its binding energy reaches quite quickly the constant level (see the plateau shown in Fig. 3a), which occurs most likely due to the formation of PO islands. After some PO nuclei consisting of a few PO molecules are formed on the surface, the following incoming PO molecules would diffuse to the boundary of these nuclei, where they will adsorb with a constant binding energy. In this case, the energy would be constant because all incoming molecules experience a very similar environment, consisting of a saturated PO island on one side and the clean Pt surface on the other side. Only after the PO surface coverage increases to such an extent that neighboring PO islands will start to coalesce, the binding energy of the lastly incoming PO molecules will decrease and approach the value characteristic for PO adsorption in the second layer.
When NEA is present on the surface, this plateau-like behavior vanishes even for the smallest NEA coverage, at which formally 50% of the surface exposes metal atoms. This observation suggests that PO island formation does not occur on this NEA-modified surface. Most likely, there is a preferred adsorption of PO in the vicinity of NEA, which prevents PO island formation on the still available metal patches. This type of adsorption behavior suggests a preferred interaction between a PO molecule and the adsorbed NEA modifier, which might be indicative of the formation of 1:1 complex between these molecules. This type of enantiospecific interaction for this modifier/probe molecule pair was also put forward in the recent work of Zaera.26 However, based on our experimental data, we cannot rule out other possible scenarios. Further microscopic investigations on the structure of these adsorbates are required in order to confirm the formation of the modifier/probe molecule complex.
In contrast to the strong modification of the PO adsorption properties induced by NEA chiral modifiers, MBA almost does not affect the adsorption behavior of PO, neither in terms of energy nor in terms of the plateau-like character of the adsorption curve. Indeed, even at relatively high MBA coverage, the plateau-like character of PO adsorption is preserved, suggesting that PO preferentially adsorbs on the free metal patches still available on the MBA-modified surface. This observation suggests a relatively weak affinity of PO to MBA surface species. Interestingly enough, even at relatively high MBA coverages PO adsorption is not affected by the presence of MBA. This effect can be rationalized based on the assumption that adsorbed MBA forms compact patches on Pt(111) and leaves relatively large areas of Pt surface atoms free, at which PO adsorbs in the same way as on the free Pt(111) surface. These observations point to a rather low strength of interaction between the PO and MBA and inability of MBA to strongly modify the adsorption behavior of PO. In Fig. 8 shows a model capturing the main points of this discussion.
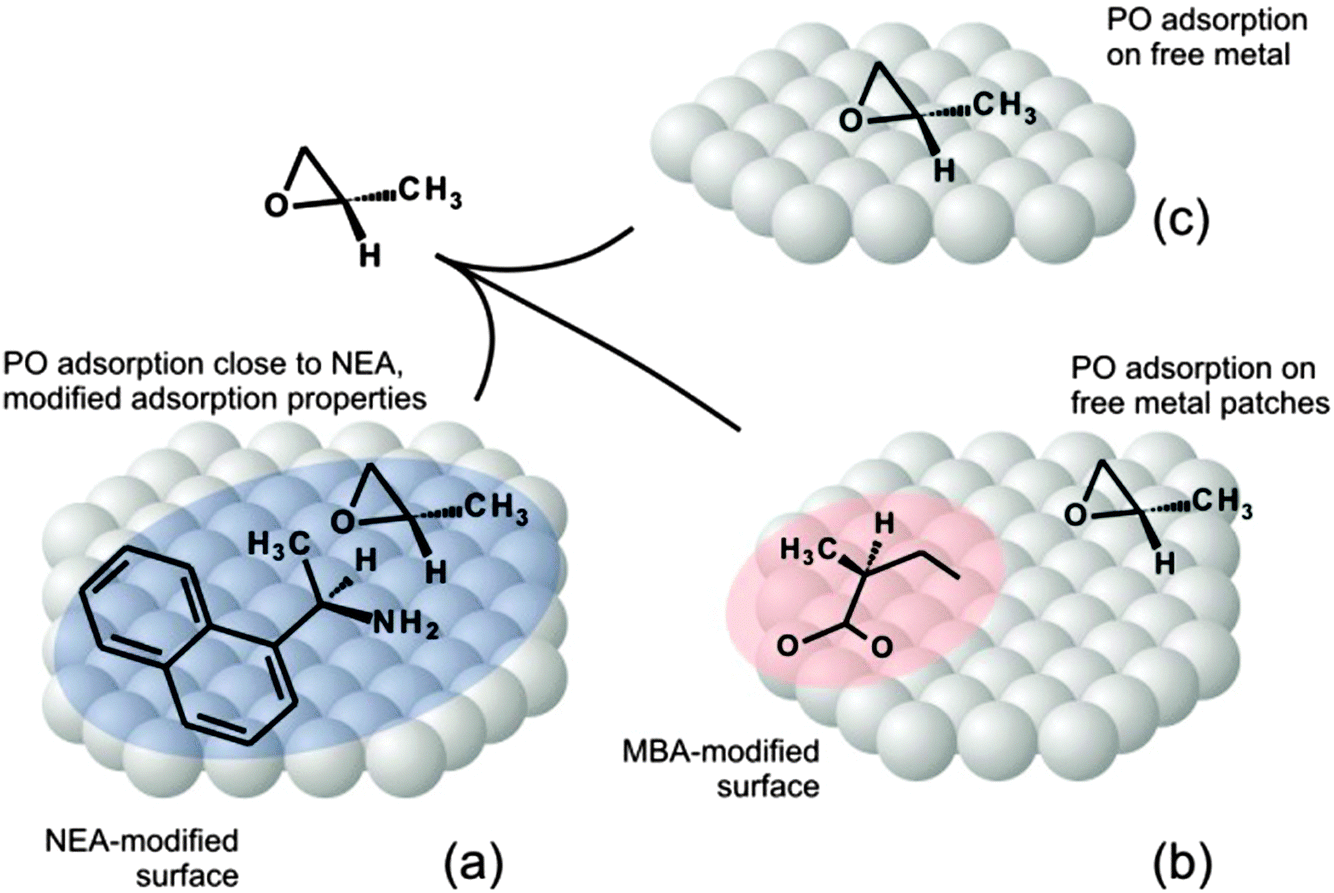 | ||
| Fig. 8 Adsorption model. (a) NEA noticeably modifies PO adsorption properties of Pt(111): PO forms a mixed layer with NEA modifier molecules; the PO binding energies are strongly affected. (b) MBA nearly does not affect the adsorption behavior of PO; PO adsorbs preferentially at the free metal patches on Pt(111) similarly to the adsorption on the clean Pt(111) surface (c). | ||
In the present study, both adsorption systems investigated by SCAC showed no enantioselective effects within our experimental accuracy either in terms of the adsorption energy or in terms of the absolute PO uptake. On the contrary, the same systems were previously reported to exhibit pronounced enantioselectivity in desorption studies: larger uptakes in homochiral probe-modifier pairs were reported with the enantiomeric ratios amounting to 2.3 and 1.24 for NEA and MBA, respectively.25,26 The apparent discrepancy between our results and the reported data originates most likely from the differences in the applied experimental methods and correspondingly in the underlying physicochemical processes. In a SCAC experiment, a direct adsorption process was investigated by measuring the adsorption energies. In addition, the absolute amounts of the adsorbed PO molecules were determined via sticking coefficient measurements. Both processes were addressed under isothermal and non-reactive adsorption conditions. The adsorption temperatures applied in this study (110–120 K) were too low to induce any further chemical transformation of PO on the chirally modified surface, so that only the adsorption process was probed. In contrast, in the above discussed experimental studies reporting enantiospecific effects the opposite process was probed – desorption of PO at elevated temperatures from chirally modified surfaces – by employing temperature programmed desorption. A number of accompanying surface processes can occur upon heating that might impart enantioselectivity to a surface process. For example, upon heating the surface modifier and/or the probe molecules and/or the modifier – probe molecule pair can decompose or undergo other structural transformations. These processes can in principle depend on the particular geometric orientation of the adsorbates with respect to the chiral modifier. Alternatively, the adsorbate and/or modifier overlayers can undergo thermal restructuring giving the adsorbates a possibility to find an energetically more favorable site for desorption (i.e. exhibiting lower activation barrier for desorption) prior to the onset of desorption. Indeed, as pointed out by Zaera and coworkers,25,26 the TPD yield of PO, firstly, does not change much in the whole range of MBA surface coverage and, secondly, shows a pronounced deviation from the monotonic decay depending on NEA coverage. Both effects seem to be inconsistent with a simple co-adsorption mechanism and might be indicative of the rearrangement of the modifier species in a desorption process.
In the very general case, the desorption process might be a very complex surface reaction consisting of a number of elementary steps, e.g. competing desorption, decomposition and diffusion to a different surface site, from where it may desorb with a lower desorption barrier. In principle, each of these elementary and competing steps might induce enantioselectivity into the overall desorption process. Therefore caution is required when referring the differences observed in a desorption experiment to the adsorption process. Both of the chiral modifiers investigated in this study show no greater adsorption capacity in homochiral modifier–adsorbate pairs as compared to heterochiral pairs as directly determined via sticking coefficient measurements. These observations are in sharp contrast to previous studies based on temperature programmed desorption reporting large variation in the amount of desorbing adsorbates for the particular chirality of the modifier and PO. Further atomistic level investigations on the observed differences between the adsorption and the desorption processes might shed some light on this problem.
Conclusions
Adsorption of S- and R-PO enantiomers onto pristine and chirally modified Pt(111) surfaces was investigated under isothermal conditions in a combined study including the direct calorimetric measurement of adsorption energies by single crystal adsorption calorimetry (SCAC) as well as measurements of sticking coefficients for determining the absolute amounts of adsorbed molecules. As chiral modifiers, NEA and MBA were employed. These chiral modifiers were chosen as potential proxies for two different mechanisms of imparting chirality to achiral metal surfaces: a templating mechanism (MBA) based on the formation of chiral overlayers and a docking mechanism (NEA) based on the formation of 1:1 complexes between the chiral modifier and the adsorbate molecules. Detailed quantitative information on adsorption energies as a function of PO coverage and the content of chiral modifiers was obtained for the first time using a direct calorimetric method for the measurement of adsorption energies on well-defined surfaces. On a pristine Pt(111) surface, S- and R-PO were found to adsorb with an initial adsorption energy of 50 ± 1 kJ mol−1 and to exhibit a very specific adsorption behavior characterized by the presence of a plateau/small peak at a PO coverage lying in the range between 4 and 5 × 1014 molecules cm−2. This specific plateau-like behavior might be explained by PO island formation. In the presence of 0.5 ML of NEA, the initial PO adsorption energy of both enantiomers does not change but the dependence of the adsorption energy on the PO coverage changes dramatically. NEA adsorbates result in the complete elimination of a plateau region in the adsorption curve and induce a strictly monotonic decrease of the PO binding energy with growing PO coverage. This observation suggests a high affinity of incoming PO molecules to adsorbed NEA, probably due to a strong attractive intermolecular interaction, which might be consistent with the formation of a 1:1 docking complex. In contrast to NEA, the interaction of S- and R-PO with the MBA-modified surfaces was found to be very similar to PO adsorption on the clean metal. Particularly, the specific plateau-like dependency of the adsorption energy on the PO coverage was observed to be preserved even at relatively high MBA coverage. PO appears to have a very low affinity for the co-adsorbed MBA and to prefer adsorption on the still available free metal sites/patches of free metal. With this, the MBA surface species seem to be rather incapable of strong intermolecular bonding with PO, at least at low PO coverages, and appear to only sterically affect adsorption on metal sites.
No significant difference was found in the adsorption of two different enantiomers of PO on both chirally-modified surfaces, either in terms of the adsorption energy or in terms of the absolute amount of PO being able to adsorb on the chiral surface of the same vs. the opposite chirality. With this, our findings turned out to be in sharp contrast to several literature studies reporting different adsorption energies and different adsorption capacities of chiral surfaces for adsorbates of different chirality. We attribute the observed discrepancy to the differences in the methodologies applied in the present and in the earlier studies: while direct methods of measurement of adsorption energies (SCAC) and the amounts of adsorbed PO (sticking coefficient measurement) were applied in this study, earlier studies were based on the investigation of the desorption processes from chirally-modified surfaces induced by heating. We discuss the possible reasons for this discrepancy.
Acknowledgements
The authors thank Andrew J. Gellman and Wilfred T. Tysoe for fruitful discussions. S.S. acknowledges support from the European Research Council (ERC Starting Grant 335205 ENREMOS) and the Verband der Chemischen Industrie for the Dozentenstipendium.References
- T. Mallat, E. Orglmeister and A. Baiker, Asymmetric catalysis at chiral metal surfaces, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS PubMed.
- H. U. Blaser and M. Studer, Cinchona-modified platinum catalysts: from ligand acceleration to technical processes, Acc. Chem. Res., 2007, 40, 1348–1356 CrossRef CAS PubMed.
- L. D. Pachon, I. Yosef, T. Z. Markus, R. Naaman, D. Avnir and G. Rothenberg, Chiral imprinting of palladium with cinchona alkaloids, Nat. Chem., 2009, 1, 160–164 CrossRef PubMed.
- M. C. Holland, F. Meemken, A. Baiker and R. Gilmour, Chiral imidazolidinone and proline-derived surface modifiers for the Pt-catalysed asymmetric hydrogenation of activated ketones, J. Mol. Catal. A: Chem., 2015, 396, 335–345 CrossRef CAS PubMed.
- G. Kyriakou, S. K. Beaumont and R. M. Lambert, Aspects of Heterogeneous Enantioselective Catalysis by Metals, Langmuir, 2011, 27, 9687–9695 CrossRef CAS PubMed.
- F. Meemken, A. Baiker, J. Dupre and K. Hungerbuhler, Asymmetric Catalysis on Cinchonidine-Modified Pt/Al2O3: Kinetics and Isotope Effect in the Hydrogenation of Trifluoroacetophenone, ACS Catal., 2014, 4, 344–354 CrossRef CAS.
- S. Tan and C. T. Williams, An In Situ Spectroscopic Study of Prochiral Reactant-Chiral Modifier Interactions on Palladium Catalyst: Case of Alkenoic Acid and Cinchonidine in Various Solvents, J. Phys. Chem. C, 2013, 117, 18043–18052 CAS.
- F. Meemken, K. Hungerbuhler and A. Baiker, Monitoring Surface Processes During Heterogeneous Asymmetric Hydrogenation of Ketones on a Chirally Modified Platinum Catalyst by Operando Spectroscopy, Angew. Chem., Int. Ed., 2014, 53, 8640–8644 CrossRef CAS PubMed.
- F. Meemken, N. Maeda, K. Hungerbuhler and A. Baiker, Platinum-Catalyzed Asymmetric Hydrogenation: Spectroscopic Evidence for an O–H–O Hydrogen-Bond Interaction between Substrate and Modifier, Angew. Chem., Int. Ed., 2012, 51, 8212–8216 CrossRef CAS PubMed.
- F. Zaera, Chiral Modification of Solid Surfaces: A Molecular View, J. Phys. Chem. C, 2008, 112, 16196–16203 CAS.
- K. H. Ernst, Molecular chirality at surfaces, Phys. Status Solidi B, 2012, 249, 2057–2088 CrossRef CAS PubMed.
- R. Raval, Creating chiral architectures at metal surfaces, J. Phys.: Condens. Matter, 2002, 14, 4119–4132 CrossRef CAS.
- S. K. Beaumont, G. Kyriakou, D. J. Watson, O. P. H. Vaughan, A. C. Papageorgiou and R. M. Lambert, Influence of Adsorption Geometry in the Heterogeneous Enantioselective Catalytic Hydrogenation of a Prototypical Enone, J. Phys. Chem. C, 2010, 114, 15075–15077 CAS.
- S. Baldanza, A. Cornish, R. E. J. Nicklin, Z. V. Zheleva and G. Held, Surface chemistry of alanine on Cu{111}: adsorption geometry and temperature dependence, Surf. Sci., 2014, 629, 114–122 CrossRef CAS PubMed.
- J. A. Boscoboinik, Y. Bai, L. Burkholder and W. T. Tysoe, Structure and Distribution of S-alpha-(1-Naphthyl)-ethylamine on Pd(111), J. Phys. Chem. C, 2011, 115, 16488–16494 CAS.
- M. Garvey, Y. Bai, J. A. Boscoboinik, L. Burkholder, T. E. Sorensen and W. T. Tysoe, Identifying Molecular Species on Surfaces by Scanning Tunneling Microscopy: Methyl Pyruvate on Pd(111), J. Phys. Chem. C, 2013, 117, 4505–4514 CAS.
- A. G. Trant and C. J. Baddeley, Surface Chemistry Underpinning Enantioselective Heterogeneous Catalysis: Supramolecular Self-Assembly of Chiral Modifiers and Pro-Chiral Reagents on Ni{111}, J. Phys. Chem. C, 2011, 115, 1025–1030 CAS.
- K. E. Wilson, A. G. Trant and C. J. Baddeley, Interaction of the Pro-Chiral Molecule, Methylacetoacetate, with (S)-Aspartic Acid Modified Ni{111}, J. Phys. Chem. C, 2012, 116, 1092–1098 CAS.
- J. Greenwood, H. A. Fruchtl and C. J. Baddeley, Self-Assembly of Upright, Partially Dehydrogenated Melamine on Pd(111), J. Phys. Chem. C, 2013, 117, 22874–22879 CAS.
- W. Y. Cheong and A. J. Gellman, Enantiospecific Desorption of R- and S-Propylene Oxide from D- or L-Lysine Modified Cu(100) Surfaces, Langmuir, 2012, 28, 15251–15262 CrossRef CAS PubMed.
- Y. J. Yun and A. J. Gellman, Enantioselective Separation on Naturally Chiral Metal Surfaces: d,l-Aspartic Acid on Cu(3,1,17)(R&S) Surfaces, Angew. Chem., Int. Ed., 2013, 52, 3394–3397 CrossRef CAS PubMed.
- G. Goubert, M. N. Groves, Y. Dong, J.-C. Lemay, P. H. McBreen and B. Hammer, Isolating a Reaction Intermediate in the Hydrogenation of 2,2,2-Trifluoroacetophenone on Pt(111), J. Phys. Chem. C, 2015, 119, 7319–7326 CAS.
- V. Demers-Carpentier, G. Goubert, F. Masini, R. Lafleur-Lambert, Y. Dong, S. Lavoie, G. Mahieu, J. Boukouvalas, H. L. Gao, A. M. H. Rasmussen, L. Ferrighi, Y. X. Pan, B. Hammer and P. H. McBreen, Direct Observation of Molecular Preorganization for Chirality Transfer on a Catalyst Surface, Science, 2011, 334, 776–780 CrossRef CAS PubMed.
- V. Demers-Carpentier, A. M. H. Rasmussen, G. Goubert, L. Ferrighi, Y. Dong, J. C. Lemay, F. Masini, Y. Zeng, B. Hammer and P. H. McBreen, Stereodirection of an alpha-Ketoester at Sub-molecular Sites on Chirally Modified Pt(111): Heterogeneous Asymmetric Catalysis, J. Am. Chem. Soc., 2013, 135, 9999–10002 CrossRef CAS PubMed.
- I. Lee and F. Zaera, Chiral templating of surfaces: adsorption of (S)-2-methylbutanoic acid on Pt(111) single-crystal surfaces, J. Am. Chem. Soc., 2006, 128, 8890–8898 CrossRef CAS PubMed.
- I. Lee, Z. Ma, S. Kaneko and F. Zaera, 1-(1-Naphthyl)Ethylamine Adsorption on Platinum Surfaces: On the Mechanism of Chiral Modification in Catalysis, J. Am. Chem. Soc., 2008, 130, 14597–14604 CrossRef CAS PubMed.
- S. Karakalos, T. J. Lawton, F. R. Lucci, E. C. H. Sykes and F. Zaera, Enantiospecific Kinetics in Surface Adsorption: Propylene Oxide on Pt(111) Surfaces, J. Phys. Chem. C, 2013, 117, 18588–18594 CAS.
- A. J. Gellman, W. T. Tysoe and F. Zaera, Surface Chemistry for Enantioselective Catalysis, Catal. Lett., 2015, 145, 220–232 CrossRef CAS.
- D. Stacchiola, L. Burkholder and W. T. Tysoe, Enantioselective chemisorption on a chirally modified surface in ultrahigh vacuum: adsorption of propylene oxide on 2-butoxide-covered palladium(111), J. Am. Chem. Soc., 2002, 124, 8984–8989 CrossRef CAS PubMed.
- D. Stacchiola, L. Burkholder and W. T. Tysoe, Probing enantio selective chemisorption in ultrahigh vacuum, J. Mol. Catal. A: Chem., 2004, 216, 215–221 CrossRef CAS PubMed.
- D. Stacchiola, L. Burkholder, T. Zheng, M. Weinert and W. T. Tysoe, Requirements for the formation of a chiral template, J. Phys. Chem. B, 2005, 109, 851–856 CrossRef CAS PubMed.
- F. Gao, Y. L. Wang, L. Burkholder and W. T. Tysoe, Enantioselective chemisorption of propylene oxide on a 2-butanol modified Pd(111) surface: the role of hydrogen-bonding interactions, J. Am. Chem. Soc., 2007, 129, 15240–15249 CrossRef CAS PubMed.
- F. Gao, Y. L. Wang and W. T. Tysoe, Enantioselective chemisorption and reactions on model chirally modified surfaces: 2-butanol on L-proline templated Pd(111) surfaces, J. Phys. Chem. C, 2008, 112, 6145–6150 CAS.
- I. Lee and F. Zaera, Enantioselectivity of adsorption sites created by chiral 2-butanol adsorbed on Pt(111) single-crystal surfaces, J. Phys. Chem. B, 2005, 109, 12920–12926 CrossRef CAS PubMed.
- E. M. Karp, T. L. Silbaugh and C. T. Campbell, Energetics of Adsorbed CH3 and CH on Pt(111) by Calorimetry: Dissociative Adsorption of CH3I, J. Phys. Chem. C, 2013, 117, 6325–6336 CAS.
- M. Peter, J. M. F. Camacho, S. Adamovski, L. K. Ono, K. H. Dostert, C. P. O'Brien, B. R. Cuenya, S. Schauermann and H. J. Freund, Trends in the Binding Strength of Surface Species on Nanoparticles: How Does the Adsorption Energy Scale with the Particle Size?, Angew. Chem., Int. Ed., 2013, 52, 5175–5179 CrossRef CAS PubMed.
- J. H. Fischer-Wolfarth, J. Hartmann, J. A. Farmer, J. M. Flores-Camacho, C. T. Campbell, S. Schauermann and H. J. Freund, An improved single crystal adsorption calorimeter for determining gas adsorption and reaction energies on complex model catalysts, Rev. Sci. Instrum., 2011, 82, 024102 CrossRef PubMed.
- D. A. King and M. G. Wells, Reaction-Mechanism in Chemisorption Kinetics – Nitrogen on (100) Plane of Tungsten, Proc. R. Soc. London, Ser. A, 1974, 339, 245–269 CrossRef CAS.
- J. Chao, K. R. Hall, K. N. Marsh and R. C. Wilhoit, Thermodynamic Properties of Key Organic Oxygen Compounds in the Carbon Range C1 to C-4.2. Ideal-Gas Properties, J. Phys. Chem. Ref. Data, 1986, 15, 1369–1436 CrossRef CAS PubMed.
- J. M. Bonello, F. J. Williams and R. M. Lambert, Aspects of enantioselective heterogeneous catalysis: structure and reactivity of (S)-(−)-1-(1-naphthyl)ethylamine on Pt{111}, J. Am. Chem. Soc., 2003, 125, 2723–2729 CrossRef CAS PubMed.
- Y. Y. Yeo, L. Vattuone and D. A. King, Calorimetric heats for CO and oxygen adsorption and for the catalytic CO oxidation reaction on Pt{111}, J. Chem. Phys., 1997, 106, 392–401 CrossRef CAS PubMed.
- J.-H. Fischer-Wolfarth, J. Hartmann, J. A. Farmer, J. M. Flores-Camacho, C. T. Campbell, S. Schauermann and H.-J. Freund, An improved single crystal adsorption calorimeter for determining gas adsorption and reaction energies on complex model catalysts, Rev. Sci. Instrum., 2011, 82, 024102 CrossRef PubMed.
| This journal is © the Owner Societies 2015 |