
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen bonding in the ethanol–water dimer†
Ian A.
Finneran
,
P. Brandon
Carroll‡
,
Marco A.
Allodi‡
 and
Geoffrey A.
Blake
*
and
Geoffrey A.
Blake
*
Division of Chemistry and Chemical Engineering, California Institute of Technology, 1200 E California Blvd., Pasadena, CA 91125, USA. E-mail: gab@gps.caltech.edu
First published on 26th August 2015
Abstract
We report the first rotational spectrum of the ground state of the isolated ethanol–water dimer using chirped-pulse Fourier transform microwave spectroscopy between 8–18 GHz. With the aid of isotopic substitutions, and ab initio calculations, we identify the measured conformer as a water-donor/ethanol-acceptor structure. Ethanol is found to be in the gauche conformation, while the monomer distances and orientations likely reflect a cooperation between the strong (O–H⋯O) and weak (C–H⋯O) hydrogen bonds that stabilizes the measured conformer. No other conformers were assigned in an argon expansion, confirming that this is the ground-state structure. This result is consistent with previous vibrationally-resolved Raman and infrared work, but sheds additional light on the structure, due to the specificity of rotational spectroscopy.
1 Introduction
Hydrogen bonds play a critical role in many chemical and biochemical processes. In the Earth's atmosphere, hydrogen bonds control cluster formation and influence reaction rates, while in the condensed phase they guide protein folding and solvent–solute interactions. Their importance to molecular processes on Earth is due in part to their moderately low bond energies of 4–40 kcal mol−1, which allow them to freely associate and dissociate under ambient conditions, as well as their directional preferences, which influence the structure of molecular assemblies.1Since the first descriptions of prototypical hydrogen bonds at the beginning of the 20th century a second class of “weak” hydrogen bonds has emerged. As the name suggests, weak hydrogen bonds are generally defined by interactions of <4 kcal mol−1, which opens the door for many new bonding partners.2 Structurally, all hydrogen bonds are generally defined as interactions of X–H⋯A, where H carries a partial positive charge, and A a partial negative charge. Weak hydrogen bonds stretch this definition to include C–H⋯O, O–H⋯π, and C–H⋯π interactions, among others. Although they are similar in energy to van der Waals interactions, weak hydrogen bonds retain a distinct directional preference. In condensed phase chemistry, these weak interactions have been shown to be both ubiquitous and influential in drug–receptor recognition, molecular crystallization, and macromolecular structure.1,3
The ethanol–water dimer is a excellent model system for hydrogen bonding, as it exhibits both a strong O–H⋯O hydrogen bond, as well as a weak C–H⋯O hydrogen bond. The energy landscape of the dimer is thus an interplay between the relative donor/acceptor strengths of water and ethanol, as well as the gauche/trans conformations of the ethanol monomer.4 In the condensed phase, ethanol–water mixtures have been studied extensively, due to their broad applications as well as their abnormal behavior. They exhibit many thermodynamic anomalies, such as a negative entropy of mixing, for example, which are believed to originate from incomplete mixing on the microscopic scale.5 Elucidating the structure and dynamics of hydrogen bonding of the ethanol–water clusters, especially the dimer, may inform such studies.
The Raman and infrared spectrum of the ethanol–water dimer were previously reported in the literature.6,7 Two ethanol acceptor conformers were identified, with the ground state corresponding to a gauche-ethanol conformation. We report a combined experimental and computational study of the ethanol–water dimer using microwave rotational spectroscopy and ab initio calculations. Microwave spectroscopy is unmatched for structure determination of small hydrogen bonded clusters in the gas phase, as spectra directly reveal the moments of inertia of various species.8–10 Indeed, microwave spectroscopy provided some of the first direct structural evidence for the O–H⋯π weak hydrogen bond in the benzene–water dimer,8 as well as unambiguous assignments of the relative energies of the water hexamer conformers.9
In this work, we definitively assign the ground state structure of the ethanol–water dimer to a water-donor structure, with ethanol in the gauche conformation. With the use of three Kraitchman substitution coordinates and ab initio calculations, we find evidence of cooperativity between both the strong and weak hydrogen bond interactions in this structure.
2 Experimental methods
The spectrum of the ethanol–water dimer was collected using the Caltech chirped-pulse Fourier transform microwave (CP-FTMW) spectrometer between 8–18 GHz.11 The mixed dimer was prepared by loading water into a reservoir pulsed valve, and flowing a backing gas (argon) over a separate external reservoir containing ethanol. The mixture of ethanol, water, and 2 atmospheres of backing gas (typically Ar or He) was supersonically expanded into a vacuum chamber at 10−5 Torr with a pulse repetition rate of 5 Hz. The CP-FTMW spectrometer has been described in a previous publication,11 so only a brief overview will be given here. A 0.5–1 μs duration chirped pulse of 2 GHz bandwidth was generated, heterodyned with the output of a microwave synthesizer, amplified, and broadcast into the vacuum chamber with a waveguide horn. The resulting molecular free induction decay was amplified, heterodyned back down with the same synthesizer and digitized on a high speed 4 Gs/s analog-to-digital converter. Double resonance measurements were carried out using a chirped pulse followed by a second single tone 0.5–1 μs sinc pulse. Samples of ethanol (99.5% purity), ethan(ol-d) (99% purity) and D2O (99% purity) were purchased from Sigma-Aldrich and used without further purification.The measured spectra from the instrument are dual sideband, and so cover a total of 4 GHz for each local oscillator (LO) setting (2 GHz each in the upper and lower sideband). Thus, care must be taken in assigning molecular rest frequencies. We use a python data acquisition script to cyclically shift the LO every 3 minutes to cover 8–18 GHz and to discriminate upper and lower sideband peaks. For every LO setting, we shift by +10 MHz and deconvolve with a separate python script.
Spectral fitting was performed with the Watson-S Hamiltonian in SPFIT/SPCAT12 and a newly written python graphical interface. Due to the high line density of the spectra, we used AUTOFIT, an automated broadband fitting program, to generate initial quantum number assignments and fits to the various species.13 Kraitchman substitution coordinates were calculated with the KRA program.14
All ab initio calculations were performed using Gaussian 09.15 Geometry optimizations were carried out with second-order Møller–Plesset (MP2) perturbation theory16 and the augmented-correlation consistent polarized Valence-only Triple-Zeta (aug-cc-pVTZ) basis set.17 All optimized structures were confirmed to be true minima on the potential energy surface via harmonic frequency calculations at the same level of theory as the optimization. We subsequently performed single-point coupled cluster energy calculations with singles, doubles, and perturbative triples (CCSD(T))18 with the aug-cc-pVTZ basis set on the MP2 optimized structures. The zero point vibrational energy (ZPVE) contribution to the relative energies was evaluated from the previous harmonic frequency calculation using MP2/aug-cc-pVTZ. We computed equilibrium rotational constants (Be) for all conformers using the molecular geometries from the MP2/aug-cc-pVTZ optimization. Next, we calculated the anharmonic cubic and semi-diagonal quartic force constants in a normal mode representation at the MP2/aug-cc-pVTZ level, and from this determined the ground state rotational constants (B0) of the WE-g+ conformer using second-order vibrational perturbation theory.19 We did not perform anharmonic calculations of the other conformers due to the limited computational resources available.
3 Results and discussion
3.1 Computational results
The ab initio energy landscape, including the zero point vibrational energy (ZPVE), of the ethanol–water dimer is shown in Fig. 1, and the associated equilibrium rotational constants (Be) of each conformer are given in Table 1. We use nomenclature from the literature to label the various structures; WE indicates a water-donor geometry, EW a ethanol-donor motif, while-g and -t specify the hydroxyl conformation of the ethanol subunit.7 The two hydrogen bonding sites of the WE-g conformer are further differentiated with + and −.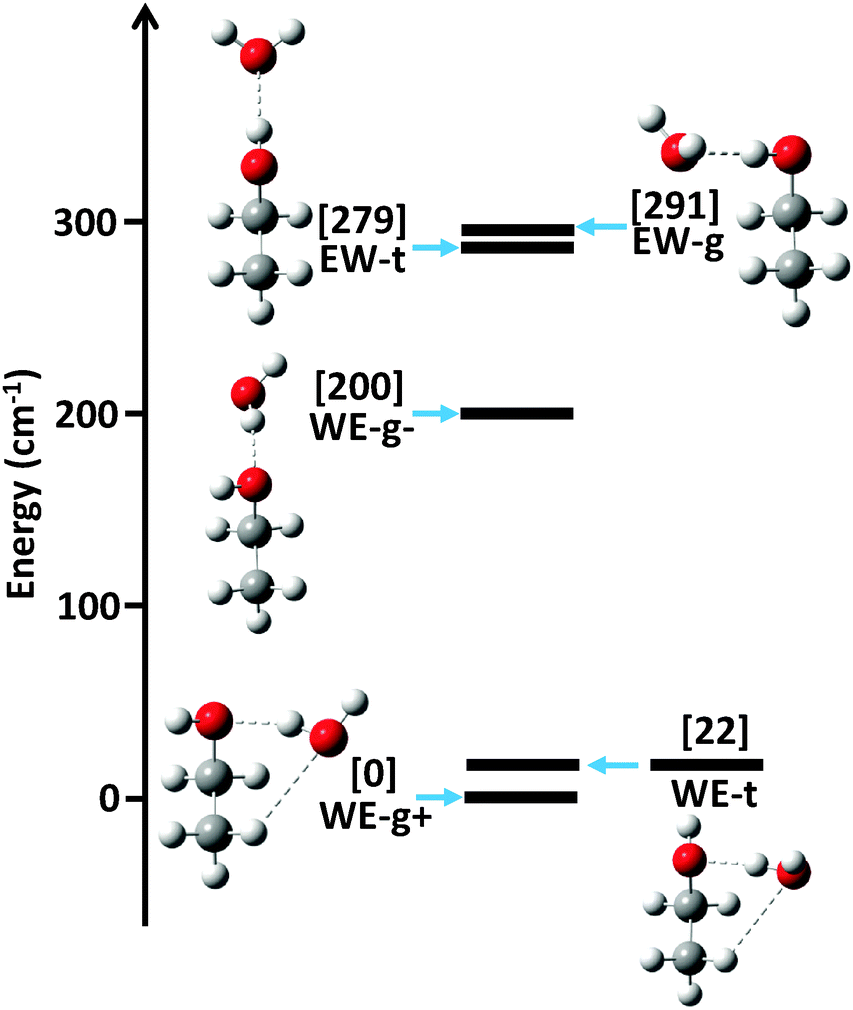 | ||
| Fig. 1 The ab initio relative energy landscape, including ZPVE, of the ethanol–water dimer. Geometry optimizations and harmonic force field calculations were performed at the MP2/aug-cc-pVTZ level of theory, followed by single point energy calculations using CCSD(T)/aug-cc-pVTZ. | ||
| WE-g+ | WE-t | WE-g− | EW-t | EW-g | |
|---|---|---|---|---|---|
| A/MHz | 8882 | 9163 | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2081 2081 |
27305 |
9772 |
| B/MHz | 3676 | 3502 | 2309 | 2071 | 3062 |
| C/MHz | 2888 | 2799 | 2164 | 1991 | 2525 |
| μ a/D | 1.8 | 1.7 | 2.5 | −2.8 | −2.6 |
| μ b/D | 0.9 | 1.3 | 0.9 | 0.5 | 1.3 |
| μ c/D | 0.4 | −0.2 | −0.1 | 0 | 0.3 |
| Energy/cm−1 | 0 | 22 | 200 | 279 | 291 |
The strong hydrogen bond between ethanol and water makes the largest contribution to the relative energies of the structures. Since ethanol is a better hydrogen bond acceptor than donor, the water donor structures are lower in energy than the water acceptor structures.4 The ethanol molecule can exist in the trans or gauche conformation, which dictates the position of the strong hydrogen bond.
Weaker interactions in the dimer lead to further energy separations between conformers. The lowest three conformations all exhibit water donor structures, as expected, yet the WE-g+ and WE-t conformers are significantly lower in energy than the WE-g− structure. As Fig. 1 shows, however, the WE-g+ and WE-t structures are both compact and exhibit a second weak C–H⋯O hydrogen bond interaction, while the WE-g− structure is elongated and contains no secondary interaction(s). We posit that the weak hydrogen bond stabilizes these conformers by ∼200 cm−1 (0.5 kcal mol−1), which is consistent with previous estimates of C–H⋯O binding energies.1
Interestingly, the predicted energies of the two water acceptor conformers do not exhibit a clear preference for compact or elongated structures. We attribute this to cooperativity between the weak and strong hydrogen bond interactions.20 In the WE-g+ and WE-t structures, the hydrogen bonds are cooperative, as the water acts as a donor in the strong hydrogen bond and acceptor in the weak hydrogen bond. The EW-g structure shows an anti-cooperative interaction, in which the water acts as an acceptor for both interactions, increasing the energy of the structure relative to EW-t.
3.2 Experimental results
The microwave spectrum of ethanol and water with argon as the backing gas is shown in Fig. 2. Peaks from the ethanol dimer, water dimer, and ethanol monomer are all present, as can be seen by the predictions included from the literature.21–23 A series of unknown strong peaks were identified when ethanol and water were present that disappeared in expansions of ethanol/argon and water/argon. Accordingly, the peaks were assumed to originate from a cluster containing both molecules.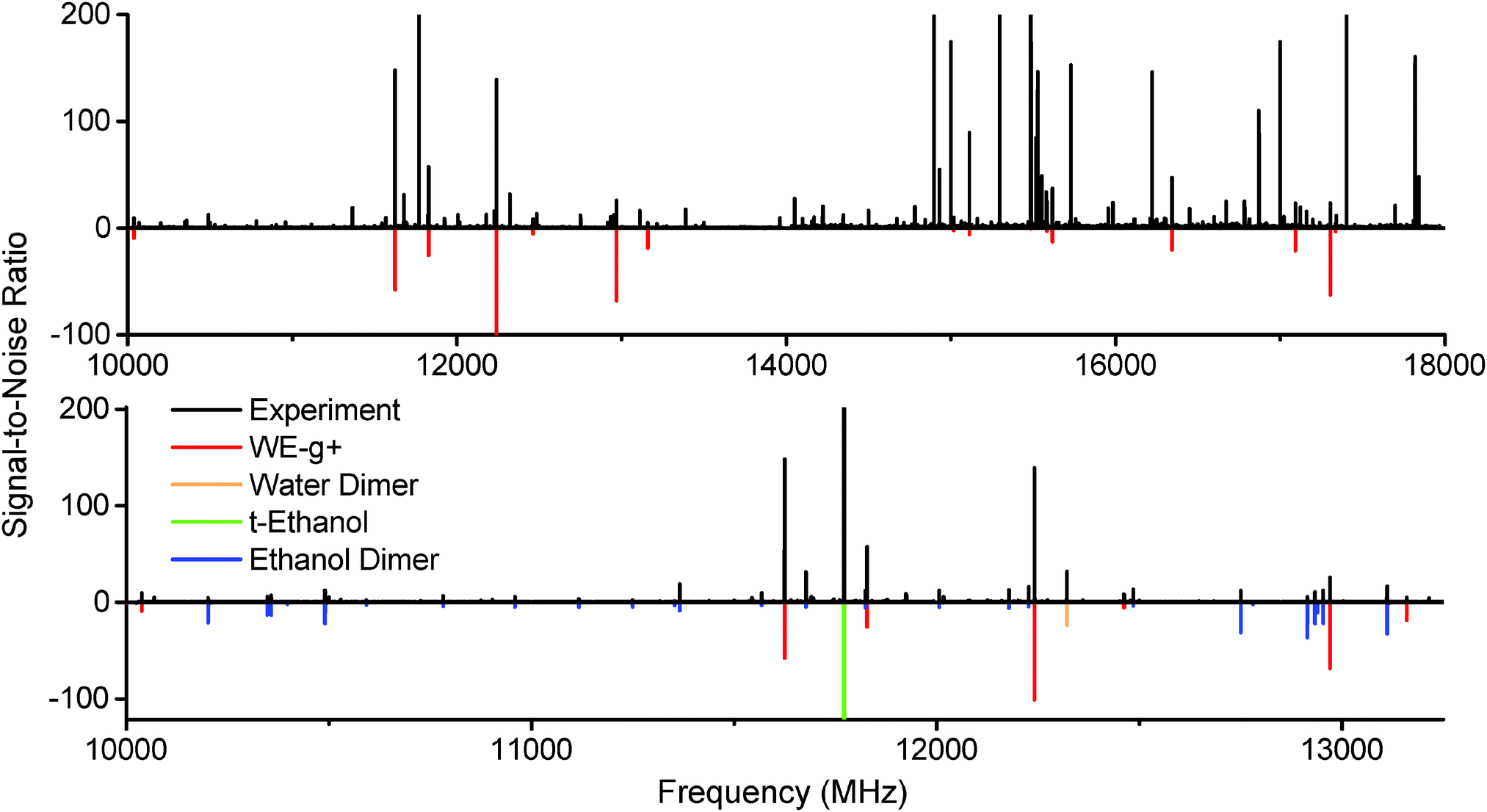 | ||
| Fig. 2 Top: The spectrum of ethanol and water in an argon expansion between 10–18 GHz (15 million averages, 40 hour acquisition at 4 LO settings). A series of strong unassigned peaks have been fit and assigned to the WE-g+ conformer of the ethanol–water dimer (red). Bottom: A 3 GHz inset of the ethanol + water expansion spectrum. Literature data for ethanol dimer,21trans-ethanol,22 and water dimer23 have been plotted in various colors, along with WE-g+, using a rotational temperature of 2 K. | ||
Using AUTOFIT13 and SPFIT,12 we fit 21 a-type and b-type transitions to a microwave rms of 14 kHz, commensurate with the line center uncertainty of the instrument (Table 2). The assignments were further confirmed with double resonance measurements (Fig. 3). The rotational constants and dipole components (μa > μb) rule out all possible conformers except WE-t and WE-g+. Both conformers have similar structures and dipole moments; the main distinguishing feature is the orientation of the hydroxyl hydrogen of the ethanol, and the dihedral of the water molecule along the strong hydrogen bond.
| WE-g+ | Ab initio | EtOH:H2O | EtOD:D2O | EtOH:D2O | EtOD:HOD | EtOD:H2O |
|---|---|---|---|---|---|---|
| A/MHz | 8940 | 9089.862(10) | 8610.610(20) | 9046.626(36) | 8614.300(39) | 8648.244(40) |
| B/MHz | 3485 | 3410.8841(35) | 3163.8418(85) | 3191.064(14) | 3203.203(17) | 3381.274(13) |
| C/MHz | 2777 | 2737.9705(28) | 2568.4509(73) | 2593.615(15) | 2594.324(18) | 2710.510(10) |
| D J/kHz | 15.0 | 20.33(15) | 14.94(19) | 16.67(83) | 15.88(94) | * |
| D JK/kHz | −13.8 | −31.35(19) | −18.50(62) | −26.9(17) | * | * |
| D K/kHz | 48.5 | 137.4(25) | 96.2(39) | * | * | * |
| d 1/kHz | −4.2 | −5.957(16) | −4.03(12) | * | * | * |
| d 2/kHz | −0.40 | −0.6016(46) | * | * | * | * |
| N | 21 | 15 | 12 | 9 | 8 | |
| rms/kHz | 14 | 15 | 37 | 41 | 41 |
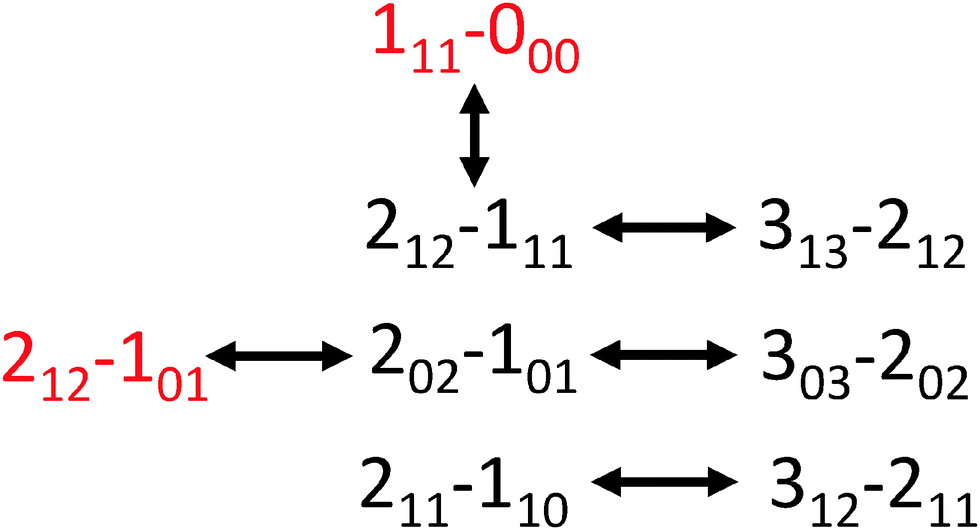 | ||
| Fig. 3 The transition connectivities confirmed with double resonance measurements (a-types in black, and b-types in red). | ||
To differentiate the identity of the conformer, we measured the spectra of several deuterated species of ethanol and water. Again, double resonance measurements were performed on each isotopomer to confirm assignments. The rotational constants for four isotopomers are given in Table 2, and the singly-substituted Kraitchman structure is shown in Fig. 4. The position of the hydroxyl H atom of ethanol was calculated by substitution of EtOD:H2O into the normal species, the outer H atom of water was calculated with substitution of EtOD:HOD into EtOD:H2O, while the hydrogen bonded H atom was calculated with substitution of EtOD:D2O into EtOD:HOD. We doubly confirmed the position of the ethanol hydroxyl H with substitution of EtOD:D2O into EtOH:D2O. For each substitution, the H-atom coordinate in the principal axis system was converted to internal coordinates and plotted on the normal species result. The deuterated spectra have lower signal-to-noise and higher spectral density than the non-deuterated spectra so definitive assignments of EtOH:HOD, EtOH:DOH, and EtOD:DOH were not possible. Finally, we have recently identified the spectrum of WE-t in a helium expansion (not present in argon), and will report a structural analysis of this conformer in a forthcoming publication.
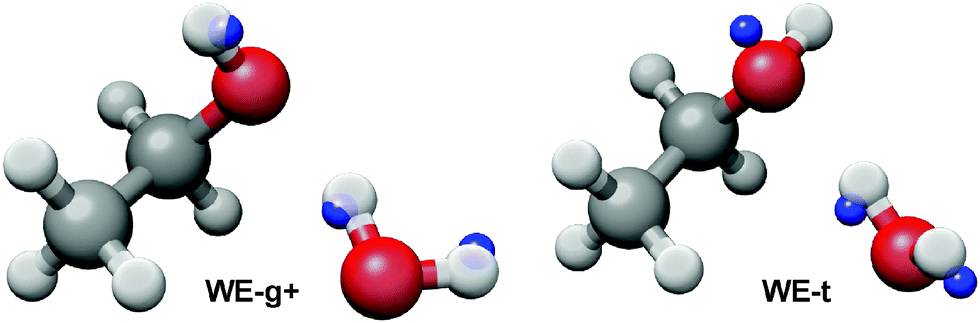 | ||
| Fig. 4 Kraitchman substitution positions of three hydrogens in the ethanol water dimer are shown as blue spheres, superimposed on the ab initio structures of WE-g+ (left) and WE-t (right). Based on the substitution positions, it is clear that WE-g+ is the measured conformer. | ||
The measured geometry of the dimer observed in the argon expansion is clearly consistent with WE-g+, confirming its identity as the lowest energy conformer. This is in contrast to the ethanol monomer, which has a trans-hydroxyl ground state structure. Thus, the hydrogen bond interaction between water and ethanol stabilizes the gauche structure, an example of adaptive aggregation. This is consistent with previous infrared and Raman work.6,7 Furthermore, the measured rotational constants are in good agreement with the ground state rotational constants (B0) determined in the ab initio anharmonic calculation. The large difference between the calculated B0 and Be constants indicates significant vibrational averaging of the dimer geometry even in the ground state. Large vibrational corrections to rotational constants have been reported in other hydrogen bonded clusters.9
We hypothesize that the stabilization of the gauche conformation comes from the weak hydrogen bond of the dimer, as the strength of a C–H⋯O interaction is dependent on the bond angle and C–H⋯O distance. The Kraitchman structure in Fig. 4 indicates that the outer hydrogen of the water is collinear with the weak hydrogen bond, which may enable a larger overlap of the oxygen lone pair with the C–H, rather than the perpendicular H–O⋯C angle of the t-WE structure. Furthermore, in the ab initio structures the WE-g+ conformer has a C–H⋯O distance of 2.66 Å, while the length is 2.79 Å in WE-t. Additional microwave measurements of isotopically substituted dimers are needed to confirm the length of the weak hydrogen bond in WE-g+.
4 Conclusions
We have measured the pure rotational spectrum of the ethanol–water dimer in supersonic expansions with argon. Transitions from three isotopomers of one conformer were identified and assigned to a compact water-donor structure with ethanol in the gauche configuration. These first fully rotationally resolved measurements confirm that ethanol is a better hydrogen-bond donor than acceptor, and adaptive aggregation of ethanol, as the interaction with water stabilizes the gauche conformer relative to the trans geometry. Weak hydrogen bond interactions likely play a role in this process, as the overlap between the oxygen of the water with the C–H is more optimal in the WE-g+ conformer than the WE-t conformer.To date, ethanol–water dimer is the second alcohol–water cluster for which rotationally resolved spectra have been reported, the first being the methanol–water dimer in 1997.24 It is clear that while both alcohols act as better hydrogen bond acceptors than donors, weak hydrogen bond interactions seem to play a larger role in the ethanol–water structure. Further studies of mixed water–alcohol clusters in the gas phase will shed additional light on the fascinating influence of both strong and weak hydrogen bonds in these mixtures.
Acknowledgements
The authors acknowledge the National Science Foundation (Grant No. CHE-1214123 and the Graduate Research Fellowship Program) for financial support.References
- G. Debrus and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press Inc., New York, 1999 Search PubMed.
- G. Desiraju, et al. , Chem. Commun., 1998, 891–892 Search PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg and P. Hobza, et al. , Pure Appl. Chem., 2011, 83, 1637–1641 CAS.
- M. Masella and J. P. Flament, J. Chem. Phys., 1998, 108, 7141–7151 CrossRef CAS PubMed.
- I. Juurinen, K. Nakahara, N. Ando, T. Nishiumi, H. Seta, N. Yoshida, T. Morinaga, M. Itou, T. Ninomiya and Y. Sakurai, Phys. Rev. Lett., 2011, 107, 197401 CrossRef CAS.
- M. Nedić, T. N. Wassermann, Z. Xue, P. Zielke and M. A. Suhm, Phys. Chem. Chem. Phys., 2008, 10, 5953–5956 RSC.
- M. Nedić, T. N. Wassermann, R. W. Larsen and M. A. Suhm, Phys. Chem. Chem. Phys., 2011, 13, 14050–14063 RSC.
- S. Suzuki, P. G. Green, R. E. Bumgarner, S. Dasgupta, W. A. Goddard and G. A. Blake, Science, 1992, 257, 942–945 CAS.
- C. Pérez, M. T. Muckle, D. P. Zaleski, N. A. Seifert, B. Temelso, G. C. Shields, Z. Kisiel and B. H. Pate, Science, 2012, 336, 897–901 CrossRef PubMed.
- G. G. Brown, B. C. Dian, K. O. Douglass, S. M. Geyer, S. T. Shipman and B. H. Pate, Rev. Sci. Instrum., 2008, 79, 053103 CrossRef PubMed.
- I. A. Finneran, D. B. Holland, P. B. Carroll and G. A. Blake, Rev. Sci. Instrum., 2013, 84, 083104 CrossRef PubMed.
- H. M. Pickett, J. Mol. Spectrosc., 1991, 148, 371–377 CrossRef CAS.
- N. A. Seifert, I. A. Finneran, C. Perez, D. P. Zaleski, J. L. Neill, A. L. Steber, R. D. Suenram, A. Lesarri, S. T. Shipman and B. H. Pate, J. Mol. Spectrosc., 2015, 312, 13–21, DOI:10.1016/j.jms.2015.02.003.
- Z. Kisiel, PROSPE KRAitchman's substitution coordinates, 2015, [http://www.ifpan.edu.pl/kisiel/struct/struct.htm, online; accessed 19-June-2015].
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision D.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618 CrossRef.
- R. A. Kendall, T. H. Dunning Jr and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS PubMed.
- J. A. Pople, M. Head-Gordon and K. Raghavachari, J. Chem. Phys., 1987, 87, 5968–5975 CrossRef CAS PubMed.
- V. Barone, J. Chem. Phys., 2005, 122, 014108 CrossRef PubMed.
- R. Kumar and J. Skinner, J. Phys. Chem. B, 2008, 112, 8311–8318 CrossRef CAS.
- J. P. I. Hearn, R. V. Cobley and B. J. Howard, J. Chem. Phys., 2005, 123, 134324 CrossRef.
- F. Lovas, J. Phys. Chem. Ref. Data, 1982, 11, 251–312 CrossRef CAS PubMed.
- T. R. Dyke and J. Muenter, J. Chem. Phys., 1974, 60, 2929–2930 CrossRef CAS PubMed.
- P. A. Stockman, G. A. Blake, F. J. Lovas and R. D. Suenram, J. Chem. Phys., 1997, 107, 3782–3790 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp03589a |
| ‡ Indicates equal author contribution. |
| This journal is © the Owner Societies 2015 |