
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical-shift tensors of heavy nuclei in network solids: a DFT/ZORA investigation of 207Pb chemical-shift tensors using the bond-valence method†
Fahri
Alkan
and
C.
Dybowski
*
Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716-2522, USA. E-mail: dybowski@udel.edu
First published on 2nd September 2015
Abstract
Cluster models are used in calculation of 207Pb NMR magnetic-shielding parameters of α-PbO, β-PbO, Pb3O4, Pb2SnO4, PbF2, PbCl2, PbBr2, PbClOH, PbBrOH, PbIOH, PbSiO3, and Pb3(PO4)2. We examine the effects of cluster size, method of termination of the cluster, charge on the cluster, introduction of exact exchange, and relativistic effects on calculation of magnetic-shielding tensors with density functional theory. Proper termination of the cluster for a network solid, including approximations such as compensation of charge by the bond-valence (BV) method, is essential to provide results that agree with experiment. The inclusion of relativistic effects at the spin–orbit level for such heavy nuclei is an essential factor in achieving agreement with experiment.
1. Introduction
Lead has a rich and diverse co-ordination chemistry, ranging from organometallic compounds1 to inorganic salts.2 Solid lead materials often show a variety of structural motifs, e.g. the structural features of the various oxides of lead. The large NMR chemical-shift range (∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm) of 207Pb,3,4 and the strong dependence of the NMR chemical shift on co-ordination geometry and electronic structure, makes the study of the 207Pb nucleus in solid materials an attractive means to analyze them, and reports have appeared in which the experimental shifts are empirically correlated with structure.5–15 Aside from the isotropic shift, the principal components of the chemical-shift tensor in the solid are sensitive to the local electronic structure.16 The full chemical-shift tensor provides more details on structural motifs in a solid than a measure of the isotropic chemical shift alone.
000 ppm) of 207Pb,3,4 and the strong dependence of the NMR chemical shift on co-ordination geometry and electronic structure, makes the study of the 207Pb nucleus in solid materials an attractive means to analyze them, and reports have appeared in which the experimental shifts are empirically correlated with structure.5–15 Aside from the isotropic shift, the principal components of the chemical-shift tensor in the solid are sensitive to the local electronic structure.16 The full chemical-shift tensor provides more details on structural motifs in a solid than a measure of the isotropic chemical shift alone.
The magnetic shielding, as specified by the chemical shift, is a quantum mechanical property of the electronic system, and a characterization of experimental solid-state NMR (SSNMR) results requires calculation of the magnetic shielding for models of the solid state. It is known, from calculations in the literature, that relativistic quantum chemistry methods including spin–orbit (SO) corrections are necessary to produce meaningful theoretical predictions of 207Pb magnetic-shielding parameters.17 For a solid, one must also include intermolecular effects in calculating magnetic shielding, which requires a model of the extended local structure of the system.18,19 For light nuclei such as 13C or 29Si, the gauge included periodic augmented wave (GIPAW) method20–22 has been shown to produce very good agreement with experiment.23 However, for systems containing heavy nuclei such as 119Sn, 207Pb, and 199Hg, the GIPAW method is not as useful due to the lack of inclusion of relativistic spin–orbit effects in the current implementations of the GIPAW algorithm, and the basis sets used have not been optimized for these heavy nuclei.
With the cluster model, one may calculate the magnetic-shielding tensor at various levels of approximation, up to and including spin–orbit effects.19 Comparison allows one to determine which effects are most important. Several recipes have been proposed to employ clusters in magnetic-shielding calculations.13–15,24–38 By calculation with clusters of different sizes, the components of the magnetic-shielding tensor tend towards a limit as the size of the cluster is increased. Because the magnetic-shielding tensor depends most strongly on the electronic structure near the nucleus,39–42 agreement with experiment is obtained with calculations on moderate-size clusters, as discussed below. A combination of a sufficiently large cluster and proper treatment of spin–orbit effects yields good agreement between theory and experiment.19
The main disadvantage of the use of cluster models occurs when one attempts to extend these methods to ionic or covalent network solids. In that case, termination of the periodic structure yields dangling bonds and uncompensated positive or negative charge in the model cluster. Quantum chemical modelling of such systems usually yields either non-convergence during the self-consistent-field (SCF) cycle or a difficult convergence, which signals that the model has a non-physically small HOMO–LUMO gap. The resulting NMR parameters cannot be trusted, and one must seek alternative ways to model the structure.
The high charge or dangling bonds on a model cluster results in incorrect placement of electron density, a situation that would not be found in the extended solid. One solution to this practical problem is to add hydrogens to outer atoms of the cluster (maintaining proper symmetry) to stabilize the cluster. This method has been employed to obtain converged solutions with meaningful NMR parameters in solids for a variety of systems.24,25,43 Another method to account for this effect is to terminate the cluster with pseudo-atoms generated by changing the nuclear charges (Znuc) of the cluster's outer atoms that have missing co-ordination. The aim of this procedure is to stabilize the system by reducing the non-compensated charge on the edges of the cluster. Different versions of this methodology have been applied to systems to study energetics of surface reactions computationally.44–47 To our knowledge, there is no computational study of NMR parameters that has employed this method.
In this contribution, we report calculations of the 207Pb magnetic-shielding tensor by using clusters extracted from X-ray or neutron diffraction geometries of the corresponding solid. We investigate effects of terminating the cluster, of cluster size, of charge, and of symmetry on calculated magnetic-shielding tensors. We discuss the importance of various relativistic effects on the 207Pb magnetic-shielding tensor at different levels of relativistic correction. We briefly discuss effects of various methodological differences on the accuracy of calculated 207Pb magnetic-shielding tensors.
2. Computational methods
All computations were performed using the Amsterdam Density Functional (ADF v2014)48–50 program package. NMR parameters were computed with the NMR module by employing the GIAO formalism.51–55 Calculations were carried out at the BP86 level.56,57 For the NMR nuclei and for the first co-ordination shell, the TZ2P basis set was employed at the all-electron level. For the remainder of the cluster, the DZ basis set and the frozen core approximation (FCA)52,55 were employed to reduce the computational time. Details of the FCA for each individual nucleus, cluster structural information, and results of calculations are given in the ESI.† To include relativistic effects, the zeroth order regular approximation (ZORA)58–61 was employed at the spin–orbit level, unless otherwise indicated.Cluster definition
X-ray or neutron diffraction parameters were used to generate input geometries for the molecular clusters.62–71 For each solid, the space group and the corresponding Inorganic Crystal Structure Database6 reference code are given in Table 1. The clusters were created by defining a central NMR-active atom, and successively adding atoms to form clusters with one, three, or five layers of surrounding atoms. Terminal atoms of a cluster were treated either by coordinating additional hydrogens to the cluster or by changing the nuclear charge of terminal atoms to ensure SCF convergence. For the hydrogen-addition (HA) method, the hydrogen atoms were coordinated to outer oxygen atoms such that the O–H bond length was 0.96 Å and the Pb–O–H angle was 180°.| Crystal system | Reference codea | Space group | Unique Pb(II) sites by symmetry |
|---|---|---|---|
| a Codes from the Inorganic Crystal Structure Database, or structures are from the literature where no code is given. b There is also a unique Pb(IV) site for this system. | |||
| α-PbO | 15466 | P4/nmm | 1 |
| β-PbO | 40180 | Pbcm | 1 |
| Pb3O4 | 4106 | P42/mbc | 1b |
| Pb2SnO4 | 31482 | Pbam | 2 |
| PbF2 | 154994 | Pnma | 1 |
| PbCl2 | 27736 | Pnma | 1 |
| PbBr2 | 202134 | Pnma | 1 |
| PbClOH | 404572 | Pnma | 1 |
| PbBrOH | 404573 | Pnma | 1 |
| PbIOH | Lutz et al.69 | Pnma | 1 |
| PbSiO3 | 26812 | P2/n | 3 |
| Pb3(PO4)2 | 14247 | C2/c | 2 |
For clusters where Znuc was modified for the outer atoms with missing co-ordination, two different methodologies were employed. In the first method, the nuclear charges of the terminal atoms in the cluster were increased by +1. This method we call valence modification of the terminal atoms (VMTA). In this scheme, the total charge on the cluster is the same as in the HA method.
In the second method, Znuc of a terminal atom is modified according to the sum of the bond strengths of that atom. The bond strength (S) for a terminal atom is calculated using an equation from the bond-valence model:72–75
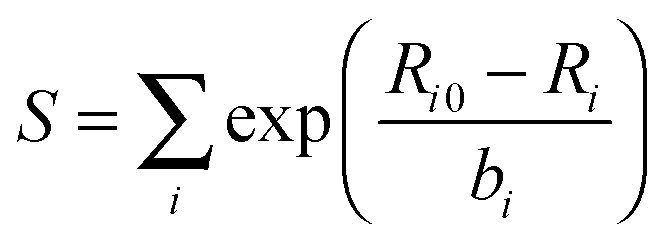 | (1) |
| Zmod = Znuc + ΔS | (2) |
| ΔS = Va − S | (3) |
As an example of the VMTA/BV method, terminal oxygen atoms with different coordination spheres are illustrated in Fig. 1a and b for the third co-ordination shell clusters of α-PbO and β-PbO, respectively. For α-PbO, there is only one unique Pb–O bond length in the crystal structure. Therefore, the bond strength and Zmod of the terminal oxygen atoms only depend on the number of lead atoms coordinated to the terminal oxygen atom. In Fig. 1a, there are two distinct terminal oxygen sites. O1 is coordinated to a single lead site, whereas O2 is coordinated to two lead sites with the same bond length. Calculated bond strengths are 0.50 and 1.00 valence units (vu)75 for O1 and O2 respectively. Because the bond strength of oxygen having complete coordination (i.e., surrounded by four lead atoms) is 2.00 vu, Zmod for each of the two types of terminal oxygen atoms in these clusters is calculated to be 9.50 and 9.00 vu for O1 and O2, respectively. For β-PbO, calculation of bond strengths is not as simple, due to lower symmetry and multiple Pb–O bond lengths in the structure. As a result, there is more variation in the number of distinct terminal oxygen sites. The calculated bond strengths are 0.37, 0.61, 0.64, 0.99, and 1.01 vu for O1 to O5, respectively.
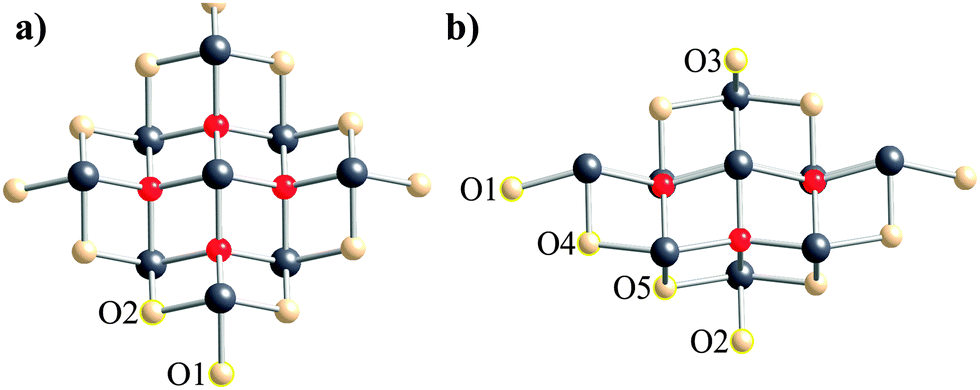 | ||
| Fig. 1 Different terminal oxygen sites according to bond valence model in (a) α-PbO and (b) β-PbO. | ||
The effect of VMTA/BV on the energy levels of a cluster is illustrated in Fig. 2 for the third co-ordination shell geometry of α-PbO. For a cluster without any treatment of the terminal atoms, there is no clear difference in energy between the HOMO and LUMO levels. As a result, convergence under SCF is difficult or impossible as the algorithm becomes oscillatory. Upon closer inspection, it is seen that frontier MOs are mostly dominated by the p orbitals of the terminal oxygen atoms. When Znuc for the atoms is adjusted as described, these levels are stabilized, due to an additional component of the nuclear potential that creates the energy difference between the HOMO and LUMO levels. As a result, SCF convergence is achieved easily for clusters modified with VMTA/BV.
 | ||
| Fig. 2 Energy levels for the 3rd co-ordination cluster of α-PbO. The occupied levels are shown in black whereas unoccupied levels are shown in red. These qualitative calculations carried out with BP86 functionals and the ZORA spin–orbit Hamiltonian. | ||
Magnetic shielding
The magnetic shielding of a nucleus is a tensor property, σ, which can be represented as a 3 × 3 matrix in a Cartesian co-ordinate system. | (4) |
In the NMR experiment, the chemical shift, the shielding relative to the (usually isotropic) resonance position of some reference material, is reported. The principal components of the magnetic-shielding tensor and the principal chemical-shift components are related by eqn (5).
 | (5) |
In principle, one defines the chemical-shift tensor experimentally with the three principal components, δ11, δ22, and δ33. There are several other descriptions in use. In one convention, the tensor is expressed in terms of its irreducible spherical-tensor components.77 Another convenient description for spectra of powders is given by the Maryland convention, a set of three NMR parameters describing the shape of the spectrum of a random powder sample, the isotropic chemical shift (δiso), the span (Ω), and the skew (κ).76 These three parameters are related to the principal components of the chemical-shift tensor by the following relations:
| δiso = ⅓(δ11 + δ22 + δ33) | (6a) |
| Ω = δ11 − δ33 | (6b) |
 | (6c) |
3. Calculations for α-PbO and β-PbO
3.1. Effects of termination method and cluster size on calculated NMR parameters
We investigate modelling of PbO clusters of different sizes, as well as the effects of cluster size on predicted principal components of the shielding tensor, by the HA, VMTA and VMTA/BV methods. Clusters up to the first, third, and fifth atomic co-ordination shells around the NMR nucleus are shown in Fig. 3. Only the odd-numbered co-ordination shells are considered, because clusters terminated with lead atoms are inherently difficult to handle computationally. The calculated NMR parameters for clusters of the two different forms of PbO (α-PbO and β-PbO) using the HA, VMTA, and VMTA/BV methods are given in Table 2. The method used in each case is given by its abbreviation and the prefix gives the maximum co-ordination shell in the cluster. In Table 2, the reduced chemical shifts are defined by the following relation:| Δδii = δii − δiso = σiso − σii | (7) |
 | ||
| Fig. 3 1st, 3rd and 5th co-ordination shell geometries of α-PbO. The terminal oxygen atoms are shown in red circles. The corresponding β-PbO clusters have the same bonding network with differences in bond lengths and angles. | ||
| σ 11 (ppm) | σ 22 (ppm) | σ 33 (ppm) | σ iso (ppm) | Δδ11 (ppm) | Δδ22 (ppm) | Δδ33 (ppm) | Ω (ppm) | Residuala | |
|---|---|---|---|---|---|---|---|---|---|
a
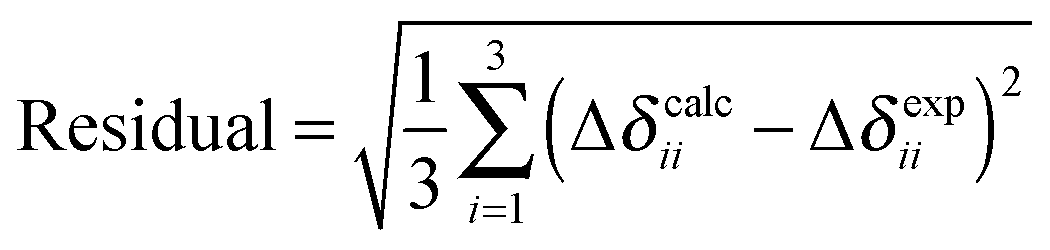 . .
|
|||||||||
| α-PbO | |||||||||
| Experiment8 | 1100 | 1100 | −2200 | 3300 | — | ||||
| 1-HA | 9400 | 9400 | 10645 |
9815 | 415 | 415 | −830 | 1244 | 969 |
| 1-VMTA | 7385 | 7385 | 9681 | 8151 | 765 | 765 | −1531 | 2296 | 473 |
| 1-VMTA/BV | 9451 | 9451 | 11269 |
10057 |
606 | 606 | −1212 | 1818 | 699 |
| 3-HA | 6204 | 6204 | 8918 | 7109 | 905 | 905 | −1809 | 2714 | 276 |
| 3-VMTA | 5861 | 5861 | 8870 | 6864 | 1003 | 1003 | −2006 | 3010 | 137 |
| 3-VMTA/BV | 5887 | 5889 | 8827 | 6868 | 981 | 979 | −1960 | 2940 | 170 |
| 5-HA | 5935 | 5935 | 8922 | 6931 | 995 | 995 | −1991 | 2986 | 148 |
| 5-VMTA | 5936 | 5936 | 8906 | 6926 | 990 | 990 | −1980 | 2970 | 156 |
| 5-VMTA/BV | 5914 | 5915 | 8900 | 6910 | 996 | 994 | −1990 | 2986 | 148 |
| β-PbO | |||||||||
| Experiment8 | 1293 | 1233 | −2527 | 3820 | — | ||||
| 1-HA | 9125 | 9533 | 10871 |
9843 | 718 | 310 | −1028 | 1746 | 1069 |
| 1-VMTA | 7109 | 7525 | 9537 | 8057 | 948 | 532 | −1480 | 2428 | 754 |
| 1-VMTA/BV | 8630 | 9283 | 11516 |
9810 | 1180 | 527 | −1706 | 2886 | 629 |
| 3-HA | 5956 | 6406 | 9270 | 7211 | 1255 | 805 | −2059 | 3314 | 367 |
| 3-VMTA | 5747 | 6228 | 9273 | 7083 | 1335 | 855 | −2190 | 3525 | 294 |
| 3-VMTA/BV | 5655 | 6197 | 9352 | 7068 | 1413 | 871 | 158 | 3697 | 261 |
| 5-HA | 6136 | 6150 | 9630 | 7305 | 1169 | 1155 | −2324 | 3493 | 144 |
| 5-VMTA | 6098 | 6172 | 9593 | 7288 | 1190 | 1115 | −2305 | 3495 | 157 |
| 5-VMTA/BV | 6100 | 6150 | 9581 | 7277 | 1177 | 1127 | −2304 | 3481 | 158 |
For α-PbO and β-PbO, the predicted values of the principal shielding components for a cluster model terminated at the first co-ordination sphere strongly depend on the termination method, with values that can be different from one another by more than 2000 ppm. On the other hand, for clusters of α-PbO and β-PbO including atoms up to the third co-ordination shell, the predicted values of the principal shielding components are significantly less dependent on the termination method. For example, the largest difference between values found with different methods (in this case, VMTA and HA) is only 343 ppm, which is the deviation for σ11 (= σ22) of α-PbO. There are only small differences in the principal components calculated by the VMTA or the VMTA/BV method, showing that these two methods are similar. The maximum difference for calculated components by these two methods is no greater than 100 ppm.
For clusters that include the fifth co-ordination shell of α-PbO or β-PbO, the principal components calculated using the three termination methods agree to within 50 ppm. This agreement reflects the fact that the various termination methods have little effect on the shielding values calculated for fifth-coordination-sphere clusters chosen to represent these network solids.
A comparison of the calculated values to experimental results8 is illustrated with the residuals of the components of the reduced chemical-shift tensor [Table 2]. These residuals are generally smaller for larger clusters. For clusters containing only the first co-ordination shell, the residuals range from 473 ppm to 1069 ppm. For clusters containing up to the third co-ordination shell, the residuals range from 137 ppm to 367 ppm. For clusters containing up to the fifth co-ordination shell, the residuals range only from 144 to 158 ppm. The larger the clusters, regardless of termination method, the closer the calculated components are to the experimental components.
3.2. Symmetry requirements for calculated principal components
The local symmetry of the electronic environment surrounding the NMR-active nucleus affects the values of the principal components of the magnetic-shielding tensor.78 We have deliberately perturbed the symmetry of cluster models of α-PbO to determine the extent of this effect. In Fig. 4 is shown an example, in which a fifth-coordination-shell cluster model is perturbed by adding up to the seventh coordination shell along the +x axis. For the calculated magnetic-shielding parameters in Table 3, we have used models extended by two coordination shells in the +x direction for the first-, third-, and fifth-coordination-shell cluster models to lower the symmetry at the site of the NMR-active nucleus. | ||
| Fig. 4 Distorted 5th co-ordination shell cluster of α-PbO. The added co-ordination in x-direction is shown in red circles whereas the central 207Pb nuclei is highlighted. | ||
| α-PbO | σ 11 (ppm) | σ 22 (ppm) | σ 33 (ppm) | σ iso (ppm) | Ω (ppm) | κ |
|---|---|---|---|---|---|---|
| a Symmetric cluster. b Perturbed cluster. | ||||||
| 1syma | 9451 | 9451 | 11269 |
10057 |
1818 | 1.00 |
| 1pertb | 8183 | 8578 | 10964 |
9241 | 2781 | 0.72 |
| Difference | 1268 | 873 | 305 | 816 | −963 | 0.28 |
| 3syma | 5887 | 5889 | 8827 | 6868 | 2940 | 1.00 |
| 3pertb | 5734 | 5875 | 8737 | 6782 | 3004 | 0.91 |
| Difference | 153 | 14 | 90 | 86 | −64 | 0.09 |
| 5syma | 5914 | 5915 | 8900 | 6910 | 2986 | 1.00 |
| 5pertb | 5906 | 5909 | 8888 | 6901 | 2982 | 1.00 |
| Difference | 8 | 6 | 2 | 9 | 4 | 0.00 |
From the X-ray crystal structure, Pb sites in α-PbO have C4v site symmetry.62 By symmetry constraints, the skew (κ) is either −1.00 (σ11 < σ22 = σ33) or +1.00 (σ11 = σ22 < σ33). Experimentally, α-PbO has a skew of +1.00.6,8,79 Calculated NMR parameters for the symmetric and perturbed clusters are presented in Table 3.
For a cluster that contains the first co-ordination shell, the differences between principal components of symmetric and perturbed clusters are 1268, 873 and 305 ppm for σ11, σ22 and σ33, respectively, with a predicted κ of 0.72 for the perturbed structure. For a cluster that includes the third co-ordination shell, κ is 0.91, only 0.09 from the ideal value. For this cluster model, the differences range only between 153 and 14 ppm for the principal components. For a model containing co-ordination shells through the fifth, the differences between the calculated principal components of the perturbed and symmetric models are very small. The calculated κ for both clusters are +1.00, within 1%. These results, along with the results in Table 2, indicate that the principal values of the magnetic-shielding tensor converge to a limit for a cluster that contains up to the fifth co-ordination shell of 207Pb. Deviations from symmetry occurring at the edges of a cluster of sufficient size seem to have minimal effects on the derived magnetic-shielding tensor and its symmetry at this level of precision. For other nuclei, the limit may depend on the nucleus and the local structure.
3.3. Effects of the charge on the terminal atoms
For α-PbO, the VMTA/BV method predicts 9.50 and 9.00 for Zmod of the two types of terminal oxygen atoms (O1 and O2 in Fig. 1). We investigate how the value of Zmod in the range of 9.30 to 9.70 and 8.80 to 9.20 for these two sites, respectively, affects the predicted NMR magnetic-shielding parameters. For these models, the total charge on a cluster depends on the size of the cluster as well as Zmod of the two terminal oxygen sites, as indicated in Table 4. As seen in Fig. 5, σiso and Ω are linearly correlated with the deviation, ΔZmod, of Zmod from the optimal values (9.50 and 9.00). For the cluster containing up to only the first co-ordination sphere, the variation of the isotropic shielding and the span with Zmod is large. In changing Zmod by 0.4, the isotropic magnetic shielding varies by over 2000 ppm and the span varies by over 1000 ppm. On the other hand, for the largest cluster (through the fifth co-ordination sphere), the isotropic shielding differs by 157 ppm and the span varies by 55 ppm, at most, showing the lack of sensitivity to Zmod in large clusters.| Z mod on O1 and O2 | Total charge on cluster | Mulliken charge on Pb | σ 11 (ppm) | σ 22 (ppm) | σ 33 (ppm) | σ iso (ppm) | Ω (ppm) |
|---|---|---|---|---|---|---|---|
| 9.30, 8.80 | −4.0 | 1.344 | 6005 | 6006 | 8959 | 6990 | 2954 |
| 9.40, 8.90 | −2.0 | 1.364 | 5957 | 5959 | 8928 | 6948 | 2971 |
| 9.50, 9.00 | 0.0 | 1.381 | 5914 | 5915 | 8900 | 6910 | 2986 |
| 9.60, 9.10 | 2.0 | 1.395 | 5874 | 5876 | 8873 | 6874 | 2999 |
| 9.70, 9.20 | 4.0 | 1.408 | 5839 | 5841 | 8848 | 6843 | 3009 |
 | ||
| Fig. 5 The effect of Zmod on (a) isotropic shielding and (b) span for models that extend through the first (blue), third (red), and fifth (black) co-ordination shell for α-PbO. ΔZmod is the deviation of Zmod from the optimal values determined by the VMTA/BV method. | ||
The modification of the charge, Zmod, on the terminal oxygen atoms is partially delocalized onto other atoms in the cluster. As an example, from Table 4, there is a small, but strong, positive correlation between the Mulliken charge on the central lead atom and Zmod of the terminal oxygen sites in this fifth-co-ordination-shell model. In addition, the magnetic-shielding components are correlated with the Mulliken charge on the central lead atom, showing that magnetic shielding reflects the delocalization of charge. The change in the principal components of magnetic shielding with the Mulliken charge demonstrates that there is a somewhat stronger effect on the two degenerate components (σ11 and σ22) than on the unique component (σ33). This difference between the unique component and the non-unique components in their dependence on delocalization of charge suggests that more electron density from delocalization ends up in orbitals the principal direction of which is in the 1–2 plane, rather than in orbitals whose orientation is perpendicular to that plane. This change is also reflected in the gradual change of Ω with total charge on the cluster. These variations of magnetic-shielding parameters with charge on the cluster are even stronger for smaller clusters, as shown in Fig. 5.
4. 207Pb magnetic shielding tensors for various systems
4.1. Cluster size
We present a comparison of experimental and calculated principal components of the 207Pb shielding tensors for a variety of materials (Table 1). The calculations are carried out with two cluster models, the first including only the first co-ordination shell and the second including atoms through the third co-ordination shell.80 Examples of the clusters are given in Fig. 6. For the terminal atoms in the clusters, VMTA/BV modelling (discussed in Section 2) is employed to reduce charge on the cluster, which also ensures SCF convergence. In this model, the bond strengths are calculated using eqn (1), with parameters, Ri0 and bi, tabulated in the ESI.† | ||
| Fig. 6 1st and 3rd co-ordination shell clusters for selective systems investigated in this work. The TZ2P/AE (all-electron) region is shown in the ball-and-stick model, whereas region where FCA/DZ is used is shown by a stick model. | ||
The relationship between experimental and calculated principal components of the 207Pb shielding tensors of these various materials is displayed in Fig. 7a for the first-co-ordination-shell model and in Fig. 7b for the third-co-ordination-shell model. Results for the first-co-ordination-shell model show a strong scatter of the data, with R2 of only 0.608 for a linear correlation. Even for qualitative predictions, NMR parameters obtained using the first-co-ordination-shell model to represent the structure cannot be trusted for these kinds of network solids, as compared to the situation for molecular solids,19 and we do not report calculations with this model in subsequent analyses.
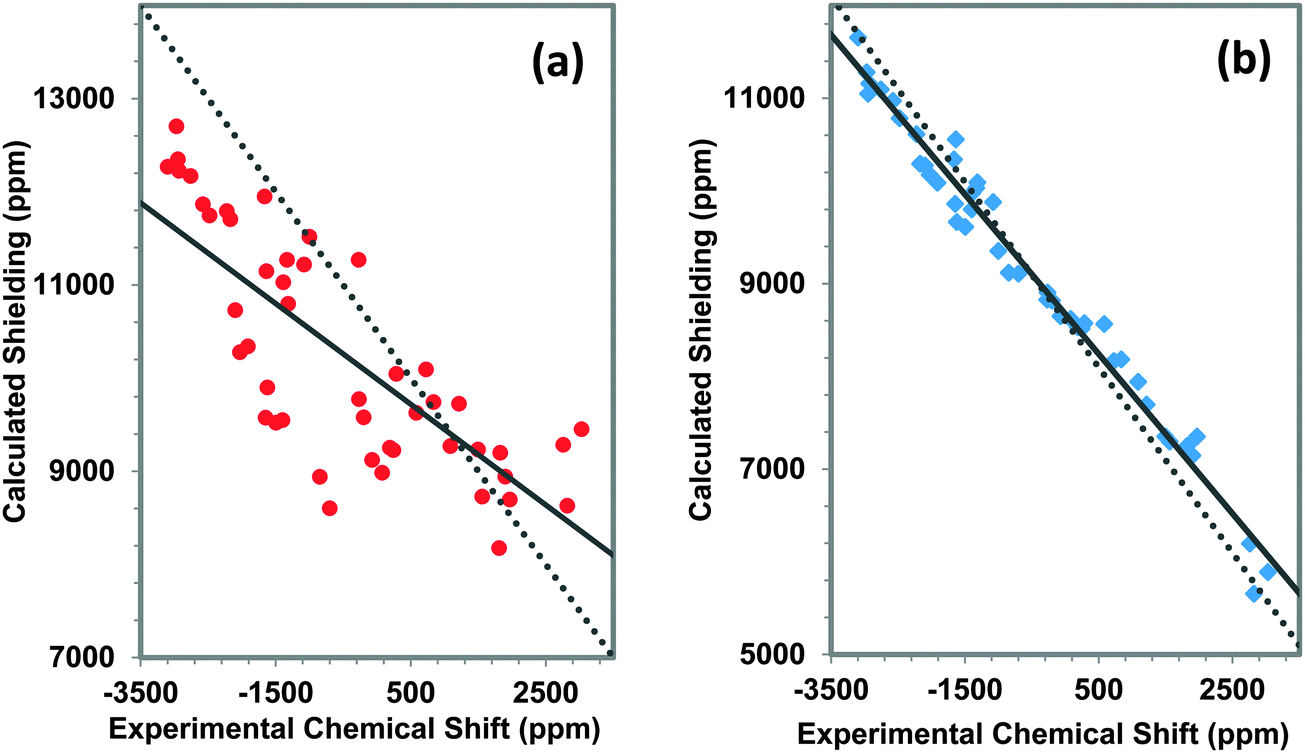 | ||
Fig. 7 The correlation between experimental and calculated principal components for (a) the first co-ordination-shell model ( ) and (b) the third co-ordination-shell model ( ) and (b) the third co-ordination-shell model ( ). The equation of the linear correlation line for the first-co-ordination-shell model is: σcal = −0.541δexp + 9990 with R2 = 0.608. For the third-co-ordination-shell model, the linear correlation is expressed as: σcal = −0.869δexp + 8643 with R2 = 0.983. The dotted line shows ideal behavior (with a slope of −1). ). The equation of the linear correlation line for the first-co-ordination-shell model is: σcal = −0.541δexp + 9990 with R2 = 0.608. For the third-co-ordination-shell model, the linear correlation is expressed as: σcal = −0.869δexp + 8643 with R2 = 0.983. The dotted line shows ideal behavior (with a slope of −1). | ||
Use of a model that includes structure through the third co-ordination shell greatly improves the correlation between experimental and calculated principal components, as can be seen in Fig. 7b. For a linear correlation, R2 = 0.983. The slope of the best-fit linear correlation line is −0.869, with an intercept of 8643 ppm. The slope of the correlation line deviates by 13% from the ideal case, which has a slope of −1.
The predicted absolute magnetic shielding of the reference material, tetramethyllead (TML), from a linear correlation is 9990 ppm for the model that includes only up to the first co-ordination shell. A similar linear correlation of the third-co-ordination-shell model gives a value of 8653 ppm. The absolute shielding of TML calculated from a model of the molecular solid based on the reported X-ray structure19 with optimized hydrogen atom positions gives a value of the isotropic shift of TML of 8136 ppm. There is a significant difference between this estimated shielding of TML and that extracted from the linear correlation of Fig. 7a. On the other hand, the value extracted from Fig. 7b is much closer to the predicted shielding of TML based on its solid-state structure.
4.2. Relativistic effects
In general, for heavy atoms, the contributions to the shielding due to the relativistic nature of the electrons are significant.19,81–85 For the third-co-ordination-shell model, we compare magnetic-shielding tensors of the suite of materials in Table 1 determined with inclusion of only scalar relativistic corrections to the magnetic-shielding tensors determined above using the full spin–orbit relativistic Hamiltonian. The treatment is at the ZORA/DFT level of theory. Correlation of experimental and theoretical principal components and spans is shown in Fig. 8. | ||
Fig. 8 (a) The correlation between principal components of the magnetic shielding and experimental chemical shift of various lead-containing solids: ( ) with inclusion of only scalar relativistic terms; ( ) with inclusion of only scalar relativistic terms; ( ) with inclusion of all relativistic corrections through spin–orbit effects. (b) The correlation of magnetic-shielding span with experimental span of the various lead-containing solids: ( ) with inclusion of all relativistic corrections through spin–orbit effects. (b) The correlation of magnetic-shielding span with experimental span of the various lead-containing solids: ( ) with inclusion of only scalar relativistic terms; ( ) with inclusion of only scalar relativistic terms; ( ) with inclusion of all relativistic corrections through spin–orbit effects. The calculations were carried out on a third-co-ordination-shell model at the ZORA/DFT level. The equation of the correlation line for the principal components is: σcal = −0.365δexp + 7060 with R2 = 0.887 for ZORA/scalar calculations, and σcal = −0.869δexp + 8643 with R2 = 0.983 for ZORA/spin–orbit calculations. For the span, the correlation is: Ωcal = 0.281Ωexp with R2 = 0.871 for ZORA/scalar calculations and Ωcal = 0.866Ωexp with R2 = 0.962 for ZORA/spin–orbit calculations. The dotted line shows ideal behavior. ) with inclusion of all relativistic corrections through spin–orbit effects. The calculations were carried out on a third-co-ordination-shell model at the ZORA/DFT level. The equation of the correlation line for the principal components is: σcal = −0.365δexp + 7060 with R2 = 0.887 for ZORA/scalar calculations, and σcal = −0.869δexp + 8643 with R2 = 0.983 for ZORA/spin–orbit calculations. For the span, the correlation is: Ωcal = 0.281Ωexp with R2 = 0.871 for ZORA/scalar calculations and Ωcal = 0.866Ωexp with R2 = 0.962 for ZORA/spin–orbit calculations. The dotted line shows ideal behavior. | ||
The slope of the correlation line for shielding principal components with experimental chemical-shift components is −0.365 when only scalar relativistic effects are included, whereas the slope of the correlation line for magnetic-shielding principal components when the full relativistic Hamiltonian is used is −0.869. Neither is the ideal value of −1, but the inclusion of spin–orbit relativistic terms gives a correlation much closer to the ideal than does the inclusion of only scalar relativistic effects in the ZORA Hamiltonian, showing that spin–orbit terms cannot be neglected in calculations of magnetic shieldings of 207Pb. We have observed a similar result for the 199Hg magnetic shielding of solids.19
The predicted magnetic shielding of TML from the correlation at the scalar relativistic level is 7060 ppm, whereas a calculation for TML at this scalar relativistic level gives a shielding of 5171 ppm, a difference of 1889 ppm. This difference of 507 ppm indicates that inclusion of the spin–orbit correction is essential to achieve better agreement with the calculated reference shielding from calculation on the reference molecule.
The span of a tensor is independent of the reference. In Fig. 8b, we show the correlation of the predicted and experimental spans for these lead-containing solids. At the ZORA/scalar level of theory, the slope of the best-fit linear correlation of 0.281 deviates significantly from the ideal value of +1. At the ZORA/spin orbit level of theory, the slope of the best-fit linear correlation is 0.866, much closer to the ideal value. This disparity again demonstrates that spin–orbit terms must be included in calculations of magnetic shielding of 207Pb solids.
One striking feature of Fig. 8b is that the predicted NMR parameters obtained by use of the scalar relativistic terms only systematically underestimate the span of the shielding tensor (σ33–σ11), as compared to values calculated at the spin–orbit level. The spin–orbit calculation also underestimates the span as compared to the experiment, but by a substantially smaller difference. Similar results have been shown for 207Pb, as well as for 199Hg.13,19
4.3. Accuracy of calculated principal components of the 207Pb shielding tensor
We have shown in the sections above that it is possible to achieve a good correlation between experimental and theoretical principal components of a wide array of lead-containing materials, provided one uses the full spin–orbit-including Hamiltonian at the ZORA level and creates clusters using the VMTA/VB model with inclusion of structure at least to the third co-ordination shell. Even at this level of approximation, the correlation between predicted and experimental results may deviate from the ideal case (in which the slope of the correlation line is exactly −1). | ||
Fig. 9 A comparison of the correlation of the principal components of the experimental chemical-shift tensor and the predicted shielding tensor for α-PbO, β-PbO, Pb2SnO4, Pb3O4 and PbSiO3 using (a) third- ( ) and (b) fifth-co-ordination-shell ( ) and (b) fifth-co-ordination-shell ( ) cluster models. The equation of the correlation line for the third-co-ordination-shell models is: σcal = −0.891δexp + 8689 with R2 = 0.983. For the fifth-co-ordination-shell model, the correlation line is σcal = −0.910δexp + 8690 with R2 = 0.986. The dotted line shows ideal behavior. ) cluster models. The equation of the correlation line for the third-co-ordination-shell models is: σcal = −0.891δexp + 8689 with R2 = 0.983. For the fifth-co-ordination-shell model, the correlation line is σcal = −0.910δexp + 8690 with R2 = 0.986. The dotted line shows ideal behavior. | ||
Cluster models, without the use of VMTA/BV theory, have been applied to calculations of 207Pb principal components in other solid systems.13,15 In those examples, the ZORA/spin–orbit Hamiltonian was applied at the BP86 level of density functional theory, and no additional treatment was applied to the terminal atoms. For all investigated systems, the span, Ω, is consistently underestimated by the model, whether in molecular13,15 or network solids. This underestimation cannot therefore be attributed to the use of VMTA/BV for termination of the cluster.
In Table 5, the principal components and spans of 207Pb magnetic-shielding tensors for five materials are evaluated, with the BP86 and B3LYP94,95 functionals, on a third-co-ordination-shell cluster. There are two characteristics of the shielding parameters determined by the calculations with B3LYP and with BP86. Firstly, the difference of the B3LYP and BP86 values of σ33 for a particular material is always larger than the difference of either σ11 or σ22. Secondly, the spans, Ω, calculated with B3LYP are always larger than those calculated with BP86. In comparing to experiment,6,8 the predicted span determined with B3LYP is always closer to the experiment than spans calculated with BP86.
| σ 11 (ppm) | σ 22 (ppm) | σ 33 (ppm) | σ iso (ppm) | Ω (ppm) | |
|---|---|---|---|---|---|
| BP86 | |||||
| α-PbO | 5887 | 5889 | 8827 | 6868 | 2940 |
| β-PbO | 5655 | 6197 | 9352 | 7068 | 3697 |
| PbSiO3 (site 1) | 7459 | 7995 | 10331 |
8595 | 2872 |
| PbSiO3 (site 2) | 7829 | 8249 | 10522 |
8867 | 2693 |
| PbSiO3 (site 3) | 7940 | 8532 | 10281 |
8918 | 2341 |
| B3LYP | |||||
| α-PbO | 5862 | 5865 | 8919 | 6882 | 3058 |
| β-PbO | 5572 | 6119 | 9587 | 7092 | 4015 |
| PbSiO3 (site 1) | 7419 | 7960 | 10551 |
8643 | 3133 |
| PbSiO3 (site 2) | 8009 | 8484 | 11030 |
9174 | 3021 |
| PbSiO3 (site 3) | 8027 | 8679 | 10710 |
9139 | 2683 |
In Fig. 10, the correlation between experimental and calculated principal components of 207Pb shielding tensors at the BP86 and B3LYP levels of theory are shown. As expected from the comparison of spans, Ω, the slope of the correlation line determined with the hybrid functional (B3LYP) is −0.985, much closer to the ideal value than the correlation line for the same parameters determined at the BP86 level of theory (−0.895), demonstrating that the use of hybrid functionals accounts for contributions to the magnetic shielding more completely than the use of GGA functionals like BP86.
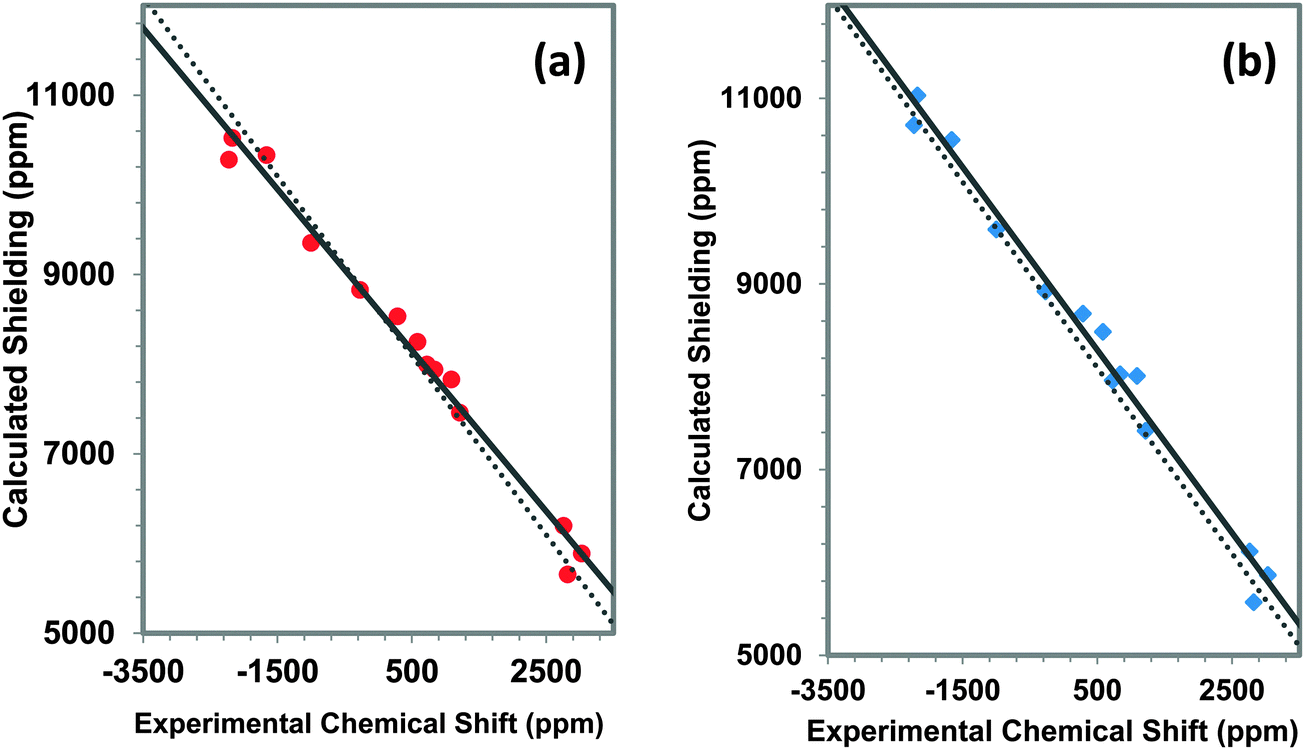 | ||
Fig. 10 The comparison of the use of (a) BP86 [ ] and (b) B3LYP [ ] and (b) B3LYP [ ] to calculate the principal components of sites in α-PbO, β-PbO and PbSiO3 for the third-co-ordination-shell model. The equation of the correlation line for the model using BP86 is: σcal = −0.895δexp + 8605 with R2 = 0.987. The equation of the correlation line for the model using B3LYP is: σcal = −0.985δexp + 8781 with R2 = 0.987. The dotted line shows ideal behavior. The values of the slope show how use of B3LYP approaches the ideal behavior. ] to calculate the principal components of sites in α-PbO, β-PbO and PbSiO3 for the third-co-ordination-shell model. The equation of the correlation line for the model using BP86 is: σcal = −0.895δexp + 8605 with R2 = 0.987. The equation of the correlation line for the model using B3LYP is: σcal = −0.985δexp + 8781 with R2 = 0.987. The dotted line shows ideal behavior. The values of the slope show how use of B3LYP approaches the ideal behavior. | ||
| δ 11 (ppm) | δ 22 (ppm) | δ 33 (ppm) | δ iso (ppm) | Ω (ppm) | |
|---|---|---|---|---|---|
| α-PbO | |||||
| Gabuda et al.8 | 3030 | 3030 | −270 | 1930 | 3300 |
| Fayon et al.6 | 2977 | 2977 | −137 | 1939 | 3114 |
| Zhao et al.79 | 2984 | 2984 | −334 | 1878 | 3318 |
| Average | 2997 ± 48 | 2997 ± 48 | −247 ± 169 | 1916 ± 55 | 3244 ± 190 |
| β-PbO | |||||
| Gabuda et al.8 | 2820 | 2760 | −1000 | 1527 | 3820 |
| Fayon et al.6 | 2945 | 2573 | −972 | 1515 | 3917 |
| Zhao et al.79 | 2953 | 2695 | −1040 | 1536 | 3993 |
| Average | 2906 ± 126 | 2676 ± 160 | −1004 ± 58 | 1526 ± 18 | 3910 ± 146 |
Apart from approximations in the computational formalism that may contribute to the uncertainty in predicted values of the principal components, the uncertainty may also reflect uncertainty in X-ray and neutron diffraction structural parameters used in the definition of the cluster. Dmitrenko et al.14,32 showed that calculated 207Pb chemical-shift parameters can vary significantly for small changes in bond length and bond angle. They also show that calculated NMR parameters may vary by as much as 200 ppm, depending on the X-ray geometry used to define the system. For these reasons, we conclude that agreement between experiment and theory for the 207Pb principal components of a chemical-shift tensor of ±5% is agreement within the current levels of combined uncertainty.
5. Conclusions
We have presented predictions of 207Pb magnetic-shielding (and chemical-shift) tensors using a cluster model for network solids. For such network solids, one must deal appropriately with termination of the cluster to obtain a reasonable SCF solution to the many-body equation. From the test calculations on various clusters, the predicted principal components are not dependent on the termination scheme when a cluster containing atoms through the fifth co-ordination geometry is employed. For all termination schemes, the agreement between experiment and calculation improves as larger clusters are employed. Most errors due to truncation of the structure are minimized if one uses clusters terminated at the third co-ordination shell or higher. Including only the first co-ordination sphere in a cluster is usually not sufficient to account for longer-range effects.For the solid systems which evince large variation in bond lengths in the structure, modification of terminal-atom nuclear charge by a bond valence model, VMTA/BV, allows one to obtain meaningful SCF solutions for clusters of network solids. In particular, in this work, principal components of the shielding tensor for various lead-containing solids are computed with reasonable accuracy. Although the current investigation is limited to the shielding tensor, we feel this method may be appropriate for computations of localized properties such as spin–spin couplings or surface reaction energetics.
For the prediction of 207Pb shielding parameters in various systems, we show that it is possible to obtain reasonably quantitative agreement with experiment by calculations with this method applied to clusters that contain at least the third co-ordination shell, provided the full spin–orbit ZORA Hamiltonian at the BP86 level of theory is used. The correlation between experiment and prediction still does not approach the ideal case, showing that other factors influence the calculation.
We have examined factors that may affect the agreement between prediction and experiment. Aside from the inclusion of spin–orbit effects, the most significant source of disagreement of predicted and experimental values arises from the use of the GGA density functional. Considerable improvement of the correlation between experimental and calculated spans results from employing the B3LYP hybrid functional.
An important consideration in the determination of the reliability of calculational techniques is the precision of experimental determinations of parameters, which is difficult for these very broad powder patterns. In one instance, independent determinations of the principal components of the chemical-shift tensor of the two forms of PbO show that there is a substantial variation of the experimental values to which the predicted values are to be compared. This experimental uncertainty is of the order of the variation of predicted values due to variation of structural parameters upon which the calculation is based.
Acknowledgements
This work was supported by the National Science Foundation under Grant CHE-0956006. The authors thank Dr Shi Bai and Mr Sean Holmes for helpful discussions about the details of these calculations, Dr Eric van Lenthe for helpful discussions of pseudo-atom properties in calculation with the ADF program, and Ms Ozlem K. Alkan for help in writing programs to evaluate bond strengths of terminal atoms which allow prediction of the modified terminal atom charges.References
- P. G. Harrison, Orgaometallic Compounds of Germanium, Tin, and Lead, Chapman and Hall, London, 1985 Search PubMed.
- A. F. Wells, Structural Inorganic Chemistry, Oxford University Press, Oxford, 5th edn, 1984 Search PubMed.
- C. Brevard and P. Granger, Handbook of High Resolution Multinuclear NMR, John Wiley & Sons, New York, 1981 Search PubMed.
- K. J. D. MacKenzie and M. E. Smith, Multinuclear Solid-State NMR of Inorganic Materials, Pergamon, Amsterdam, 2002 Search PubMed.
- G. Neue, C. Dybowski, M. L. Smith, M. A. Hepp and D. L. Perry, Solid State Nucl. Magn. Reson., 1996, 6, 241–250 CrossRef CAS.
- F. Fayon, I. Farnan, C. Bessada, J. Coutures, D. Massiot and J. P. Coutures, J. Am. Chem. Soc., 1997, 119, 6837–6843 CrossRef CAS.
- C. Dybowski, M. L. Smith, M. A. Hepp, E. J. Gaffney, G. Neue and D. L. Perry, Appl. Spectrosc., 1998, 52, 426–429 CrossRef CAS.
- S. P. Gabuda, S. G. Kozlova, V. V. Terskikh, C. Dybowski, G. Neue and D. L. Perry, Chem. Phys. Lett., 1999, 305, 353–358 CrossRef CAS.
- S. P. Gabuda, S. G. Kozlova, V. V. Terskikh, C. Dybowski, G. Neue and D. L. Perry, Solid State Nucl. Magn. Reson., 1999, 15, 103–107 CrossRef CAS.
- C. Dybowski, S. P. Gabuda, S. G. Kozlova, G. Neue, D. L. Perry and V. V. Terskikh, J. Solid State Chem., 2001, 157, 220–224 CrossRef CAS.
- C. Dybowski and G. Neue, Prog. Nucl. Magn. Reson. Spectrosc., 2002, 41, 153–170 CrossRef CAS.
- S. E. Van Bramer, A. Glatfelter, S. Bai, C. Dybowski, G. Neue and D. L. Perry, Magn. Reson. Chem., 2006, 44, 357–365 CrossRef CAS PubMed.
- G. G. Briand, A. D. Smith, G. Schatte, A. J. Rossini and R. W. Schurko, Inorg. Chem., 2007, 46, 8625–8637 CrossRef CAS PubMed.
- O. Dmitrenko, S. Bai, P. A. Beckmann, S. van Bramer, A. J. Vega and C. Dybowski, J. Phys. Chem. A, 2008, 112, 3046–3052 CrossRef CAS PubMed.
- B. J. Greer, V. K. Michaelis, M. J. Katz, D. B. Leznoff, G. Schreckenbach and S. Kroeker, Chem. – Eur. J., 2011, 17, 3609–3618 CrossRef CAS PubMed.
- J. Catalano, A. Murphy, Y. Yao, G. P. A. Yap, N. Zumbulyadis, S. A. Centeno and C. Dybowski, Dalton Trans., 2015, 44, 2340–2347 RSC.
- A. Rodriguez-Fortea, P. Alemany and T. Ziegler, J. Phys. Chem. A, 1999, 103, 8288–8294 CrossRef CAS.
- A. M. Orendt and J. C. Facelli, Annu. Rep. NMR Spectrosc., 2007, 62, 115–178 CrossRef CAS.
- F. Alkan and C. Dybowski, Phys. Chem. Chem. Phys., 2014, 16, 14298–14308 RSC.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 13 CrossRef.
- D. Sebastiani and M. Parrinello, J. Phys. Chem. A, 2001, 105, 1951–1958 CrossRef CAS.
- D. Sebastiani, G. Goward, I. Schnell and M. Parrinello, Comput. Phys. Commun., 2002, 147, 707–710 CrossRef CAS.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azais, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS PubMed.
- J. A. Tossell, J. Magn. Reson., 1997, 127, 49–53 CrossRef CAS.
- G. Valerio, A. Goursot, R. Vetrivel, O. Malkina, V. Malkin and D. R. Salahub, J. Am. Chem. Soc., 1998, 120, 11426–11431 CrossRef CAS.
- J. A. Tossell, Chem. Phys. Lett., 1999, 303, 435–440 CrossRef CAS.
- J. A. Tossell, Phys. Chem. Miner., 1999, 27, 70–80 CrossRef CAS.
- G. Valerio and A. Goursot, J. Phys. Chem. B, 1999, 103, 51–58 CrossRef CAS.
- Y. Zhang and E. Oldfield, J. Phys. Chem. B, 2004, 108, 19533–19540 CrossRef CAS.
- M. Body, G. Silly, C. Legein and J. Y. Buzare, J. Phys. Chem. B, 2005, 109, 10270–10278 CrossRef CAS PubMed.
- D. Stueber, Concepts Magn. Reson., Part A, 2006, 28, 347–368 CrossRef PubMed.
- O. Dmitrenko, S. Bai and C. Dybowski, Solid State Nucl. Magn. Reson., 2008, 34, 186–190 CrossRef CAS PubMed.
- R. E. Taylor, C. T. Carver, R. E. Larsen, O. Dmitrenko, S. Bai and C. Dybowski, J. Mol. Struct., 2009, 930, 99–109 CrossRef CAS PubMed.
- J. Weber and J. Gunne, Phys. Chem. Chem. Phys., 2010, 12, 583–603 RSC.
- A. J. Rossini, A. W. Macgregor, A. S. Smith, G. Schatte, R. W. Schurko and G. G. Briand, Dalton Trans., 2013, 42, 9533–9546 RSC.
- R. E. Taylor, F. Alkan, D. Koumoulis, M. P. Lake, D. King, C. Dybowski and L. S. Bouchard, J. Phys. Chem. C, 2013, 117, 8959–8967 CAS.
- Y. Zhang, S. Mukherjee and E. Oldfield, J. Am. Chem. Soc., 2005, 127, 2370–2371 CrossRef CAS PubMed.
- S. Adiga, D. Aebi and D. L. Bryce, Can. J. Chem., 2007, 85, 496–505 CrossRef CAS.
- N. F. Ramsey, Phys. Rev., 1950, 78, 699–703 CrossRef CAS.
- R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS PubMed.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- V. G. Malkin, O. L. Malkina, M. E. Casida and D. R. Salahub, J. Am. Chem. Soc., 1994, 116, 5898–5908 CrossRef CAS.
- D. H. Brouwer and G. D. Enright, J. Am. Chem. Soc., 2008, 130, 3095–3105 CrossRef CAS PubMed.
- V. M. Bermudez, J. Phys. Chem. C, 2010, 114, 3063–3074 CAS.
- M. Casarin, C. Maccato and A. Vittadini, Inorg. Chem., 2000, 39, 5232–5237 CrossRef CAS.
- M. Casarin, C. Maccato and A. Vittadini, J. Phys. Chem. B, 2002, 106, 795–802 CrossRef CAS.
- M. Casarin, D. Falcomer, A. Glisenti and A. Vittadini, Inorg. Chem., 2003, 42, 436–445 CrossRef CAS.
- ADF2014, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, 2014, http://www.scm.com Search PubMed.
- C. F. Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS PubMed.
- G. Schreckenbach and T. Ziegler, J. Phys. Chem., 1995, 99, 606–611 CrossRef CAS.
- G. Schreckenbach and T. Ziegler, Int. J. Quantum Chem., 1996, 60, 753–766 CrossRef CAS.
- G. Schreckenbach and T. Ziegler, Theor. Chem. Acc., 1998, 99, 71–82 CrossRef CAS.
- S. K. Wolff, T. Ziegler, E. van Lenthe and E. J. Baerends, J. Chem. Phys., 1999, 110, 7689–7698 CrossRef CAS PubMed.
- J. Autschbach and E. Zurek, J. Phys. Chem. A, 2003, 107, 4967–4972 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- E. Van lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS PubMed.
- E. Vanlenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS PubMed.
- E. van Lenthe, R. van Leeuwen, E. J. Baerends and J. G. Snijders, Int. J. Quantum Chem., 1996, 57, 281–293 CrossRef CAS.
- E. vanLenthe, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1996, 105, 6505–6516 CrossRef CAS PubMed.
- J. Leciejewicz, Acta Crystallogr., 1961, 14, 1304 CrossRef CAS.
- P. Garnier, J. Moreau and J. R. Gavarri, Mater. Res. Bull., 1990, 25, 979–986 CrossRef CAS.
- J. R. Gavarri and D. Weigel, J. Solid State Chem., 1975, 13, 252–257 CrossRef CAS.
- J. R. Gavarri, J. P. Vigouroux, G. Calvarin and A. W. Hewat, J. Solid State Chem., 1981, 36, 81–90 CrossRef CAS.
- Y. Z. Nozik, L. E. Fykin and L. A. Muradyan, Kristallografiya, 1976, 21, 76–79 CAS.
- M. Lumbreras, J. Protas, S. Jebbari, G. J. Dirksen and J. Schoonman, Solid State Ionics, 1986, 20, 295–304 CrossRef CAS.
- L. Ehm, K. Knorr, F. Madler, H. Voigtlander, E. Busetto, A. Cassetta, A. Lausi and B. Winkler, J. Phys. Chem. Solids, 2003, 64, 919–925 CrossRef CAS.
- H. D. Lutz, K. Beckenkamp, T. Kellersohn, H. Moller and S. Peter, J. Solid State Chem., 1996, 124, 155–161 CrossRef CAS.
- M. L. Boucher and D. R. Peacor, Z. Kristallogr., Kristallgeom., Kristallphys., Kristallchem., 1968, 126, 98–111 CrossRef.
- U. Keppler, Z. Kristallogr., Kristallgeom., Kristallphys., Kristallchem., 1970, 132, 228–235 CrossRef CAS.
- I. D. Brown and R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1973, 29, 266–282 CrossRef CAS.
- I. D. Brown and K. K. Wu, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 1957–1959 CrossRef.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1985, 41, 244–247 CrossRef.
- I. D. Brown, Chem. Rev., 2009, 109, 6858–6919 CrossRef CAS PubMed.
- J. Mason, Solid State Nucl. Magn. Reson., 1993, 2, 285–288 CrossRef CAS.
- U. Haeberlen, High Resolution NMR in Solids: Selective Averaging, Academic Press, New York, 1976 Search PubMed.
- J. C. Facelli, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 58, 176–201 CrossRef PubMed.
- P. D. Zhao, S. Prasad, J. Huang, J. J. Fitzgerald and J. S. Shore, J. Phys. Chem. B, 1999, 103, 10617–10626 CrossRef CAS.
- Calculations through the fifth co-ordination shell are carried out in Section 4.3 for several systems. The results demonstrate that calculations through the third co-ordination shell account for most of the magnetic shielding with reasonable computational time.
- J. Autschbach and S. Zheng, Annu. Rep. NMR Spectrosc., 2009, 67, 1–95 CrossRef CAS.
- V. Arcisauskaite, J. I. Melo, L. Hemmingsen and S. P. A. Sauer, J. Chem. Phys., 2011, 135, 11 CrossRef PubMed.
- J. Roukala, A. F. Maldonado, J. Vaara, G. A. Aucar and P. Lantto, Phys. Chem. Chem. Phys., 2011, 13, 21016–21025 RSC.
- A. Wodynski, M. Repisky and M. Pecul, J. Chem. Phys., 2012, 137, 11 CrossRef PubMed.
- J. Autschbach, Philos. Trans. R. Soc., A, 2014, 372, 39 CrossRef PubMed.
- P. Pyykko, Chem. Phys., 1983, 74, 1–7 CrossRef CAS.
- R. Fukuda, M. Hada and H. Nakatsuji, J. Chem. Phys., 2003, 118, 1027–1035 CrossRef CAS PubMed.
- R. Fukuda, M. Hada and H. Nakatsuji, J. Chem. Phys., 2003, 118, 1015–1026 CrossRef CAS PubMed.
- J. Autschbach, Principles and Applications of Density Functional Theory in Inorganic Chemistry I, 2004, vol. 112, pp. 1–48 Search PubMed.
- A. Bagno, G. Casella and G. Saielli, J. Chem. Theory Comput., 2006, 2, 37–46 CrossRef CAS.
- J. Autschbach, Theor. Chem. Acc., 2004, 112, 52–57 CrossRef CAS.
- S. T. Holmes, R. J. Iuliucci, K. T. Mueller and C. Dybowski, J. Chem. Phys., 2014, 141, 12 CrossRef PubMed.
- B. E. G. Lucier, K. E. Johnston, W. Q. Xu, J. C. Hanson, S. D. Senanayake, S. Y. Yao, M. W. Bourassa, M. Srebro, J. Autschbach and R. W. Schurko, J. Am. Chem. Soc., 2014, 136, 1333–1351 CrossRef CAS PubMed.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp03348a |
| This journal is © the Owner Societies 2015 |