
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The low frequency modes of solvated ions and ion pairs in aqueous electrolyte solutions: iron(II) and iron(III) chloride
Fabian
Böhm
a,
Vinay
Sharma
b,
Gerhard
Schwaab
*a and
Martina
Havenith
a
aDepartment of Physical Chemistry II, Ruhr-Universität Bochum, Germany. E-mail: gerhard.schwaab@rub.de; Fax: +49 234 321 4183; Tel: +49 234 322 4256
bInstitute of Chemistry, The Hebrew University, Jerusalem, Israel
First published on 1st July 2015
Abstract
We have investigated the hydration dynamics of solvated iron(II) and iron(III) chloride. For this, THz/FIR absorption spectra of acidified aqueous FeCl2 and FeCl3 solutions have been measured in a frequency range of 30–350 cm−1 (≈1–10 THz). We observe a nonlinear concentration dependence of the absorption, which is attributed to the progressive formation of chloro-complexes of Fe(II) and Fe(III), respectively. By principal component analysis of the concentration dependent absorption spectra, we deduced the molar extinction spectra of the solvated species Fe2+ + 2Cl− and FeCl+ + Cl−, as well as FeCl2+ + 2Cl− and FeCl2+ + Cl−. In addition, we obtain ion association constants log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KFeCl2 = −0.88(5) and logKFeCl3 = −0.32(16) for the association of Fe2+ and Cl− to FeCl+ and the association of FeCl2+ and Cl− to FeCl2+, respectively. We performed a simultaneous fit of all the effective extinction spectra and their differences, including our previous results of solvated manganese(II) and nickel(II) chlorides and bromides. Thereby we were able to assign absorption peaks to vibrational modes of ion–water complexes. Furthermore, we were able to estimate a minimum number of affected water molecules, ranging from ca. 7 in the case of FeCl+ + Cl− to ca. 21 in the case of FeCl2+ + Cl−.
KFeCl2 = −0.88(5) and logKFeCl3 = −0.32(16) for the association of Fe2+ and Cl− to FeCl+ and the association of FeCl2+ and Cl− to FeCl2+, respectively. We performed a simultaneous fit of all the effective extinction spectra and their differences, including our previous results of solvated manganese(II) and nickel(II) chlorides and bromides. Thereby we were able to assign absorption peaks to vibrational modes of ion–water complexes. Furthermore, we were able to estimate a minimum number of affected water molecules, ranging from ca. 7 in the case of FeCl+ + Cl− to ca. 21 in the case of FeCl2+ + Cl−.
1 Introduction
Ion hydration phenomena have attracted the attention of many researchers.1–4 Whereas macroscopically ion hydration is well understood, at the molecular level it is still part of an ongoing and sometimes controversial discussion.3–8 Recently, the solvation dynamics of alkali and alkaline earth metals have been studied in detail.2,3,6–9 Most of these studies indicate prevalently self confined cations and anions in aqueous phase at low and moderate concentrations.2,6–8,10 In certain cases, however, cooperative effects between the ions have been postulated.9The hydration properties of transition metal ions have only recently been studied.11–13 The behaviour of ferrous (Fe2+) and ferric (Fe3+) iron in aqueous solution with respect to their tendency towards formation of distinct chloro-complexes are of special interest to the fields of iron metabolism and biological function,14,15 geochemical and hydrothermal studies,16,17 isotopic fractionation18 and chemistry of natural waters.19 In spite of a broad range of technical applications, simultaneous quantitative analysis of aqueous iron(II) and iron(III) at high concentrations still remains a challenge. Calorimetry,20 isotachophoresis,21 ion chromatography19,22 and spectrophotometry17,23–27 are restricted to the millimolar concentration range, since the formation of ion associates complicates data evaluation at higher concentrations.
While the masses of Fe2+ and Fe3+ are practically the same, the ionic radii of the ions differ considerably (0.92 Å for Fe2+ and 0.79 Å for Fe3+) due to the different charge states. Fe3+ has a much higher charge density, which causes substantially different hydration behavior compared to Fe2+. Besides the octahedral hexaaqua complex [Fe(H2O)6]2+, several chloro-complex species have been identified in acidic solutions of ferrous chloride, which are: octahedral monochloro-complex [Fe(H2O)5Cl]+, dichloro-complex [Fe(H2O)4Cl2], and tetrahedral tetrachloro-complex [FeCl4]2−.28 The latter species, however, is formed exclusively at very high chloride excess29 and/or high temperatures.28,30 Similarly, the complexes formed by ferric chloride are: octahedral hexaaqua complex [Fe(H2O)6]3+, monochloro-complex [Fe(H2O)5Cl]2+, dichloro-complex trans-[Fe(H2O)4Cl2]+, trichloro-complex [Fe(H2O)3Cl3] and tetrahedral tetrachloro-complex [FeCl4]−.17,31–41 Similarly to the ferrous salt, the highest order chloro-species is only found at high chloride excess and/or high temperatures.
So far, the equilibrium formation constant of the ferrous monochloro-complex at room temperature has been determined by spectrophotometry28,42 and potentiometry43 to be 0.69 kg mol−1, 0.43 kg mol−1 and 0.75 kg mol−1, respectively. The formation constant of the dichloro-complex has been determined spectrophotometrically to be 0.018 kg2 mol−2.28
In contrast to ferrous chloride, there have been many attempts to determine equilibrium constants for the formation of ferric chloro-complexes in aqueous phase using spectrophotometry17,26,32,33,44–49 and potentiometry.50,51 Most of them, however, did not include the concept of ion activity, and thus there is a clear divergence in the values of formation constants for solutions of different ionic strength. Only a handful of publications present an estimate of equilibrium constants at zero ionic strength, either by extrapolation32,33,44,51 or by introducing activity coefficients for the ions.17,26,49 While the values for the formation constant of the monochloro-complex at room temperature are in good agreement, ranging from 19.1 to 33.1 kg mol−1,17,44,49,51 there are large inconsistencies present for the formation constant of the dichloro-complexes ranging from 2 to 14 kg mol−1.17,26,33,44,49,51 The trichloro-complex formation constant is considerably smaller, ranging from 0.04 to 0.08 kg mol−1.17,26,33,44 Note that cumulative formation constants have been converted to stepwise formation constants.
Despite these previous efforts, several questions remain unresolved. For instance, are the aforementioned methods sensitive only to the formation of first shell complexes, or could other associates also play a role? Do the water molecules incorporated in ferrous and ferric complexes exhibit a different dynamic behaviour compared to bulk water molecules? Is water beyond the first hydration shell affected by the ions and ion associates as well?
In the present study, we investigate the hydration behavior of iron(II) and iron(III) chloride salts in aqueous phase, using THz/FIR Fourier transform spectroscopy. THz/FIR absorption spectroscopy has proven to be a powerful tool to analyse electrolyte solutions at high concentrations. Using this technique, we have determined the low frequency absorption characteristics of several alkaline,7 alkaline earth,8 transition metal52 and lanthanum halides53 as well as hydrochloric and hydrobromic acid.54 The obtained frequency dependent absorption spectra exhibit distinguished ion specific bands, which are not present in the spectrum of bulk water. The spectra of these solutions can be well described by a linear superposition of (a) the absorption of bulk water and (b) the absorption of hydrated anions and cations. In case of manganese(II), nickel(II) and lanthanum(III) chloride and bromide, as well as hydrochloric and hydrobromic acid, we have shown that the nonlinear concentration dependence of absorption can be attributed to the formation of ion pairs.52–54 Thorough analysis of the experimental data allowed us to extract the single ionic features as well as the ion pair absorption spectra.
Here we investigate the concentration dependent THz/FIR absorption of iron(II) and iron(III) chloride in water with respect to the formation of ion associates. Using principal component analysis (PCA) as an unbiased mathematical procedure in conjunction with a chemical equilibrium model, we are able to dissect the experimental spectra into the absorption features of the various ions and complex species. This enables us to extract information about the vibrational properties of the solvated ions and ion associates, as well as their dynamical hydration shells. This study provides a proof of principle for the applicability of THz/FIR spectroscopy as an analytical tool for the simultaneous determination of Fe(II) and Fe(III) chloride at concentrations up to the solubility limit.
2 Materials and methods
2.1 Materials
The sample solutions were prepared by dissolving the weighed out amounts of FeCl2·4H2O and FeCl3·6H2O (Sigma Aldrich, 99% purity) in HPLC grade water. In order to prevent oxidation and hydroxide formation, all solutions were acidified with HCl to an H+ concentration of 1 M, corresponding to pH 0. For determination of water concentration in the samples, densities were measured at 20 °C using an Anton-Paar DMA58 density meter.2.2 FTIR measurements
THz/FIR absorption measurements were carried out using a Bruker Vertex 80v FTIR spectrometer equipped with a liquid helium cooled silicon bolometer from Infrared Laboratories as detector (for more specific details about the experimental setup, see Schmidt et al.7 and supporting information therein). During the complete series of measurements, the sample compartment was constantly purged with technical grade dry nitrogen to avoid humidity effects of the air. The temperature of the sample cell was kept constant at 20 ± 0.2 °C with a commercial high precision thermostat. For each single spectrum, 256 scans with a resolution of 2 cm−1 were averaged. The sample solutions were placed in a standard Bruker liquid cell between two parallel TPX windows of 4 mm thickness with a Kapton spacer of ca. 25 μm thickness. The exact sample layer thickness was determined prior to each measurement using the Fabry–Pérot etalon effect of the empty cell.Using Lambert–Beer's law, the frequency dependent absorption coefficient α(![[small upsilon, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e131.gif) ) of an aqueous solution is expressed as
) of an aqueous solution is expressed as
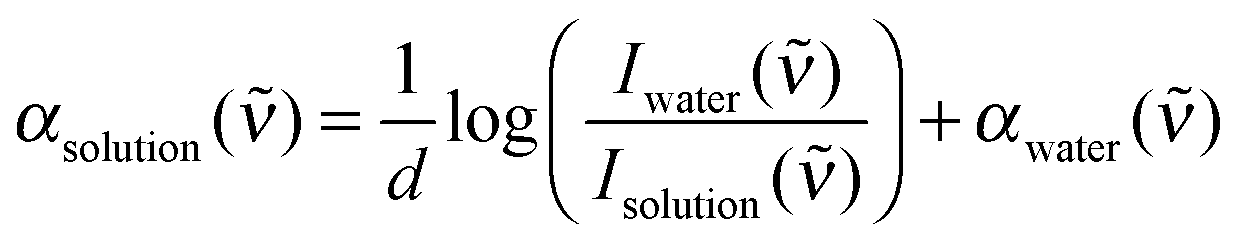 | (1) |
) and Isolution() are the transmitted intensities of the water reference and the sample, respectively, and αwater() is a fit of the absorption spectrum of water.52,55 In this way, artefacts due to reflections at the cell windows are minimized.
The contribution of HCl to the absorption was determined in an independent measurement and subtracted from all subsequent measurements, assuming a weak interaction with the coexisting ions.
For further data evaluation, we subtract the expected absorption of water in the sample to get the effective ionic absorption αeffion:
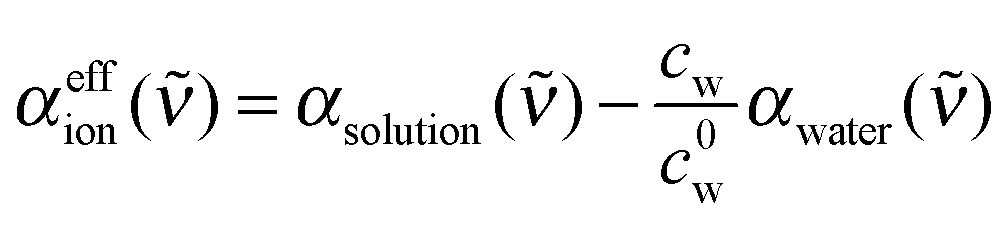 | (2) |
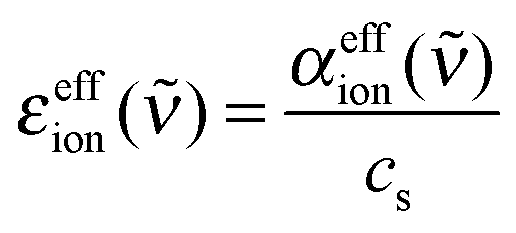 | (3) |
It is important to mention here that any changes in solvation water absorption induced by the ions are inherent in the effective ionic extinction εeffion. We assume that the interaction between each ion and surrounding water molecules can be understood by taking into account two contributions.7 Since the absorption properties of water are modified in the vicinity of ions, subtraction of the bulk water absorption from the sample absorption results in a negative contribution with the line shape of the water spectrum. The hydrated ion, however, has additional low frequency modes which can be described in terms of rattling modes and/or vibrational modes of the ion–water complexes.7,8
3 Experimental results
Sample solutions with concentrations ranging from 0.5 M to 3.5 M of FeCl2 and FeCl3, respectively, were measured using FTIR spectroscopy. The absorption spectra and the effective molar extinction spectra of both salts, respectively, are presented in Fig. 1 and 2. The positive value of the extinction over the entire frequency range can be attributed to the solvated ions. By examination of Fig. 1B and 2B it is obvious that the two iron chlorides have distinct nonlinear absorption features (marked with black arrows).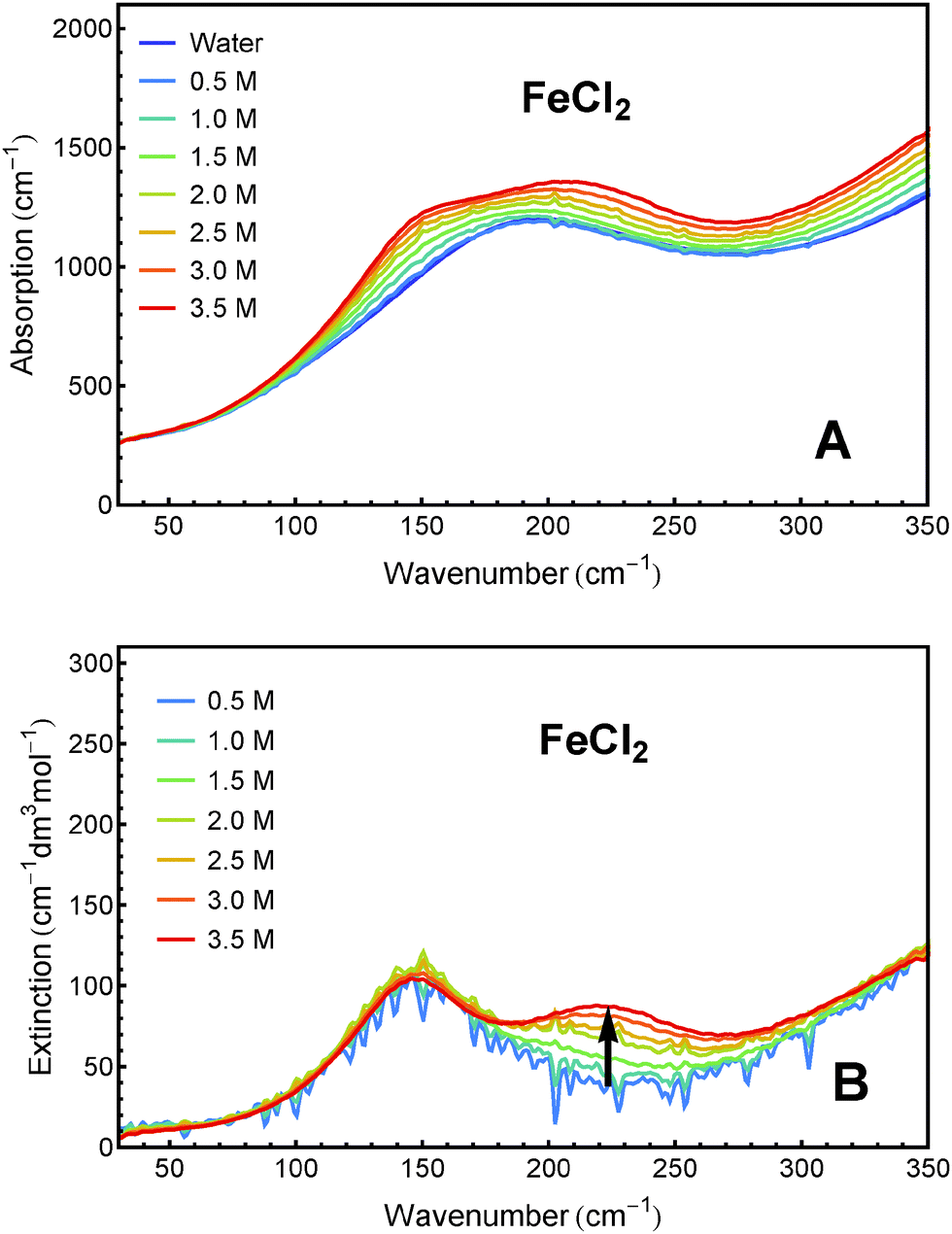 | ||
| Fig. 1 THz/FIR absorption (A) and effective molar extinction spectra (B) of aqueous FeCl2 solutions at 20 °C, as obtained from FTIR absorption measurements. | ||
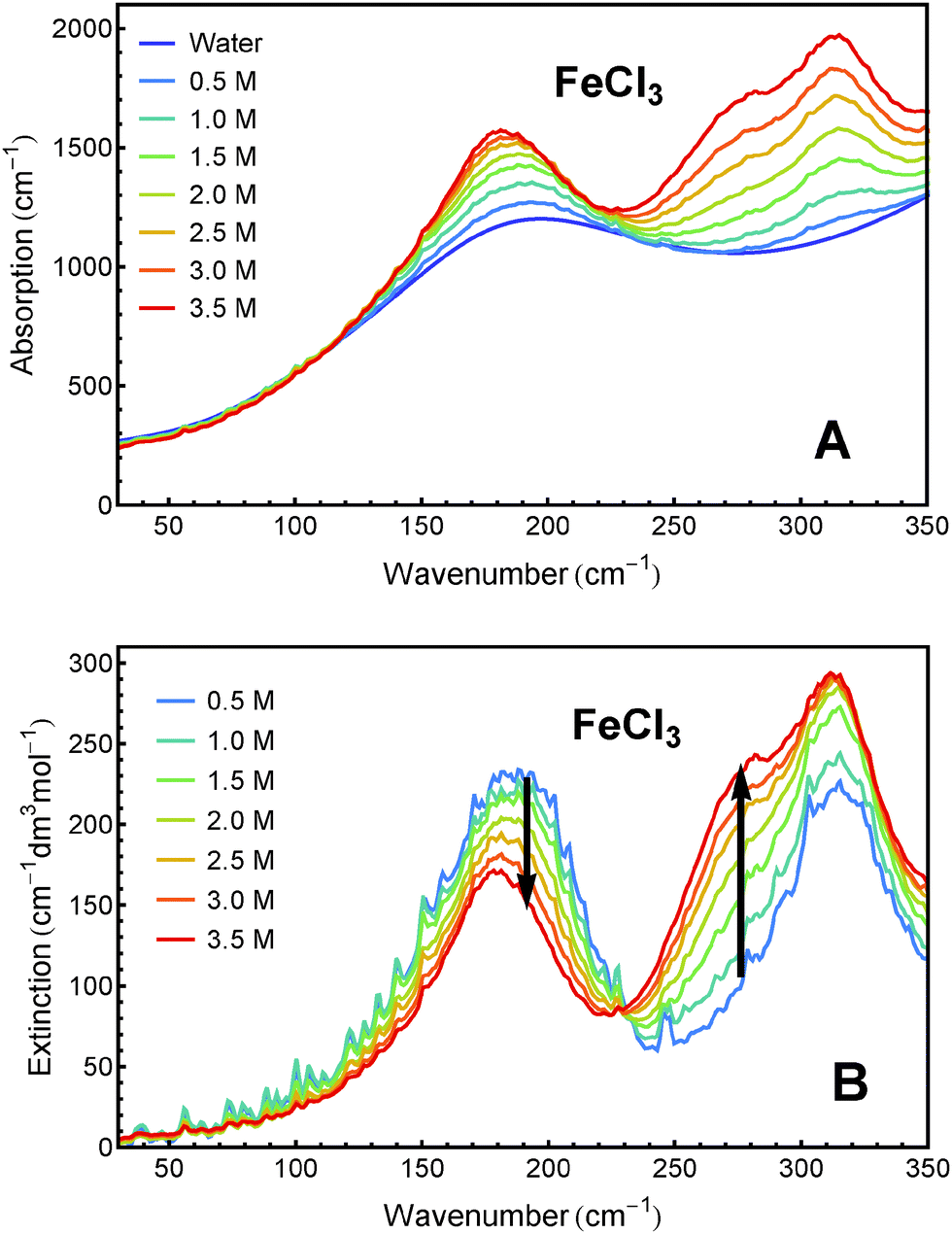 | ||
| Fig. 2 THz/FIR absorption (A) and effective molar extinction spectra (B) of aqueous FeCl3 solutions at 20 °C, as obtained from FTIR absorption measurements. | ||
4 Spectral dissection
In order to deconvolute the experimental spectra into distinct components, we performed a principal component analysis (PCA) on the αeffion spectra. Using this method (which has been described in detail before52), we were able to reduce both data sets of FeCl2 and FeCl3 to linear combinations of two components each, which include more than 96% of the information in both cases. The loading vectors and concentration dependent scores of the principal components of both salts are shown in Fig. 3.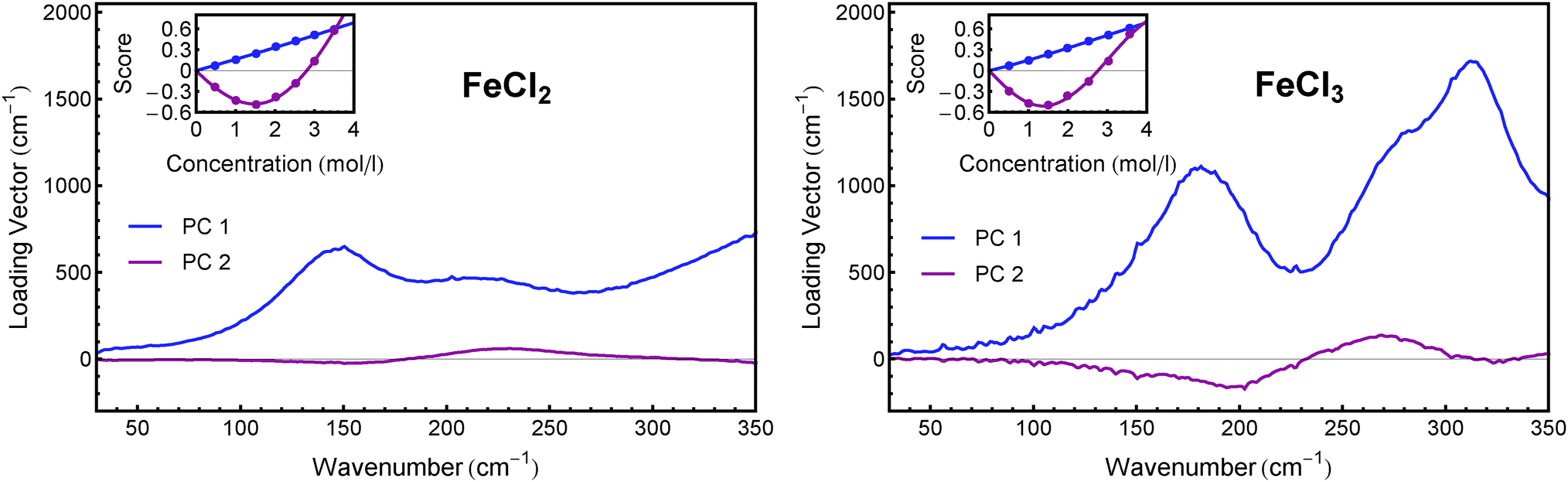 | ||
| Fig. 3 First two principal components of FeCl2 (left) and FeCl3 (right). The insets show the associated scores as obtained from PCA. | ||
Although for both salts the results of the PCA are rather similar, the underlying mechanism seems to be different. Comparing different sets of association constants from literature (cf. Section 2) it becomes evident that in the case of FeCl2, the solvated ions and the first ion associate FeCl+ dominate over the whole concentration range, while in the case of FeCl3 the first and second associates, FeCl2+ and FeCl2+, dominate the solution's composition.
For the further evaluation of our data we make the assumption that next to Fe2+, FeCl+, FeCl2+, FeCl2+ and Cl− the contribution of other species to the observed absorption is negligible. Any changes in the concentration dependent absorption spectrum are attributed to a shift in the equilibrium:
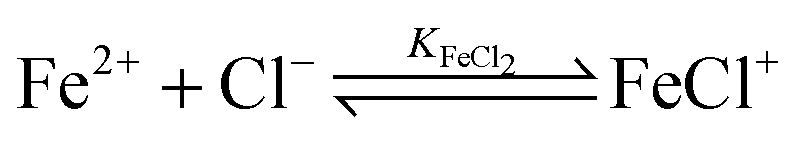 | (4) |
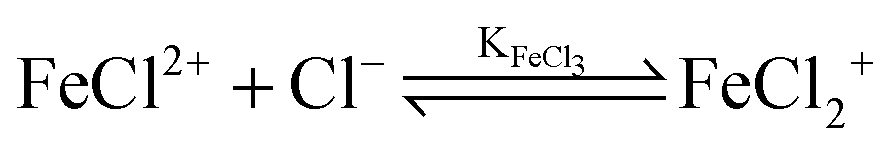 | (5) |
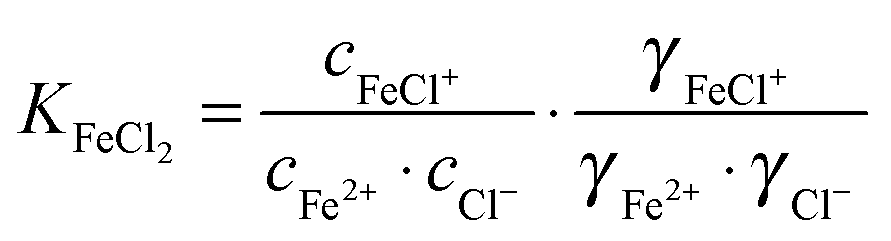 | (6) |
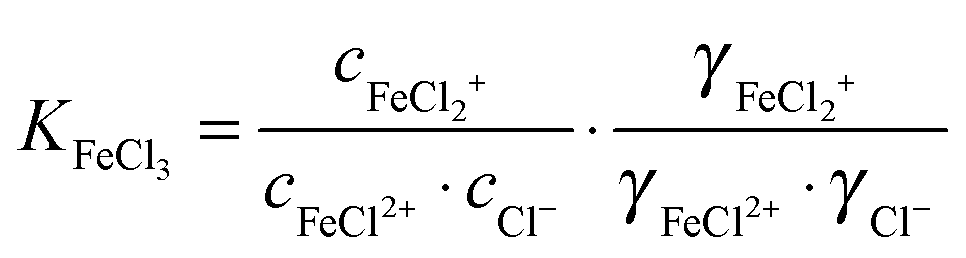 | (7) |
| Compound | β (0) | β (1) | C ϕ |
|---|---|---|---|
| FeCl2 | 0.339 ± 0.005 | 1.48 ± 0.06 | −0.019 ± 0.001 |
| FeCl3 | 0.42 ± 0.01 | −0.028 ± 0.002 | 7.0 ± 0.4 |
The respective underlying models for the effective ionic absorption are the following:
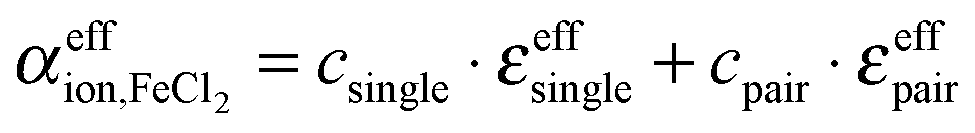 | (8) |
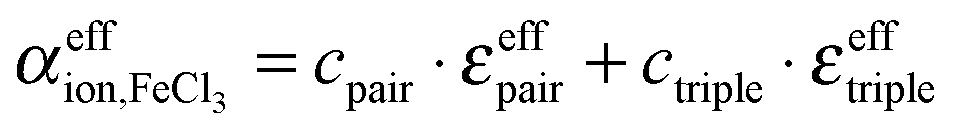 | (9) |
We performed a fit of the scores of the first two principal components with KFeCl2 and KFeCl3, respectively, as parameters, as has been described before.52 Clearly the fitted lines match the scores quite well (see insets in Fig. 3, solid lines). The resulting values for the equilibrium constants are presented in Table 2.
In addition to the equilibrium constants, we were also able to deduce the molar extinction spectra of the different solvated species. The result is displayed in Fig. 4. Note that each of these spectra contains the anion contribution corresponding to the stoichiometry of the salt 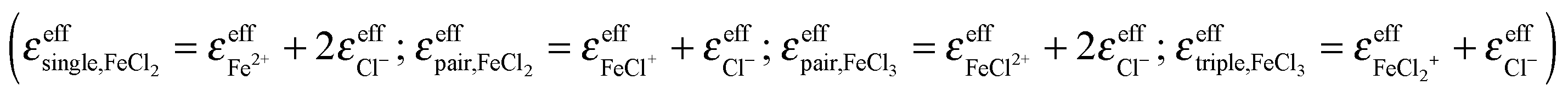 .
.
| K (M−1) | logK |
|
|---|---|---|
| FeCl2 | 0.13(1) | −0.89(4) |
| FeCl3 | 0.49(8) | −0.32(8) |
The extinction spectrum of Fe2+ + 2Cl− in Fig. 4A is dominated by one broad band at 150 cm−1 and the tail of a feature that peaks at >350 cm−1. The spectrum of FeCl+ + Cl− shows two resonances centered at 145 cm−1 and 230 cm−1, as well as a high frequency wing.
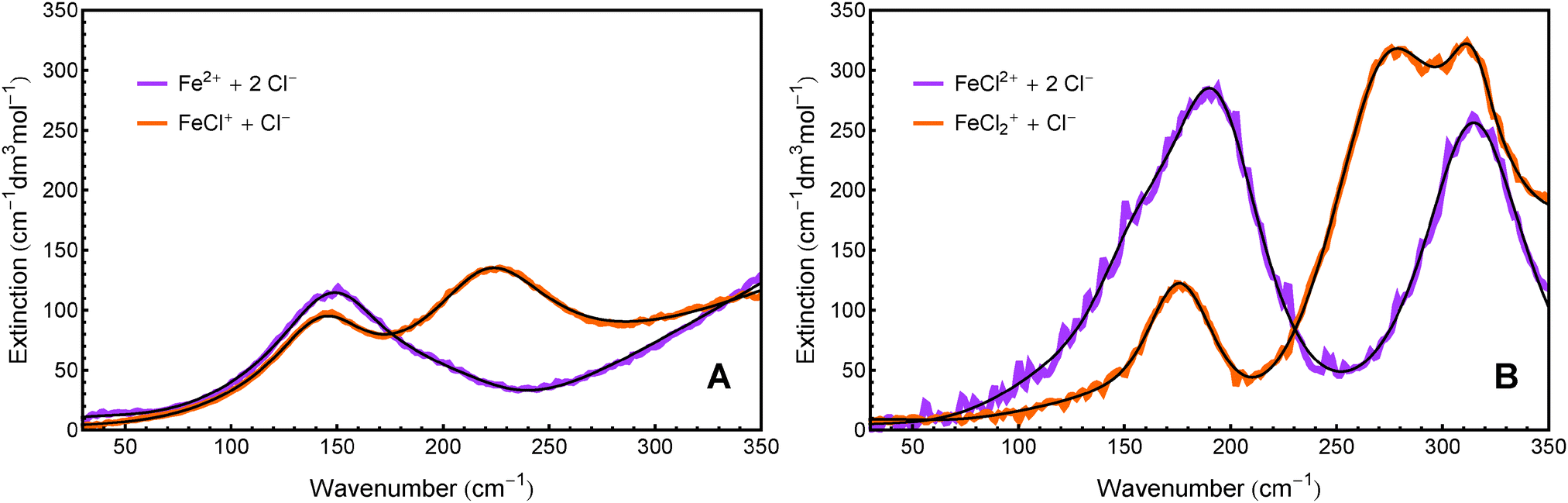 | ||
| Fig. 4 Molar extinction spectra of the respective two main components in the FeCl2 (A) and FeCl3 solutions (B), as obtained by fitting the score functions. The black lines represent the spectral fit described in Section 6. | ||
The extinction spectrum of FeCl2+ + 2Cl− in Fig. 4B displays two strong peaks at 190 cm−1 and 315 cm−1. The spectrum of FeCl2+ + Cl−, however, shows a clear resonance around 170 cm−1 and at least two overlapping spectral features between 250 cm−1 and 350 cm−1.
5 Global fit
In our previous studies, we have performed a global fit of the aqueous ionic spectra of MnCl2, NiCl2, MnBr2 and NiBr2 and their differences, in order to pin down precise peak positions of spectral features as well as to determine the effect of the ions on the water spectrum.52In the global fit, the anion and cation bands are modeled by a modified damped harmonic oscillator function:
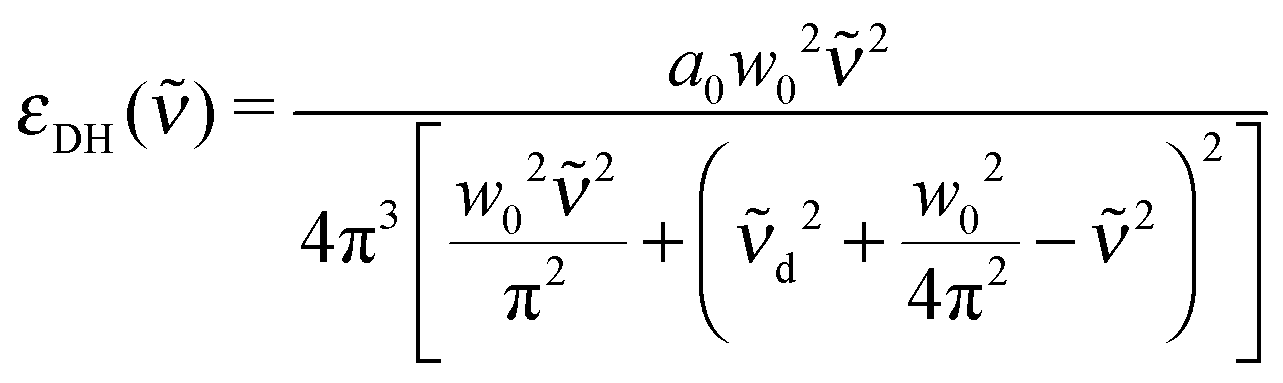 | (10) |
d the center frequency of the mode. The corresponding center frequency of an unperturbed Brownian harmonic oscillator is given as 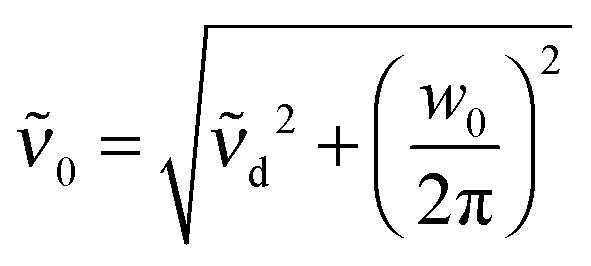 . The low and high frequency part of the spectra are modeled using the corresponding modes of the water spectrum52 scaled by factors nLF and nHF. In addition to this, a negative contribution is present in each spectrum (cf. Section 3.2), which can be modeled by the water extinction spectrum scaled by a factor nhydration.
. The low and high frequency part of the spectra are modeled using the corresponding modes of the water spectrum52 scaled by factors nLF and nHF. In addition to this, a negative contribution is present in each spectrum (cf. Section 3.2), which can be modeled by the water extinction spectrum scaled by a factor nhydration.
Here we extend the model to include the four spectra of FeCl2 and FeCl3. The inclusion of these in the global fit does not affect the previously determined parameters for the manganese and nickel bromide spectra significantly. Therefore the results of the bromide spectra are omitted here, and only the results for MnCl2 and NiCl2 are reported for comparison. The determined fit parameters are listed in Table 3.
d and 0 (cm−1), amplitude a0 (cm−1 dm3 mol−1) and linewidth w0 (cm−1) of the water mode (WM), the modes of the cationic complexes (CM) and the anion modes (AM), minimum number of affected water molecules nhyd, and scaling factors of the low and high frequency water modes nLF and nHF. The 2σ standard error is given in brackets in units of the last digit. Arrows indicate the restriction of a parameter to the same value for several bands. Arrows with an asterisk denote parameters, that have been fixed to the indicated value prior to fitting
| Mn2+ + 2Cl− | Ni2+ + 2Cl− | Fe2+ + 2Cl− | FeCl+ + Cl− | FeCl2+ + 2Cl− | FeCl2+ + Cl− | |
|---|---|---|---|---|---|---|
| n hyd | 14.5(5) | ← | ← | 7.3(6) | 21.4(7) | 16.9(5) |
| n LF | 17.0(5) | ← | ← | 8.4(7) | 23.2(7) | 20.3(4) |
| n HF | 20.0(5) | ← | 20.8(4) | 12.5(6) | 25.0(7) | 18.7(4) |
| WM | ||||||
|
d
|
117(2) | ← | ← | ← | ← | ← |
|
0
|
136(9) | ← | ← | ← | ← | ← |
| a 0 | 382(32) | ← | ← | 297(45) | 1072(92) | 781(68) |
| w | 426(19) | ← | ← | ← | ← | ← |
| CM 1 | ||||||
|
d
|
135(1) | 161(1) | 148(1) | 142(1) | 154(1) | 142(2) |
|
0
|
140(1) | 165(1) | 153(1) | 145(1) | 157(1) | 147(2) |
| a 0 | 1561(29) | 1513(30) | 1926(38) | 1080(56) | 2181(48) | 597(45) |
| w 0 | 231(1) | ← | ← | 195(7) | 205(1) | Mn2+← |
| CM 2 | ||||||
|
d
|
214(1) | 262(1) | 210(1) | 219(1) | 192(1) | 175(1) |
|
0
|
217(1) | 265(1) | 213(1) | 223(1) | 195(1) | 177(1) |
| a 0 | 555(13) | 218(5) | 535(24) | 1544(55) | 4142(39) | 2269(52) |
| w 0 | CM 1 ↑ | ← | ← | 248(5) | CM 1↑ | 158(3) |
| CM 3 | ||||||
|
d
|
358(3) | — | 0 | — | 313(1) | 271(1) |
|
0
|
360(3) | — | 37 | — | 315(1) | 273(1) |
| a 0 | 322(24) | — | 46(4) | — | 3643(25) | 3582(14) |
| w 0 | CM 1↑ | — | Mn2+← | — | CM 1↑ | Mn2+← |
| CM 4 | ||||||
|
d
|
— | — | — | — | — | 313(1) |
|
0
|
— | — | — | — | — | 315(1) |
| a 0 | — | — | — | — | — | 1482(15) |
| w 0 | — | — | — | — | — | 98(2) |
| AM | ||||||
|
d
|
184(1) | ← | ← | ←* | ←* | ←* |
|
0
|
188(1) | ← | ← | ←* | ←* | ←* |
| a 0 | 1343(12) | 1260(7) | 852(19) |
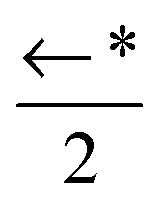
|
←* |
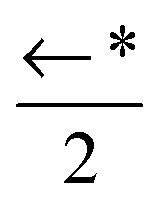
|
| w 0 | CM 1 ↑ | ← | ← | ←* | ←* | ←* |
We observe that several species have identical parameters, e.g. the linewidth of ionic bands, reducing the complexity of the fit. As an example for the various contributions to a fitting curve of one experimental spectrum, the spectrum of Fe2+ + 2Cl− is displayed in Fig. 5.
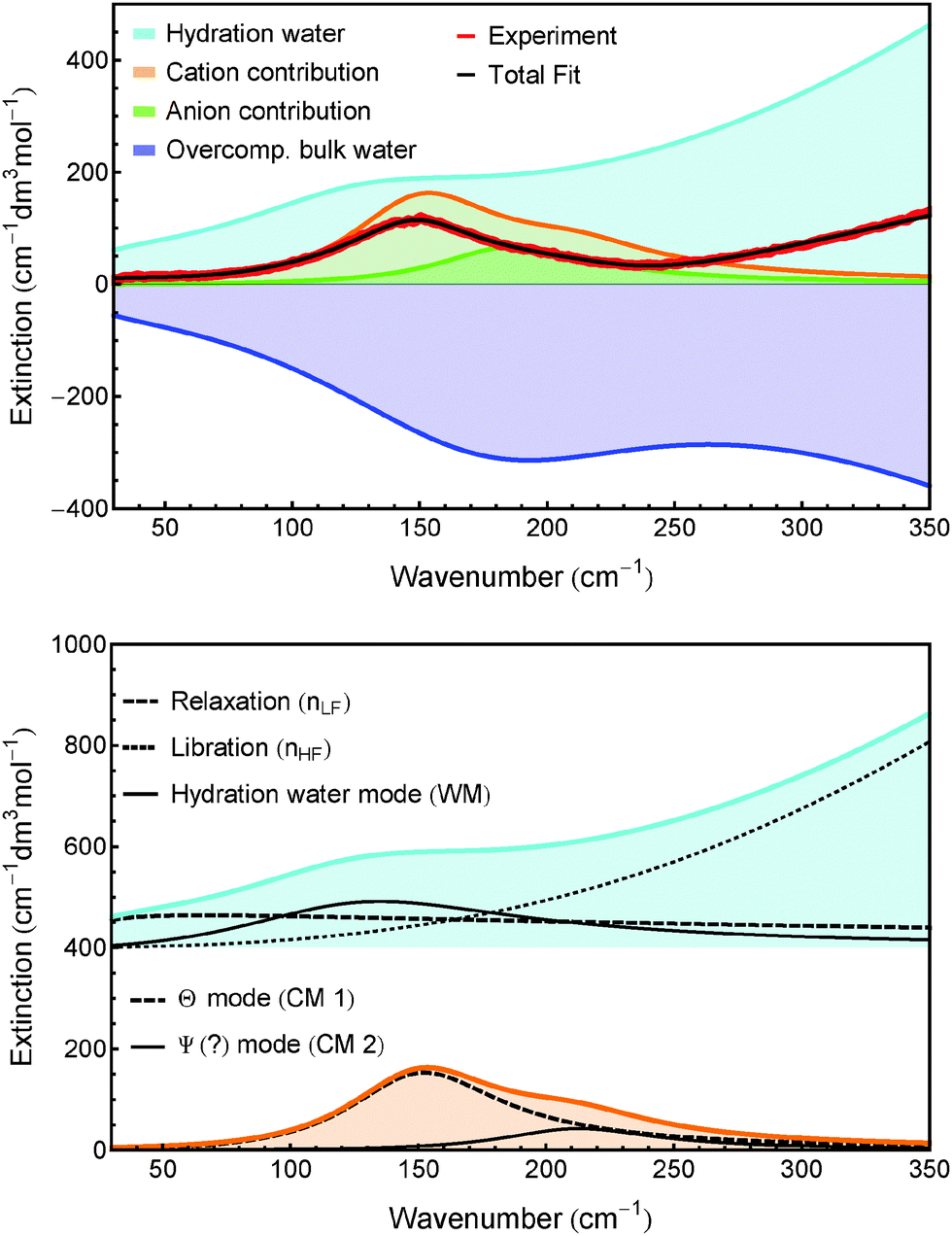 | ||
| Fig. 5 Top: Shown are the distinct contributions to the fit of the spectrum of Fe2+ + 2Cl−. Bottom: Hydration water spectrum (offset vertically for better display) and cation bands. nLF, nHF, WM, CM 1 and CM 2 are defined in the caption of Table 3. | ||
While trying to fit the spectra of the ion associates, it is possible that overlapping bands of anionic and cationic species cause an ambiguity in the assignment. Sharma et al. previously have overcome this challenge by including the difference spectra of each metal chloride and bromide into the fit, thereby uncoupling the anion from cation contributions.52 Since in the present work we lack the spectra of ferrous and ferric bromides for a direct comparison, we proceeded in an iterative approach: As a first step, we included only the spectrum of Fe2+ + 2Cl− into the fit of the manganese(II) and nickel(II) chloride and bromide spectra, thereby determining the Cl− mode. In a second step, this mode was then fixed in the spectra of FeCl+ + Cl−, FeCl2+ + 2Cl− and FeCl2+ + Cl− prior to inclusion of these spectra into the fit. After the global fit, the parameters of the chloride mode were fixed to the slightly changed new values. This was repeated iteratively until the change in the chloride fit parameters was smaller than the uncertainty. In Table 3, the fixed parameters are marked with an asterisk (*).
6 Discussion
6.1 Association constants
Prior to describing and comparing different values of ion-association constants, it is essential to emphasize that the results are method dependent. One reason for this is the different degrees of experimental sensitivity towards other ion associates besides contact ion pairs. In fact when it comes to ion association, the real number of different species is much larger than the simple one-step reaction scheme suggests. As described by Marcus and Hefter,59 the ions undergo a series of reaction steps from (a) solvent separated ion pairs, in which both ions still have their full hydration sphere, over (b) solvent shared ion pairs, in which the hydration spheres are overlapping, to (c) contact ion pairs, in which one ion penetrates the hydration sphere of the other. The system becomes even more complex if a third ion is included. For example, an equilibrium constant found by the anion exchange method33 may be smaller than the one determined using potentiometry,51 since the first is only sensitive towards contact ion pairs, while the latter is also sensitive to the presence of other ion pairs.Here we performed a principal component analysis of our concentration dependent THz/FIR absorption spectra of FeCl2 and FeCl3 solutions, and fitted the scores with association constants KFeCl2 and KFeCl3, respectively, as fitting parameters. The logarithm of the determined parameters KFeCl2 and KFeCl3 is −0.89(4) and −0.32(8), respectively. These numbers are well below the values determined by other methods ranging from −0.37 to −0.12 in the case of FeCl228,42,43 and 0.32 to 1.16 in the case of FeCl3.17,26,33,44,49,51 This indicates that the THz/FIR absorption is especially sensitive to the formation of contact ion pairs, while the other methods might be more susceptible to solvent shared and solvent separated ion pairs.
6.2 Band assignment
By exchanging a water ligand by chloride, the reduced mass for the Θ vibration of the octahedral Fe2+ complex is increased to 43.27 g mol−1 (Cl− axial) or to 45.08 g mol−1 (Cl− equatorial). In absence of other effects, this increase would cause a red-shift of the Θ mode by ca. 4–6% or 6–9 cm−1. In good agreement with this, we observe a red-shift of 6 cm−1 to 142 cm−1.
Moving from Fe2+ to Fe3+, the reduced mass of the monochloro complex remains the same, while the bond strength increases due to the higher charge of the metal center. Accordingly, the Θ mode is blue-shifted by 12 cm−1 to 154 cm−1. Exchanging another water molecule (in trans-position) by Cl−, the reduced mass for the Θ vibration is increased to 45.68 g mol−1 (Cl− axial) or to 48.98 g mol−1 (Cl− equatorial). In absence of any other effect, this increase is expected to lead to a red-shift of 3–4% or 5–6 cm−1. In fact, we observe a much larger red-shift of 12 cm−1. We attribute this to a weakening of the bond strength by the partial charge compensation of the metal center by the negatively charged ligands.
For Fe2+ we fitted a small amplitude mode at fixed center frequency of 0 cm−1, which we attribute to a relaxational process.
We observe only one band which can be attributed to the chloride anion in the spectra of Mn2+ + 2Cl−, Fe2+ + 2Cl− and Ni2+ + 2Cl− at 184 cm−1, which is close to the chloride band around 190 cm−1 found for other salts.7,8 Due to the increased complexity of the spectra of FeCl+ + Cl−, FeCl2+ + 2Cl− and FeCl2+ + Cl−, we decided to fix the values of the anion band to the values deduced for Fe2+ + 2Cl− prior to the fit.
The linewidth of the modes attributed to the chloro-complexes of Fe2+ and Fe3+ varies. The first and third band of FeCl2+ have the same width as the free ion bands (231 cm−1), while the width of the second band of FeCl+ is slightly larger (248 cm−1). For all the other bands we observe a smaller width, ranging from 98 cm−1 to 205 cm−1. Distinct linewidths indicate that these modes are either connected to a different set of thermal bath states, or connected to the same bath, but with a different coupling parameter.
At this point, we can only speculate about the underlying molecular mechanism. We propose that the width of modes of solvated ions depends on the librational motions of the surrounding water molecules. These librations act as a random force causing line broadening. According to calculations by Vila Verde et al., water molecules between two ions (in case of a solvent-shared ion pair) or close to their point of contact (in case of a contact ion pair), are slowed down cooperatively.61 We therefore interpret the reduced linewidth as a consequence of inhibited librational motion of water close to the ion associates.
6.3 Hydration water
The low frequency mode at 117 cm−1 (called WM in Table 3) is attributed to a hydration water mode. It is most likely either due to a shifted hindered translational mode of the first shell hydration water, or due to an increased line strength of a second shell mode, which is not IR active in a bulk water environment. In addition to this, there are high and low frequency components attributed to the relaxational and librational modes of hydration water, scaled by factors nLF and nHF. Finally, there is a negative contribution to the effective ionic extinction in the shape of the bulk water spectrum scaled by a factor nhyd. Since the relaxational and librational parts are compensated by nLF and nHF, the negative contribution is dominated by the shifted translational band of hydration water. The scaling factor nhyd provides an estimate of the minimum number of water molecules affected by the ions.For the extinction spectra of Mn2+, Fe2+ and Ni2+ we can use the same fitting parameters for nhydration and nLF, while the effect on the librational mode of water (nHF) is slightly higher for Fe2+ compared to Mn2+ and Ni2+. nhydration is in the range of 14–15. In a previous study we found that HCl affects ca. 5 water molecules and attributed this effect mainly to the anion.54 If we transfer this result to the present study, a minimum number of 4–5 water molecules are affected by the metal ions Mn2+, Fe2+ and Ni2+, which is in good agreement with a value of six water molecules of an octahedral geometry.62
For the ion-associate FeCl+ + Cl−, the value obtained for the minimum number of affected water molecules is nhydration = 7.3, which is distinctly lower than for all other species. Assuming again a number of 5 water molecules affected by Cl−, the chloro-complex affects only a minimum number of 2–3 water molecules. This can only be explained if both the ferrous and chloride ions lose part of their influence on hydration water upon ion pairing.
For FeCl2+ + 2Cl− we obtain a lower limit of nhydration = 21.4 affected water molecules, with similar values for the high and low frequency components (nLF = 23.2 and nHF = 25.0). Subtracting the proposed effect of the anions, we deduce that 11–12 water molecules are affected by FeCl2+. This is surprisingly large, especially in comparison to FeCl+, in which the Cl− ligand loses most of its effect on water upon ion pairing. We speculate that this indicates an effect beyond the first hydration shell of Fe3+.
The minimum number of affected water molecules (nhydration = 16.9) as well as the low and high frequency components (nLF = 20.3 and nHF = 18.7) are smaller in the case of FeCl2+ + Cl− compared to FeCl2+ + 2Cl−. This can be explained by a compensation, or an effective shielding, of the positive charge of the central metal ion by the two axial chloride ligands.
7 Quantitative analysis of FeCl2/FeCl3 mixtures
As described in the previous sections, the absorption characteristics of FeCl2 and FeCl3 are distinctly different in the THz/FIR region. Their spectral signatures can therefore be used to distinguish ferrous and ferric iron species in aqueous solution.Using Pitzer's equations58 and the complex formation constants KFeCl2 and KFeCl3, the distribution of different ions and ion complex species can be predicted for FeCl2 and FeCl3 solutions of any concentration and even for any mixture of both salts (cf. Section 5). For a given water concentration, the extinction spectrum of water and the extinction spectra of the different complex species displayed in Fig. 4 can be used to predict the total absorption spectrum of any solution of FeCl2 and/or FeCl3 according to the following equation:
| αtot = cwaterεwater + cFe2+(εFe2+ + 2εCl−) + cFeCl+(εFeCl+ + εCl−) + cFeCl2+ (εFeCl2+ + 2εCl−) + cFeCl2+(εFeCl2+ + εCl−) | (11) |
The concentrations cx can be calculated as functions of density ρ and initial salt concentrations 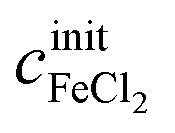 and
and 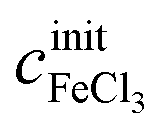 of a given solution. It is therefore possible to determine the composition of an unknown mixed solution of FeCl2 and FeCl3 by fitting eqn (11) to the experimental absorption spectrum, using
of a given solution. It is therefore possible to determine the composition of an unknown mixed solution of FeCl2 and FeCl3 by fitting eqn (11) to the experimental absorption spectrum, using 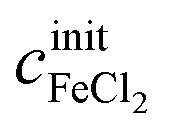 and
and 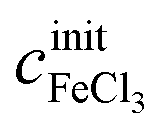 as fitting parameters.
as fitting parameters.
We have tested this method for a quantitative analysis of five sample solutions containing FeCl2 and FeCl3 in concentration ratios of 1 M/0 M, 0.75 M/0.25 M, 0.5 M/0.5 M, 0.25 M/0.75 M and 0 M/1 M. Each solution was acidified with 1 M HCl to prevent hydroxide formation and oxidation. The absorption spectra of these solutions after subtraction of the HCl contribution are presented in Fig. 6. The real concentrations (known from sample preparation) and measured concentrations (obtained from fitting) of FeCl2 and FeCl3 are plotted in Fig. 7.
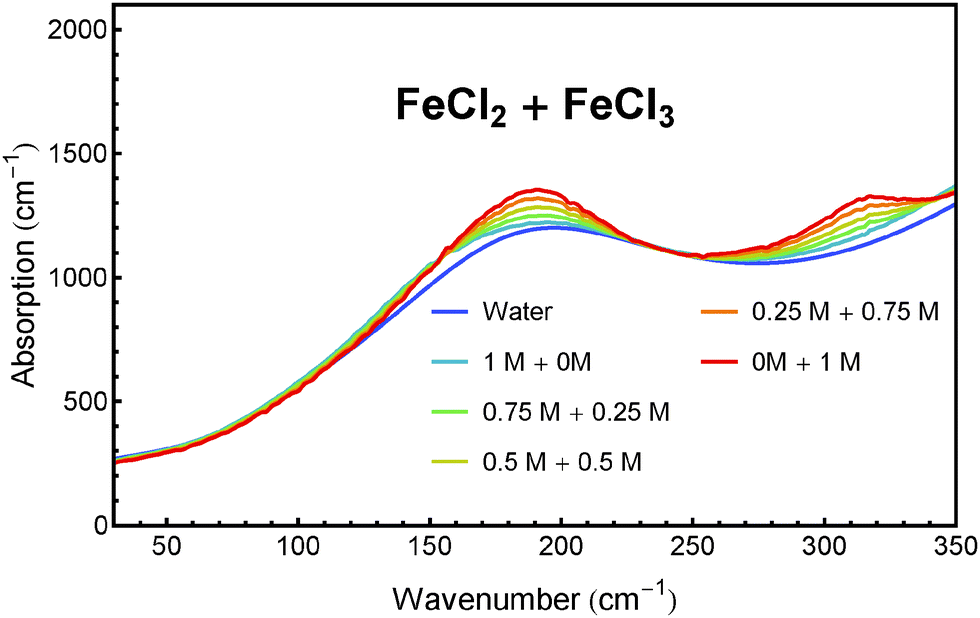 | ||
| Fig. 6 Determined THz/FIR absorption of water and mixed solutions of FeCl2 and FeCl3 at 20 °C. | ||
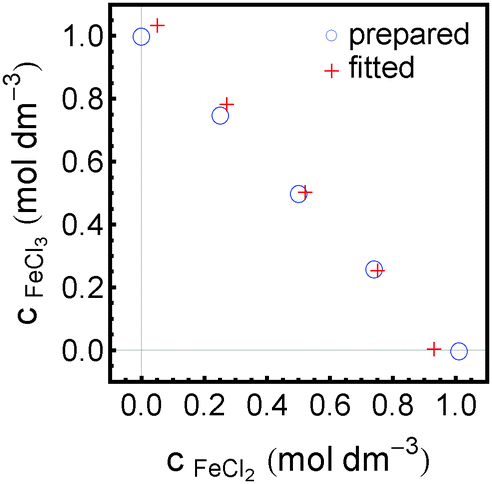 | ||
| Fig. 7 Comparison of concentrations as prepared and as spectroscopically retrieved of FeCl2 and FeCl3 in five mixed sample solutions. | ||
8 Summary and conclusion
We have measured the THz/FIR absorption spectra of acidified aqueous FeCl2 and FeCl3 solutions at concentrations of 0.5 to 3.5 M in a frequency range of 30–350 cm−1. We observed a non-linear concentration dependence of the effective ionic extinction for both salts. This can be rationalized in terms of the formation of chloro-complexes of Fe(II) and Fe(III), respectively. We were able to dissect the spectra of FeCl2 into the contributions of single ions and ion pair and the spectra of FeCl3 into the contributions of ion pair and ion triplet using principal component analysis. In addition, we were able to determine the ion association constants for both salts.The logarithm of the formation constant of the ion pair FeCl+ determined this way is −0.89(4); the logarithm of the formation constant of the ion triplet FeCl2+ is −0.32(8). Both values are considerably lower than the values found in literature.17,26,28,33,42–44,49,51 However, we have to keep in mind that our measurements are susceptible mainly to the formation of contact ion pairs, while other methods might also be sensitive towards the formation of solvent separated or solvent shared ion pairs.
From the PCA of the FeCl2 spectra we extracted the molar extinction spectra of Fe2+ + 2Cl− and FeCl+ + Cl−; from the PCA of the FeCl3 spectra we extracted the molar extinction spectra of FeCl2+ + 2Cl− and FeCl2+ + Cl−. We performed a global fit of all the extinction spectra and their differences, including the previous results for manganese(II) and nickel(II) chlorides and bromides.52
For all cations we observe a peak centered around 150 cm−1, which we assign to the Θ vibration of the octahedral aqua- and chloro-complexes. This peak shifts for different cations, which can be explained by changes in reduced mass and bond strength of the complexes.
Mn2+ + 2Cl−, Ni2+ + 2Cl− and Fe2+ + 2Cl− affect approximately the same amount of water, with a minimum number of affected water molecules of 14–15. The ion pair FeCl+ + Cl− affects only half as much water, which we attribute to the charge compensation of the paired ions. The ion pair FeCl2+ + 2Cl−, on the other hand, affects a much larger number of water molecules (ca. 21), which is an indication for a hydration effect extending beyond the first hydration shell of Fe3+. Again, the minimum number of affected water molecules is reduced upon association of another chloride ion to ca. 17 in the case of FeCl2+ + Cl−.
Furthermore, our analysis could be used successfully for a quantitative determination of concentrations of FeCl2 and FeCl3 in mixed solutions of total salt concentration of 1 M with errors of less than 0.08 M. Thus we have provided a proof of principle that this technique can be used as an analytical tool for highly concentrated salt solutions.
Acknowledgements
This work is supported by the Cluster of Excellence RESOLV (EXC1069) funded by the Deutsche Forschungsgemeinschaft.References
- H. Ohtaki and T. Radnai, Chem. Rev., 1993, 93, 1157 CrossRef CAS.
- I. A. Heisler and S. R. Meech, Science, 2010, 327, 857 CrossRef CAS PubMed.
- H. J. Bakker, Chem. Rev., 2008, 108, 1456 CrossRef CAS PubMed.
- Y. Marcus, Chem. Rev., 2009, 109, 1346 CrossRef CAS PubMed.
- A. S. Merbach, L. Helm and E. Toth, The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, John Wiley and Sons, Ltd, 2nd edn, 2013 Search PubMed.
- D. Laage and J. T. Hynes, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11167 CrossRef CAS PubMed.
- D. A. Schmidt, Ö. Birer, S. Funkner, B. Born, R. Gnanasekaran, G. Schwaab, D. M. Leitner and M. Havenith, J. Am. Chem. Soc., 2009, 131, 18512–18517 CrossRef CAS PubMed.
- S. Funkner, G. Niehues, D. A. Schmidt, M. Heyden, G. Schwaab, K. M. Callahan, D. J. Tobias and M. Havenith, J. Am. Chem. Soc., 2012, 134, 1030 CrossRef CAS PubMed.
- K. J. Tielrooij, N. Garcia-Araez, M. Bonn and H. J. Bakker, Science, 2010, 328, 1006 CrossRef CAS PubMed.
- A. W. Omta, M. F. Kropman, S. Woutersen and H. J. Bakker, Science, 2003, 301, 347 CrossRef CAS PubMed.
- P. Giaquinta, M. Tosi and N. March, Phys. Chem. Liq., 1983, 13, 1 CrossRef CAS.
- I. Persson, Pure Appl. Chem., 2010, 82, 1901 CrossRef CAS.
- J. E. Enderby, Annu. Rev. Phys. Chem., 1983, 34, 155–185 CrossRef CAS.
- F. W. Outten and E. C. Theil, Antioxid. Redox Signaling, 2009, 11, 1029 CrossRef CAS PubMed.
- T. S. Koskenkorva-Frank, G. Weiss and W. H. Koppenol, Free Radical Biol. Med., 2013, 65, 1174 CrossRef CAS PubMed.
- A. C. Lasaga, J. Geophys. Res., 1984, 89, 4009 CrossRef CAS.
- A. Stefánsson, K. H. Lemke and T. M. Seward, 15th ICPWS Proceedings, 2008.
- C. M. Johnson, J. L. Skulan, B. L. Beard, H. Sun, K. H. Nealson and P. S. Braterman, Earth Planet. Sci. Lett., 2002, 195, 141 CrossRef CAS.
- S. Schnell, S. Ratering and K.-H. Jansen, Environ. Sci. Technol., 1998, 32, 1530 CrossRef CAS.
- M. J. Verschoor and L. A. Molot, Limnol. Oceanogr.: Methods, 2013, 22, 113 CrossRef.
- P. Praus and M. Blahutová, Fresenius' J. Anal. Chem., 2001, 369, 466 CrossRef CAS PubMed.
- C. O. Moses, A. T. Herlihy, J. S. Herman and A. L. Mills, Talanta, 1988, 35, 15 CrossRef CAS PubMed.
- S. O. Pehkonen, Y. Erel and M. R. Hoffmann, Environ. Sci. Technol., 1992, 26, 1731 CrossRef CAS.
- D. Y. Yegorov, A. V. Kozlov, O. A. Azizova and Y. A. Vladimirov, Free Radical Biol. Med., 1993, 15, 565 CrossRef CAS PubMed.
- A. A. Ensafi, M. A. Chamjangali and H. R. Mansour, Anal. Sci., 2004, 20, 645 CrossRef CAS PubMed.
- W. Liu, B. Etschmann, J. Brugger, L. Spiccia, G. Foran and B. McInnes, Chem. Geol., 2006, 231, 326 CrossRef CAS.
- J. Kozak, N. Jodlowska, M. Kozak and P. Koscielniak, Anal. Chim. Acta, 2011, 702, 213 CrossRef CAS PubMed.
- R. Zhao and P. Pan, Can. J. Chem., 2001, 79, 131 CAS.
- L. V. Koplitz, D. S. McClure and D. A. Crerar, Inorg. Chem., 1987, 26, 308 CrossRef CAS.
- W. Liu, B. Etschmann, G. Foran, M. Shelley and J. Brugger, Am. Mineral., 2007, 92, 761 CrossRef CAS.
- G. S. Pokrovski, J. Schott, F. Farges and J.-L. Hazemann, Geochim. Cosmochim. Acta, 2003, 67, 3559 CrossRef CAS.
- G. A. Gamlen and D. O. Jordan, J. Chem. Soc., 1953, 1435 RSC.
- Y. Marcus, J. Inorg. Nucl. Chem., 1960, 12, 287 CrossRef CAS.
- C. L. Standley and R. F. Kruh, J. Chem. Phys., 1961, 34, 1450 CrossRef.
- M. D. Lind, J. Chem. Phys., 1967, 46, 2010 CrossRef CAS.
- M. Magini and T. Radnai, J. Chem. Phys., 1979, 71, 4255 CrossRef CAS.
- D. L. Wertz and M. D. Luter, Inorg. Chem., 1981, 20, 3118 CrossRef CAS.
- H. Kanno and J. Hirashi, J. Raman Spectrosc., 1982, 12, 224 CrossRef CAS.
- G. Giubileo, M. Magini, G. Licheri, G. Paschina, G. Piccaluga and G. Pinna, Inorg. Chem., 1983, 22, 1001 CrossRef CAS.
- K. Asakura, M. Nomura and H. Kuroda, Bull. Chem. Soc. Jpn., 1985, 58, 1543 CrossRef CAS.
- K. Murata, D. E. Irish and G. E. Toogood, Can. J. Chem., 1989, 67, 517 CrossRef CAS.
- C. A. Heinrich and T. M. Seward, Geochim. Cosmochim. Acta, 1990, 54, 2207 CrossRef CAS.
- D. A. Palmer and K. E. Hyde, Geochim. Cosmochim. Acta, 1993, 57, 1393 CrossRef CAS.
- E. Rabinowitch and W. H. Stockmayer, J. Am. Chem. Soc., 1942, 64, 335 CrossRef CAS.
- R. N. Heistand and A. Clearfield, J. Am. Chem. Soc., 1963, 85, 2566 CrossRef CAS.
- J. K. Rowley and N. Sutin, J. Phys. Chem., 1970, 74, 2043 CrossRef CAS.
- R. H. Byrne and D. R. Kester, J. Solution Chem., 1981, 10, 51 CrossRef CAS.
- G. R. Brubaker and R. A. Peterson, Inorg. Chim. Acta, 1989, 155, 139 CrossRef CAS.
- R. J. Millero, W. Yao and J. Aicher, Mar. Chem., 1995, 50, 21 CrossRef.
- R. H. Byrne and D. R. Kester, Mar. Chem., 1976, 4, 275 CrossRef CAS.
- B. R. Tagirov, I. I. Diakonov, O. A. Devina and A. V. Zotov, Chem. Geol., 2000, 162, 193 CrossRef CAS.
- V. Sharma, F. Böhm, G. Schwaab and M. Havenith, Phys. Chem. Chem. Phys., 2014, 16, 25101 RSC.
- V. Sharma, F. Böhm, M. Seitz, G. Schwaab and M. Havenith, Phys. Chem. Chem. Phys., 2013, 15, 8383 RSC.
- D. Decka, G. Schwaab and M. Havenith, Phys. Chem. Chem. Phys., 2015, 17, 8295 RSC.
- J. E. Bertie and Z. Lan, Appl. Spectrosc., 1996, 50, 1047 CrossRef CAS.
- R. N. Goldberg, R. L. Nuttall and B. R. Staples, J. Phys. Chem. Ref. Data, 1979, 8, 923 CrossRef CAS.
- W. Kangro and A. Groeneveld, Z. Phys. Chem., 1962, 32, 110 CrossRef.
- C. E. Harvie and J. h. Weare, Geochim. Cosmochim. Acta, 1980, 44, 981 CrossRef CAS.
- Y. Marcus and G. Hefter, Chem. Rev., 2006, 106, 4585 CrossRef CAS PubMed.
- A. Nitzan, Chemical Dynamics in Condensed Phases, Oxford University Press Inc., New York, 2006 Search PubMed.
- A. V. Verde and R. Lipowsky, J. Phys. Chem. B, 2013, 36, 10556 CrossRef PubMed.
- H. Ohtaki, T. Yamaguchi and M. Maeda, Bull. Chem. Soc. Jpn., 1976, 49, 701 CrossRef CAS.
| This journal is © the Owner Societies 2015 |