
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into colour-tuning of chlorophyll optical response in green plants†
Joaquim
Jornet-Somoza
*a,
Joseba
Alberdi-Rodriguez
bc,
Bruce F.
Milne
bd,
Xavier
Andrade
e,
Miguel A. L.
Marques
fg,
Fernando
Nogueira
d,
Micael J. T.
Oliveira
h,
James J. P.
Stewart
i and
Angel
Rubio
*bj
aUniversitat de Barcelona, Martí i Franquès 1, 08028 Barcelona, Spain. E-mail: j.jornet.somoza@gmail.com
bNano-Bio Spectroscopy Group and ETSF Scientific Development Centre, Department of Materials Physics, University of the Basque Country, CFM CSIC-UPV/EHU-MPC and DIPC, Tolosa Hiribidea 72, E-20018 Donostia-San Sebastián, Spain. E-mail: angel.rubio@ehu.es
cDepartment of Computer Architecture and Technology, University of the Basque Country UPV/EHU, M. Lardizabal, 1, 20018 Donostia-San Sebastián, Spain
dCFísUC, Department of Physics, University of Coimbra, Rua Larga, 3004-516 Coimbra, Portugal
eLawrence Livermore National Laboratory, Livermore, CA 94550, USA
fInstitut für Physik, Martin-Luther-Universität Halle-Wittenberg, D-06099 Halle, Germany
gInstitut Lumière Matière, UMR5306 Université Lyon 1-CNRS, Université de Lyon, F-69622 Villeurbanne Cedex, France
hDepartment of Physics, University of Liège, B-4000, Liège, Belgium
iStewart Computational Chemistry, 15210 Paddington Circle, Colorado Springs, CO 80921, USA
jMax Planck Institute for the Structure and Dynamics of Matter and Center for Free-Electron Laser Science, Luruper Chaussee 149, 22761 Hamburg, Germany. E-mail: angel.rubio@mpsd.mpg.de
First published on 17th July 2015
Abstract
First-principles calculations within the framework of real-space time-dependent density functional theory have been performed for the complete chlorophyll (Chl) network of the light-harvesting complex from green plants, LHC-II. A local-dipole analysis method developed for this work has made possible the studies of the optical response of individual Chl molecules subjected to the influence of the remainder of the chromophore network. The spectra calculated using our real-space TDDFT method agree with previous suggestions that weak interaction with the protein microenvironment should produce only minor changes in the absorption spectrum of Chl chromophores in LHC-II. In addition, relative shifting of Chl absorption energies leads the stromal and lumenal sides of LHC-II to absorb in slightly different parts of the visible spectrum providing greater coverage of the available light frequencies. The site-specific alterations in Chl excitation energies support the existence of intrinsic energy transfer pathways within the LHC-II complex.
1 Introduction
Central to the continued existence of life on Earth, the conversion of solar energy into a chemical form that can be stored until needed, transported to different parts of an organism and released as required in order to drive the fundamental processes of biology is one of the most highly studied of all biochemical phenomena. Developing a detailed understanding of the microscopic physical processes involved in photosynthesis has the potential to directly impact upon a wide range of technological applications. These range from optimisation of food crop production1,2 to aiding attempts to utilise aspects of photosynthetic light harvesting to generate usable energy either directly in a conventional solar cell apparatus3,4 or through secondary processes such as hydrogen combustion in photosynthesis-driven (bio)reactor systems.5–8More than 50% of the light captured by green plants for use in photosynthesis is absorbed by the major light harvesting complex (LHC-II).9 LHC-II is a trimeric protein assembly with three-fold symmetry and contains over 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms. Complexed within the protein framework, each monomer unit contains 14 chlorophyll (Chl) molecules (both types a and b) that are the key functional units in the light-harvesting process in LHC-II (Fig. 1).4,10 Additionally, each monomer contains four secondary carotenoid chromophores (lutein (×2), neoxanthin and violoxanthin) that play important roles in photoprotection and energy transport within LHC-II.11–13 Approximately 7000 of the LHC-II atoms belong to the chromophores (Fig. 1). Usually bound within cell membranes in vivo, LHC-II possesses a robust structure and maintains structural integrity during purification and crystallisation meaning that the detailed structure has been obtained from crystallographic studies and that it has become possible to perform experimental studies of isolated LHC-II.14–16
000 atoms. Complexed within the protein framework, each monomer unit contains 14 chlorophyll (Chl) molecules (both types a and b) that are the key functional units in the light-harvesting process in LHC-II (Fig. 1).4,10 Additionally, each monomer contains four secondary carotenoid chromophores (lutein (×2), neoxanthin and violoxanthin) that play important roles in photoprotection and energy transport within LHC-II.11–13 Approximately 7000 of the LHC-II atoms belong to the chromophores (Fig. 1). Usually bound within cell membranes in vivo, LHC-II possesses a robust structure and maintains structural integrity during purification and crystallisation meaning that the detailed structure has been obtained from crystallographic studies and that it has become possible to perform experimental studies of isolated LHC-II.14–16
 | ||
| Fig. 1 LHC-II trimer, showing locations of protein, carotenoid and Chl molecules (Chl a in blue and Chl b in green) of one monomeric unit. For improved visability hydrogen atoms, Chl phytyl chains and primary structure of the protein are not shown. | ||
After absorption of light, excitation energy is transferred from the LHC-II antenna to a photosynthetic reaction center complex. At low light levels the quantum efficiency of this process approaches unity.4 It is interesting to note that at chromophore concentrations three orders of magnitude smaller than those found in biological light harvesting systems, solutions of chlorophyll display negligible radiance due to concentration quenching of the fluorescence process.17 The electronic structure of the chromophores within LHC-II clearly must undergo considerable modification relative to the same group of chlorophyll molecules in bulk solution.14
Various models based on excitonic coupling between the Chl units and/or modulation of the chromophores' energy levels by the protein environment have been invoked to explain the high efficiency of excitation energy transfer (EET) in photosynthesis.18–22 In bacteria the excitonic mechanism dominates, however there is evidence supporting a more micro-environment driven mechanism being most important in plant LHC-II.9,15,16
The electronic structure of the Chl antenna molecules of LHC-II is well understood as is the nature of the excitations involved in photo-absorption by Chl in the relevant regions of the solar spectrum. Whilst the effect of the protein environment on the optical response of the Chl chromophores is accepted as being minimal, the role of chromophore packing remains less well studied but can be expected to have a greater impact on the absorption spectra due to the interaction of electronic states of very similar energies. Due to difficulties in applying computational electronic structure methods to systems of this size, previous theoretical studies have tended to focus on the individual chromophores in isolation with some simple model of the complex micro-environment or by a small explicit portion of the complex.23
Here, we report a first-principles study of the electronic absorption spectrum of the full LHC-II Chl chromophore network in which all atoms were treated at the same theoretical level using real-space time-dependent density functional theory (TDDFT)24 as implemented in the Octopus code.25 Some of the TDDFT calculations performed in this work are at the edge of what is feasible with today's computer resources and require extensive optimisations of Octopus. These involve not only the parallel execution,26 but also issues related to memory management,27 the Poisson solver,28 the initialisation,29 input/output efficiency30 and post-processing. To the best of our knowledge the geometry optimisation of the full 17000 atom LHC-II complex performed in this work using the Mopac electronic structure package is the largest performed to date using semi-empirical electronic structure methods.
Using a newly implemented charge density decomposition method we have obtained results which highlight alterations in individual Chl spectra within the complete Chl network which contribute to broadening of the coverage of the optical spectrum by LHC-II in green plants. Similarly, our decomposition analysis indicates that the Chl chromophores located on the stromal and lumenal sides of the thylakoid membrane provide distinctly different contributions to the total LHC-II absorption spectrum. Furthermore, relative shifts of the stromal/lumenal Chl spectra suggest possible intrinsic directions for the flow of excitation energy within the complex.
2 Methods
2.1 Preparation of LHC-II geometry
The initial geometry of LHC-II was extracted from the crystal structure of the spinach major light-harvesting complex (PDB accession code 1RWT).31 Several lipid molecules originating in the experimental crystallisation mixture that remained in the crystal structure, such as β-nonylglucoside and digalactosyl diacyl glycerol, were removed as these were not part of the biological unit. Furthermore, these were expected to have little or no impact on the excited state electron dynamics of the LHC-II chromophores.Hydrogen atoms were added using the Chimera molecular modeling package.32 At this stage it was necessary to select the protonation state of acidic residues located at the stromal and lumenal membrane/aqueous interfaces of LHC-II. It was decided to protonate all acidic side chains at these interfaces since it was considered that in the cellular environment the charges associated with these side chains would be effectively shielded by counter-ions in solution or by association with other charged bio-molecular species. Since accurate modeling of this shielding was beyond the scope of the present study the simplified approach of capping these side chains with protons was considered most likely to provide a reasonable model of the charge environment in these regions of the LHC-II structure. The initial structure prepared in this way possessed a net charge of +30. The total number of atoms in this structure was 17280.
Following the initial placement of the hydrogen atoms on the LHC-II structure it was necessary to refine their positions, and since there was a strong possibility that protonation states could have been misassigned it was decided to use a method that would permit the migration of protons away from the unfavourable sites and/or towards more favourable ones. This requirement meant that approaches that used classical force fields, and therefore had fixed bonding connectivities, could not be employed. A solution to this problem lies in quantum mechanical electronic structure methods where no bonds are enforced and atomic nuclei are free to move in the potential created by the electronic structure of the system being studied.
Due to the size of the LHC-II system the only feasible choice in this case was to use a semi-empirical electronic structure method. The recently developed PM7 Hamiltonian as implemented in the Mopac semi-empirical electronic structure package was selected as this has been shown to closely reproduce molecular geometries calculated using high level quantum chemical methods and also provides a good model of weaker interactions such as hydrogen-bonding and dispersion forces.33
The initial step in the refinement of the crystal structure at the PM7 level involved optimisation of the positions of only the hydrogen nuclei with all other atoms fixed in space. In addition to refining the hydrogen positions this highlighted some cases where the initial guess for the protonation states of ionisable residues had been incorrect. In these cases occurrences such as the formation of salt bridges or the presence of very long bonds between acidic groups and protons were taken as indicators that the local protonation state had been incorrectly assigned. This hydrogen optimisation procedure was repeated in an iterative fashion each time correcting the protonation assignments until no further change was observed. This procedure resulted in a reduction of the total charge to zero giving an overall neutral structure.
Next, a full geometry optimisation was carried out on all ligand molecules including water. The rationale for this was that the ligand molecules (including all important chromophores in the LHC-II system) would be the most important for the later calculations of the optical absorption processes and these should be optimised as fully as possible. During this process it was found that the parameterisation of the PM7 Hamiltonian for the interaction between nitrogen and Mg2+ was incorrect and gave rise to overly long internuclear distances that resulted in the Mg2+ ions being displaced out of the plane of the chlorophyll macrocycle by up to 1.4 Å. Re-optimisation of the necessary PM7 parameters was performed so as to reproduce a chlorophyll geometry calculated at the DFT/PW91D/6-31G(d) level of theory. The improved PM7 description of this interaction produced out of plane displacements closer to 0.3 Å. The improved PM7 parameters were used in all subsequent semi-empirical calculations in the present work and have been incorporated into all distributions of Mopac released since the beginning of 2014. An overall gradient cutoff of 25 kcal mol−1 Å−1 was employed and this gave final gradients for the individual chromophores and ligands of less than 4 kcal mol−1 Å−1, which is not much above that which could be expected to be achieved in the optimisation of small to medium sized molecules in isolation.
The final stage was to refine the crystallographic structure of the full system of proteins, ligands and water to attempt to produce a total structure that was as well optimised as possible. Given the complexity of the biological setting of LHC-II where the system exists embedded in a lipid bilayer and has numerous interactions with other proteins and biochemical structures it was not feasible to attempt to replicate the packing effects of this environment. Similarly, the crystal environment was too complex for any attempt to model its effects. For this reason it was decided to perform a constrained refinement of the total LHC-II system that would permit local optimisation of bond lengths and angles but incorporate sufficiently strong restrains that larger movements involving e.g. torsional rotations would not occur. The reason for this being that test optimisations where no such restraints had been applied had shown that the protein structure began to slowly unravel in the absence of any environment (limitations in the accuracy of the PM7 method may also be expected to play a part in this process).
A series of these constrained refinements were performed with restraint values of 10, 5, 1, 0.5 and 0.1 kcal mol−1 Å−2. With values of 10, 5 and 1 kcal mol−1 Å−2 convergence was achieved and low total RMSD values with respect to the crystal structure of the protein were obtained (see Table 1). The lower restraint values did not converge to a steady RMSD even after several hundreds of refinement steps. Thus, it was decided that the LHCII geometry obtained from the refinement using a restraint strength of 1 kcal mol−1 Å−2 (final RMSD = 0.151 Å) was the best possible structure using this methodology and this was chosen to be used in the subsequent TDDFT calculations.
| Restraint strength/kcal mol−1 Å−2 | Final RMSD/Å |
|---|---|
| 10 | 0.044 |
| 5 | 0.064 |
| 1 | 0.151 |
| 0.5 | Not converged |
| 0.1 | Not converged |
2.2 Real-space time-propagation TDDFT calculations
All absorption spectrum calculations of the LHC-II complex were performed using the real-space/time-propagation TDDFT code Octopus.25,34,35 In Octopus, all the relevant functions are discretised in a real-space regular rectangular grid. To make the calculations computationally affordable, the core electrons were treated using norm-conserving Troullier–Martins pseudopotentials. These pseudopotentials were filtered using the scheme proposed by Tafipolsky and Schmid to remove the high frequency components that cannot be described accurately in the grid.36 The grid parameters that control the numerical convergence are the spacing between the grid points and the size of the simulation box. We found that a grid spacing of 0.2 Å and simulation boxes constructed from a union of spheres of radius 4 Å centered at the nuclei were necessary to converge the position of the main spectral peaks within 0.1 eV in all our calculations. All calculations were performed using the Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional corrected using the averaged-density self-interaction (ADSIC) method proposed by Legrand and co-workers, which reduces the self-interaction error of the semi-local DFT approach and restores the correct −1/r asymptotic behavior of the exchange functional.37,38Absorption spectra were computed using time-propagation TDDFT as implemented in the Octopus code.25,34,35 In this approach the time-independent Schrödinger equation for the system, eqn (1), is initially solved using the Kohn–Sham ansatz to obtain the electronic wavefunctions ψi(r) and the N-electron ground-state charge density n(r) of the ground-state:
| ĥKSψi(r) = εiψi(r) | (1) |
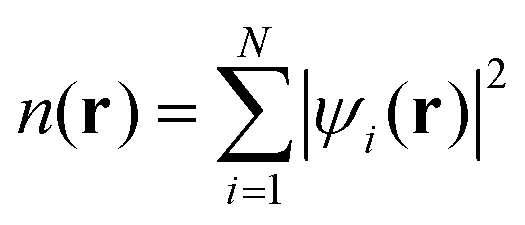 | (2) |
An instantaneous electric field perturbation κ is applied to the Kohn–Sham states ψi(r) in one of the Cartesian directions so as to introduce a small momentum to the electrons and the states are then repeatedly propagated for short time steps δt
| ψi(r,δt) = eiκzψi(r,0) | (3) |
 | (4) |
 | (5) |
The time-propagation after the perturbation was continued until 60 ℏ eV−1 (39.49 fs) for the isolated chromophores and chromophore networks. A total propagation time of 60 ℏ eV−1 was also used for the study of the protein environment. This simulation time allows a good peak resolution (being the full width at the half maximum 2π/T = 0.105 eV) for both Q- and Soret-bands. The largest time-step that ensured the numerical stability of the time propagation in all calculations was found to be 0.003 ℏ eV−1 (0.00197 fs).
The contribution of each individual chromophore to the optical response of the whole system was obtained by performing an a posteriori decomposition of the electronic density. The local electronic density corresponding to each chromophore has been obtained by performing a division of the ground-state density based on the Bader charge-topological approach.39 The dynamical polarisability tensor assigned to each chromophore can then be obtained by applying the corresponding Bader volume to the time-dependent electronic densities of the entire system. Fig. S1 (ESI†) shows the convergence of the simulated spectra while increasing the size of the chromophore Bader volume with respect to the one obtained for the chlorophyll molecule 607b (numbering from PDB file 1RWT). These tests indicated that the phytyl chain does not play an important role in the absorption process, and these were therefore removed when computing the spectral decomposition of the chlorophyll network. Fig. S2 (ESI†) shows the selected Bader volumes for the chlorophyll network of the monomer for which the local dynamical polarisability tensors have been computed.
Although alternative schemes for partitioning of the charge density exist, methods such as those of Voronoi40 or Hirshfeld41,42 require that assumptions must be made concerning the partitioning based on pre-computed promolecular/atomic densities whereas the Bader method relies purely on the features of the density in question, removing the need for such approximations. Similarly, testing convergence of the calculated spectra with respect to the selected density volumes in a first-principles manner as described above would be complicated due to the approximations involved in these other methods and so they were not considered for the present work.
3 Results and discussion
3.1 Individual LHC-II chromophores
 | ||
| Fig. 2 Distribution of peak energies in Chl TDDFT spectra. Q-band excitations occur between 1.9 and 2.0 eV and those of the Soret-band between 2.5 and 2.9 eV. Chl numbering taken from PDB file 1RWT. Solid lines correspond to the isolated Chl spectra and dashed lines to the spectra of the individual Chl within the LHC-II monomer Chl network, calculated using our local-dipole method. Vertical lines indicate the position of the maxima of the Q- (1.86 eV) and Soret-bands (2.62 and 2.85 eV) found experimentally.43 | ||
:Soret (1:10 in vacuo) also changed on inclusion of the protein environment to 1:2, the same as seen for the experimental spectrum in Fig. 4. The appearance of the small peak at 1.65 eV is most likely due to transfer of oscillator strength from the red-shifted Soret-band peaks. Similar promotion of dark states was also observed in the local-dipole analysis of the individual Chls (Fig. S4, ESI†).
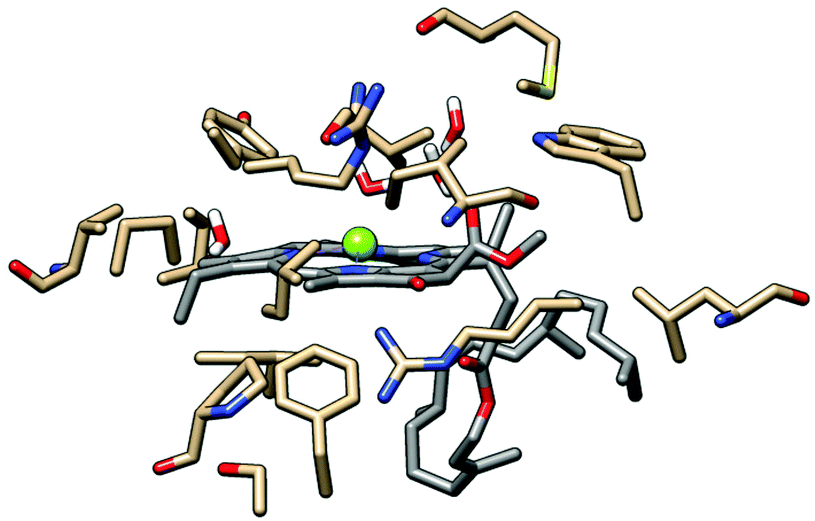 | ||
| Fig. 3 Model system used in the evaluation of the effect of the protein environment on the chlorophyll absorption spectrum. Chlorophyll 608b carbon atoms shown in grey, amino acid side chain carbon atoms in brown. Except for water molecules, all hydrogen atoms are omitted to aid visualisation. This Chl was selected because of its more complete protein surroundings as compared with other Chl which overlapped significantly with neighboring chromophores (Fig. S9, ESI†). | ||
 | ||
| Fig. 4 Effect of inclusion of surrounding amino acid residues on the spectrum of Chl 608b. Vertical lines indicate the experimental position of the Q-band (1.86 eV) and the Chl b component of the Soret-band (2.62 eV).43 | ||
Based on the data obtained from the Chl 608b/protein study it was decided to remove the majority of the protein from the optimised LHC-II structure in order to reduce the dimension of the electronic structure calculation. Amino acid side chains and water directly complexed to Chl Mg2+ ions were retained as were charged species lying close to the chlorin ring (Fig. S3, ESI†) because these were expected to play a major role in modulating the details of the absorption spectrum.
The main roles of the carotenoids, besides providing coverage of the green region of the optical spectrum, are accepted as being involved with photoprotection and energy transfer.50–52 The location of the carotenoid absorption maxima means that their influence on the absorption spectrum of the Chl network should be minimal and this is supported by the calculated spectra in Fig. S7 (ESI†). It was therefore decided to also remove the carotenoid molecules so as to facilitate the analysis of the calculated Chl spectra.
The resulting simplified LHC-II trimer chromophore network contained 6075 atoms corresponding to 15600 explicitly calculated electronic states (after the removal of core electrons by the pseudopotential approximation).
3.2 Chlorophyll network in the LHC-II monomer, dimer and trimer
The calculated spectra of the LHC-II monomer, dimer and trimer Chl networks are shown in Fig. 5. The experimental LHC-II absorption spectrum (filled grey curve) displays the Chl Q- and Soret-bands at approximately 1.85 and 2.6/2.8 eV for the b and a forms, respectively.43 Our calculated spectra show good agreement with the experimental one; very little deviation was observed for the Q-band (approximately 0.05 eV), while the Soret-band was found to display a larger deviation of 0.35 eV.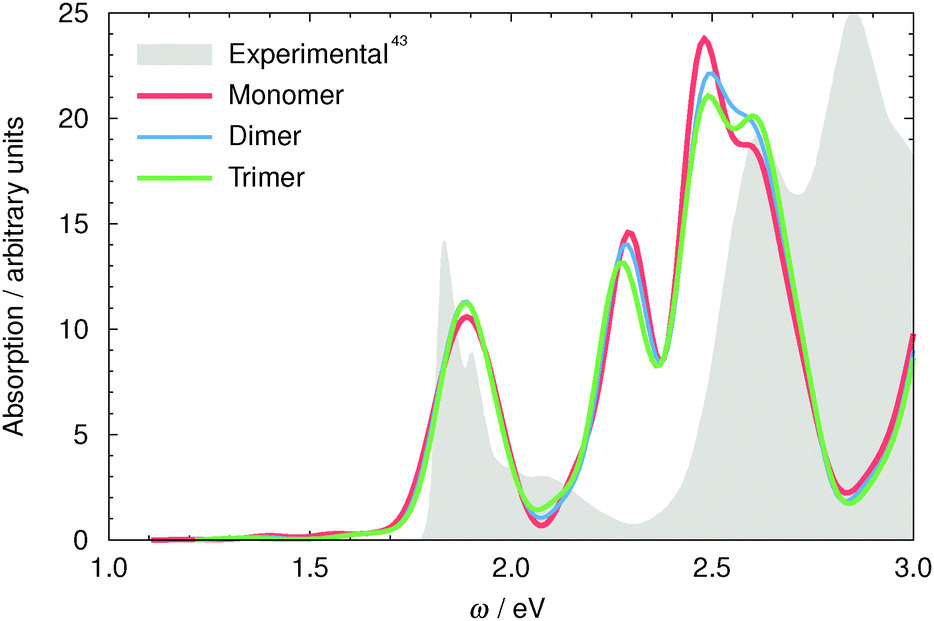 | ||
| Fig. 5 LHC-II monomer (red), dimer (blue) and trimer (green) TDDFT spectrum. The absorption intensities of the dimer have been scaled by half and the trimer by one third to aid in comparison of the spectra. The filled gray curve is the experimental absorption spectrum of LHC-II.43 Here the lower absolute accuracy of TDDFT for the predominantly multi-electron Soret-band excitation energies (discussed above) can be seen. | ||
This Soret-band shift relative to the independent Chl molecules is similar to that seen for Chl 608b in the tests of the effect of the protein environment (see above) and suggests that a similar non-specific environmental effect on the Chl Soret-bands can be expected due to the presence of adjacent Chl chromophores.
From these spectra, and the inter-monomer local-dipole analysis (Fig. S8, ESI†), it appears that the contribution of each monomer to the total spectrum is essentially independent and only minor inter-monomer perturbations exist.
Fig. 2 shows the variation in the location of the peaks for each individual Chl molecule within the chromophore network. Overall, the inclusion of interactions with the other chromophores leads to a red-shift in the peak energies. In general, the major influence of the chromophore network on the individual chlorophylls appears to be similar to that found previously for the protein environment effect, i.e., an electrostatic interaction is sufficient to explain the observed spectral shifts. The largest observed effect was a strong red-shift (∼0.2 eV) on the Soret-band of the Chl b molecules chlorophylls on the lumenal side of the LHC-II complex. Contrary to the non-specific electrostatic red-shift, this larger alteration in absorption energy can be attributed to an electronic interaction due to a close overlap between 605b, 606b and 607b. This is highlighted in Fig. S9 (ESI†) which shows schematically the spatial distribution of the Chl molecules within a single LHC-II monomer.
Fig. 6 shows the contributions of the Q- and Soret- bands of different chlorophylls obtained from the local Bader analysis of the total monomer charge density. This approach facilitates the separation of the chromophore contributions to the photo-absorption profile of the LHC-II system. These data indicate that the LHC-II Q-band can be attributed mainly to Chl a. Furthermore, this analysis suggests that the small Q-band shoulder observed experimentally is due to differences in absorption energy between lumenal and stromal chromophores. Given that energy transfer pathways should be determined in our static model system by excitation frequency gradients alone, our results can be considered to indicate that the most probable energy transfer after an excitation in the Q-band region should take place following an energy descent path from lumen to stroma, in agreement with data obtained using femtosecond transient absorption spectroscopy.53
 | ||
| Fig. 6 LHC-II monomer TDDFT spectrum. Contribution from Chl on the stromal side (left) and the lumenal side (right) of the membrane. Chl a are in red and Chl b in green, the gray line shows the calculated spectrum of the full monomer chlorophyll network. Individual Chl contributions are summed to give the curves shown here. | ||
Two key observations were made regarding the Soret-band in the present work. Our simulations indicate that the Chl a molecules located on the lumenal side are subjected to a blue-shift relative to the Chl a on the stromal side of LHC-II, broadening the LHC-II complex Soret-band. This enables LHC-II to absorb light across a broader range of frequencies. In addition, our calculations show that the peak at approximately 2.3 eV (2.6 eV experimentally) is due to the Chl b molecules. As a result of the local-dipole analysis it was found that lumenal chlorophylls (605b–606b–607b) cause broadening of the Soret-band to the red. These results, which display a significant difference in Soret-band energy in the chlorophylls in different locations within the complex, suggest a route for the LHC-II system to absorb blue light on the stromal side and then transfer the excitation energy down the energy gradient to the center of the complex where 607b is located.
These spectral alterations are most pronounced for a small subset of the chromophores studied; the majority of the Chl spectra do not undergo large alterations when the influence of the chromophore network is taken into account (Fig. S4, ESI†). This suggests that the overall electronic structure of the Chl is not altered significantly when the entire network is included, and the small observed red-shift can be attributed to a Coulombic effect. Thus, most of the excitation energy transfer (EET) ought to be reasonably described using the electric dipole moment coupling scheme proposed by Förster.54 However, due to the specific relative orientation of lumenal Chl b molecules, a strong red-shift and change of the shape of the Soret-band are observed, which suggest that non-electrostatic effects are relevant and Förster's theory is not sufficient to describe the EET between Chl chromophores on the lumenal side of the LHC-II trimer.
4 Conclusions
The absorption spectrum of the full Chl network of the major light harvesting complex, LHC-II, has been simulated using first-principles electronic structure methodology. We have demonstrated that the electrostatic and non-electrostatic effects of the environment as well as specific Chl–Chl interactions are essential for theoretical investigations of systems of this type and that these effects combine to produce significant modulation of the LHC-II absorption spectrum. Local analysis of the contribution of each Chl has been introduced and shown to be a powerful tool for use in studying inter-chromophore interactions and site-specific contributions to the total absorption spectrum.This work indicates that after light absorption in the Q-band region of the LHC-II spectrum the most probable excitation energy propagation will take place from lumen to stroma (Fig. 6 and Fig. S10, ESI†). The major source of absorption in the Soret-band region can be attributed to the Chls situated on the stromal side of the thylakoid membrane. Thus, light energy of around 2.5 eV absorbed in the stromal half of LHC-II should be transferred to the low-energy lumenal grouping of Chl b in the center of the complex. Ongoing studies using our real-space TDDFT method on the Chl network aim to quantify the proposed coupling between chromophores and distinguish its different components in order to clarify the EET mechanisms within LHC-II.
Acknowledgements
The authors acknowledge financial support from the European Research Council Advanced Grant DYNamo (ERC-2010-AdG-267374), European Commission Project CRONOS (grant number 280879-2, CRONOS CP-7P7), Spanish Grant (FIS2013-46159-C3-1-P), Grupos Consolidados UPV/EHU del Gobierno Vasco (IT578-13 and IT395-10), UPV/EHU (grant UFI11/45), COST Actions CM1204 (XLIC) and MP1306 (EUSpec). We also thank PRACE and RES for awarding access to MareNostrum III at BSC-CNS, Barcelona, Spain (2013081486). We acknowledge RZG for the computing time in Hydra. JJS gratefully acknowledges the Comissionat per a Universitats i Recerca del Departament dInnovació, Universitats i Empresa, de la Generalitat de Catalunya for financial support, and the Marie Curie Cofund Action (FP7) of the European Union for the Beatriu de Pinós (2010 BP-A2-00024) postdoctoral fellowship. JAR acknowledges the fellowship of the UPV/EHU. JJPS acknowledges financial support from the National Institute Of General Medical Sciences of the National Institutes of Health (Award Number 1R44GM108085-01A1). BFM acknowledges financial support from the Portuguese Foundation for Science and Technology (CONT_DOUT/11/UC/405/10150/18/2008), the Donostia International Physics centre and the Centro de Fisica de Materiales, UPV/EHU, for financial support. BFM also thanks the Laboratory for Advanced Computing of the University of Coimbra, Portugal the provision of computer resources, technical support and assistance.References
- S. P. Long, Plant, Cell Environ., 2014, 37, 19–21 CrossRef PubMed.
- D. K. Ray, N. D. Mueller, P. C. West and J. A. Foley, PLoS One, 2013, 8, e66428 CAS.
- A. V. Ruban, M. P. Johnson and C. D. P. Duffy, Energy Environ. Sci., 2011, 4, 1643–1650 CAS.
- G. R. Fleming, G. S. Schlau-Cohen, K. Amarnath and J. Zaks, Faraday Discuss., 2012, 155, 27–41 RSC.
- S. I. Allakhverdiev, V. Thavasi, V. D. Kreslavski, S. K. Zharmukhamedov, V. V. Klimov, S. Ramakrishna, D. A. Los, M. Mimuro, H. Nishihara and R. Carpentier, J. Photochem. Photobiol., C, 2010, 11, 101–113 CrossRef CAS PubMed.
- H. Krassen, A. Schwarze, B. Friedrich, K. Ataka, O. Lenz and J. Heberle, ACS Nano, 2009, 3, 4055–4061 CrossRef CAS PubMed.
- S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- S. Y. Searle and C. J. Malins, Biomass Bioenergy, 2014, 65, 3–12 CrossRef PubMed.
- M. A. Palacios, F. L. de Weerd, J. A. Ihalainen, R. van Grondelle and H. van Amerongen, J. Phys. Chem. B, 2002, 106, 5782–5787 CrossRef CAS.
- G. D. Scholes, T. Mirkovic, D. B. Turner, F. Fassioli and A. Buchleitner, Energy Environ. Sci., 2012, 5, 9374–9393 CAS.
- H. A. Frank and R. J. Cogdell, Photochem. Photobiol., 1996, 63, 257–264 CrossRef CAS PubMed.
- A. Young and G. Britton, Carotenoids in photosynthesis, Chapman and Hall Ltd, 1993 Search PubMed.
- T. Polvka and V. Sundström, Chem. Rev., 2004, 104, 2021–2072 CrossRef PubMed.
- H. Lee, Y.-C. Cheng and G. R. Fleming, Science, 2007, 316, 1462–1465 CrossRef CAS PubMed.
- T. R. Calhoun, N. S. Ginsberg, G. S. Schlau-Cohen, Y.-C. Cheng, M. Ballottari, R. Bassi and G. R. Fleming, J. Phys. Chem. B, 2009, 113, 16291–16295 CrossRef CAS PubMed.
- E. Collini, C. Y. Wong, K. E. Wilk, P. M. G. Curmi, P. Brumer and G. D. Scholes, Nature, 2010, 463, 644–647 CrossRef CAS PubMed.
- G. S. Beddard and G. Porter, Nature, 1976, 260, 366–367 CrossRef CAS PubMed.
- R. Clapp, Theor. Chim. Acta, 1982, 61, 105–133 CrossRef CAS.
- K. Broess, G. Trinkunas, C. D. van der Weij-de Wit, J. P. Dekker, A. van Hoek and H. van Amerongen, Biophys. J., 2006, 91, 3776–3786 CrossRef CAS PubMed.
- A. Ishizaki, T. R. Calhoun, G. S. Schlau-Cohen and G. R. Fleming, Phys. Chem. Chem. Phys., 2010, 12, 7319–7337 RSC.
- R. van Grondelle and V. I. Novoderezhkin, Phys. Chem. Chem. Phys., 2006, 8, 793–807 RSC.
- V. Novoderezhkin, A. Marin and R. van Grondelle, Phys. Chem. Chem. Phys., 2011, 13, 17093–17103 RSC.
- C. Konig and J. Neugebauer, Phys. Chem. Chem. Phys., 2011, 13, 10475–10490 RSC.
- E. Runge and E. K. Gross, Phys. Rev. Lett., 1984, 52, 997 CrossRef CAS.
- X. Andrade, D. Strubbe, U. De Giovannini, A. H. Larsen, M. J. T. Oliveira, J. Alberdi-Rodriguez, A. Varas, I. Theophilou, N. Helbig, M. J. Verstraete, L. Stella, F. Nogueira, A. Aspuru-Guzik, A. Castro, M. A. L. Marques and A. Rubio, Phys. Chem. Chem. Phys., 2015 10.1039/C5CP00351B.
- X. Andrade, J. Alberdi-Rodriguez, D. A. Strubbe, M. J. T. Oliveira, F. Nogueira, A. Castro, J. Muguerza, A. Arruabarrena, S. G. Louie, A. Aspuru-Guzik, A. Rubio and M. A. L. Marques, J. Phys.: Condens. Matter, 2012, 24, 233202 CrossRef PubMed.
- J. Alberdi-Rodriguez, M. J. T. Oliveira, P. García-Risueño, F. Nogueira, J. Muguerza, A. Arruabarrena and A. Rubio, Computational Science and Its Applications – ICCSA 2014, Springer International Publishing, 2014, vol. 8582, pp. 607–622 Search PubMed.
- P. García-Risueño, J. Alberdi-Rodríguez, M. J. T. Oliveira, X. Andrade, M. Pippig, J. Muguerza, A. Arruabarrena and A. Rubio, J. Comput. Chem., 2014, 35, 427–444 CrossRef PubMed.
- J. Alberdi-Rodriguez, A. Rubio, M. Oliveira, A. Charalampidou and D. Folias, PRACE white paper, PRACE, 2015 Search PubMed.
- J. Alberdi-Rodriguez, PhD thesis, University of the Basque Country UPV/EHU, 2015.
- Z. Liu, H. Yan, K. Wang, T. Kuang, J. Zhang, L. Gui, X. An and W. Chang, Nature, 2004, 428, 287–292 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- J. J. P. Stewart, J. Mol. Model., 2013, 19, 1–32 CrossRef CAS PubMed.
- M. A. L. Marques, A. Castro, G. F. Bertsch and A. Rubio, Comput. Phys. Commun., 2003, 151, 60–78 CrossRef CAS.
- A. Castro, H. Appel, M. Oliveira, C. A. Rozzi, X. Andrade, F. Lorenzen, M. A. L. Marques, E. K. U. Gross and A. Rubio, Phys. Status Solidi B, 2006, 243, 2465–2488 CrossRef CAS PubMed.
- M. Tafipolsky and R. Schmid, J. Chem. Phys., 2006, 124, 174102 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- C. Legrand, E. Suraud and P.-G. Reinhard, J. Phys. B: At., Mol. Opt. Phys., 2002, 35, 1115 CrossRef CAS.
- R. Bader, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 13348–13356 CrossRef CAS.
- C. Fonseca Guerra, J. Handgraaf, E. J. Baerends and F. M. Bickelhaupt, J. Comput. Chem., 2004, 25, 189–210 CrossRef PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- P. Bultinck, C. van Alsenoy, P. W. Ayers and R. Carbó-Dorca, J. Chem. Phys., 2007, 126, 144111 CrossRef PubMed.
- D. Siefermann-Harms, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 325–355 CrossRef CAS.
- B. F. Milne, A. Rubio and S. Brøndsted Nielsen, Angew. Chem., Int. Ed. Engl., 2015, 54, 2170–2173 CrossRef CAS PubMed.
- M. H. Stockett, L. Musbat, C. Kjær, J. Houmøller, Y. Toker, A. Rubio, B. F. Milne and S. Brønsted Nielsen, Phys. Chem. Chem. Phys., 2015 10.1039/C5CP01513H.
- N. T. Maitra, F. Zhang, R. J. Cave and K. Burke, J. Chem. Phys., 2004, 120, 5932 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, J. Am. Chem. Soc., 2004, 126, 4007–4016 CrossRef CAS PubMed.
- A. Dreuw, J. Phys. Chem. A, 2006, 110, 4592–4599 CrossRef CAS PubMed.
- M. Macernis, J. Sulskus, C. D. P. Duffy, A. V. Ruban and L. Valkunas, J. Phys. Chem. A, 2012, 116, 9843–9853 CrossRef CAS PubMed.
- C. D. P. Duffy, J. Chmeliov, M. Macernis, J. Sulskus, L. Valkunas and A. V. Ruban, J. Phys. Chem. B, 2013, 117, 10974–10986 CrossRef CAS PubMed.
- J. P. Götze, D. Kröner, S. Banerjee, B. Karasulu and W. Thiel, ChemPhysChem, 2014, 15, 3392–3401 CrossRef PubMed.
- J. Chmeliov, W. P. Bricker, C. Lo, E. Jouin, L. Valkunas, A. V. Ruban and C. D. P. Duffy, Phys. Chem. Chem. Phys., 2015 10.1039/C5CP01905B.
- J. Linnanto, J. Martiskainen, V. Lehtovuori, J. Ihalainen, R. Kananavicius, R. Barbato and J. Korppi-Tommola, Photosynth. Res., 2006, 87, 267–279 CrossRef CAS PubMed.
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional spectra of LHC-II chromophores, decomposition of LHC-II spectra into regional contributions, images of complex chlorophyll geometries and inter-chromophore distances. See DOI: 10.1039/c5cp03392f |
| This journal is © the Owner Societies 2015 |