
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nonlinear optical response of photochromic azobenzene-functionalized self-assembled monolayers†
Michael
Schulze
*ab,
Manuel
Utecht
c,
Thomas
Moldt
b,
Daniel
Przyrembel
b,
Cornelius
Gahl
b,
Martin
Weinelt
b,
Peter
Saalfrank
c and
Petra
Tegeder
*a
aPhysikalisch-Chemisches Institut, Ruprecht-Karls-Universität Heidelberg, 69120 Heildeberg, Germany. E-mail: tegeder@uni-heidelberg.de; Fax: +49 6221 546199; Tel: +49 6221 548475
bFachbereich Physik, Freie Universität Berlin, 14195 Berlin, Germany
cInstitut für Chemie, Universität Potsdam, 14476 Potsdam, Germany
First published on 18th June 2015
Abstract
The combination of photochromic and nonlinear optical (NLO) properties of azobenzene-functionalized self-assembled monolayers (SAMs) constitutes an intriguing step towards novel photonic and optoelectronic devices. By utilizing the second-order NLO process of second harmonic generation (SHG), supported by density-functional theory and correlated wave function method calculations, we demonstrate that the photochromic interface provides the necessary prerequisites en route towards possible future technical applications: we find a high NLO contrast on the order of 16% between the switching states. These are furthermore accessible reversibly and with high efficiencies in terms of cross sections on the order of 10−18 cm2 for both photoisomerization reactions, i.e., drivable by means of low-power LED light sources. Finally, both photostationary states (PSSs) are thermally stable at ambient conditions.
1 Introduction
Molecular switches are molecules which can be switched reversibly between two or more (meta-) stable states. Prominent examples are azobenzenes1–4 as well as fulgides5 and fulgimides,6 spiropyrans7 and diarylethenes,8,9 which all undergo large changes in their conformation and/or the electronic structure upon switching. Regarding possible future technological applications of this class of molecules, promising concepts are conceivable if the molecular conversion can efficiently be triggered by an external stimulus. Among the various possible excitation mechanisms, such as using electric fields, electrons or mechanical force,3 light often provides the most convenient means due to availability and tuneability of sources.1,2,4,8–19 For instance the light-inducible ring-opening/-closure reaction in diaryethenes has been demonstrated to be accompanied by a switching of the conductivity through the molecules,9,20,21 which renders them prototypal optoelectronic molecular transistors. Equally intriguing is the utilization of molecular switches in all-optical signal processing units11,12 or for three-dimensional optical data storage modules.8,10,13,14Regarding the actual implementation of photonic and optoelectronic devices based on molecular switches, the common requirement for accessing the respective molecular functionality is an immobilization strategy which preserves the photochromism of the molecules. Among the various existing strategies regarding this goal,22 physisorbed systems are well-studied but of rather academic interest. In these systems the switching ability is often suppressed and very small cross sections (i.e., low switching efficiencies) have been observed.2,4,23 The more robust chemical immobilization of chromophore units into polymer matrices,8,24 Langmuir-25,26 or Langmuir–Blodgett-films27,28 might be a promising alternative. Additionally, incorporation of photochromic molecules into self-assembled monolayers (SAMs) has proven to preserve the switching ability of the molecules, if (a) the photochromic units are successfully electronically decoupled from the substrate using sufficiently long linker units and (b) steric hindrance17 and excitonic coupling between the chromophores29–31 can be mitigated. This has been achieved using on-surface synthesis routes and preparation strategies avoiding the formation of a close-packed chromophore layer.15,16,18,32 As shown in Fig. 1, in the present study we follow the latter approach by intermixing ‘empty’ spacers, i.e., linker units which do not carry a chromophore.
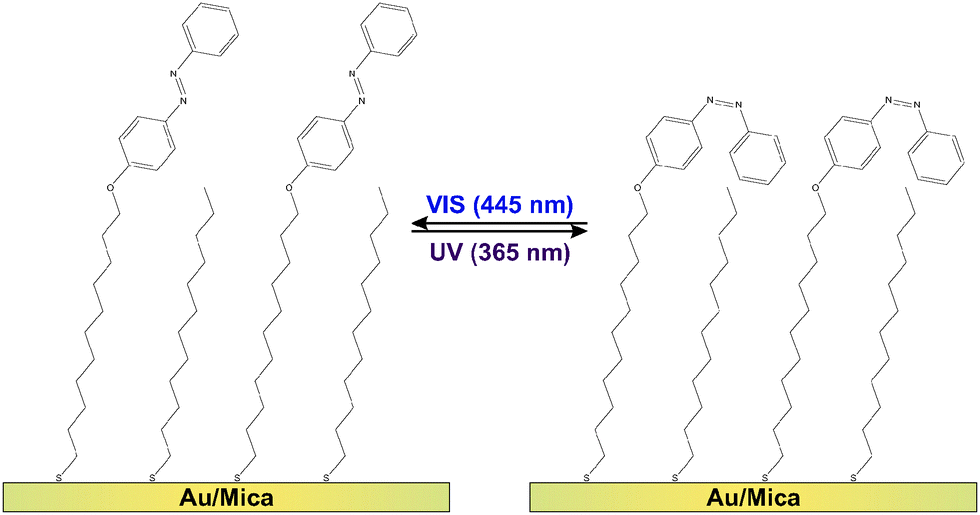 | ||
| Fig. 1 The diluted azobenzene SAM can be switched from the trans-PSS (left) to the cis-PSS (right) and vice versa, by illumination with UV and visible light at wavelengths of 365 nm and 445 nm, respectively. | ||
The azobenzene-functionalized SAMs are formed by immersing the Au/mica substrate into a methanolic solution containing a total concentration of 0.1 mM alkanethiols. The chromophore-functionalized 11-(4-(phenyldiazenyl)phenoxy)undecane-1-thiol (Az11) and the empty spacer unit 1-dodecanethiol statistically intermix at the surface yielding a chromophore packing density that is strongly dependent on the ratio between the two alkanethiols in the immersion solution. Details on the sample preparation and the verification of the chromophore packing densities by means of X-ray photoelectron spectroscopy (XPS), conducted on simultaneously prepared twin-samples, are described elsewhere.18 By following this preparation strategy the azobenzene chromophores can be sufficiently decoupled from their neighbors, allowing the photoinduced isomerization reactions between photostationary states (PSSs) enriched with azobenzene in its trans- and its cis-form, respectively (see Fig. 1).
In the present study, we access the switching state of the system via the interfacial nonlinear optical (NLO) response. This approach has already successfully applied for fulgimide-functionalized SAMs on Si(111). Thereby fulgimides undergo a reversible photoinduced ring-opening/closure reaction.19 Second harmonic generation (SHG) probes the second-order nonlinear susceptibility χ(2) of the sample. For the inversion-symmetric substrate, the second-order NLO response solely originates from the symmetry-breaking interface. The absence of an interfering bulk-contribution to the SHG signal results in a very high NLO contrast between the respective PSSs on the order of 16% for chromophore coverages between 18% and 84%. The identification of the observed contrast with the switching state of the sample is done based upon electronic structure calculations on the molecular geometries and hyperpolarizabilities β (the molecular equivalent to the macroscopic χ(2)), viz. by density functional theory (DFT) and correlated wave function method calculations. The agreement between experimental and theoretical results is sufficient to even conclude on a predominantly perpendicular interfacial molecular alignment of the chromophore in its trans-form. We furthermore determine the efficiency of the switching processes in terms of the cross sections. These are on the order of 10−18 cm2 for both isomerization reactions. The switching of the azobenzene SAM is fully reversible over many switching cycles and both PSSs are found to be thermally stable at ambient conditions. Hence, we possess a two-dimensional NLO switching system with high potential regarding emerging photonic and optoelectronic technologies.
2 Results and discussion
2.1 Photoinduced switching of the NLO response, coverage dependency
Fig. 2A shows the SHG signal intensity measured for an azobenzene-functionalized SAM with a chromophore coverage of 65 ± 5%, as a function of the alternate illumination using UV and visible light, indicated by the data marked in purple and blue, respectively.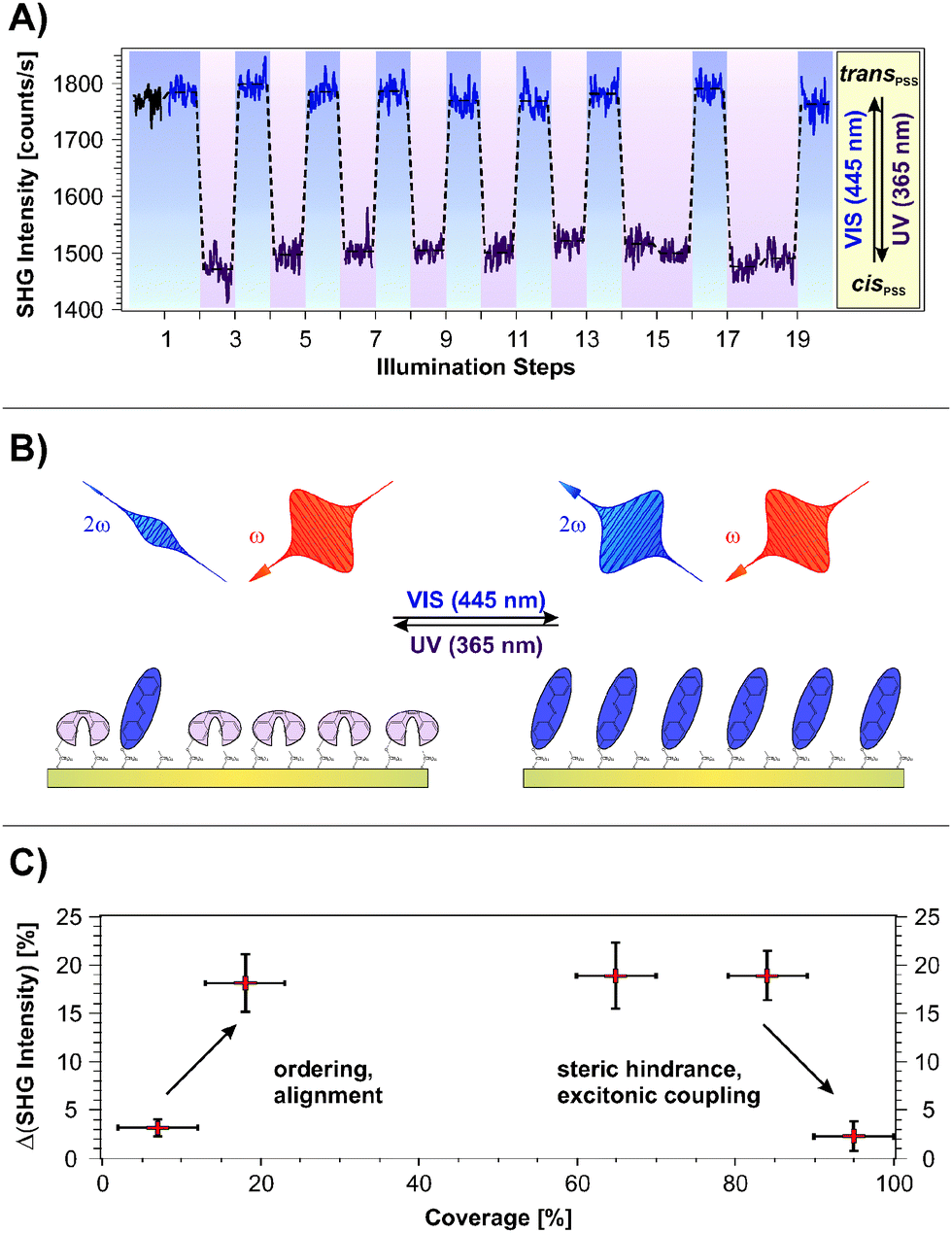 | ||
| Fig. 2 (A) SHG measurements on a sample with an azobenzene coverage of 65%. The data labeled in purple and blue denote measurements conducted subsequently to illumination with UV (365 nm, photon dose nUV = 1.3 × 1020 cm−2) and visible (445 nm, nvis = 4.4 × 1019 cm−2) light, respectively. The average SHG signal amplitude change of 16 ± 3% is attributed to the trans ↔ cis isomerization. (B) Scheme of the correlation between the NLO contrast measured in the SHG experiment and the rearrangement of the π-electron system during isomerization. (C) Change in the SHG signal intensity upon switching between the PSSs ΔISHG = (Icis − Itrans)/Itrans as a function of the chromophore coverage. | ||
Two clearly distinguishable SHG signal levels are observed: a lower one subsequent to light exposure at 365 nm and a higher level measured after illumination at 445 nm. These are reproducible over 20 illumination steps without any sign of fatigue, i.e., no decrease in the NLO contrast is observed.
The applied UV and visible light is suitable for addressing the pronounced S2 and S1 optical absorption bands of the trans- and the cis-form, respectively,1 and a switching of diluted azobenzene-functionalized alkanethiols on gold has been demonstrated.18 Therefore we assign the observed reversible, light-induced changes in the SHG signal amplitudes to the switching of the photochromic interface between the respective PSSs. Interestingly, the SHG signals of the trans-PSS and of the freshly prepared sample (i.e., containing only trans-isomers) are equal within the given experimental accuracy. The signal-to-noise ratio for the SHG signal amplitude is on the order of 10%. This result is thus in contrast to measurements on similar azobenzene-functionalized alkanethiols in solution, for which cis-fractions of 25 to 30% in the trans-PSS have been found.33 However, recent results point towards an energetic stabilization of the trans-chromophore in the SAM due to π-stacking of the elongated molecule which fosters the isomerization yield upon visible light absorption.18
The scheme depicted in Fig. 2B illustrates the correlation between the switching state and the NLO response measured in the SHG experiment. As we recently demonstrated,19 the SHG probe is highly sensitive to the degree of conjugation and the orientation of the π-electron system at the interface. Thus, we conclude for the rather densely packed sample that the high SHG signal intensity measured for the trans-PSS (using a p-polarized probe beam, vide infra) indicates a well-ordered and -aligned SAM in which the π-electron-system formed by the two phenyl rings is oriented predominantly perpendicular to the interface. For the bent cis-form this conjugation is interrupted and the lower SHG signal level is measured. As demonstrated in the following section, DFT and correlated wave function method calculations provide the suitable means to obtain an understanding of the correlation between the measured NLO responses and the electronic structures at the interface.
Fig. 2C depicts the measured NLO contrast ΔISHG = (Itrans − Icis)/Itrans as a function of the chromophore packing density in the range between 7% and 95%. The NLO contrast has been determined by conducting 20 illumination steps as shown in Fig. 2A for each sample and by subsequently averaging the data of the two respective SHG signal levels Itrans and Icis. Going from 7% to 18% chromophore coverage, a pronounced increase in the NLO contrast is observed. This increase can be attributed to the rising amount of switches contributing to the contrast. Also the expected ordering and aligning effect of the chromophores with decreasing free space per molecule is supposed to enhance the SHG signal intensity.34–36
For a wide range of coverages (20–85%) the SHG contrast remains constant, which is explained as follows. While the number of chromophores rises, also their average packing density increases and they orientate more upright. In principle this enhances the SHG contrast. However, switching is more and more suppressed due to increased steric hindrance and excitonic coupling. The consequence is a constant SHG contrast.
At highest coverages, finally, a pronounced decrease in the NLO contrast is observed which can to be attributed to excitonic coupling (i.e. the dissipation of an excitation among adjacent molecules due to aggregation, in this case before an isomerization can occur29–31) and steric hindrance. This contribution might play an increasingly important role, starting already at intermediate coverages. The sample with the chromophore concentration of 65 ± 5% has been chosen for more detailed investigations, as it exhibits the highest NLO contrast of all investigated samples. Additionally, a well-ordered molecular alignment can be expected for this rather densely packed SAM.
It has been shown that reversible photochemical reactions in polymers and Langmuir–Blodgett films containing photochromic dyes as well as in organic photochromic crystals resulted in switching of SHG signals. However, these approaches suffer from switching-induced irreversible changes in the initial chromophore alignment, which drastically limits the number of switching cycles.37–40 Comparing our results, the NLO contrast between the respective PSSs of 16%, with the before mentioned studies: in a photochrome-doped polymer thin film of a spiropyran and a furylfulgide a change of 15% and 60%, respectively, between the open and closed form have been found. But already after a few switching cycles the differences in the NLO response of both systems dropped below 10%.37 In aniline polycrystalline films photoinduced changes in the SHG intensity of around 20% have been obtained.38 We studied recently the reversible photoswitching of the NLO response of fulgimide-functionalized SAMs on Si(111) and observed an intensity change of 20%.19 Further molecular engineering of the photochromic switch may even increase the NLO contrast upon switching.
2.2 Calculated molecular geometries and hyperpolarizabilities, polarization-resolved SHG
In order to verify the aforementioned interpretation on the observed NLO responses of the two PSSs of the photochromic interface, we performed electronic structure calculations of the trans- and the cis-form of the Az11 molecule, using DFT as well as correlated wave function methods (for details of the computational methods, see the ESI†).In a first step, hybrid density functional theory on the B3LYP/6-311++G** level was utilized to optimize the structures of the molecular models.41–43 We found distinct minima for both configurations of the azobenzene-functionalized alkanethiol. The derived structures are shown in Fig. 3. The coordinate system is chosen such that the oxygen and the para carbon atom of the ‘lower’ phenyl ring of the trans-isomer span the z-axis and ‘lower’ phenyl-ring defines the xz-plane. This alignment results in a tilt angle of the alkyl chain with respect to the surface normal of 24° which does not reflect the exact tilting of the chromophores but gives a reasonable description of their average orientation in the SAM and allows for a descriptive discussion of the obtained results.
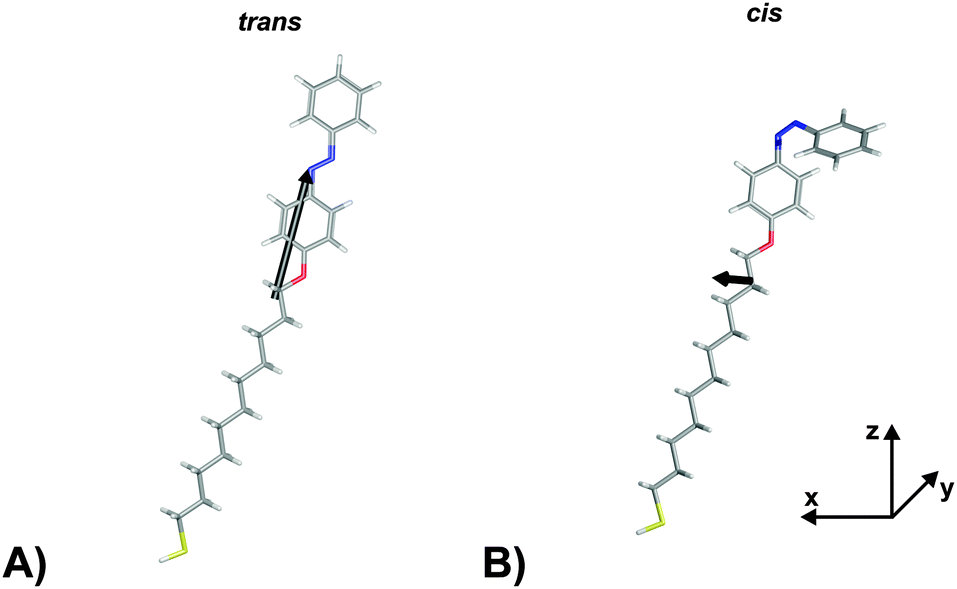 | ||
| Fig. 3 Hyperpolarizabilities of the Az11 (A) trans- and (B) cis-form models studied in this work, optimized at the B3LYP/6-311++G** level of theory. The coordinate system is chosen such that the oxygen and the para carbon atom of the ‘lower’ phenyl ring of the trans-isomer span the z-axis and its ‘lower’ phenyl-ring defines the xz-plane. Carbon, hydrogen, nitrogen, oxygen and sulfur atoms are gray, white, blue, red and yellow, respectively. Also indicated in black are the ‘hyperpolarizability vectors’ β = (βx, βy, βz) according to eqn (1), arbitrarily originating in the center of mass of the respective molecule. | ||
In a second step, the dynamical hyperpolarizabilities β (2ω; ω, ω) (the first hyperpolarizability β is the molecular equivalent to the macroscopic second-order nonlinear susceptibility χ(2)) at frequencies ω = 2πc/λ corresponding to an excitation wavelength of λ = 800 nm as adopted in the SHG measurements were calculated for both molecular configurations. The dynamic hyperpolarizabilities were determined at a MP2 (Møller–Plesset perturbation theory at second order) level at the B3LYP geometries, according to an established procedure44 (see the ESI† for further details). The NLO response can be analyzed by means of the averaged hyperpolarizability β0, which is defined as:45
| β0 = (βx2 + βy2 + βz2)½ | (1) |
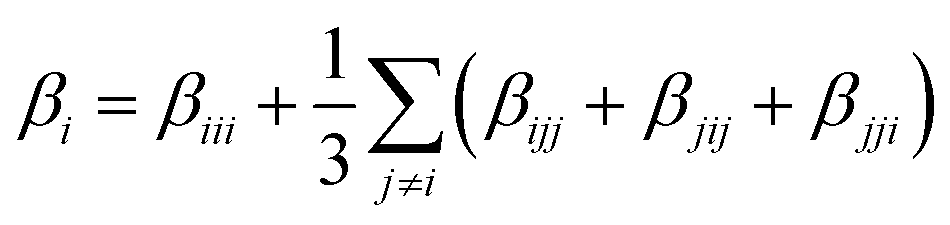 | (2) |
The components of the vector β = (βx, βy, βz) are shown in Table 1. The underlying tensor elements βjji are subject to the ESI.† Another profound way to distinguish between trans- and cis-isomers is to compare their hyperpolarizabilities orthogonal to the one parallel to the surface. The corresponding asymmetry value a is expressed by:
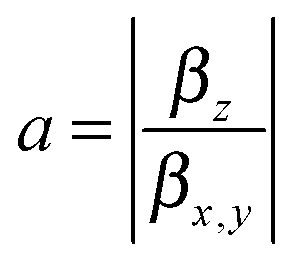 | (3) |
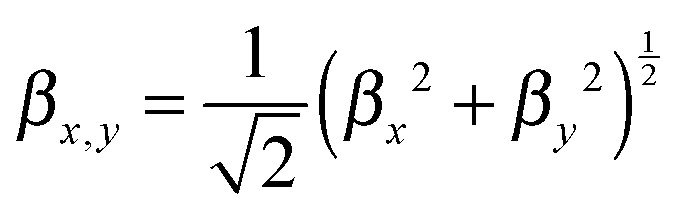
| Form | β 0 | β x | β y | β z | a | μ |
|---|---|---|---|---|---|---|
| a All values given in atomic units, where 1 au for β is 8.641 × 10−33 esu or 3.62 × 10−42 m4 V−1 and 1 au for μ is 2.54 Db or 8.478 × 10−30 Asm and a is dimensionless. | ||||||
| trans | 7846 | −1887 | 33 | 7607 | 5.70 | 1.02 |
| cis | 1825 | 1452 | −996 | 842 | 0.68 | 2.62 |
The computational results shown in Table 1 reveal pronounced changes of β in the isomerization reaction. First of all, the average hyperpolarizability β0 reduces by approximately a factor of four when switching from the elongated trans- to the bent cis-form. Second, the hyperpolarizability vector shown in Fig. 3 rotates from almost parallel to the alkanethiol (i.e., rather perpendicular to the interface) towards a direction with significant contribution in y, as the βz-component reduces significantly while a βy-component emerges for the cis-isomer due to the realignment of the (formerly interface-averted) ‘upper’ phenyl-ring. The asymmetry value a shows an equivalent picture. It is 0.68 for the cis-form, which means that the hyperpolarizabilities parallel and orthogonal to the surface exhibit similar absolute values. In contrast, a value of 5.70 for the trans-isomer expresses a clear dominance of the hyperpolarizability in z over the one in x or y.
A quantitative comparison of the calculated molecular first hyperpolarizabilities to the measured macroscopic second-order nonlinear susceptibility is generally difficult due to uncertainties in both, the experimental and the theoretical considerations. Regarding the experimental results, the linker system apparently adds an equal unknown background contribution to the SHG signal intensities measured for both PSSs and therefore reduces the apparent NLO contrast. Furthermore, as the phase information46 is not accessed in our experiment, a negative interference between the chromophore and background contributions cannot be excluded. However, due to the low film thickness we expect only a small phase shift, which does not lead to destructive interference.47
Considering the computational results, influences of the substrate and of the aggregation between adjacent molecules need to be discussed. As recently demonstrated by Nénon et al. in an extensive computational approach, the presence of the surface potentially has a non-neglectable influence on the absolute values of the calculated hyperpolarizabilities, depending primarily on the chromophore and the applied linker chain.48 Fortunately, the surface-induced effect diminishes for increasing dimension of the linker unit and a substantial influence has solely been found for chromophores with an ionic character. Furthermore, the preservation of the NLO contrast upon chemisorption has been found for each of the systems they investigated. As the distance between surface and chromophore in the SAM investigated here is large and Az11 molecules show no ionic character, we assume that the presence of the surface induces no significant alterations to our calculated hyperpolarizabilities.
The influence of adjacent molecules is difficult to access due to the enormous computational costs. Both H- and J-aggregation found in densely packed azobenzene SAMs18,29 can be assumed to influence the hyperpolarizability depending on the intermolecular distance between adjacent chromophores which thus highly depends on the packing density. Nevertheless, the qualitative agreement between experiment and theory remains and the calculations nicely confirm the pronounced differences in the hyperpolarizability of the isomers and explain the strong decrease of the NLO response upon switching from the elongated trans- to the bent cis-form observed in the SHG experiments.
Finally, we conducted polarization-resolved measurements to resolve the susceptibility of the photochromic interface perpendicular and parallel to the surface by varying the polarization of the probe beam.17 The results are shown in Fig. 4A and B, respectively. As depicted in the respective schemes in Fig. 4C and D, the p-polarized beam possesses an electric field-vector component perpendicular to the interface and is thus particularly sensitive to the trans-form of the chromophore with its π-electron system elongated predominantly along the z-axis. In contrast, in the s-polarization the sample is probed solely parallel to the surface. The polarization of the outgoing SHG beam is additionally analyzed, in order to access different elements of the susceptibility tensor.
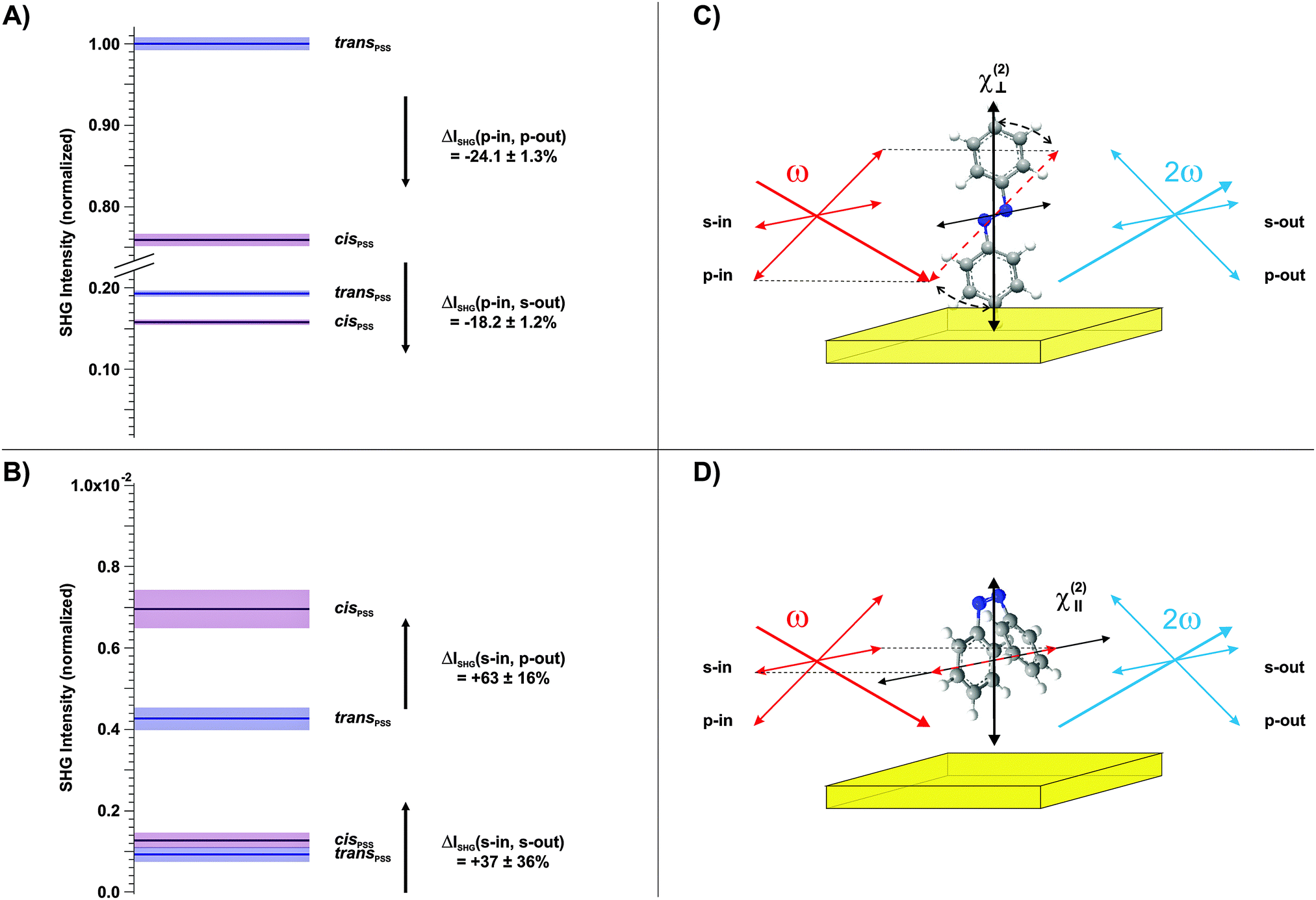 | ||
| Fig. 4 Polarization-resolved SHG data on the Az11 SAM (65% coverage) obtained using (A) a p-polarized and (B) an s-polarized probe beam. The outgoing SHG polarization is analyzed, as well. Results measured for the trans- and the cis-PSS are indicated in blue and purple, respectively. The black arrows indicate the SHG signal changes in the trans → cis-isomerization and the respective relative signal changes ΔISHG = (Icis − Itrans)/Itrans for each of the four polarization combinations is stated. The colored bars indicate the experimental uncertainty. Note that the intensity axis in (B) is scaled by a factor of 100 with respect to the axis in (A). The schemes in (C) and (D) illustrate the experimental finding that using a p- or an s-polarized probe beam results in higher SHG signal intensities for the elongated trans- or the bent cis-form, respectively, due to the different orientations of the π-electron systems for both isomers. | ||
Three observations can be derived from this data set. (1) The susceptibility perpendicular to the interface is significantly larger for the trans-isomer (Fig. 4A). (2) The susceptibility parallel to the interface is considerably larger for the cis-isomer (Fig. 4B). (3) The highest SHG signal intensities are measured for the p-in/p-out polarization combination, followed by p-in/s-out, s-in/p-out and finally s-in/s-out. As discussed above, a quantitative comparison to the calculated hyperpolarizabilities is difficult. However, findings (1) and (2) are in good agreement with the calculated rotation of the hyperpolarizability vector from mainly perpendicular to the surface towards an orientation which has significant parallel components. Finally, the increasing amplitude of the SHG signal intensities with increasing contribution of the dominating tensor elements with z-components (point (3)) is in agreement with the computational results (see the ESI†).
2.3 Switching efficiency: cross sections
An important property of a photoswitchable interface regarding a possible technological application are the efficiencies of the isomerization reactions. The possibility to access the system's functionality using low-power LED light sources is desired. In order to obtain a measure for the efficiency, we performed illumination experiments applying very low photon doses with excitation intensities on the order of 1 mW cm−2. In doing so, the photochromic interface was switched stepwise from the cis- to the trans-PSS and vice versa while the intermediate NLO responses were measured. The results are shown in Fig. 5A and B, respectively. Assuming a linear correlation between the relative number of switched chromophores and the change in the SHG signal level ΔISHG, the cross sections σ of the isomerization reactions can be obtained by fitting the data according to the relation ΔISHG = ISHG,PSS·[1 − exp(−σ·nP)], where nP is the applied photon dose.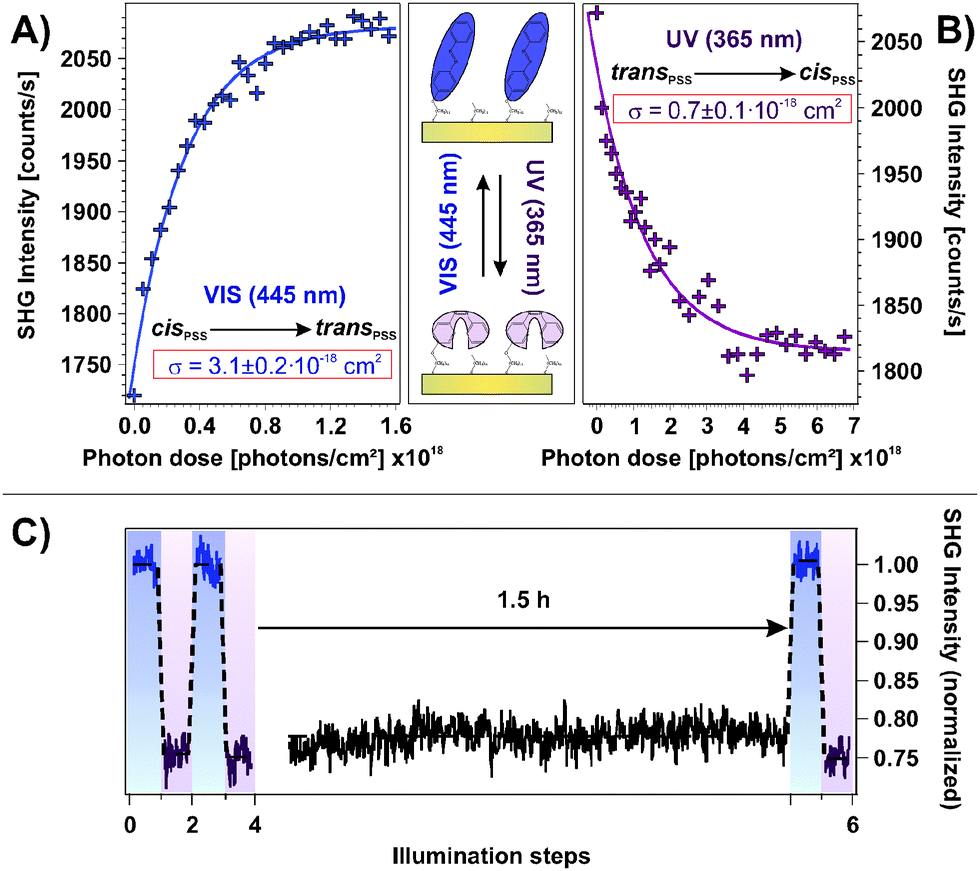 | ||
| Fig. 5 Cross sections of (A) the cis → trans and (B) the trans → cis isomerization reactions of the Az11 SAM (65% coverage) were determined by assuming a linear correlation between the number of switched chromophores and the illumination-induced changes in the SHG signal intensity ΔISHG, yielding mono-exponential fits to the intermediate signal levels. For these experiments low excitation intensities on the order of 1 mW cm−2 have been used. (C) To exclude a thermally assisted back-reaction, illumination experiments with a waiting time of 1.5 h between UV light exposure and measurement have been conducted. On this time scale, the cis-PSS is thermally stable at ambient conditions. Results measured after UV and visible light exposure are indicated in blue and purple, respectively. | ||
As shown in Fig. 5, the assumed mono-exponential behavior describes both isomerization reactions well and the derived cross sections are σcis→trans = 3.1 ± 0.2 × 10−18 cm2 and σtrans→cis = 0.7 ± 0.1 × 10−18 cm2 for the visible and the UV light induced switching process, respectively. These observations allow for two important conclusions. First, the possibility to describe the observed processes with a mono-exponential relation strongly indicates that cooperative effects on the photoisomerization can be neglected. Second, both isomerization reactions are very efficient which indicates unhindered switching triggered by the direct intramolecular photoexcitation.
The latter is best understood by means of a comparison of the derived cross sections to those observed for related systems. On the one hand, for the azobenzene derivative tetra-tert-butyl-azobenzene (TBA) physisorbed to the Au(111) surface, a substrate-mediated isomerization process has been observed to occur with cross section lower by four orders of magnitude.2 On the other hand, for different azobenzene-functionalized SAMs using large triazatriangulenium (TATA) platforms49 or bulky tripodal linker systems16 to prevent an inter-chromophore aggregation, cross sections on the same order of magnitude have been found as in the present study.
It is remarkable, that the cross section of the UV-induced trans → cis reaction is smaller than for the reverse process, triggered by visible light. This is in contrast to the findings by Wagner et al. for a similar system16 and contradicts the expectations derived from the pronounced UV absorption band of the trans-form in opposition to the weak visible light absorbance of the cis-isomer found for azobenzene in solution.1 However, this finding might be explained by the pronounced hypsochromic shift of the UV absorption band of the trans-form of the SAM investigated here. This shift is a function of the chromophore coverage and has thus been identified to originate from the π-system interaction between nearest neighbors.18 It simply reduces the absorbance around 365 nm. Alternatively, as has also been hypothesized, solely the chromophores with largest distances to neighboring molecules in the trans-form in the densely packed SAM are capable of absorbing at the applied wavelength and of switching to the bent cis-isomer. Both effects would explain the unexpected relation between the observed isomerization cross sections.
To ensure that the cis → trans back-reaction is not thermally assisted, we conducted an illumination experiment similar to the ones shown in Fig. 2A in which a waiting time of 1.5 h between UV light exposure and measurement has been applied, as shown in Fig. 5C. We found no sign of any thermal back-reaction at ambient conditions. Hence, apparently a large thermal activation barrier for the cis → trans isomerization of the azobenzene chromophore in the SAM exists.
3 Conclusions
We conducted SHG experiments and performed DFT and correlated wave function method calculations, to access the NLO response of a photochromic azobenzene-functionalized SAM. Regarding this NLO response, two different states of the sample were found to be addressable by illumination using UV and visible light. These states were identified with the trans- and the cis-PSS, respectively. The NLO contrast measured in terms of the SHG signal change between the isomers is 16%. Moreover, the switching process is fully reversible as demonstrated over 20 illumination steps and the system shows no illumination- or laser-aging, i.e. neither writing- nor reading-induced fatigue.A further, thorough investigation of the isomerization kinetics using very low excitation intensities on the order of 1 mW cm−2 did not only reveal the high efficiencies of both reactions in terms of cross sections on the order of 10−18 cm2. These measurements also demonstrated that the NLO contrast is sufficient to resolve intermediate signal levels, i.e., besides the two PSSs also intermediate switching states are accessible. Finally, the thermal stability of the cis-PSS at ambient conditions has been proven by introducing waiting times on the timescale of hours between illumination and measurement.
In summary, the present work demonstrates the intriguing potential of combining the photochromic with the nonlinear optical properties of a chromophore-functionalized interface regarding a possible future application in novel photonic and optoelectronic devices. This has been done by demonstrating that the investigated system provides the desired properties in this regard: (1) a high contrast between the PSSs and a full reversibility of the isomerization reactions, (2) high switching efficiencies, i.e., switching processes which can be driven using low-power LED light sources and (3) thermal stability at ambient conditions. The subsequent steps in this concern are (a) to also consider intermolecular interactions between adjacent chromophores which might pave the way for a theoretical modeling of the SHG signal and thus for a detailed insight on the geometric molecular arrangement and (b) to find chemical modifications to the chromophore which might increase the NLO contrast even further.
4 Experimental section
SHG experiment
A 300 kHz regenerative amplified Ti:sapphire laser system produces pulses of 50 fs duration centered at a wavelength of 800 nm with a spectral width of 30 nm. By combining a Glan–Thomson polarizer with an achromatic half-wave plate the pulse energy is set to 133 nJ. The applied polarization combination is ‘p-in’/‘all-out’ with the sample mounted under an angle of 45° with respect to the incident probe beam, where ‘in’ and ‘out’ denote the polarization of the 800 nm fundamental and the 400 nm SHG beam, respectively. The SHG beam is focused into a monochromator and detected by a photo-multiplier tube. The analogous signal is converted by a counting unit and acquired using a PCI counting device. All SHG experiments were carried out at ambient conditions.Illumination
Samples were illuminated using monochromatic LEDs emitting at 365 nm and 445 nm to drive the trans → cis and the cis → trans isomerization, respectively. Spot sizes were 4 × 5 mm2 and 2 × 10 mm2 for UV and visible diodes, respectively. Samples were illuminated for 10 minutes at normal incidence, applying UV and visible powers of 23.2 mW and 6.6 mW, yielding photon doses of 1.3 × 1020 cm−2 and 4.4 × 1019 cm−2, respectively, to ensure that the respective photo stationary states are reached. Cross sections were determined with UV and visible powers of 1.44 mW and 0.48 mW yielding photon fluxes of 1.3 × 1016 s−1 cm−2 and 5.4 × 1016 s−1 cm−2, respectively.Computational methods
Density functional theory (B3LYP), Møller–Plesset perturbation theory (MP2) and coupled perturbed Hartree Fock calculations were used in the GAUSSIAN09 program package.50 For details see the ESI.†Acknowledgements
Funding by the Deutsche Forschungsgemeinschaft through the collaborative research center Sfb 658 is gratefully acknowledge.References
- T. Nägele, R. Hoche, W. Zinth and J. Wachtveitl, Chem. Phys. Lett., 1997, 272, 489–495 CrossRef.
- S. Hagen, P. Kate, F. Leyssner, D. Nandi, M. Wolf and P. Tegeder, J. Chem. Phys., 2008, 129, 1–8 CrossRef PubMed.
- K. Morgenstern, Prog. Surf. Sci., 2011, 86, 115–161 CrossRef CAS PubMed.
- C. Bronner, B. Priewisch, K. Rück-Braun and P. Tegeder, J. Phys. Chem. C, 2013, 117, 27031–27038 CAS.
- H. Stobbe, Ber. Dtsch. Chem. Ges., 1905, 38, 3673–3682 CrossRef CAS PubMed.
- H. G. Heller, K. Koh, C. Elliot and J. Whittall, Mol. Cryst. Liq. Cryst., 1994, 246, 79–86 CrossRef CAS PubMed.
- A. Plaquet, M. Guillaume, B. Champagne, F. Castet, L. Ducasse, J.-L. Pozzo and V. Rodriguez, Phys. Chem. Chem. Phys., 2008, 10, 6223–6232 RSC.
- A. Toriumi, S. Kawata and M. Gu, Opt. Lett., 1998, 23, 1924–1926 CrossRef CAS.
- A. C. Whalley, M. L. Steigerwald, X. Guo and C. Nuckolls, J. Am. Chem. Soc., 2007, 129, 12590–12591 CrossRef CAS PubMed.
- S. Kawata and Y. Kawata, Chem. Rev., 2000, 100, 1777–1788 CrossRef CAS PubMed.
- F. M. Raymo and S. Giordani, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4941–4944 CrossRef CAS.
- S. D. Straight, P. A. Liddell, Y. Terazono, T. A. Moore, A. L. Moore and D. Gust, Adv. Funct. Mater., 2007, 17, 777–785 CrossRef CAS PubMed.
- Y. C. Liang, A. S. Dvornikov and P. M. Rentzepis, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8109–8112 CrossRef CAS PubMed.
- E. Walker and P. M. Rentzepis, Nat. Photonics, 2008, 2, 406–408 CrossRef CAS PubMed.
- K. Rück-Braun, M. A. Petersen, F. Michalik, A. Hebert, D. Przyrembel, C. Weber, S. A. Ahmed, S. Kowarik and M. Weinelt, Langmuir, 2013, 29, 11758–11769 CrossRef PubMed.
- S. Wagner, F. Leyssner, C. Kördel, S. Zarwell, R. Schmidt, M. Weinelt, K. Rück-Braun, M. Wolf and P. Tegeder, Phys. Chem. Chem. Phys., 2009, 11, 6242–6248 RSC.
- D. T. Valley, M. Onstott, S. Malyk and A. V. Benderskii, Langmuir, 2013, 29, 11623–11631 CrossRef CAS PubMed.
- T. Moldt, D. Brete, D. Przyrembel, J. R. Goldman, R. Klajn, C. Gahl and M. Weinelt, Langmuir, 2015, 31, 1048–1057 CrossRef CAS PubMed.
- M. Schulze, M. Utecht, A. Herbert, K. Rück-Braun, P. Saalfrank and P. Tegeder, J. Phys. Chem. Lett., 2015, 6, 505–509 CrossRef CAS.
- Y. Kim, T. J. Hellmuth, D. Sysoiev, F. Pauly, T. Pietsch, J. Wolf, A. Erbe, T. Huhn, U. Groth, U. E. Steiner and E. Scheer, Nano Lett., 2012, 12, 3736–3742 CrossRef CAS PubMed.
- T. Sendler, K. Luka-Guth, M. Wieser, Lokamani, J. Wolf, M. Helm, S. Gemming, J. Kerbusch, E. Scheer, T. Huhn and A. Erbe, Adv. Sci., 2015, 2, 1500017 Search PubMed.
- J. S. Prauzner-Bechcicki, S. Godlewski and M. Szymonski, Phys. Status Solidi A, 2012, 209, 603–612 CrossRef CAS PubMed.
- P. Tegeder, J. Phys.: Condens. Matter, 2012, 24, 1–34 CrossRef PubMed.
- R. Loucif-Saïbi, K. Nakatani, J. A. Delaire, M. Dumont and Z. Sekkat, Chem. Mater., 1993, 5, 229–236 CrossRef.
- X. Zhuang, H. S. Lackritz and Y. R. Shen, Chem. Phys. Lett., 1995, 246, 279–284 CrossRef CAS.
- C. Manzo, A. Savoia, D. Paparo and L. Marrucci, Mol. Cryst. Liq. Cryst., 2006, 454, 91–100 Search PubMed.
- G. G. Roberts, Adv. Phys., 1985, 34, 475–512 CrossRef CAS PubMed.
- T. Seki, M. Sakuragi, Y. Kawanishi, Y. Suzuki, T. Tamaki, R. i. Fukuda and K. Ichimura, Langmuir, 1993, 9, 211–218 CrossRef CAS.
- C. Gahl, R. Schmidt, D. Brete, E. R. McNellis, W. Freyer, R. Carley, K. Reuter and M. Weinelt, J. Am. Chem. Soc., 2010, 132, 1831–1838 CrossRef CAS PubMed.
- M. Utecht, T. Klamroth and P. Saalfrank, Phys. Chem. Chem. Phys., 2011, 13, 21608–21614 RSC.
- G. Floß, T. Klamroth and P. Saalfrank, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 1–9 CrossRef.
- B. Baisch, D. Raffa, U. Jung, O. M. Magnussen, C. Nicolas, J. Lacour, J. Kubitschke and R. Herges, J. Am. Chem. Soc., 2009, 131, 442–443 CrossRef CAS PubMed.
- W. Freyer, D. Brete, R. Schmidt, C. Gahl, R. Carley and M. Weinelt, J. Photochem. Photobiol., A, 2009, 204, 102–109 CrossRef CAS PubMed.
- T. F. Heinz, M. M. T. Loy and W. A. Thompson, Phys. Rev. Lett., 1985, 54, 63–66 CrossRef CAS.
- Y. R. Shen, Nature, 1989, 337, 519–525 CrossRef CAS PubMed.
- J. I. Dadap, B. Doris, Q. Deng, M. C. Downer, J. K. Lowell and A. C. Diebold, Appl. Phys. Lett., 1994, 64, 2139–2141 CrossRef CAS PubMed.
- J. A. Delaire and K. Nakatani, Chem. Rev., 2000, 100, 1817–1845 CrossRef CAS PubMed.
- M. Silwa, et al. , Chem. Mater., 2005, 17, 4727–4735 CrossRef.
- L. Boubekeur-Lecaque, et al. , J. Am. Chem. Soc., 2008, 130, 3286–3287 CrossRef CAS PubMed.
- A. Priimagi, et al. , Adv. Mater., 2012, 24, 6410–6415 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS PubMed.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- A. Plaquet, B. Champagne, F. Castet, L. D. E. Bogdan, V. Rodriguez and J.-L. Pozzo, New J. Chem., 2009, 33, 1349–1356 RSC.
- P. Song, A.-H. Gao, P.-W. Zhou and T.-S. Chu, J. Phys. Chem. A, 2012, 116, 5392–5397 CrossRef CAS PubMed.
- C. A. Nelson, J. Luo, A. K.-Y. Jen, R. B. Laghumavarapu, D. L. Huffaker and X.-Y. Zhu, J. Phys. Chem. C, 2014, 118, 27981–27988 CAS.
- X. Wu, H. Park and X.-Y. Zhu, J. Phys. Chem. C, 2014, 118, 10670–10676 CAS.
- S. Nénon and B. Champagne, J. Chem. Phys., 2013, 138, 1–11 CrossRef PubMed.
- U. Jung, C. Schütt, O. Filinova, J. Kubitschke, R. Herges and O. Magnussen, J. Phys. Chem. C, 2012, 116, 25943–25948 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev. B. 01, Gaussian Inc., Wallingford CT, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp03093e |
| This journal is © the Owner Societies 2015 |