
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharting the known chemical space for non-aqueous lithium–air battery electrolyte solvents†
Tamara
Husch
and
Martin
Korth
*
Institute for Theoretical Chemistry, Ulm University, Albert-Einstein-Allee 11, 89069 Ulm, Germany. E-mail: martin.korth@uni-ulm.de
First published on 27th July 2015
Abstract
Li–air batteries are very promising candidates for powering future mobility, but finding a suitable electrolyte solvent for this technology turned out to be a major problem. We present a systematic computational investigation of the known chemical space for possible Li–air electrolyte solvents. It is shown that the problem of finding better Li–air electrolyte solvents is not only – as previously suggested – about maximizing Li+ and O2− solubilities, but also about finding the optimal balance of these solubilities with the viscosity of the solvent. As our results also show that trial-and-error experiments on known chemicals are unlikely to succeed, full chemical sub-spaces for the most promising compound classes are investigated, and suggestions are made for further experiments. The proposed screening approach is transferable and robust and can readily be applied to optimize electrolytes for other electrochemical devices. It goes beyond the current state-of-the-art both in width (considering the number of compounds screened and the way they are selected), as well as depth (considering the number and complexity of properties included).
Introduction
Li-ion batteries have enabled the success of mobile electronic devices, but are not yet suited for competitive application in electric vehicles. Recent years have accordingly seen a tremendous amount of work devoted to go beyond standard Li-ion intercalation technology. In particular, the Li–air battery holds great promise, as it has the highest theoretical energy density of all lithium-based alternatives, and many researchers all over the world now investigate its chemistry.1–3 In the Li–air battery, O2 enters the cathode on discharge, where it is reduced and reacts with Li+ ions to form Li2O2. Upon charging, Li2O2 is oxidized to evolve oxygen. Though the Li–air technology still faces many different challenges, the selection of a suitable electrolyte for the reactive environment of the oxygen cathode has been identified as one of the key obstacles.4 Very recently, Luntz and coworkers as well as Bruce and coworkers identified the solubilites of Li+ and O2− as crucial parameters on the basis of a new detailed insight into the mechanisms causing Li–air batteries to die prematurely.5,6 Dimethylsulfoxide (DMSO) and Methyl-Imidazol (MeIm) were shown to offer substantial improvement over conventional electrolytes, but a search for better choices was encouraged in both studies, as for instance DMSO is not stable as a long term electrolyte.7The identification of new Li–air electrolyte solvents was targeted in the past (see below for details and references). These studies investigated compounds or compound families that were hand-picked based on previously reported desirable properties or chemical intuition. As the chemical space of possible polar-aprotic, organic liquids is tremendously large, a rational decision-making model which investigates compounds experimentally is highly desirable. The mere vastness of the space under consideration does make it seem very likely, that improving upon the current solvents is possible. At least today it seems impossible to test compounds experimentally in a magnitude that allows systematic investigations in this sense, but we will show in the following that computational high-throughput screening now offers a way to probe the full known chemical space and make systematic investigations of full sub-spaces possible. Screening should be seen as complementary to detailed experimental and computational investigations, as it first requires a detailed insight into the relevant processes to identify suitable screening parameters, but offers then a way to transfer insight into innovation by reducing effort on trial-and-error procedures through rational pre-selection.
Large-scale computational screening in battery research was first applied to identify new inorganic materials for cathodes by Ceder and coworkers within the Materials Project.8 Other fields of renewable energy research have seen similar investigations. A prominent example is the Harvard Clean Energy Project that strives to identify organic molecules for photovoltaics.9 The scope of electronic structure theory based screening projects in renewable energy research reaches from a few thousands to a few millions (Materials Project: 60K,8 Harvard Clean Energy Project: 2.3 Mio9). The utilization of screening techniques to optimize battery electrolytes is still in its early stages. Several exploratory studies with a strong focus on redox stabilities were published in the past.10 Only this year the groups of Korth and shortly afterwards Curtiss published larger scale studies based on more properties than just redox stabilities.11,12
Another noticeable feature of all published studies is the choice of structural pool. The structural pool consists of known electrolyte molecules or the candidates that are derived from a given motif, which is identified from experimental insight or chemical intuition. The chemical space under investigation thus suffers from a ‘selection bias’,13 and the question of how to navigate chemical space needs to be addressed to alleviate the effects of this bias. First steps in this direction were made by us when evaluating computational methods at different theoretical levels for the identification of new battery electrolyte solvents.11,14 Here we chart the known chemical space represented by the largest publicly available database, to identify promising candidates and relevant structural motifs for new Li–air battery electrolyte solvents. As a second step we systematically investigate full sub-spaces for the most promising compound classes.
Our screening methodology itself goes beyond the current state-of-the-art by including computational estimates for all properties reported as relevant so far. By evaluating our data with respect to multiple properties at the same time, many false predictions are avoided, which is of utmost importance when making suggestions for subsequent experimental work. A reasonably large amount of knowledge on Li–air electrolyte solvents is available from both experimental4 and theoretical15–21 investigations. This allows us to validate our screening results for the known chemical space in the first part, thereby giving support to our suggestions for new compounds in the second part.
Screening protocol
Relevant screening parameters have been collected by analyzing the literature on Li–air battery electrolyte solvents. Some requirements for a suitable electrolyte are inherited from Li-ion technology: high electrochemical stabilities, suitable melting and boiling points, high flash points, low viscosities/high ion conductivities, and high ion solubilities (as well as low toxicity and cost).22 Estimates for these properties can be computed using quantum chemical methods and the COSMOtherm model.23 Recently, the performance of COSMOtherm for the relevant properties was evaluated on a set of standard electrolyte solvents and typical errors of about 5–10% were found.11 More importantly for our case, Pearson R values for the correlation of theoretical predictions with experimental measurements are very high, thus indicating that COSMOtherm is very well suited for ranking compounds with respect to these properties. Additional criteria have to be met in the case of Li–air batteries, like high oxygen solubilities and diffusivities. Especially Khetan et al.20,21 and Bryantsev et al.15–19 have contributed greatly to identifying suitable descriptors for Li–air battery electrolyte solvents. Their work emphasizes the importance of the chemical stability of the solvent in the rough oxygen cathode environment, where it is subjected to strong bases and nucleophiles like the superoxide anion O2˙−. Bryantsev et al. showed that the pKa of the solvent is a reasonable estimator for the stability towards superoxide,15,17–19 which additionally mediates autooxidation,17 so that solvents with high pKas should also be more unlikely to undergo autoxidation.Very recent results by Johnson et al. highlight the importance of good Li+ solubilities, as they are related to changes in the morphology of the Li2O2 discharge product:5 solvents with poor Li+ solubility lead to Li2O2 film growth that is associated with low capacity, decaying rates and early cell death. In contrast, solvents with good Li+ solubility lead to particle growth, a higher capacity and sustained discharge. Shortly afterwards Luntz and coworkers reached the same conclusion, additionally emphasizing the importance of the O2− solubility.6 According to these studies, the problem is thus (to first approximation) two-dimensional: solvents with high solubilities for both Li+ and O2− should allow for high-capacity Li–air batteries. Both studies are based on quantifying ion solubility with Gutman donor (for cations) and/or acceptor (for anions) numbers (AN and DN). The COSMOtherm model allows us to compute solubilities based on input from quantum chemical calculations, thus providing an alternative that is suitable for large scale computational screenings. As high solubilities are indicated by zero by the COSMOtherm model, we turn to the chemical potential as defined in the COSMO-RS theory23 for quantification. We found the correlation between the donor number and the Li+ chemical potential to be high (R = 0.85), with substantial deviations only for very high donor numbers, which in turn do not correlate well with experimental data (cf. ESI† Section S1).
Structures were obtained from the PubChem Compound database.24 It comprised 67 million compounds at the time of retrieval. The PubChem database started in 2004 as a United States Government initiative and is maintained by the National Center for Biotechnology Information. Initially it was designed to collect information on (biological) activities of small molecules, but was since extended and many journals today automatically contribute to the extension. The PubChem Compound database is the closest image of the known chemical space that is publicly available. The only larger database in this field is the fee-based CAS registry (currently 91 million), which is commonly seen as a direct competitor.
For the first stage of the screening process we propose a hierarchical down-selection strategy as illustrated in Fig. 1. (Arguments for the validity of this strategy are given in ESI† Section S2.) The first steps at this stage are based on global criteria for organic, molecular electrolyte materials, i.e. compounds are discarded that are unlikely for application in any lithium battery technology. In the next steps, criteria specific for Li–air battery electrolytes are applied.
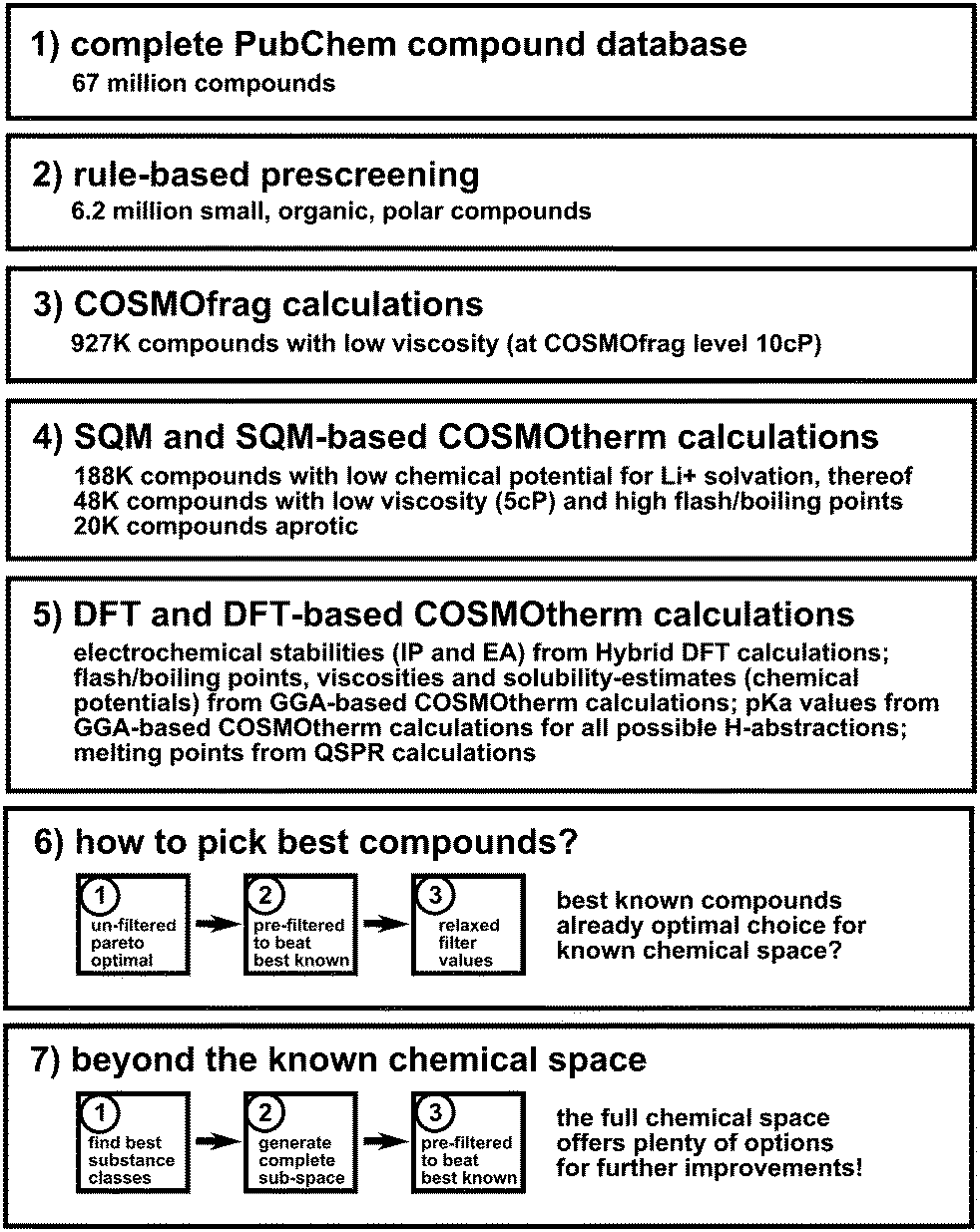 | ||
| Fig. 1 Schematic overview of the screening protocol, see the text for details. | ||
After retrieving and converting structures from the database (step 1), compounds were pre-screened based on simple rules (step 2): candidates with more than 18 heavy atoms or elements other than 1st and 2nd row elements, as well as pure hydrocarbons were excluded. The remaining 6.2 million structures were subjected to very fast COSMOfrag calculations25 to evaluate their viscosity (step 3). All compounds with a viscosity greater than 10.0 cP (at the COSMOfrag level) were discarded. The threshold for the viscosity was selected generously to not discard any compounds prematurely. The remaining 927 thousand polar, organic liquids were evaluated with respect to the chemical potential for Li+ within the bulk compound based on fast semiempirical quantum mechanical (SQM) PM6-DH+26 and SQM-based COSMOtherm calculations, leaving 188 thousand electrolyte solvent candidates after discarding all candidates with a chemical potential larger than 10 kcal mol−1 at this level of theory. These compounds were screened with respect to their ionization potential (IP), electron affinity (EA), viscosity, boiling point, flash point and the free energies of solvation and chemical potentials of Li+, O2− and O2 in the bulk compound at the SQM/COSMOtherm level. 48 thousand compounds with a viscosity below 5.0 cP, boiling points above 373 K and flash points above 343 K were optimized at the GGA-DFT level. (High boiling points are of importance also because the Li–air battery is open to ambient pressure, so that long-term stability can be problematic in the case of high vapor pressures.) All before mentioned collective properties have been re-evaluated using COSMOtherm on the basis of the BP86/TZVP calculations. Additionally, free energies of solvation and chemical potentials of water and carbon dioxide in the bulk compound were included, as the solubility of water and carbon dioxide should be very low to promote the use of air or less refined oxygen and minimize parasitic reactions.6 IP and EA values have been computed at higher levels of theory (Hybrid-DFT, CEPA27) to ensure sufficient accuracy. The final set of molecules comprises about 20 thousand potential polar, non-protic organic liquids that may be good candidates for application in Li–air batteries. Additional calculations of pKa values in DMSO using DFT/COSMOtherm were performed for this dataset, to estimate the chemical stability against nucleophilic attacks, H-abstraction reactions and autooxidation. We found DFT/COSMOtherm pKa predictions to be highly correlated (R = 0.99) with results from the best methods available, but computationally much cheaper (cf. ESI† Section S3). For each compound all possible proton abstractions were considered, and the lowest pKa was picked as the descriptor. A substantial change in molecular geometry after proton abstraction (e.g. ring opening) was taken as a sign of unsatisfactory electrochemical and chemical stability. Finally, QSPR melting point predictions were checked for selected compounds.
Our choice of screening parameters does not include estimators for every thinkable property, but every parameter previously identified as substantially important is included. Detailed follow-up investigations can be carried out subsequently for the most promising compounds. For this purpose, and to allow other researchers to try out different strategies for picking best compounds, the whole dataset will be made available on our project web page.28
Our following analysis will very much concentrate on taking into account the above-mentioned experimental results on the importance of the role of the different ion solubilities. Other experimentally working groups might want to question the significance of these results for the further development of Li–air batteries, but we will show below that also competing experimentally derived hypotheses can easily be incorporated as selection criteria within our screening approach.
Computational details
Ionization potentials (IPs) were calculated at PM6-DH+,26 BP8629,30-D331/TZVP,32 B3LYP33,34-D3/TZVP and LPNO-CEPA27/aug-def2-TZVPP levels, using MOPAC2012,35 Turbomole 6.436 and ORCA 3.0.3,37 electron affinities (EAs) were extrapolated from IPs and orbital eigenvalues according to Tozer.38 Melting points of selected compounds are estimated using a QSPR model of A. Lang.39 Viscosities, boiling and flash points, pKa values in DMSO, free energies of solvation and chemical potentials of various ions and molecules in bulk candidate compounds were computed using COSMOfrag25 and COSMOtherm23 using SQM and GGA-DFT level inputs. For flash point calculations we use a constant COSMO area of 39.23 A2 to enforce a better agreement of absolute values with experimental reference data. The overall computational effort for this study was about 2 million CPU hours. Here we mostly relied on standard compute cluster resources, but work on integrating our Volunteer Computing resources more closely is in progress.Screening results
We then tested several strategies to analyze our screening results and arrive at good suggestions for subsequent experiments, putting special emphasis on multi-dimensional evaluation to pay tribute to the underlying multi-dimensional problem. The most obvious approach is to pick Pareto-optimal candidates out of the final set, which gives 37 candidates (cf. ESI,† Section S4). DMSO is among the final candidates, which is a clear success for our screening strategy as this well-performing compound is successfully picked out of several million others. The other candidates comprise a large variety of N-heterocycles, but when checking melting points they are found to be too high for the majority. Other structural motifs include imines, amides and ureas, which all share the drawback of high melting points or show low O2− solubilities. The most promising suggestions are different sulfoxides, but the calculated pKa values are lower than for DMSO, which additionally has the lowest chemical potential for O2−. All candidates share low IP values and good oxygen, water and carbon dioxide solubilities, leading to the conclusion that water and carbon dioxide have to be excluded from the cells in other ways.As a second strategy we tested pre-filtering compounds to remove cases with poor viscosities and Li+/O2− chemical potentials. Candidates are sorted out that do not beat MeIm (which itself is beaten by DMSO) with respect to Li+ and O2− solubility. This only leaves 7 structures of which 5 are Pareto-optimal (cf. ESI,† Section S5). Among the suggestions is an amine oxide that most likely has a very high melting point. The next two hits, a dihydro-thiophene oxide and a phosphinic acid ester, are most likely reactive and cannot withstand nucleophilic attacks, which disappointingly leaves us only with DMSO and MeIm.
This surprising result that the best known compounds are already optimal (or at least very close to optimal) choices within the known chemical space (as pre-selected by the filtering) clearly merits further investigation. A key issue is a low chemical potential for Li+ in combination with a low chemical potential for O2−, which is the two-dimensional problem very recently identified by Luntz and co-workers. Our screening results now show that (even in first approximation) a third dimension needs to be considered: low chemical potentials for both ions, i.e. high donor and acceptor numbers are connected to strong intermolecular interactions in the pure bulk compound, i.e. a high viscosity and a high melting point. The problem is illustrated in Fig. 2.
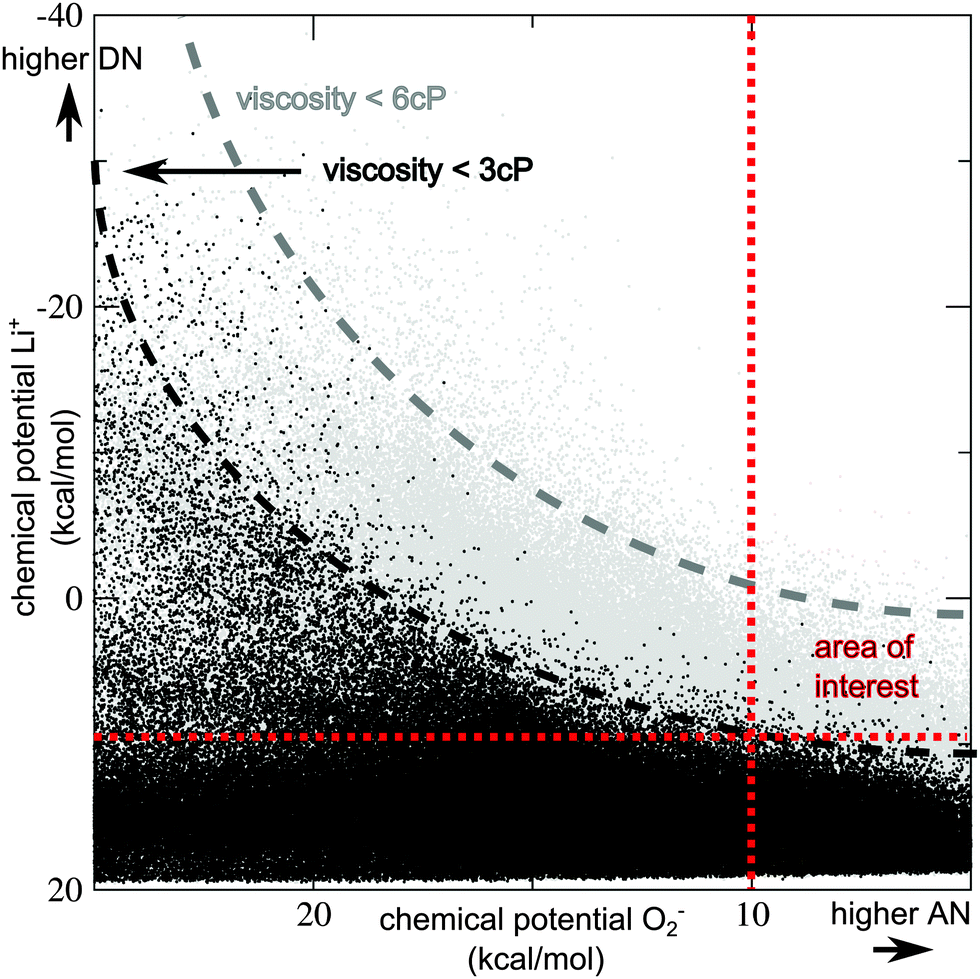 | ||
Fig. 2 Illustration of the problem by optimizing both donor and acceptor numbers (DN/AN): chemical potentials for Li+vs. chemical potentials for O2− in the bulk candidate compound are plotted for all 927![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds treated at the SQM level. (μ(Li+) is systematically underestimated in comparison to higher-level DFT-based data, but trends are similar, see ESI,† Section S1 for details.) Black dots indicate compounds with a viscosity below 3 cP, grey dots indicate compounds with a viscosity below 6 cP, and the upper right part is empty because of the (pre-)screening step 3. Black and grey lines are added to guide the eye. Lower μ(Li+) and μ(O2−) values indicate higher DN and AN, the search is for compounds with both chemical potentials lower than (approximately) 10 kcal mol−1, indicated by the red dotted lines. Almost all compounds in the ‘area of interest’ have high viscosities (i.e. above 3 cP), due to strong intermolecular interactions in the bulk solvent. 000 compounds treated at the SQM level. (μ(Li+) is systematically underestimated in comparison to higher-level DFT-based data, but trends are similar, see ESI,† Section S1 for details.) Black dots indicate compounds with a viscosity below 3 cP, grey dots indicate compounds with a viscosity below 6 cP, and the upper right part is empty because of the (pre-)screening step 3. Black and grey lines are added to guide the eye. Lower μ(Li+) and μ(O2−) values indicate higher DN and AN, the search is for compounds with both chemical potentials lower than (approximately) 10 kcal mol−1, indicated by the red dotted lines. Almost all compounds in the ‘area of interest’ have high viscosities (i.e. above 3 cP), due to strong intermolecular interactions in the bulk solvent. | ||
Candidates that clearly beat DMSO and MeIm with respect to both Li+ and O2− chemical potentials are highly viscous or solid at room temperature. The search for new Li–air battery electrolyte solvents should therefore not focus on maximum DN and AN numbers, but on finding the optimum balance between the two ion solubilities and viscosity. If a lower performance for one property can be tolerated, room is given to optimize the two other ones.
Given this directive, also compounds with somewhat higher Li+ or O2− chemical potentials become interesting. As a third strategy we therefore used relaxed filter thresholds, adding 10% to the respective values of MeIm. The results can be seen in ESI,† Section S6. The biggest share is again N-heterocycles with a too high melting point. Aromatic heterocycles with more than one nitrogen in the aromatic rings are especially interesting. Also sulfoxides are again among the hits, but they again show higher reactivity than DMSO going by the pKa. The suggestions also comprise phosphine oxides and phosphinic acid esters.
Our study shows impressively how good the choice of DMSO and MeIm is and how strong chemical intuition actually is. The PubChem database covers, on the other hand, only a tiny fraction of the relevant chemical space (which we estimate to be at least 1010 times larger), though due to its relative homogenity far less diversity is available than this number suggests.
Beyond the known chemical space
In the last part of our study we therefore turned to screening full sub-spaces for the most promising compound classes. These classes were identified by analyzing the average performance of compounds with the same functional groups. We use Checkmol40 to analyze molecules for the presence of various functional groups, of which 200 different ones are currently implemented in the program. The most important results of this compound class analysis can be seen in Table 1 (and with more detail in ESI,† Section S7).| Compound classb | μ(Li+) [kcal mol−1] | μ(O2−) [kcal mol−1] | Comment | |
|---|---|---|---|---|
| a Values averaged over all entries for each compound class, listed are classes with μ(Li+) > MeIm + 10%, for more details see ESI Section S7. b According to Checkmol classification. | ||||
| Phosphine oxide |
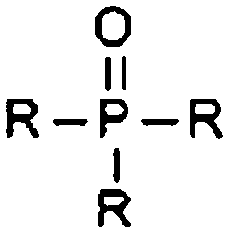
|
5.85 | 13.43 | Selected |
| Phosphine |

|
6.34 | 13.55 | Assumed to be reactive |
| Carboxylic acid amidine |
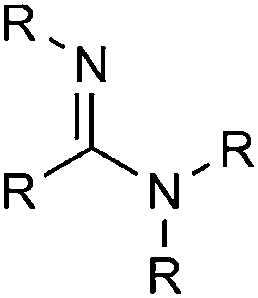
|
8.13 | 15.44 | |
| Secondary aliphatic–aromatic amine alkylarylamine | N-rings | 8.91 | 12.65 | Selected |
| Guanidine |
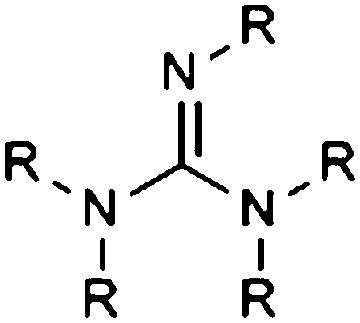
|
9.16 | 16.32 | |
| Sulfoxide |
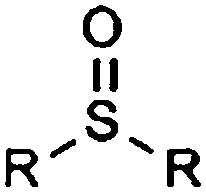
|
9.41 | 12.82 | Low pKas |
| Imine |

|
10.27 | 15.39 | Low pKas |
| Carboxylic acid hydrazide |
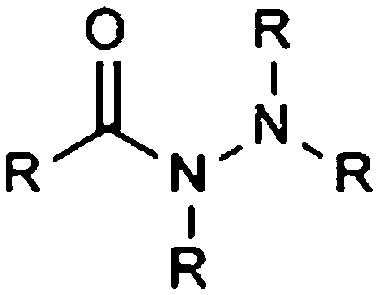
|
10.33 | 14.75 | Low pKas |
| Lactam |
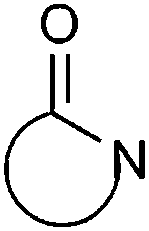
|
10.42 | 14.80 | |
| Secondary amine |
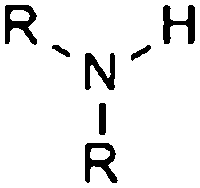
|
10.46 | 15.29 | Selected |
| Urea |

|
10.46 | 15.42 | |
| Oxohetarene |
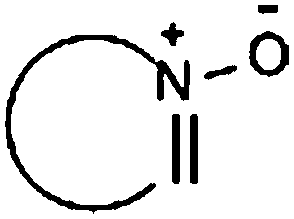
|
10.48 | 12.96 | High viscosities |
| Secondary aliphatic amine dialkylamine | N-rings | 10.61 | 15.53 | |
| Tertiary aliphatic–aromatic amine alkylarylamine | N-rings | 10.65 | 13.76 | High viscosities |
| Azide | R–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N+N− N+N− |
10.65 | 14.85 | Low pKas |
| Tertiary carboxylic acid amide |
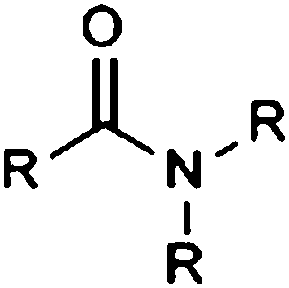
|
10.84 | 14.98 | |
Phosphine oxides show a very good overall performance, with excellent Li+ solubilities, but are known to have high melting points. This compound class was selected for further investigations to look for a candidate with a good balance between the Li+/O2− solubility and viscosity. Amidines, guanidines and imines are interesting suggestions, but though they show a good Li+ solubility, the balance with O2− is not very promising and the non-aromatic CN bond is likely reactive. The second compound class we chose for further investigations was heterocycles, especially those containing nitrogen. Various functional groups according to Haider fall in this category, for example numerous amines, lactames, and oxohetarenes. Their properties are overall very promising aside from somewhat high viscosities. Sulfoxides show as well a very good balance for the chemical potentials, but the average pKa is worrying. As within the PubChem no sulfoxide better than DMSO could be found and a large amount of sulfoxides was already included in the PubChem database, this compound class was not chosen for further studies here. (The properties of DMSO may nevertheless be succeeded by sulfoxides with additional functional groups of different types.) Derivates from carboxylic acids and urea may also be interesting for further investigation, but are not included in this study.
A comparison with the literature supports the validity of our screening results: several compound classes have been identified to fail as Li–air battery electrolyte solvents, for example organic carbonates, sulfonates, pure esters, lactones and ethers. Only one compound class that was previously excluded due to experimental or theoretical findings is found in our list of best performing ones. The compounds incorrectly listed are phosphinic acid esters, which were shown to be susceptible to nucleophilic attacks by Bryantsev et al.19 Though we do not cover this type of reactivity, pKas were lower than those for DMSO and MeIm and thus lower chemical stabilities are predicted. Our study is further supported by the fact that many compound classes previously identified as promising are among our hits. Examples are lactams, amides, phosphine oxides and N-heterocycles.4,16,19 We do not list nitriles, as we find comparably poor Li+ solubilities on average, but otherwise reasonably good properties. Our findings also indicate that when screening existing databases, further reactivity estimates beyond the pKa need to be included. Additional estimates may for example take the reactivity of double or ester bonds or highly strained ring structures into account. These findings can on the other hand easily be incorporated as rules for structure generation when generating new databases, as we will show in the following.
Based on our analysis of the average performance of compound classes, phosphine-oxides and heterocycles mainly containing nitrogen, but also oxygen and sulfur were chosen for further investigations. Turning away from the known chemical space of the PubChem database, we screened the full chemical sub-spaces of these compound classes within certain structural constraints. The Molgen algorithm41 was used to construct all possible structures for the relevant sub-spaces. To keep the number of structures manageable, structure generation was broken down into parts. For phosphine-oxides for instance we first looked at aliphatic structures and had to keep the overall number of atoms low, while in the second step we looked at cyclic phosphine-oxides, where a much larger overall number of atoms was possible, because many atoms were bound to end up in the enforced ring motif. Turning to heterocycles we first looked at mono- and bi-cycles, but constrained to aromatic systems and only considering nitrogen heteroatoms. As bi-cycles did not give promising results, we turned to 5–6 membered mono-cycles, still only considering nitrogen but now also non-aromatic systems. To investigate also oxygen and sulfur systems and N/O/S mixed ones we had to turn to constructing simple N/O/S 5- and 6-ring heterocycles first and add aliphatic rests to these core rings later on.
Overall, five different investigations were carried out: first, all phosphine-oxides P1O1C3–6 and their sulfur analogs P1S1C3–6 were constructed with no rings other than 5- to 7-membered ones allowed. We then applied the screening protocol outlined above for the PubChem database, starting at the DFT level (step 5). Out of 362 compounds we identified 10 Pareto-optimal structures with a promising balance of the chemical potential of Li+ and O2− (cf. ESI,† Section S8). Unfortunately all structures with a low melting point are most likely reactive, because they incorporate double/triple bonds or allene structures. The most promising structures, an aliphatic phosphinan-oxide and an aromatic phosphol-oxide (ID po138, po922), have an outstanding balance of the chemical potential of Li+ and O2− and good safety features, but are barely liquid. In a second run, all cyclic phosphine-oxides P1O1C5–10 were constructed with one ring enforced, only 5–6 membered rings allowed, and double or triple bonds except aromatic ones forbidden. Screening was again started at the DFT level with 926 compounds, but all were showing high μ(O2−) values. As a third step, all aromatic N-heterocycles N1–3C2–12 were constructed with the same constraints as for the cyclic phosphine-oxides, i.e. all aromatic N-based mono- and bi-cycles. It should be noted that the structure generator does only count ring-wise fully conjugated double bonds as aromatic, so that structures like MeIm are not included in this set. Screening was started at the DFT level (step 5) with 28356 structures, but no compound turned out to be competitive. As a forth step all (including non-aromatic) N-heterocycles Ni=1–3C3–(10−i) were constructed with one ring enforced, only 5–6 membered rings allowed, but now also double bonds other than aromatic ones allowed. 113140 structures were evaluated at the SQM level (step 4), and 1290 at the DFT level (step 5). 102 candidates are competitive to MeIm, of which 39 are Pareto-optimal, and 18 of the latter are ‘simple’ in the sense that they contain no reactive binding motifs (like double or triple bonds outside the ring). As a fifth step, all simple, unsubstituted 5- and 6-ring heterocycles containing up to 3 nitrogen, oxygen or sulfur atoms were constructed (865 structures) and then all possibilities of attaching up to three carbon atoms to these core rings were evaluated (204695 structures). 458 candidates are competitive to MeIm, of which 74 are Pareto-optimal, and 13 of the latter are ‘simple’ in the sense that they contain no reactive binding motifs and only one heteroatom species. (Compounds with only one heteroatom species should be more easily accessible to the experiment.) We list all 31 ‘simple’ heterocycle hits in ESI,† Section S9, but estimated melting points indicate that most of these compounds are again not very likely to be liquid at room temperature.
Further experiments are clearly necessary to find out if higher μ(O2−) can be tolerated to allow for lower μ(Li+) values at low viscosities. To make suggestions for the systematic experimental investigation into the optimal balance of Li+/O2− solubility and viscosity, we again turn to the PubChem database, thereby making sure that suggested compounds are (more or less) readily available. Section S10 (ESI†) gives a compilation of low-viscosity compounds with very different balances of the two relevant chemical potentials. From all compounds of the final set with a viscosity lower than 2 cP and a chemical potential for Li+ lower than 10 kcal mol−1, compounds with the lowest chemical potential for O2− are given for each 1 kcal mol−1 interval of μ(Li+).
The PubChem and heterocycle data are readily available for re-evaluation with adjusted filter thresholds if higher μ(O2−) can indeed be tolerated. Raw data for all screening runs will accordingly be made available on our project web page to encourage further investigations also by other researchers. As a first example we give a list of the most promising motifs for the case that the solubility of the negative species is actually of lesser importance (i.e. looking for compounds with a viscosity lower than 2 cP in combination with a lithium cation chemical potential below 10 kcal mol−1, as well as pKas higher than 25 and melting points lower than 10 °C) in ESI,† Section S11. Very different compounds are found in this analysis in comparison to the previous ones, now with an emphasis on amide and amine motifs, thus nicely illustrating how important the choice of selection criteria (and therefore an input from the experiment) is.
Conclusions
Our systematic investigation of the known chemical space represented by the PubChem database indicates that the problem of finding better Li–air electrolyte solvents is not only about maximizing donor and acceptor numbers, but also about finding the optimal balance of the relevant ion solubilities with the viscosity of the solvent. The PubChem results and the subsequent exploration of full sub-spaces for the most promising compound classes delivered a list of compounds for the experimental investigation of this balance. Our results imply that further trial-and-error investigations of commercially available chemicals are most likely doomed to failure. Instead the exploration of unknown substances should be pursued, using both computational and experimental screening techniques. We did for instance not investigate compounds with multiple functional groups apart from those in the PubChem database, as well as the opportunities offered by mixtures of known and/or unknown solvents, but both tasks can be well-handled on the computational side using the screening approach proposed here. We have thus good hopes that supplementing experimental battery research using a theory-based, rational decision model will help to speed up the transfer of insight into innovation in this field.Acknowledgements
The authors would like to thank Sylvain Brimaud, Gerhard Maas and Andreas Klamt for helpful discussions and gratefully acknowledge financial support from the Barbara Mez-Starck Foundation. This work was supported in part by the bwHPC initiative and the bwHPC-C5 project provided through associated compute services of the JUSTUS HPC facility at the University of Ulm. bwHPC and bwHPC-C5 are funded by the Ministry of Science, Research and the Arts Baden-Württemberg and the Germany Research Foundation.References
- M. D. Bhatt, H. Geaney, M. Nolan and C. O'Dwyer, Phys. Chem. Chem. Phys., 2014, 16, 12093–12130 RSC.
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114, 11721–11750 CrossRef CAS PubMed.
- J. Lu, L. Li, J.-B. Park, Y.-K. Sun, F. Wu and K. Amine, Chem. Rev., 2014, 114, 5611–5640 CrossRef CAS PubMed.
- M. Balaish, A. Kraytsberg and Y. Ein-Eli, Phys. Chem. Chem. Phys., 2014, 16, 2801–2822 RSC.
- L. Johnson, C. Li, Z. Liu, Y. Chen, S. A. Freunberger, P. C. Ashok, B. B. Praveen, K. Dholakia, J.-M. Tarascon and P. G. Bruce, Nat. Chem., 2014, 6, 1091–1099 CrossRef CAS PubMed.
- N. B. Aetukuri, B. D. McCloskey, J. M. Garca, L. E. Krupp, V. Viswanathan and A. C. Luntz, Nat. Chem., 2014, 7, 50–56 CrossRef PubMed.
- D. G. Kwabi, T. P. Batcho, C. V. Amanchukwu, N. Ortiz-Vitoriano, P. Hammond, C. V. Thompson and Y. Shao-Horn, J. Phys. Chem. Lett., 2014, 5, 2850–2856 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef PubMed.
- J. Hachmann, R. Olivares-Amaya, A. Jinich, A. L. Appleton, M. A. Blood-Forsythe, L. R. Seress, C. Roman-Salgado, K. Trepte, S. Atahan-Evrenk, S. Er, S. Shrestha, R. Mondal, A. Sokolov, Z. Bao and A. Aspuru-Guzik, Energy Environ. Sci., 2014, 7, 698–704 CAS.
- M. Korth, Chemical Modelling, 2014, 11, 57 CrossRef PubMed.
- T. Husch, N. D. Yilmazer, A. Balducci and M. Korth, Phys. Chem. Chem. Phys., 2015, 17, 3394–3401 RSC.
- L. Cheng, R. S. Assary, X. Qu, A. Jain, S. P. Ong, N. N. Rajput, K. Persson and L. A. Curtiss, J. Phys. Chem. Lett., 2015, 6, 283–291 CrossRef CAS.
- M. Korth and S. Grimme, J. Chem. Theory Comput., 2009, 5, 993–1003 CrossRef CAS.
- M. Korth, Phys. Chem. Chem. Phys., 2014, 16, 7919–7926 RSC.
- V. S. Bryantsev, Chem. Phys. Lett., 2013, 558, 42–47 CrossRef CAS PubMed.
- V. S. Bryantsev and M. Blanco, J. Phys. Chem. Lett., 2011, 2, 379–383 CrossRef CAS.
- V. S. Bryantsev and F. Faglioni, J. Phys. Chem. A, 2012, 116, 7128–7138 CrossRef CAS PubMed.
- V. S. Bryantsev, V. Giordani, W. Walker, M. Blanco, S. Zecevic, K. Sasaki, J. Uddin, D. Addison and G. V. Chase, J. Phys. Chem. A, 2011, 115, 12399–12409 CrossRef CAS PubMed.
- V. S. Bryantsev, J. Uddin, V. Giordani, W. Walker, D. Addison and G. V. Chase, J. Electrochem. Soc., 2013, 160, A160–A171 CrossRef CAS PubMed.
- A. Khetan, H. Pitsch and V. Viswanathan, J. Phys. Chem. Lett., 2014, 5, 1318–1323 CrossRef CAS.
- A. Khetan, H. Pitsch and V. Viswanathan, J. Phys. Chem. Lett., 2014, 5, 2419–2424 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- A. Klamt, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 699–709 CrossRef CAS PubMed.
- E. E. Bolton, Y. Wang, P. A. Thiessen and S. H. Bryant, in PubChem: Integrated Platform of Small Molecules and Biological Activities, ed. R. A. Wheeler and D. C. Spellmeyer, Elsevier, 2008, ch. 12, vol. 4 Search PubMed.
- M. Hornig and A. Klamt, J. Chem. Inf. Model., 2005, 45, 1169–1177 CrossRef CAS PubMed.
- M. Korth, J. Chem. Theory Comput., 2010, 6, 3808–3816 CrossRef CAS.
- F. Neese, A. Hansen, F. Wennmohs and S. Grimme, Acc. Chem. Res., 2009, 42, 641 CrossRef CAS PubMed.
- cleanmobility.now, http://www.qmcathome.org/clean_mobility_now.html, accessed March 17, 2015.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- OPENMOPAC, http://www.openmopac.net, accessed March 1, 2015.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73 CrossRef CAS PubMed.
- F. De Proft, N. Sablon, D. J. Tozer and P. Geerlings, Faraday Discuss., 2007, 135, 151 RSC.
- A. S. I. D. Lang, MeltingPointModel010, http://onschallenge.wikispaces.com/MeltingPointModel010, accessed Jul. 15, 2014.
- N. Haider, Molecules, 2010, 15, 5079–5092 CrossRef CAS PubMed.
- A. Kerber, R. Laue, T. Grüner and M. Meringer, MATCH Commun. Math. Comput. Chem., 1998, 37, 205–208 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c5cp02937f. Additionally, raw data will be made available as a web-accessible database on http://qmcathome.org/clean_mobility_now.html. |
| This journal is © the Owner Societies 2015 |