
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Retinal shows its true colours: photoisomerization action spectra of mobility-selected isomers of the retinal protonated Schiff base†
N. J. A.
Coughlan
,
B. D.
Adamson
,
L.
Gamon
,
K.
Catani
and
E. J.
Bieske
*
School of Chemistry, The University of Melbourne, Melbourne, Australia. E-mail: evanjb@unimelb.edu.au; Tel: +61 3 8344 7082
First published on 5th August 2015
Abstract
Retinal is one of Nature's most important and widespread chromophores, exhibiting remarkable versatility in its function and spectral response, depending on its protein environment. Reliable spectroscopic and photochemical data for the isolated retinal molecule are essential for calibrating theoretical approaches that seek to model retinal's behaviour in complex protein environments. However, due to low densities and possible co-existence of multiple isomers, retinal is a challenging target for gas-phase investigations. Here, the photoisomerization behaviour of the trans isomer of the retinal protonated Schiff base (RPSB) is investigated in the gas phase by irradiating mobility-selected RPSB ions with tunable light in a tandem ion mobility spectrometer. trans RPSB ions are converted to single cis isomers and also more compact isomers through irradiation with visible light. The S1 ← S0 photoisomerization action spectrum of trans RPSB, obtained by monitoring production of cis isomers as a function of wavelength, exhibits a single well-defined peak with a maximum at 618 ± 5 nm. Corresponding action spectra of cis RPSB isomers exhibit broader peaks, conclusively demonstrating an isomeric dependence for the RPSB spectrum in the gas phase.
1 Introduction
Harvesting light to perform cellular functions is a keystone of biological systems, which normally combine three-dimensional protein structures and chromophore sub-units that function with remarkable efficiency, specificity and tunability. Retinal is widespread as a chromophore in light sensitive cells, where it is hosted by opsin-type proteins, anchored as RPSB to a lysine residue (see Fig. 1), and is responsible through its photoisomerization for initiation of cellular signalling processes including animal vision and for photosynthesis in bacteria.1 Two features of RPSB stand out – the tunability of its spectral response and the specificity of its isomerization pathways. In the visual receptors of animals, retinal is converted from the 11-cis isomer to the trans isomer, whereas in photosynthetic bacteria and archaea it is converted from the trans isomer to the 13-cis isomer. The absorption characteristics of retinal-containing opsins vary widely between different species, as well as between members of the same species, despite sharing the same chromophore. For example, the scotopic pigment of Euphasia pacifica, a deep water fish, has an absorption at 462 nm, whereas similar pigments found in surface-dwelling fish have an absorption maximum close to 500 nm.2 In comparison, humans possess three distinct rhodopsin pigments associated with colour vision, with absorption maxima at 425, 530 and 560 nm.1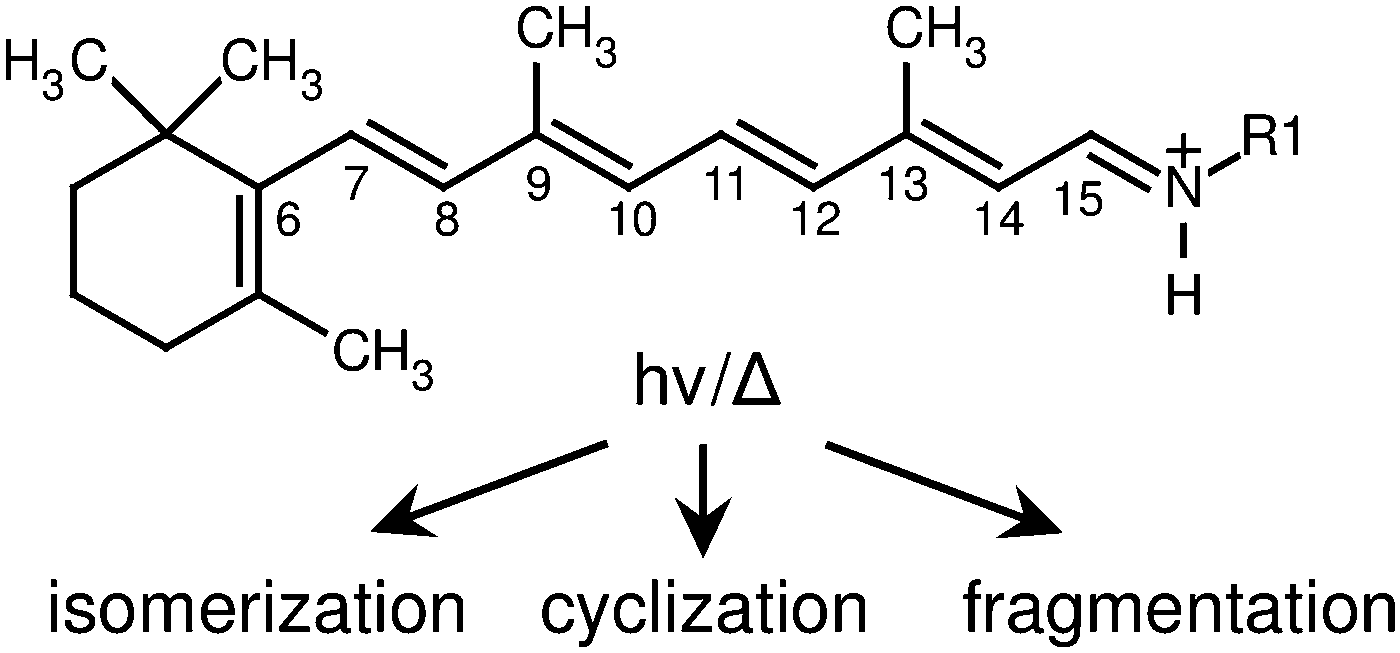 | ||
| Fig. 1 All-trans retinal protonated Schiff base undergoes structural rearrangement following absorption of a photon or thermal excitation (in this study R1 = n-Bu, in vivo R1 = Lys). | ||
The tunability of RPSB's spectral response in biological systems stems from several factors. The quaternary structure of the protein scaffolding surrounding the chromophore dictates the nature of electrostatic interactions between protein residues and the chromophore, which in turn influence the absorption wavelength. Furthermore, the ground electronic state of RPSB can be stabilized by a counter-ion neighbouring the protonation site, causing a blue shift in the absorption. Conversely, a counter-ion situated near the β-ionone ring stabilizes the excited state leading to a red shift.1 The protonation state of the chromophore also influences the pigment's absorption profile; the unprotonated retinal Schiff base absorbs light in the UV region (λmax = 360 nm), whereas RPSB absorbs visible light (λmax = 450 nm).1 Finally, the absorption wavelength of RPSB depends subtly on its isomeric form. For example, in a hexane solution 9-cis RPSB absorbs at 441 nm, whereas the all-trans isomer absorbs at 458 nm.3
A crucial step in understanding the protein's influence on the spectral response of RPSB and explaining the specificity for isomeric conversion lies in thoroughly characterizing the isolated chromophore. For reasons of tractability the RPSB chromophore is usually modelled as the retinal protonated Schiff base with a butyl group replacing the lysine chain (Fig. 1). Early studies focused on the photophysical properties of different RPSB isomers in solution. Spectroscopic and HPLC experiments revealed that in solution all-trans RPSB photoisomerizes mainly to the 11-cis and 13-cis forms, with traces of the 7-cis and 9-cis isomers, whereas 11-cis RPSB photoisomerizes exclusively to the all-trans isomer.3,4 Subsequent NMR studies largely confirmed these isomerization pathways; the main photoisomer from the all-trans isomer was found to be 11-cis, followed by 9-cis.5 After continued excitation, the NMR spectra became complicated and difficult to assign. HPLC analysis confirmed that the primary photoproduct of the all-trans RPSB was the 11-cis isomer, and that all cis isomers reverted exclusively back to the all-trans isomer following absorption of a photon.6
The visible absorption spectrum of RPSB isolated in vacuo was recorded first using photodissociation action spectroscopy in an ion storage ring.7 The original spectrum displayed an asymmetric band in the visible region with a maximum at 610 nm, and with a steep onset on the long wavelength side.7 In contrast, the most recent photodissociation spectrum exhibited a flat-topped peak with a plateau extending from 610 to 530 nm that was explained in terms of contributions from RPSB molecules in thermally accessible 6s-cis and 6s-trans conformations, which were predicted to have absorptions separated by 70 nm.8 The CASSCF calculations upon which this interpretation was based were subsequently contested by Valsson and Filippi who predicted instead a minor dependence of the absorption wavelength on the configuration of the 6s bond.9 The controversy emphasizes the importance of understanding and preferably controlling the isomeric constitution of the RPSB sample before photoexcitation.
In order to characterise the photoisomerization behaviour of selected RPSB isomer ions in the gas phase we have exploited a combination of ion mobility spectrometry and laser spectroscopy, separating RPSB isomers in a drift-tube ion mobility spectrometer (IMS) and using the photoinduced change in drift mobility as the basis for PISA spectroscopy.10 Originally, we observed at least 5 different RPSB isomers, some of which could be interconverted through exposure to visible light. The RPSB PISA spectrum displayed a maximum at 615 nm and possessed a similar band shape to the original photodissociation spectrum described in ref. 7 (i.e. no plateau extending from 530 to 610 nm). However, there was some ambiguity in the investigation because the irradiated ion packet contained different RPSB isomers, all of which possibly contributed to the measured spectrum. Here, we reduce uncertainties by probing mobility-selected RPSB isomers using a tandem IMS-photo-IMS arrangement, measuring the wavelength dependence for photoisomerization, and branching ratios for formation of different product isomers. Most importantly, through appropriate sample preparation and mobility selection we are able to isolate trans RPSB and record its PISA spectrum, the first occasion that a specific RPSB isomer has been spectroscopically probed in the gas phase.
The use of tandem IMS-photo-IMS to probe photoisomerization of RPSB complements recent collisional studies by Toker et al.11 in which the isomerization of mobility-selected RPSB isomers was investigated using the tandem IMS-collision-IMS approach of Clemmer and colleagues.12 Isomerization barriers extracted from the analysis ranged from 38 to 96 kJ mol−1, with the barrier between the two dominant isomers estimated to be 62 kJ mol−1.11
2 Experimental strategy
The experimental approach involves irradiating RPSB in a custom built tandem, IMS-photo-IMS apparatus, described in ref. 13–15. Briefly, ions propelled by an electric field through a neutral buffer gas are separated on the basis of their collision cross sections. The drift velocity (vd) of an ion moving under the influence of a weak electric field is proportional to the applied electric field (E),16| vd = K × E. | (1) |
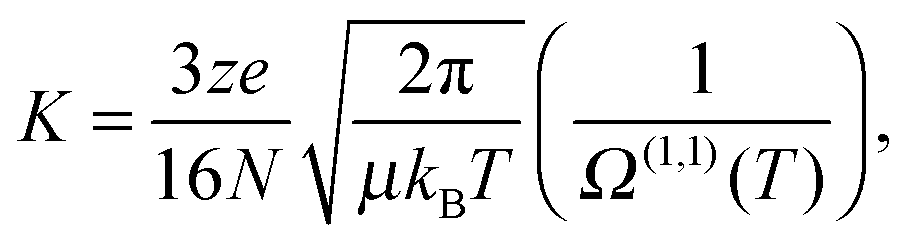 | (2) |
The experimental layout is shown in Fig. 2. RPSB cations were electrosprayed from a 10−5 M solution of trans RPSB in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of methanol and H2O with 0.2% acetic acid. Care was taken to shield the solution from light prior to electrospray. The ions entered the tandem IMS–IMS machine through a heated desolvation capillary (T = 400 K) and were accumulated in an ion funnel (IF1) before being launched in a 100 μs pulse through an electrostatic ion gate (IG1) into a 0.9 m drift tube filled with N2 buffer gas (P ≈ 6 torr, E = 44 V cm−1). Approximately halfway along the drift region is a Bradbury–Nielsen ion gate (IG2) that was opened fully to use the machine as a standard IMS apparatus, or pulsed open (typically for 100 μs) to operate the machine as a tandem IMS–IMS apparatus. At the end of the drift tube, the ions were gathered radially using a second ion funnel (IF2) before passing through a 0.35 mm orifice into an octopole ion guide from which they exited through a 3 mm orifice into a quadrupole mass filter (QMF) set to m/z = 340 Da. Ions exiting the QMF were sensed using an electron multiplier connected to a discriminator and a multichannel scaler. The apparatus was run at 20 Hz and the ions' ATD was built up as a histogram of ion counts versus time. The mobility resolution for the RPSB ions is typically tf/Δtf ≈ 60. Under usual operating conditions, the effective temperature of the drifting ions is predicted to be 314 K.17
:1 mixture of methanol and H2O with 0.2% acetic acid. Care was taken to shield the solution from light prior to electrospray. The ions entered the tandem IMS–IMS machine through a heated desolvation capillary (T = 400 K) and were accumulated in an ion funnel (IF1) before being launched in a 100 μs pulse through an electrostatic ion gate (IG1) into a 0.9 m drift tube filled with N2 buffer gas (P ≈ 6 torr, E = 44 V cm−1). Approximately halfway along the drift region is a Bradbury–Nielsen ion gate (IG2) that was opened fully to use the machine as a standard IMS apparatus, or pulsed open (typically for 100 μs) to operate the machine as a tandem IMS–IMS apparatus. At the end of the drift tube, the ions were gathered radially using a second ion funnel (IF2) before passing through a 0.35 mm orifice into an octopole ion guide from which they exited through a 3 mm orifice into a quadrupole mass filter (QMF) set to m/z = 340 Da. Ions exiting the QMF were sensed using an electron multiplier connected to a discriminator and a multichannel scaler. The apparatus was run at 20 Hz and the ions' ATD was built up as a histogram of ion counts versus time. The mobility resolution for the RPSB ions is typically tf/Δtf ≈ 60. Under usual operating conditions, the effective temperature of the drifting ions is predicted to be 314 K.17
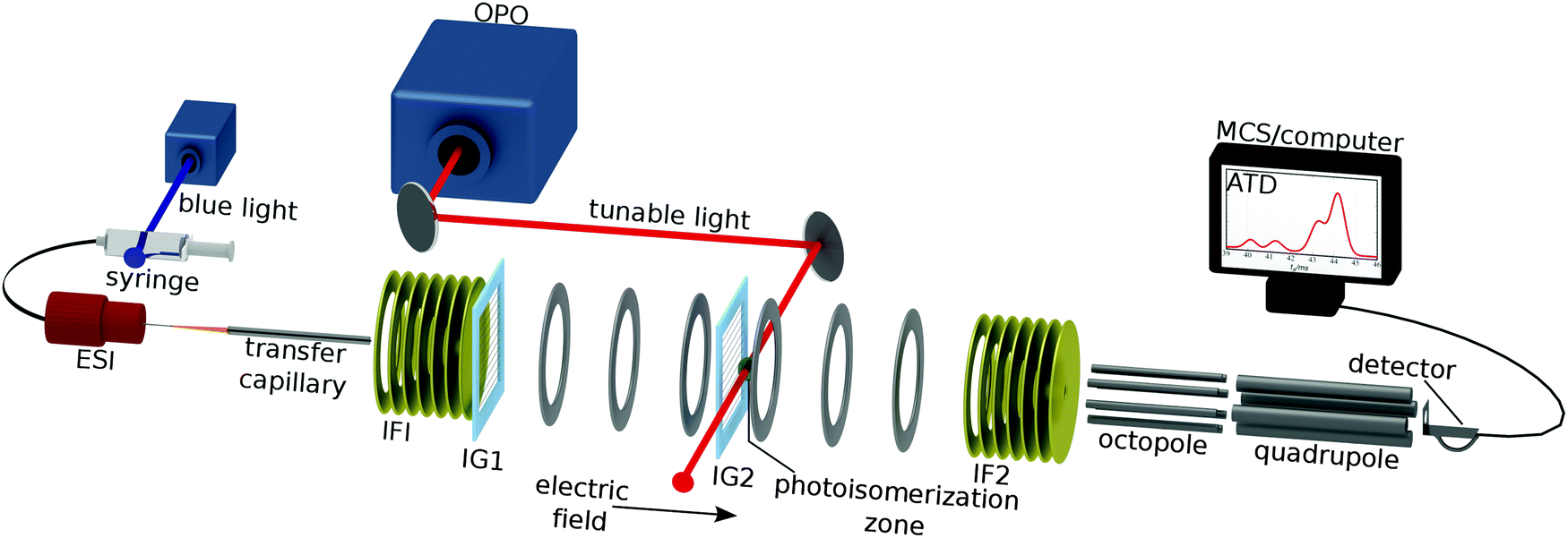 | ||
| Fig. 2 Ion mobility spectrometer for investigating photoisomerization of RPSB ions. Electrosprayed RPSB ions pass through a heated desolvation capillary (T ≈ 400 K) and into the first ion funnel (IF1) from where they are injected through an electrostatic ion gate (IG1) into the drift region (N2 gas, P ≈ 6 Torr, E ≈ 44 V cm−1). After travelling 45 cm through the drift region the ions meet a second ion gate (IG2) that is opened to transmit the desired isomer. Shortly after IG2 the ions are exposed to the pulsed output of an OPO. After passing through the drift region, ions are collected by a second ion funnel (IF2), and pass through a 0.35 mm orifice into an octopole ion guide and then a quadrupole mass filter. The sample solution can be irradiated with 460 nm light to induce RPSB isomerization prior to electrospray. | ||
Nitrogen rather than helium was used as a buffer gas because of its higher threshold for electrical breakdown, allowing the drift tube to be operated at higher drift voltage improving the mobility resolution and isomer separation.
For photoisomerization studies, mobility-selected ion packets were irradiated 15 mm after passing through IG2, with the 10 Hz output of a pulsed, tunable optical parametric oscillator (OPO, λ = 410–710 nm, 5 ns pulse width, ≈5 cm−1 bandwidth, ≈1 mJ per cm2 per pulse energy flux). Every second packet of ions was intercepted by a laser pulse. Laser-induced changes in the isomer populations are reflected in the difference between laser-on and laser-off ATDs. By recording difference ATDs at different wavelengths, it is possible to chart the wavelength dependence for depletion of the initially selected isomer and photoisomer formation. These photoisomerization action (PISA) spectra were normalized by the photon number at each wavelength.
All-trans RPSB was synthesised by exclusion of light in an N2 atmosphere as described previously.18 The sample purity was checked using electrospray time-of-flight mass spectrometry. Isomeric purity was established by comparison of 1H NMR data with literature data (see ESI†).19
Collision cross sections (Ω) for RPSB isomers were determined by measuring the arrival time (tf) for mobility-selected ions while varying the potential difference (Vd) between the second ion gate and entrance to the second ion funnel over the 1000–2000 V range. The plot of tfversus 1/Vd yields a straight line whose slope is proportional to Ω (see ESI†). Cross sections measured in this way are 1–2% larger than our previously reported values,10 which were determined from the ion flight time over the entire drift tube, second ion funnel, octopole and quadrupole, and which made allowance for counterflow of N2 gas near the first ion gate.
3 Results and discussion
3.1 DFT calculations and predicted mobilities
Relative energies and collision cross sections of RPSB isomers were calculated to help interpret the experimental ATDs. There are 29 = 512 possible combinations of cis and trans configurations for the single and double bonds along the polyene chain from C7 to N (see Fig. 1). Furthermore, the potential energy curve corresponding to torsion about the C6–C7 bond has 3 minima [6s-cis(+), 6s-cis(−), 6s-trans forms],9 giving a total 512 × 3 = 1536 possible geometric isomers, although because of steric interference many of these isomers are unstable. Previously we used density functional theory (DFT) with the M06-2X/functional and the cc-pVDZ basis set to calculate energies and collision cross sections for ≈100 lower energy isomers (including almost all single and double cis isomers), and also estimated selected isomerization barriers.10 We have extended these computations by calculating the energies and structures of the remaining single and double cis isomers, along with selected triple cis and cyclic isomers. The calculations were undertaken using Gaussian 09.20 Collision cross sections and mobilities were predicted using the classical trajectory method (TM) in the Mobcal program.21,22 As explained in ref. 10 and the ESI,† collision cross sections for the different isomers were averaged over the three configurations for the 6s bond. Atom–atom interaction parameters for collisions between N2 and RPSB were taken from ref. 23. Selected energies and collision cross sections are presented in Table 1, while a comprehensive data set is provided in the ESI.†| Isomer | ΔE (kJ mol−1) | V b (kJ mol−1) | Ω calc (Å2) |
|---|---|---|---|
| All-trans | 0 | — | 219.9 |
| 7-cis | 13.2 | 117 | 215.7 |
| 8-cis | 11.4 | 31 | 218.4 |
| 9-cis | 6.0 | 131 | 216.5 |
| 10-cis | 23.6 | 60 | 218.0 |
| 11-cis | 20.2 | 113 | 217.6 |
| 12-cis | 10.7 | 68 | 220.3 |
| 13-cis | 5.5 | 100 | 219.2 |
| 14-cis | 17.9 | 81 | 219.0 |
| 15-cis | 2.4 | 102 | 217.4 |
| Double-cis | 4.2–47.8 | — | 203.4–223.8 |
| Triple-cis | 22.0–49.4 | — | 196.3–211.0 |
| Cyclic | 45.7–70.4 | — | 190.7–205.8 |
3.2 Selective preparation of RPSB isomers
The main goal of the current study was to investigate the photophysical properties of selected RPSB isomers, particularly trans RPSB. Therefore, we systematically studied the influence on the isomer populations of pre-irradiating the electrosprayed solution, and also on the role of collisional excitation of the ions prior to their injection into the drift tube. In our previous study we identified 5 distinct peaks in the RPSB ATD.10 Although it is tempting to associate each of the observed ATD peaks with a specific RPSB isomer, it is possible that each peak contains contributions from several isomers with similar arrival times. Nevertheless, bearing this in mind, it is convenient in the following discussion to refer to each ATD peak as a specific isomer with the tacit understanding that they may actually represent several isomers.In the first experiments, we electrosprayed a solution of trans RPSB that had been shielded from light, and with the RF drive voltage for IF1 switched off to avoid collisional excitation of the ions prior to their injection into the drift tube. The resulting ATD, shown in Fig. 3, is dominated by peak A but also features the faster, less intense peak C, and two very weak peaks, B and D. These peaks were identified in our previous study.10 If the solution was cooled and protected from light for several days, peaks B–D disappeared almost entirely, leaving only peak A (see Fig. S3 in the ESI†). In contrast, as shown in Fig. 3, if the sample solution was exposed to 460 nm light, peak A was progressively depleted, whereas peaks B and C were enhanced, and a new peak E appeared. Peak D was unaffected by radiative pretreatment of the solution. The process was largely reversed if the blue light was blocked and the solution was returned to darkness, with peaks B, C and E diminishing and peak A being replenished (Fig. 3).
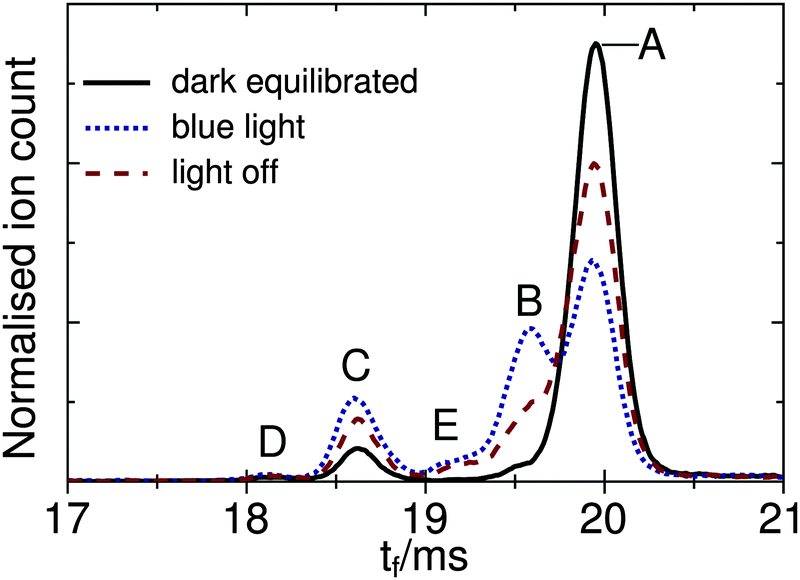 | ||
| Fig. 3 ATDs for RPSB for different pre-treatments of the electrosprayed solution. Dark equilibrated: RPSB solution in the dark at −20 for 2 hours prior to electrospray ionization. Blue light: RPSB solution exposed to 460 nm light for 1 h. Light off: 2 h after 460 nm light was blocked. The RF drive voltage to IF1 was switched off, to avoid collision induced isomerization. | ||
In a second set of experiments we investigated the effect of progressively increasing the RF drive voltage for IF1, promoting collisional activation and isomerization of the ions before they were launched into the drift tube. As noted above, at low IF1 RF drive voltage and when dark-adapted RPSB solution was electrosprayed, the ATD was dominated by isomer A [Fig. 4(a)]. However as the RF drive voltage was increased [Fig. 4(b)], isomers B and D were progressively generated, presumably through collisional activation of isomer A. On the other hand, as explained above, if the RPSB solution was irradiated with 460 nm light, the ATD obtained at low IF1 drive voltage showed evidence for isomers A–C and E [Fig. 5(a)]. As the IF1 drive voltage was raised, isomer A was again converted to isomer B, while the population of isomer D also increased. Interestingly, isomer E was destroyed by collisional activation in IF1, perhaps an indication that it is separated by a low barrier from a more stable isomer. Furthermore, at higher IF1 drive voltage, isomer C arrives at slightly longer time than at low drive voltage, giving a new peak labelled C′. The slight difference in arrival times for isomers C and C′ is discernible in Fig. S2 in the ESI.† As explained below, isomers C and C′ have distinct photochemical properties.
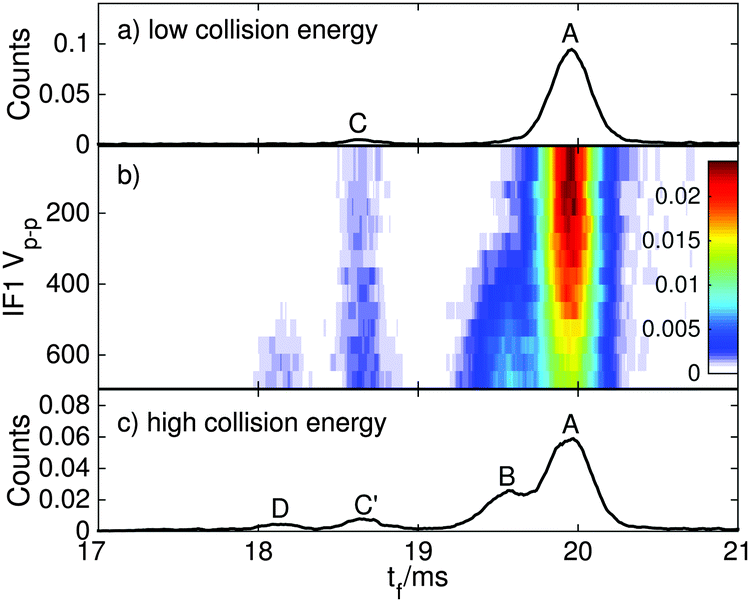 | ||
| Fig. 4 ATDs for RPSB generated from dark-equilibrated solution for different RF drive voltages in IF1. (a) At low RF drive voltage, the ATD is dominated by isomer A, with minor amounts of B and C. (b) As the drive voltage is increased, the population of isomers B, C′ and D increase, while the relative population of A decreases. (c) At high drive voltage, isomers A–D are observed. | ||
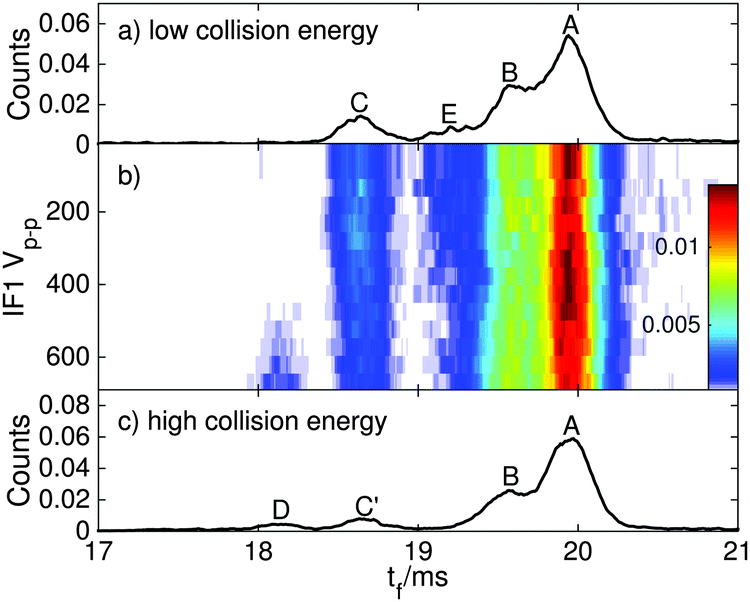 | ||
| Fig. 5 ATDs for RPSB ions electrosprayed from a solution pre-irradiated with 460 nm light for different RF drive voltages in IF1. (a) At low RF drive voltage, isomers A, B, E and C are present. (b) As the drive voltage is increased, the population of isomers C′ and D increases. (c) At high drive voltage, isomers A–D are observed. | ||
Overall, the transformations of the ion populations caused by collisions in IF1 are consistent with the tandem IMS collisional activation experiments of Toker et al.11 which showed that isomer A is mainly converted by collisions to isomer B, whereas at higher energy two other more compact isomers (labelled X and Y in their paper) were also formed. There are differences in the two experiments that make detailed comparisons difficult, including use of different buffer gases (N2versus He), preparation conditions (isomer B is more abundant than isomer A in the experiments of Toker et al.), and lack of any obvious correspondence between isomers C, C′, D and E in our ATDs, and isomers X and Y in the ATDs of Toker et al. Note that the isomerization energy estimated by Toker et al. (60 kJ mol−1) is comparable to the calculated single bond isomerization energies (30–80 kJ mol−1) but is less than the calculated double bond isomerization energies (100–130 kJ mol−1).
3.3 Photoisomerization of mobility-selected RPSB isomers
:0.15:0.09 (averaged over the 500–650 nm range). Notably, the distribution of photoisomers generated from isomer A in the gas phase was very similar to the one produced when the solution of trans RPSB was pre-irradiated with 460 nm light – production of isomers B, C and E, whereas isomer D was not formed. The sum of the B, C and E signals matches the depletion of A within 1%, proving that photodissociation was unimportant at the power levels used in these experiments. Photoisomerization data are summarized in Table 2.
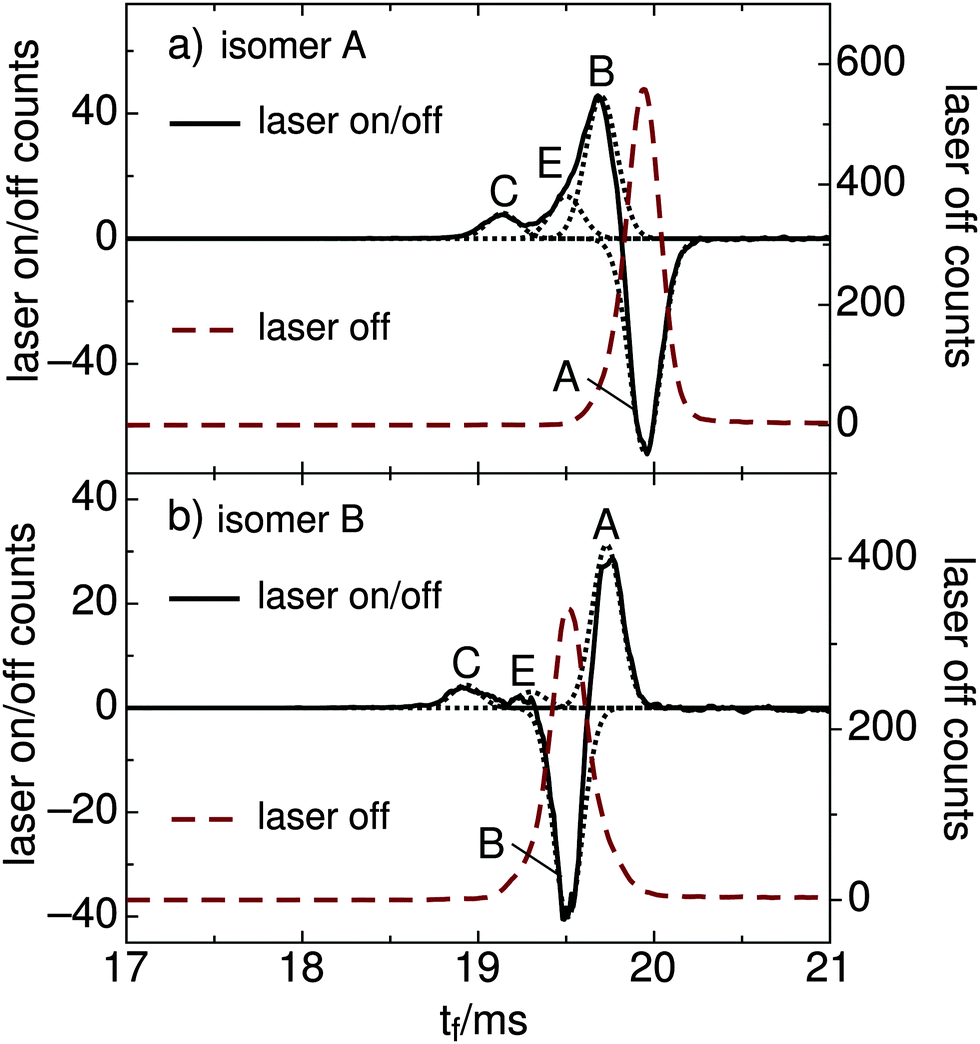 | ||
| Fig. 6 (a) ATD of RPSB cations produced from a trans RPSB solution pre-irradiated with 460 nm light. Laser-off ATD for mobility-selected isomer A (dashed red curve) and laser-on/off difference ATD (solid curve) showing depletion of A and formation of B, E, and C. (b) Laser-off ATD for mobility-selected isomer B (dashed red curve) and laser-on/off difference ATD (solid curve) showing depletion of B and formation of A, E, and C. Fitted Gaussian functions are indicated as dotted curves. The data are averaged over the 500–650 nm wavelength range. | ||
| Isomer | Ω m (Å2) | λ max (nm) | Isomer assignment | Branching ratio |
|---|---|---|---|---|
| a Measured collision cross sections have estimated absolute errors of ±5 Å2 and relative errors of ±0.3 Å2. b No discernible photoisomerization response to 410–710 nm light. | ||||
| A | 231.9 | 618 | All-trans | B:E:C |
| 0.76:0.15:0.09 |
||||
| B | 226.9 | 615 | 11-cis, 9-cis | A:E:C |
| 0.83:0.07:0.10 |
||||
| C | 213.2 | 590 | Double or triple cis | |
| C′ | 214.6 | Cyclic | ||
| D | 207.9 | Cyclic | ||
| E | 221.6 | Vis | Double-cis | |
By plotting the photoisomer signal as a function of wavelength, one can generate a PISA spectrum corresponding to the selected isomer's absorption spectrum convoluted with the wavelength dependent photoisomerization yield. The spectrum of mobility-selected isomer A obtained by monitoring production of isomers B, C, and E is shown in Fig. 7(a). The observed band, which corresponds to the S1 ← S0 transition, has a steep onset at 670 nm, a maximum at 618 ± 5 nm, and gradually declines at 470 nm. The onset of the weaker S2 ← S0 transition is apparent below 450 nm. Spectra obtained for different product channels and by monitoring the depletion of isomer A are all similar to the spectrum shown in Fig. 7(a). These spectra are provided in the ESI.†
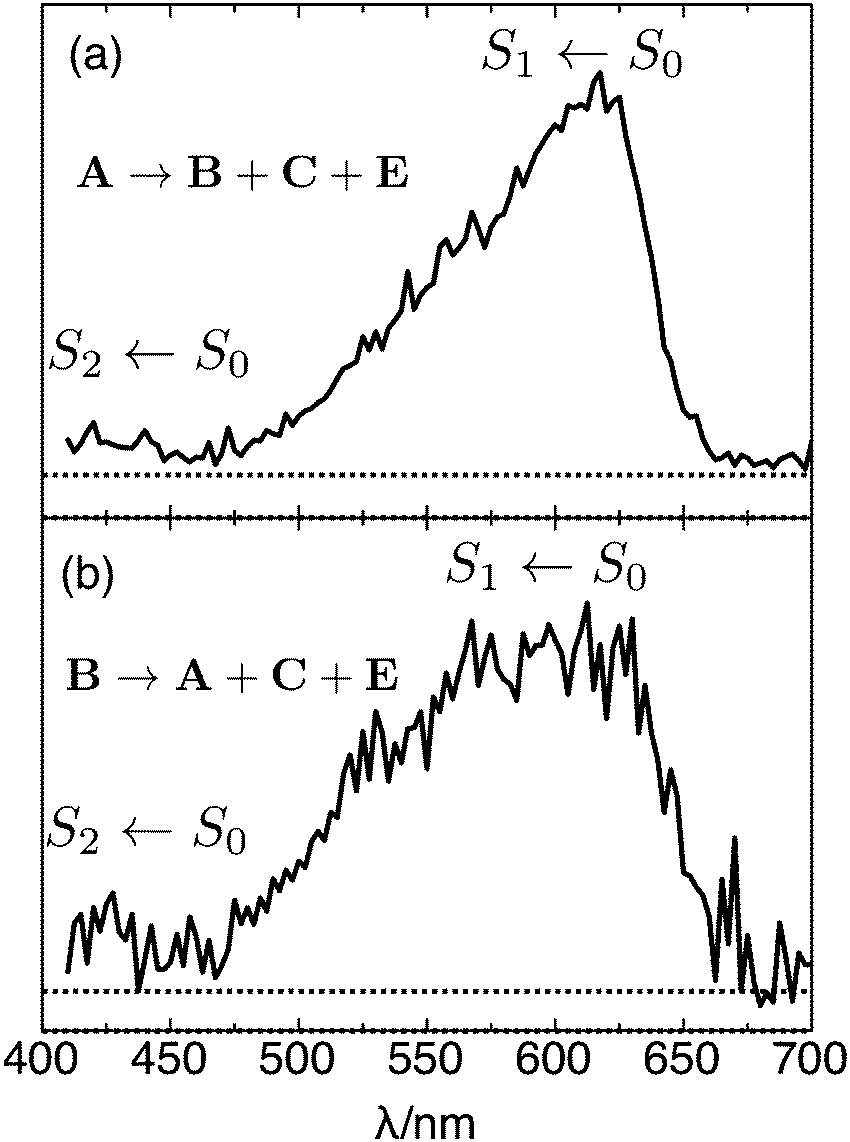 | ||
| Fig. 7 (a) PISA spectrum of isomer A (trans RPSB) obtained by monitoring total production of isomers B, C, and E as a function of excitation wavelength. (b) PISA spectrum of isomer B (cis RPSB) obtained by monitoring total production of isomers A, C and E as a function of excitation wavelength. In both cases, the signal was normalized with respect to photon number. | ||
The fact that peak A dominates the ATD when the ions were generated from a solution of trans RPSB and were not subject to violent collisions in IF1 strongly suggests that it corresponds to trans RPSB, although it is possible that the 13-cis isomer, which is known to form thermally from the trans isomer, makes a minor contribution.5 The trans RPSB spectrum is very similar to the previously reported PISA spectrum for a mixed RPSB isomer population (which was dominated by isomer A),10 and also to the original photodissociation spectrum.7 Naturally, the question arises as to how faithfully the PISA spectrum follows the RPSB absorption spectrum and whether the photoisomerization yield depends on wavelength. At this stage it is impossible to answer these questions definitely. Certainly there are no energetic constraints to photoisomerization, with estimated barriers for isomerization about single and double bonds (30–130 kJ mol−1, see Table 1 and ref. 24) being much lower than the photon energies (170–290 kJ mol−1).
:0.07:0.10 (averaged over the 500–650 nm range). The resulting difference ATD depicted in Fig. 6(b) clearly shows that isomer A is the main photoproduct. The PISA spectrum for isomer B, obtained by monitoring the total production of isomers A, C and E [Fig. 7(b)], is similar to the trans RPSB spectrum with a maximum at 615 ± 10 nm, but exhibits a poorer signal to noise ratio and a somewhat broader peak. At this stage, because it is difficult to cleanly select isomer B without some contamination from either isomer A or isomer E, the isomer B PISA spectrum and photoisomer branching ratios should be regarded as provisional.
Peak B presumably corresponds mainly to 11-cis or 9-cis RPSB, which are the chief photoisomers produced in solution from trans RPSB following visible irradiation,3,5 but may include contributions from other single cis isomers. Comparisons of measured and calculated collision cross sections support assignment of B to one or more single cis isomers. The measured collision cross section for isomer B is 2% less than that of isomer A, whereas in comparison, calculated cross sections for 11-cis and 9-cis RPSB are respectively 1% and 2% less than the calculated cross section for trans RPSB (see Table 1). The relatively broad PISA band obtained when isomer B is selected [Fig. 7(b)] may also be an indication that the ATD peak represents several different single cis isomers, each of which has a slightly different absorption band profile as found for RPSB in solution. For example, absorption maxima of 9-cis, 11-cis, 13-cis and trans retinal in hexane are reported to be 441, 458, 443 and 458 nm, respectively.3 It is interesting to note that the flat-topped photodissociation spectrum of RPSB reported by Rajput et al.8 resembles the isomer B PISA spectrum better than the isomer A PISA spectrum, suggesting that the ion population probed in the photodissociation experiment contained a mixture of trans and cis RPSB isomers.
The position and width of peak B in the ATD depends slightly on the ions' preparation (collisions in IF1 or photoexcitation of trans RPSB) with a slight broadening and shift of the peak maximum to shorter times as the IF1 RF voltage is increased. This is apparent in Fig. S2 (ESI†) which shows the influence of IF1 drive voltage on the ATD for RPSB pre-irradiated with blue light. Some change in the peak is expected given that in solution photoisomerization is a selective process, transforming trans RPSB primarily to 11-cis or 9-cis RPSB,3,5 whereas collisional excitation should generate a broader range of energetically accessible single and double cis isomers (7-cis, 9-cis, 11-cis, 13-cis) leading to overlapping ATD peaks that probably all contribute to peak B.
As noted in Section 3.2, collisional excitation of RPSB in the first ion funnel generates isomer C′, which has a slightly larger collision cross section than isomer C. Because isomer C′ does not photoisomerize following absorption of visible light it is suspected to be a form in which the polyene chain has cyclized, disrupting the conjugation.
To this point we have presumed that isomer C rather than isomer C′ is the photoisomerization product from isomers A and B in the drift tube, the justification being that because isomer C is formed through photoexcitation of RPSB in solution prior to electrospray it is also more likely to be the gas-phase photoproduct. Distinguishing the putative photoproducts may be possible through experiments in which isomer A is excited by visible light at the beginning of the drift tube, with mobility selection of the resulting product isomer by the second ion gate, followed by a PISA spectroscopy step. Isomer C should photoisomerize to give A and B whereas isomer C′ would be unaffected by light.
Isomer D is generated through energetic collisions in the first ion funnel, but is not a photoproduct of A, B or C, and does not photoisomerize following exposure to visible light. These observations together with its relatively small collision cross section are consistent with D being a cyclic form with disrupted conjugation. Candidate cyclic structures for isomers C′ and D include the protonated dihydropyridine isomer shown in Fig. 8(a), the unprotonated version of which has previously been prepared through electrocyclization of 13-cis retinal,25 and the structure shown in Fig. 8(b) which has been proposed as an intermediate in the fragmentation of RPSB.26 Identifying these cyclic isomers may be possible through synthesis of candidate compounds that are then introduced into the IMS apparatus to verify matches between ATD peaks. It is also possible, although unlikely, that C and D are isomers in which the proton resides on a carbon atom in the polyene chain. These protomer forms are predicted to lie ≥100 kJ mol−1 higher in energy than all-trans RPSB.10
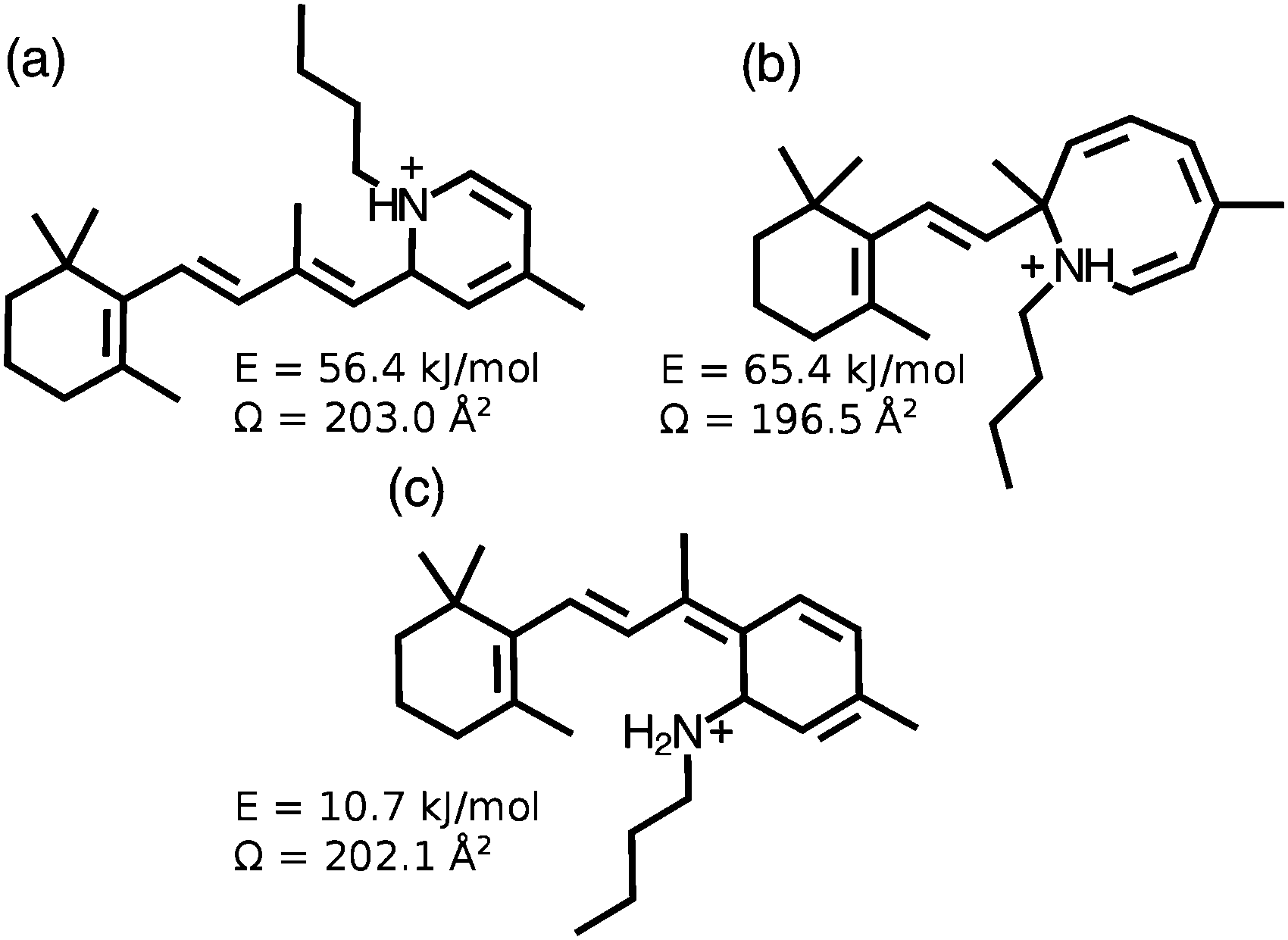 | ||
| Fig. 8 Possible cyclic isomers generated from RPSB through collisions or photo excitation. Energies of the different isomers are given with respect to that of 6s-cis(−) conformer of trans RPSB. Collision cross sections are calculated values as described in Section 3.1. | ||
Considering isomer E, we note that it is generated through irradiation of the RPSB solution before electrospray [see Fig. 5(a)], and also through photoexcitation of isomers A and B in the drift tube, that it photoisomerizes with visible light, is destroyed by energetic collisions in IF1, and has a smaller collision cross section than isomers A and B. These observations are consistent with E being a double or triple cis isomer (or family of isomers) which should be more compact than single cis isomers, yet should still absorb visible light.
3.4 RPSB S1 ← S0 transition − experiment and theory
Accurately forecasting the absorption wavelength of the isolated RPSB molecule is a necessary precursor for more challenging predictions for the absorptions of RPSB in solution or protein environments and for understanding the effects of counterions, non-covalent interactions and conformation.27–31 The peak of the trans RPSB spectrum at 618 ± 5 nm (2.01 ± 0.02 eV) presumably corresponds to the S1 ← S0 origin transition (i.e. the adiabatic excitation) with the declining signal to shorter wavelength associated primarily with unresolved vibronic structure due to excitation of Franck–Condon active vibrational modes, particularly those associated with C–C stretch vibrations, which are known to undergo changes in equilibrium separation upon electronic excitation.31 There may also be some contribution to the band's profile from absorptions of different 6s-cis(−), 6s-cis(+) and 6s-trans conformers, which, because they have similar energies and are separated by relatively low barriers (≤10 kJ mol−1), should interconvert in the drifting ion bunch.9,24 In the future, it may be possible to distinguish the spectral contributions of the different 6s conformers using double resonance or hole burning strategies for a cooled RPSB sample.There are several theoretical predictions for the S1 ← S0 transition energy of trans RPSB that can be compared with the new experimental results. It should be remembered that the calculated values usually represent vertical transition energies that will be somewhat higher than the adiabatic energies. Rajput et al. computed a vertical excitation wavelength of 620 nm for the 6s-trans isomer, and a vertical excitation wavelength of 547 nm for the slightly lower energy 6s-cis(−) isomer,8 well to the blue of the trans RPSB PISA spectrum maximum (618 nm) and of its centre of gravity (583 nm). The photodissociation spectrum accompanying the calculations, which exhibits a plateau extending from 530 to 610 nm, was deemed to reflect the absorptions of 6s-cis(−) and 6s-trans conformers present in the ion population. However, the PISA spectrum of trans RPSB shows no sign of this plateau despite the fact that the different 6s-cis(−), 6s-cis(+) and 6s-trans conformers should be present in the drifting ion bunch. As noted above, the photodissociation spectrum reported in ref. 8 better resembles the PISA spectrum of Peak B (Fig. 7(b)), and probably reflects contributions from a broad range of trans and cis isomers.
Valsson and Filippi calculated vertical excitation wavelengths of 551, 530 and 523 nm for the 6s-trans, 6-cis(−) and 6-cis(+) isomers using the NEVPT2 method (and comparable wavelengths using the CASPT2 approach).9 The similarity of the vertical transition wavelengths for the three 6s isomers is more consistent with our observation of a relatively narrow S1 ← S0 band. However, the predicted vertical transition energies probably overestimate the actual value − the predicted vertical transition wavelengths are at least 60 nm to the blue of the observed band's maximum and at least 30 nm to the blue from its centre of gravity (583 nm).
Altun et al. calculated the absorption wavelengths of 6s-trans and 6s-cis(−) isomers using several different methods with the closest match with experiment being for MRCISD+Q(6/6) vertical energies calculated using B3LYP ground state geometries with vertical excitation wavelengths of 604 nm for both 6s-trans and 6s-cis(−) isomers.28 These calculations provide perhaps the best agreement with experiments. As well, Zhou et al. recently predicted a transition wavelength for the 6s-cis(−) isomer of 620 nm (2.00 eV) using the XMC-QDPT2/cc-pVDZ vertical excitation on M06-2X/cc-pVDZ ground state geometry, which closely matches the peak of the trans RPSB PISA spectrum.32
3.5 Photoisomerization mechanism
Here we briefly discuss the photoisomerization mechanism for RPSB in the gas phase. Although the current study demonstrates conclusively that photoisomerization occurs in the gas phase and favours specific isomers, details of the process are unclear. When cradled in a protein or in solution, RPSB follows specific isomerization pathways dictated by the system's evolution on the S1 potential energy surface (PES) followed by passage through a conical intersection and onto the S0 PES.30–36 For example, computational studies of 11-cis retinal suggest that isomerization proceeds on the S1 surface by a path involving skeletal deformations followed by torsion about the 11–12 double bond ending at a twisted conical intersection through which it accesses the S0 surface.31 In the condensed phase, rapid collisional quenching removes excess vibrational energy stabilizing the nascent isomer on the S0 PES. Two conditions must be satisfied for a similar process to occur in the gas phase. First, there must be a negligible barrier between the Franck–Condon region and the conical intersection (because collisions are too infrequent to provide energy for surmounting a barrier on the S1 surface). Second, survival of the nascent isomer in the gas phase depends on the efficacy of collisional quenching. At higher pressure, if collisional quenching of vibrational energy is sufficiently rapid, the RPSB isomer resulting from passage through the conical intersection(s) should be preserved. On the other hand, at low pressure or in vacuum, with no efficient way to dissipate vibrational energy, isomerization, rearrangement and eventually dissociation (if energetically feasible) will continue on the S0 PES. It is difficult to decide which of these regimes applies in the drift tube where the collision rate is 108–109 s−1. The fact that photodissociation is relatively unimportant compared to photoisomerization (≤1%), proves that collisional quenching is sufficiently rapid to thwart a process that recent model calculations suggest has an activation energy of ≈150 kJ mol−1 with respect to trans RPSB and which should be energetically feasible following absorption of a single visible photon.26 Therefore, it seems likely that the nascent isomer distribution is at least partially preserved. Future measurements of photoproduct branching ratios as a function of buffer gas pressure may elucidate the role of collisional quenching and cast more light on the photoisomerization mechanism.4 Concluding remarks
In this study we have selectively prepared and probed different RPSB isomers in the gas phase. The dominant photoisomerization pathways involve trans–cis interconversion, although cyclic isomers, which do not appear to absorb visible light, are also formed through collisional excitation of RPSB. Photodissociation is a very minor channel (≤1%) in the drift tube environment presumably because the excited RPSB molecules undergo rapid vibrational deactivation through buffer gas collisions. Most importantly, we have recorded the S1 ← S0 photoisomerization action spectrum for trans RPSB which features a distinct peak with a maximum at 618 ± 5 nm. The new trans RPSB spectrum is very similar to the previously reported PISA spectrum for a mixture of RPSB isomers,10 and to the original photodissociation spectrum of Andersen and colleagues.7 The PISA spectrum of the other prominent isomer family (isomer B), most likely single cis isomers, features a somewhat broader peak than the trans RPSB spectrum, perhaps due to overlapping absorptions of several cis isomers. The clear difference between the trans RPSB spectrum (isomer A) and the cis RPSB spectrum (isomer B) conclusively demonstrates that the RPSB spectral properties depend on the isomeric form. This allows a rationalisation of the marked difference between the photodissociation spectra reported in ref. 7 and 8, which may have been recorded for samples with different fractions of trans and cis isomers. The current study demonstrates that isomers of RPSB are easily interconverted in solution by light, and in the gas phase by either light or collisions, emphasising the importance of carefully controlling preparation of RPSB prior to its spectroscopic interrogation. Ultimately we hope that photoisomerization studies of mobility-selected RPSB isomers (spectra and product branching ratios) will provide benchmark data for calibrating computational approaches used for predicting electronic spectra and dynamical behaviour of biochromophores.Finally, we have confirmed that ion mobility spectrometry is a useful method for assessing isomerization of RSPB in solution. Previously, photoisomerization of RPSB in various solvents has mainly been followed using either HPLC or NMR,3,5 which are somewhat cumbersome methods for on-line monitoring. In contrast, IMS can potentially be deployed to continually assess the effects of light and heat on the isomer distribution for RPSB in solution. IMS has recently been used in this way to monitor photoisomerization of other photochromic molecules in solution, including protonated spiropyran/merocyanine.37,38
Acknowledgements
This research was supported under the Australian Research Council's Discovery Project funding scheme (Project Numbers DP110100312 and DP120100100). We thank Prof. Matthew Bush for providing a modified version of the MOBCAL code with N2 collision parameters described in ref. 23.References
- O. P. Ernst, D. T. Lodowski, M. Elstner and P. Hegemann, Chem. Rev., 2013, 114, 126–163 CrossRef PubMed.
- F. Crescitelli, Prog. Retinal Res., 1991, 11, 1–32 CrossRef CAS.
- K. A. Freedman and R. S. Becker, J. Am. Chem. Soc., 1986, 108, 1245–1251 CrossRef CAS.
- R. S. Becker and K. A. Freedman, J. Am. Chem. Soc., 1985, 107, 1477–1485 CrossRef CAS.
- R. F. Childs and G. S. Shaw, J. Am. Chem. Soc., 1988, 110, 3013–3017 CrossRef CAS.
- Y. Koyama, K. Kubo, M. Komori, H. Yasuda and Y. Mukai, Photochem. Photobiol., 1991, 54, 433–443 CrossRef CAS PubMed.
- L. H. Andersen, I. B. Nielsen, M. B. Kristensen, M. O. A. El Ghazaly, S. Haacke, M. B. Nielsen and M. Å. Petersen, J. Am. Chem. Soc., 2005, 127, 12347–12350 CrossRef CAS PubMed.
- J. Rajput, D. B. Rahbek, L. H. Andersen, A. Hirshfeld, M. Sheves, P. Altoè, G. Orlandi and M. Garavelli, Angew. Chem., Int. Ed., 2010, 49, 1790–1793 CrossRef CAS PubMed.
- O. Valsson and C. Filippi, J. Phys. Chem. Lett., 2012, 3, 908–912 CrossRef CAS PubMed.
- N. J. A. Coughlan, K. J. Catani, B. D. Adamson, U. Wille and E. J. Bieske, J. Chem. Phys., 2014, 140, 164307 CrossRef CAS PubMed.
- J. Dilger, L. Musbat, M. Sheves, A. V. Bochenkova, D. E. Clemmer and Y. Toker, Angew. Chem., Int. Ed., 2015, 54, 4748–4752 CrossRef CAS PubMed.
- N. A. Pierson and D. E. Clemmer, Int. J. Mass Spectrom., 2015, 377, 646–654 CrossRef CAS PubMed.
- B. D. Adamson, N. J. A. Coughlan, P. B. Markworth, R. E. Continetti and E. J. Bieske, Rev. Sci. Instrum., 2014, 85, 123109 CrossRef CAS PubMed.
- B. D. Adamson, N. J. A. Coughlan, R. E. Continetti and E. J. Bieske, Phys. Chem. Chem. Phys., 2013, 15, 9540–9548 RSC.
- B. D. Adamson, N. J. A. Coughlan, G. da Silva and E. J. Bieske, J. Phys. Chem. A, 2013, 117, 13319–13325 CrossRef CAS PubMed.
- E. A. Mason and E. W. McDaniel, Transport Properties of Ions in Gases, WIley-VCH, 1988 Search PubMed.
- H. E. Revercomb and E. A. Mason, Anal. Chem., 1975, 47, 970–983 CrossRef CAS.
- A. K. Singh and R. V. Aruna, Biochim. Biophys. Acta, 1995, 1245, 167–172 CrossRef.
- T. Sovdat, G. Bassolino, M. Liebel, C. Schnedermann, S. P. Fletcher and P. Kukura, J. Am. Chem. Soc., 2012, 134, 8318–8320 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision A.1, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- A. A. Shvartsburg and M. F. Jarrold, Chem. Phys. Lett., 1996, 261, 86–91 CrossRef CAS.
- M. Mesleh, J. Hunter, A. Shvartsburg, G. Schatz and M. Jarrold, J. Phys. Chem., 1996, 100, 16082–16086 CrossRef CAS.
- I. Campuzano, M. F. Bush, C. V. Robinson, C. Beaumont, K. Richardson, H. Kim and H. I. Kim, Anal. Chem., 2012, 84, 1026–1033 CrossRef CAS PubMed.
- S. Zhu, M. F. Brown and S. E. Feller, J. Am. Chem. Soc., 2013, 135, 9391–9398 CrossRef CAS PubMed.
- W. H. Okamura, A. R. de Lera and W. Reischl, J. Am. Chem. Soc., 1988, 110, 4462–4464 CrossRef CAS.
- N. J. A. Coughlan, B. D. Adamson, K. J. Catani, U. Wille and E. J. Bieske, J. Phys. Chem. Lett., 2014, 5, 3195–3199 CrossRef CAS PubMed.
- A. Cembran, R. Gonzalez-Luque, P. Altoè, M. Merchán, F. Bernardi, M. Olivucci and M. Garavelli, J. Phys. Chem. A, 2005, 109, 6597–6605 CrossRef CAS PubMed.
- A. Altun, S. Yokoyama and K. Morokuma, J. Phys. Chem. B, 2008, 112, 16883–16890 CrossRef CAS PubMed.
- S. S. Sekharan and K. K. Morokuma, J. Am. Chem. Soc., 2011, 133, 19052–19055 CrossRef CAS PubMed.
- O. Valsson, P. Campomanes, I. Tavernelli, U. Rothlisberger and C. Filippi, J. Chem. Theory Comput., 2013, 9, 2441–2454 CrossRef CAS.
- I. Rivalta, A. Nenov and M. Garavelli, Phys. Chem. Chem. Phys., 2014, 16, 16865–16879 RSC.
- P. Zhou, J. Liu, K. Han and G. He, J. Comput. Chem., 2014, 35, 109–120 CrossRef CAS PubMed.
- M. Garavelli, P. Celani, F. Bernardi, M. A. Robb and M. Olivucci, J. Am. Chem. Soc., 1997, 119, 6891–6901 CrossRef CAS.
- R. Gonzalez-Luque, M. Garavelli, F. Bernardi, M. Merchan, M. A. Robb and M. Olivucci, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9379–9384 CrossRef CAS.
- B. G. Levine and T. J. Martnez, Annu. Rev. Phys. Chem., 2007, 58, 613–634 CrossRef CAS PubMed.
- W. Wang, Z. Nossoni, T. Berbasova, C. T. Watson, I. Yapici, K. S. S. Lee, C. Vasileiou, J. Geiger and B. Borhan, Science, 2012, 338, 1340–1343 CrossRef CAS PubMed.
- R. A. Rogers, A. R. Rodier, J. A. Stanley, N. A. Douglas, X. Li and W. J. Brittain, Chem. Commun., 2014, 50, 3424–3426 RSC.
- P. B. Markworth, N. J. A. Coughlan, B. D. Adamson, L. Goerigk and E. J. Bieske, Phys. Chem. Chem. Phys., 2015 10.1039/c5cp01567g.
Footnote |
| † Electronic supplementary information (ESI) available: Additional details for collision cross section measurements, arrival time distributions obtained with different injection conditions, PISA spectra for isomers A, B and C obtained by monitoring different photoproduct channels, and a compilation of calculated energies and collision cross sections for RPSB isomers. See DOI: 10.1039/c5cp03611a |
| This journal is © the Owner Societies 2015 |