
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpectral properties and isomerisation path of retinal in C1C2 channelrhodopsin†
I.
Dokukina
and
O.
Weingart
*
Institut für Theoretische Chemie und Computerchemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstr. 1, 40225 Düsseldorf, Germany. E-mail: Oliver.Weingart@hhu.de
First published on 2nd September 2015
Abstract
Structure and excited state isomerisation pathway of retinal in the channelrhodopsin chimera C1C2 have been investigated with combined quantum mechanical/molecular mechanical (QM/MM) techniques, applying CD-MS-CASPT2//CASSCF and DFT-MRCI quantum methods. The absorbing S1 state is of 1Bu-like character, and the second excited S2 state is dominated by HOMO–LUMO double excitation with small oscillator strength. Upon photoexcitation and torsion along the reactive C13![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C14 double bond we observe bond length equalisation and a two-path deactivation mechanism in positive and negative torsion directions. The computed path is barrierless in positive direction while a small barrier exists for the opposite side. Comparative protonation studies suggest a charged glutamate E162 residue, with computed resonance Raman data in valuable agreement with experimental channelrhodopsin-2 data. The two negatively charged counter-ions and a positive lysine residue close to the retinal Schiff base terminus have the largest influence on the chromophore absorption wavelength.
C14 double bond we observe bond length equalisation and a two-path deactivation mechanism in positive and negative torsion directions. The computed path is barrierless in positive direction while a small barrier exists for the opposite side. Comparative protonation studies suggest a charged glutamate E162 residue, with computed resonance Raman data in valuable agreement with experimental channelrhodopsin-2 data. The two negatively charged counter-ions and a positive lysine residue close to the retinal Schiff base terminus have the largest influence on the chromophore absorption wavelength.
Introduction
Channelrhodopsins (ChRs) mediate phototaxis and photophobic reactions in the green algae chlamydomonas reinhardtii and haematococcus pluvialis.1,2 Their primary function is the light induced regulation of cell conductivity through opening or closing ion channels of different size.3,4 Unlike G-protein coupled receptors, which involve external agonists to produce a delayed but amplified nerve stimulus,5 ChRs provide an immediate response after light excitation. Their fast regulation of cell polarisation and feasible expression in mammalian neurons6 make ChRs ideal candidates for the study of basic brain or nerve regulation functions through non-invasive external light control.7,8The initial step in the photoreaction of ChRs is the light induced isomerisation of its chromphore retinal (Fig. 1), which is bound via a Schiff base linkage to a lysine residue of the protein. Subsequent thermal reactions open the ion channel and lead to depolarisation of the cell.1–4
 | ||
| Fig. 1 All-trans retinal with isomerising double bond (in negative direction), numbering and QM areas indicated. | ||
The two known forms of ChR, ChR1 and ChR2, provide highest selectivity for Li+, Na+ and Ca2+ cations (in decreasing order9). ChR2 is able to depolarise the cell membrane within ms and has therefore been widely used in studying neuronal activity.10 Recently, the crystal structure of dark-adapted C1C2, a chimera of ChR1 and ChR2, has been reported at a resolution of 2.3 Å, together with a model of ion vestibules which may form within the protein.11 The studied chimera contains mostly structural features of ChR1, the last two helices were exchanged for those of ChR2. The resulting protein has been successfully expressed in insect cells. Stable photocurrents were measured, i.e., the chimera is fully functional.
The initial photoreaction in ChRs is not yet completely resolved. Resonance Raman and extraction experiments reveal two retinal conformers in dark adapted ChR2: 70% in all-trans and 30% in 13-cis configuration.12 Femtosecond time-resolved absorption spectroscopy indicates the formation of a bacteriorhodopsin (bR) K-like ground state intermediate through torsion around the C13C14 retinal bond.9 Since the absorption signal of 13-cis retinal is very similar to all-trans, participation of the 13-cis isomer in the initial step of the photoreaction could not be excluded.9,10
The absorbing state in retinal Schiff bases can be compared to the 1Bu excited state dominated by HOMO–LUMO excitation in symmetric polyenes. Upon excitation, a charge migration from the Schiff base nitrogen terminus towards the β-ionone ring takes place.13 The latter process however does not occur in neutral symmetric polyenes, thus the same type of excitation generates different geometrical changes in the two types: while the bond length pattern is inverted in Schiff bases,14 the bond lengths equalise in polyenes.15
Currently, two models for the ChR binding pocket are discussed in the literature. Early models propose two negatively charged counter-ions in the vicinity of the retinal Schiff base linkage, E123 and D253 (E162 and D292 in C1C2), while a recent, DFTB//Charmm based computational study of Welke et al. gives indications for a protonated E123 residue in ChR1 and C1C2.16
Computational multi-reference MP2 QM/MM studies on the C1C2 chimera structure performed by Hayashi and coworkers (computed λmax = 426 nm)17 and polarisable embedded RI-CC2 studies by Sneskov et al.18 (λmax = 459 nm) reproduce with good accuracy the experimental absorption of 470 nm. Both studies were based on the double-counter-ion model, but lack information on state composition and other states beyond the spectroscopic bright state associated with 1Bu-like HOMO–LUMO excitation. This information is of importance to correctly model and interpret transient absorption in fs-UV/Vis and 2DES spectroscopy.19
Photoreaction path analyses for retinal in C1C2 have not been pursued so far, but for similar systems as e.g. the anabaena sensory rhodopsin20,21 and bR,22 revealing unidirectional bicycle-pedal like motion23 of two neighboring double bonds during photoisomerisation.
To investigate the nature of low lying excited states in C1C2, we applied DFT-MRCI/Amber and MS-CASPT2/Amber QM/MM strategies for both, single and double counter-ion binding pocket models. We provide excitation energies for structure models of C1C2 refined at different levels and follow the C13C14 torsion path starting in the bright 1Bu-like state. We investigate the spectral characteristics of 13-cis retinal in C1C2, and provide computed resonance Raman spectra for both retinal isomers. Finally, we examine the influence of residues on the absorption of retinal.
Computational methods
QM/MM single point computations, geometry optimisations and numerical frequency analysis have been performed using COBRAMM, a QM/MM program interfacing various QM and MM codes.24 The Amber12 suite of programs25 has been used to generate a molecular mechanics setup for ChR utilising the 3UG9 PDB structure, complemented by the SBildn extension to the ff99 parameter set.26 Protonation states of protein residues have been computed with proPka.27 The configurations of backbone histidine residues were screened by visual inspection. Missing hydrogens were added and the hydrogen positions were re-optimised with all heavy atoms fixed. Side chains with missing residues in 3UG9 were saturated with ACE or NME groups to prevent charged termini. QM/MM ground state geometries were obtained within an electrostatic embedding scheme using DFT and CASSCF routines of Molpro201028 as QM part and Amber12 ff99SBildn as MM method. MM structure parameters were generated with the Antechamber toolset of Amber. Chromophore ground state charges were derived by fitting the electrostatic potential of the bare chromophore using the Merz–Kollmann Scheme in Gaussian09. To saturate dangling QM-bonds, a hydrogen link-atom scheme with MM charge-repartitioning has been used. The charges of the MM boundary atoms were set to zero and the charge was partitioned proportionally among the neighboring MM atoms. Forces of the link hydrogens were repartitioned among the QM/MM border atoms. For more details on the QM/MM setup, see also ref. 19.Geometry optimisations
A full CASSCF 12/12 π-space was employed for ground state optimisation, using the 6-31G* basis set for all computations. DFT structures were obtained using the B3LYP functional. The QM layer consisted of the whole retinal chromophore and one CH2 group of lysine. The retinal, the bound lysine residue, the two counter-ions and one close water molecule (WAT619) were free to move. The rest of the protein structure was kept fixed at the crystal structure values. Further refinement was not pursued at this stage to keep larger structural rearrangements, as e.g. observed in the dynamics computations by Hayashi and coworkers,17 where full equilibration of the system was pursued in solvent, or changes due to the missing loops in our model at a minimum. This strategy will allow to judge, whether the sole crystal structure can already provide valuable information on the photophysics of C1C2. A similar protocol has also been used in the work of Sneskov et al.18MS-CASPT2 and DFT-MRCI29 energy values for the first five electronic states and corresponding oscillator strengths were computed for the optimised structures using Molcas 8.130 and Turbomole 6.3, respectively.31 For MS-CASPT2 computations the IPEA shift has been set to 0.0, and a standard imaginary shift of 0.2 has been applied.
Relaxed excited state scan
Excited state torsion paths were obtained using the Molcas 8.1 CD-CASSCF routines as QM method with an active space of 12 electrons in 12 π-orbitals. A reduced QM layer, including the whole conjugated part and the C5 methyl group but neglecting the β-ionone alkyl part (C1–C4, see also Fig. 1), was used. The two counter-ions were fixed to their positions after CASSCF S0-optimisation. The torsion profile computations were performed within a frozen environment, because the ultrafast fs-isomerisation of the chromophore prohibits an immediate energy exchange with the surrounding. Relaxation and thermalisation of the latter will only take place in a later phase, thus additional optimisation of the binding pocket residues may significantly bias the computed retinal torsion path. The wavefunction was averaged over the ground state and the three lowest excited states (state averaging, SA). SA-CASSCF gradients were computed for the third excited CASSCF state (S3) resembling HOMO–LUMO single excitation. Gradients were corrected using the molcas mclr routine. A relaxed scan along the C12–C13C14–C15 torsion angle was performed. The torsion angle was varied in steps of 5 degrees and kept fixed, while the rest of the movable part was fully relaxed.
CD-MS-CASPT2 energy values for the four lowest states were computed at converged geometries.
Resonance Raman data
Numerical frequencies were computed to generate resonance Raman data. To reduce the computational effort in producing numerical frequencies, the models were re-optimised at the reduced CD-CASSCF(10/10)//Amberff99SBildn level of theory, deleting the highest unoccupied and lowest occupied orbital from the former CAS(12/12) space. The β-ionone ring was deleted from the QM part and all protein residues were frozen at the crystal structure values.Raman activities were computed using the approach formulated by Orlandi and coworkers,32 which has proven to yield reliable data for Schiff bases.33 In the harmonic approximation, the relative displacement parameter Bi for the transition from S0 to S1 in mode i can be approximated as the matrix product of the S1 force vector fS1 with the mass-weighted Cartesian normal coordinates for mode i, 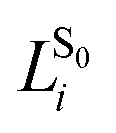 , scaled by the associated vibrational frequency v−3/2i:
, scaled by the associated vibrational frequency v−3/2i:
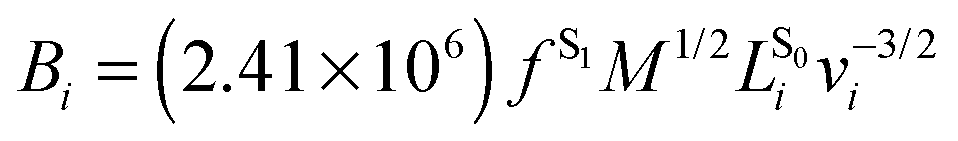
| Ii ∝ ½Bi2 |
Results and discussion
Geometry changes after optimisation
After QM/MM optimisation, we note significant geometrical changes from the crystal structure for the orientation of the retinal NH+ moiety (Fig. 2). In all ground-state optimisations this fragment moves significantly closer towards the D292 counter-ion and away from E162. Leaving also the counter-ions D292 and E162 free to move during optimisation, leads to a small shift of the E162 position, away from the NH+ moiety. The orientation towards D292 persists also, when the protein binding pocket is relaxed (see ESI†), suggesting D292 as the primary proton acceptor in the modelled structure. This result seems to be in contrast to UV/Vis and FTIR measurements34 which support formation of a salt bridge solely between the Schiff base NH+ and E162. The recent work of Bartl and coworkers however reports the same protonation characteristics for both counter-ions after illumination,35 such that both initial orientations are plausible. This picture is further supported by reaction path computations in the next chapter. The C1C2 crystal structure consequently appears more biased towards the NH+ → D292 configuration, an effect which may also result from protonation of E162 under the slightly acidic conditions during the crystallisation procedure. This effect is obviously enhanced in our QM/MM optimisations, and can be released through further refinement and equilibration of the used model. Through optimisation of the residues within 5 Å of retinal and using a lower level HF method for the QM region, we obtained a structure with hydrogen bonding to E162 and very similar absorption properties (see ESI†). It thus appears that both hydrogen bonded configurations may be independently stable.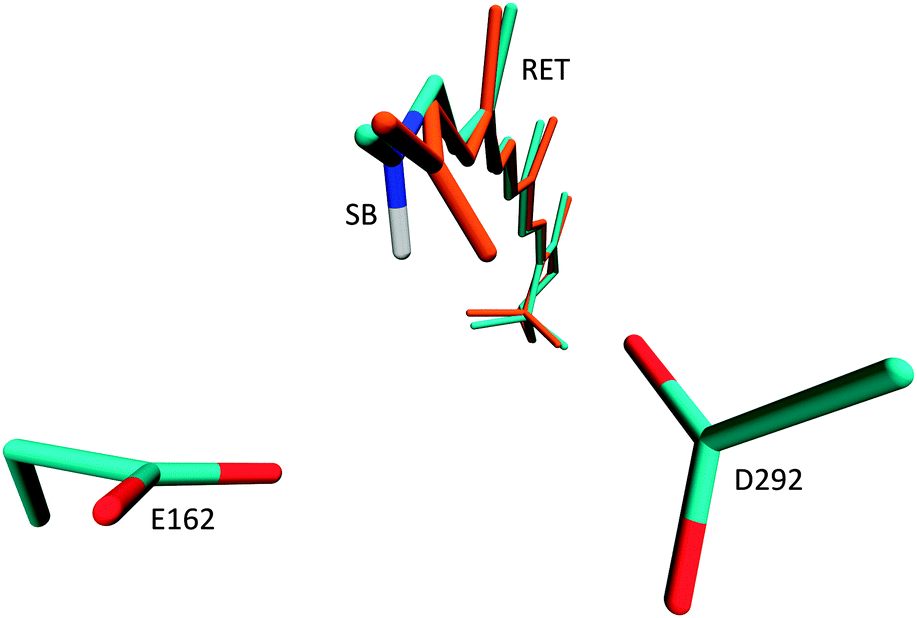 | ||
| Fig. 2 Location of the two negatively charged counter-ions E162 and D292 and geometry change from crystal structure (cyan) to QM/MM optimised ground state (orange). | ||
The conjugated chain is significantly twisted at the C13C14 double bond (−171°) and the C10–C11 (−169°) and C14–C15 (−163°) single bonds, i.e., the chromophore has overall M-helicity. The twist persists when the geometry is optimised in the excited 1Bu-like state. At the CASSCF level, this structure is a stationary point (see ESI† for an overlay of the optimised ground state and 1Bu-like structures).
Fig. 3 shows the bond length alternation (BLA) pattern of single and double bonds along the conjugated chain of retinal computed at different levels of theory in the ground state and in the relaxed CASSCF 1Bu-like geometry. A strong BLA (varying from ca. 1.35 to 1.49 Å along the carbon chain) is observed for the CASSCF-optimised structure, while DFT computes a stronger conjugation, i.e. a less alternating single/double bond pattern, along the chain (1.38 to 1.44 Å). No significant changes in the bond pattern can be observed in the single counter-ion structure (see ESI†).
 | ||
| Fig. 3 Bond lengths in crystal structure and QM/MM optimised geometries. | ||
In the relaxed bright 1Bu-like excited state, the bond length pattern changes drastically in the fragment from C9 to C15. Former double bonds become shorter and single bonds longer, with values between 1.38 and 1.42 Å. Thus, the bonds become more equivalent after excitation. This observation is somewhat different from the previously computed retinal chromophore in rhodopsin (Rh), where a complete bond order inversion is observed in the excited S1 state,14 but it is in agreement with trends in symmetric carotenoids and polyenes. These molecules show bond order inversion only in the 2Ag state due to HOMO–LUMO double excitation.15 A similar effect has also been observed in Rh with a locked retinal.36 This process of bond equalisation in ChR can be attributed to the two negatively charged counter-ions in the vicinity of the Schiff base bond, which have profound influence on the nature of the excited states. In the ground state, the counter-ions strongly stabilise the positive charge at the Schiff base tail of the chromophore, leading to a large BLA. The 1Bu-like state associated with HOMO–LUMO excitation leads to a charge transfer towards the β-ionone moiety.13,14 A complete transfer of the positive nitrogen charge, which would invert the bond ordering, is hindered by the negatively charged environment around the Schiff base. This destabilises the energy of the excited 1Bu-like state and results in an overall stronger conjugation along the C9–C14 fragment. We suspect that the two negatively charged counter-ions lead to the more “polyene-like” character of retinal in this surrounding. The short distance of the two negatively charged counter-ions is also responsible for the significant blue shift of retinal in ChR with respect to other retinal-binding proteins.
Nature of low lying excited states for different protonation states
Vertical excitation energies computed at MS-CASPT2 and DFT-MRCI levels of theory are collected in Table 1 and Fig. 4 for the CASSCF S0-optimised chromophore, counter-ions and nearby water. The corresponding data for CASSCF, HF and DFT optimisations of retinal with frozen protein environment are given in the ESI.† Generally, the additional optimisation of the counter-ion positions leads to a red-shift of the computed maximum absorption wavelength (see ESI†). We note a strong influence on the CASSCF state ordering in the charged environment of the binding pocket, which is only removed at higher levels of correlation in DFT-MRCI and MS-CASPT2. E.g., the bright HOMO–LUMO excitation is the first excited state in the latter two descriptions, while in CASSCF it corresponds to the third excited state. This deficiency of CASSCF, which lacks a major part of dynamic electron correlation, can only partly be corrected at the single state CASPT2 level. Too high S1 excitation energies are obtained with a single state perturbation strategy (see ESI†). Only the multi-state CASPT2 or DFT-MRCI levels allow for a stronger stabilisation of the S1 state, with computed excitation energies close to the experimental values.| Method | State | E (eV) | Config. | Weight | f |
|---|---|---|---|---|---|
| MS-CASPT2 | S0 | 0.0 | GS | 0.72 | — |
| S1 | 2.59 | H → L | 0.54 | 1.20 | |
| S2 | 3.95 | 2H → 2L | 0.23 | 0.13 | |
| H−1 → L | 0.15 | ||||
| S3 | 4.96 | H−2 → L | 0.17 | 0.14 | |
| H−2/H−1 → L | 0.17 | ||||
| S4 | 5.39 | H−3 → L | 0.19 | 0.01 | |
| H−1 → L,H → L+1 | 0.08 | ||||
| DFT-MRCI | S0 | 0.0 | GS | 0.93 | — |
| S1 | 2.74 | H → L | 0.82 | 1.47 | |
| S2 | 3.52 | H−1 → L | 0.37 | 0.56 | |
| H → L+1 | 0.10 | ||||
| S3 | 4.13 | H → L+1 | 0.28 | 0.25 | |
| H−1 → L | 0.15 | ||||
| H−2 → L | 0.11 | ||||
| S4 | 4.46 | H−2 → L | 0.26 | 0.08 | |
| H → L+1 | 0.22 | ||||
| H−1 → L | 0.13 | ||||
 | ||
| Fig. 4 Excitation energies to the 1Bu-like S1 state of retinal in ChR with standard protonation and E162 protonated. Values are shown for B3LYP- and CASSCF optimised geometries. | ||
Both methods, MS-CASPT2 and DFT-MRCI, show a large contribution of the HOMO–LUMO CSF in the first excited state and a large oscillatory strength for this state. The second excited state is dominated by the HOMO–LUMO double excitation and HOMO−1 → LUMO single excitation in MS-CASPT2. It has a small but non-negligible oscillatory strength. The same is true for the strongly blue-shifted S3 state, which is of mixed character. The S4 state is strongly mixed as well and essentially dark in MS-CASPT2. DFT-MRCI yields a slightly different picture for the higher excited states. S2, e.g., is more dominated by the HOMO−1 → LUMO excitation, a double excitation is missing. This results in a larger oscillatory strength for this state. The maximum absorption band is well reproduced at both levels (479 and 452 nm for MS-CASPT2 and DFT-MRCI, respectively). Higher excited states (S2–S4) appear significantly more stabilised in the DFT-MRCI description.
To investigate the effect of the alternative E162 protonated configuration on absorption and state composition, we have re-optimised the geometry of retinal and counter-ion positions using the protonated GLH residue at the corresponding position with DFT and CASSCF as QM methods. The geometry and the composition of the excited states change only slightly with respect to the previously optimised geometry in standard protonation (see ESI†). For the CASSCF optimised geometry we note a significant red shift in the maximum absorption of ca. 0.17 eV (ca. 30 nm) in MS-CASPT2 and more than 0.37 eV (70 nm) at the DFT-MRCI level (Fig. 4). The deviation in absorption energy persists, when we use a DFT-optimised geometry to determine the excited state properties. Here, the difference between MS-CASPT2 and DFT-MRCI computed absorption values becomes even more pronounced; the 1Bu state is strongly stabilised in the latter.
Properties after HOMO–LUMO single excitation
After bond relaxation in the 1Bu-like state, the properties of S1 and S2 slightly change. At the optimised 1Bu-like geometry the emission wavelength from the S1 state is 489 nm, with an oscillator strength of 1.69. The HOMO–LUMO excitation contributes ca. 50% to this state (see ESI†), while some fraction of the same CSF can now be found in S2 (ca. 8%, osc. strength of this state 0.091) – together with the HOMO–LUMO double excitation (ca. 24%). At this point, the S1 and S2 states are very close in energy, the MS-CASPT2 energy difference is only 7.3 kcal mol−1. Thus, state interaction which may lead to population of the S2 state during dynamics, similar to the proposed three-state model by Anfinrud and coworkers,37 cannot be excluded. The computed torsion path however follows the 1Bu-like excited state. The other excited states are well separated and essentially dark. Only S3 has significant oscillator strength (305 nm, 0.12).Torsion path in the 1Bu-like state
The torsion path was obtained by following the CASSCF S3 state, which until ca. 120° along the C12–C13C14–C15 dihedral angle keeps its HOMO–LUMO single excitation character (see ESI†). Fig. 5 shows the energy profiles of the four lowest retinal electronic states along negative and positive C13C14 bond twists, computed at the CD-MS-CASPT2/CASSCF(12,12) level. The paths start at the optimised 1Bu-like excited state structure with a torsion angle of −172°. The positive pathway leads, nearly barrierless, towards an S0/S1 state crossing (CI−) with ca. −100° twist. The negative twisting pathway has a ca. 3.8 kcal mol−1 barrier at 155°. This barrier is slightly higher than the available excess energy after excitation (i.e. the difference between the FC energy and the bond-length relaxed geometry, ca. 3.6 kcal mol−1). Thus, the molecule will need a small amount of additional thermal energy to overcome the barrier in the negative torsion direction. After the barrier, the pathway becomes steeper and finally results, at a C13C14 torsion value of ca. 110°, in an S0/S1 state crossing (CI+). Both optimised pathways are extremely flat, the difference in potential energy between the −172° S1 starting point and CI+ is ca. 8.8 kcal mol−1, even much smaller for the CI− point (ca. 1.25 kcal mol−1). During torsion, the nature of the first excited state smoothly changes from 45% HOMO–LUMO single excitation character up to 55% for both directions. Along both torsion paths, the bond length alternation increases. Shortly before the crossing, the HOMO–LUMO single excitation contribution drops, and the ground state configuration slowly but steadily increases in weight. At ca. −100 and +110° twist, we note a faster change in state character. Now the ground state configuration dominates the upper state, and the HOMO–LUMO excitation the lower state – a clear indication that a surface crossing point has been passed between this and the last point. It must be noted that the corresponding CASSCF-profiles show a barrier of ca. 3 kcal mol−1 on both sides in the corresponding CAS-S3 state, and all states are well separated at this level without any state crossing.
 | ||
| Fig. 5 State energy profiles for C13C14 torsion in 1Bu-like excited state. The path starts at ca. −172° twist in negative (left side of the graph) and positive directions (right side of the graph). The energy difference to the optimised CASSCF S0 geometry is given in eV. | ||
After excitation and bond relaxation, the β-ionone-part carries a charge of ca. +0.6 electrons, which is effectively transported to the Schiff base moiety during isomerisation (see ESI†).
During isomerisation in either direction, the whole fragment from C14 to the nitrogen Schiff base moves as one rigid part, i.e. the observed mechanism is somewhat different from the bicycle-pedal motion reported for other retinal proteins,21–23,38 where two double bonds rotate in different directions (Fig. 6). The reaction route shows much more similarities to the hula-twist mechanism suggested by Liu et al.,39 where torsion takes place around one single and one double bond. The corresponding single bond is the N–CH2 bond, which twists by up to 90°. The CN double bond rotates only by ca. 20° in positive direction and 40° for the negative path. In the negative path, the configuration of the N–CH2 single bond changes from syn towards anti, while the configuration is kept in the positive twisting direction.
 | ||
| Fig. 6 Evolution of torsion angles during simulated S1 torsion along C13C14 bond. | ||
In the negative torsion path, the hydrogen bonding to D292 is conserved at the crossing, while in the positive path the hydrogen bond is released, and the Schiff base hydrogen orients towards a nearby serine (S295, see ESI†).
Properties of 13-cis retinal in C1C2
To obtain the 13-cis retinal configuration in C1C2 we started a CASSCF single state optimisation in the ground state using as a starting point the ca. −120° twisted structure from the above mentioned torsion path. For comparability to the optimised all-trans structure, this optimisation was also carried out with both counter-ions movable. Fig. 7 shows an overlay of the 13-cis and all-trans optimised S0 structures. Significant changes are apparent along the conjugated chain and the orientation of the Schiff base nitrogen, which instead of pointing towards the ASP292 counter-ion, now has a weak hydrogen bond with SER295 (2.43 Å for the latter compared to 1.68 Å in all-trans and ASP292). Due to the 13-cis configuration this chromophore is slightly shorter than all-trans, which leads to a small clockwise tilt in the β-ionone ring, continuing along the chain. The C13 methyl group is strongly tilted, and the chromophore occupies slightly more volume in the fragment from C12 to lysine. This leads to enhanced van der Waals and electrostatic interactions, destabilizing the ground state QM/MM energy by ca. 31 kcal mol−1 for 13-cis at the MS-CASPT2 level. Interestingly, the chromophore changes its helicity from P to M during our simulated all-trans to 13-cis conversion. Its state compositions are very similar to the previously discussed all-trans retinal (see ESI†), with only 12 nm red shift. Thus, the two isomers will be difficult to distinguish in sole UV/Vis measurements. Better chances are provided by spectroscopic techniques considering the differences in vibrational modes in the two molecules in combination with UV/Vis excitation, i.e. resonance Raman spectroscopy, which we attempt to simulate in the following.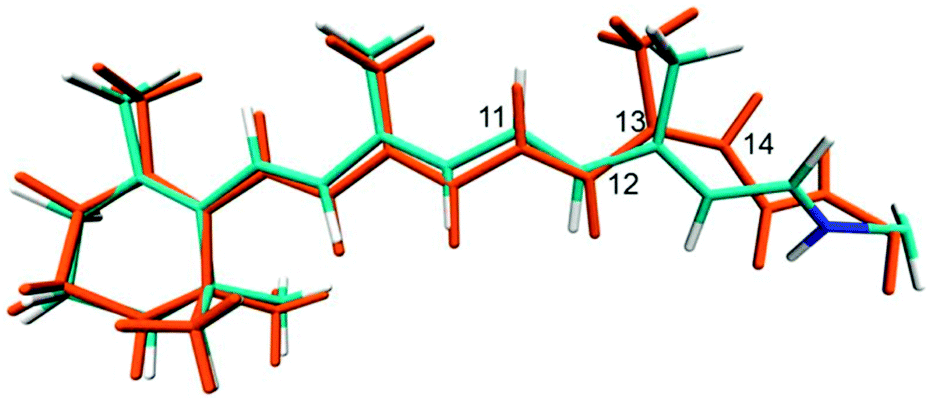 | ||
| Fig. 7 Overlay of CASSCF optimised ground state structures of all-trans retinal (cyan) and 13-cis retinal (orange) in C1C2. | ||
Resonance Raman spectra of all-trans and 13-cis C1C2
The procedure of CAS space reduction and QM/MM frontier shift (see Computational section) lead to three spurious low imaginary frequencies in both models. Further optimisation along these modes did not lead to structure relaxation and energy reduction. For generation of resonance Raman data, these modes were ignored. The obtained vibrational frequencies were scaled to match the highest intensity band in the experimental spectrum, defined by CC in plane stretching of the polyene fragment (1551 cm−1). The applied scaling factor was 0.877. Fig. 8 shows the computed resonance Raman spectra for all-trans retinal (black line) and 13-cis retinal (blue line) in comparison with the experimental spectrum obtained by Nack et al.12
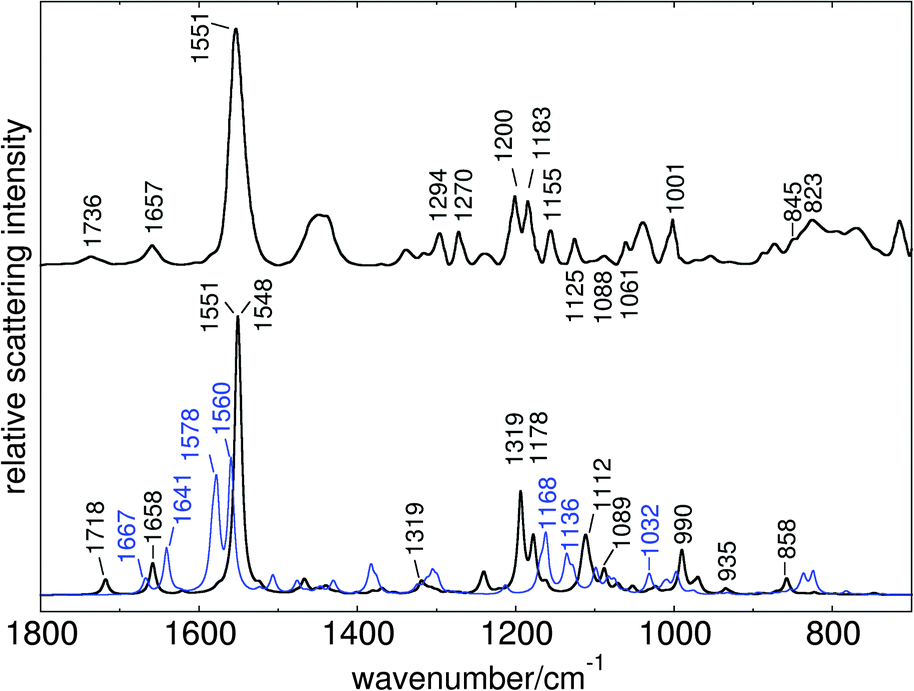 | ||
| Fig. 8 Computed resonance Raman spectra of all-trans retinal (black line) and 13-cis retinal (blue line) in C1C2, in comparison to experimental ChR2 spectrum of Nack et al. (data reproduced from Fig. 1 of ref. 12). | ||
The computed C1C2 spectrum shows good overall agreement with the experimental ChR2 spectrum, indicating distinct similarities in the geometrical and electrostatic environments of the binding pockets in C1C2 and ChR2. For both chromophores, the gradient of the 1Bu-like state (see ESI†) shows major components for bond length changes after excitation. The highest intensity band in our computed spectra is therefore produced by the double bond stretching modes of the C9–C14 fragment. In all-trans retinal this band consists of two separate close lying lines, describing C13C14 stretching (1551 cm−1) and combined C9C10 and C11C12 stretching (1548 cm−1). The 13-cis spectrum shows slightly shifted and more distinct bands for these modes (1560 and 1578 cm−1, respectively).
The two peaks left from the highest absorption band are coupled C15N stretching modes, which also appear in 13-cis retinal but significantly shifted. The in-plane bending modes of the vinyl hydrogens, which have been assigned to 1270 cm−1 by Nack et al., appear as lower intensity band at 1319 cm−1 in our spectrum. The 1240 cm−1 peak is produced by H11 in-plane bending. The two bands at 1193 cm−1 and 1178 cm−1 which are very close to the characteristic all-trans and 13-cis assigned bands of Nack et al., are due to single H14 in-plane bending and combined vinyl hydrogen in-plane bend including H10–H12. We suspect that in a combined 13-cis all-trans spectrum, the latter peak may be enhanced by combination with the shifted H10–H12 in-plane band at 1168 cm−1, because no separate band in 13-cis has enough intensity to produce a single peak at 1183 cm−1 when a 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 mixture of the two isomers is assumed.
:30 mixture of the two isomers is assumed.
Single bond stretchings, combined with CH2 twisting motions, are observed at 1089 cm−1 and around 1112 cm−1 in all-trans, and 1136 cm−1 in 13-cis. The gauche stretching bands in the latter appear at 1032 cm−1 with low intensity, again very close to the experimental assignment. Also the Me rocking band at 1001 cm−1 is well reproduced (990 cm−1). HOOP motion is observed at 858 cm−1 (mainly H14) and with very low intensity at 935 cm−1 (symmetric H11–H12 HOOP). The two small peaks at the right end of the 13-cis spectrum are due to H12 in-plane bending.
The calculated frequencies also allow the computation of the IR difference spectrum between the two species. This spectrum, which is in valuable agreement with the recent findings of Furutani et al.,40 is reported in the ESI.†
Influence of the electrostatic environment on retinal absorption
The contribution of selected protein residues to the retinal maximum absorption was investigated by computing their absorption differences to the uncharged protein pocket. In the choice of potentially contributing amino acids we follow the suggestions of Kamyia et al., who prodived a list of 12 charged and polarizable residues in the vicinity of the retinal chromophore as potential candidates for absorption-regulating mutations.17 These are: E122, E129, K132, C167, H173, D195, G220, F228, S295, N297 and finally the counter-ions E162 and D292 (see ESI† for the location of these amino acids in the retinal binding pocket).The charges of all named amino acids were initially set to zero. The computed excitation energy of this configuration is used as a reference value for the uncharged environment. To study the effect of one single amino acid, we insert the charges of the specific residue, compute the absorption with MS-CASPT2 and build the difference from the reference value. As before, the SS-CASPT2 computed values largely overestimate the contribution of the charged residues.
The computed values in eV are summarised in Fig. 9. On the right-hand side the overall effect of the remaining protein backbone (compared to the retinal vacuum absorption) becomes apparent. Its contribution is a slight blue shift with a value <0.15 eV. Only K132, situated below the Schiff base terminus, contributes, in sum with the protein backbone, a significant red shift in comparison to the vacuum absorption. All other amino acids only slightly modulate the blue shift induced by the remaining protein residues. The strongest blue shift contribution results from the two counter-ions E162 (0.260 eV) and D292 (0.261 eV), followed by S295 (0.065 eV). It should be noted that the contribution of both counter-ions results in a higher value (0.728 eV) than the sum of their single contributions.
 | ||
| Fig. 9 Electrostatic influence of protein residues suggested by Kamiya et al.17 on S0–S1 excitation energy of retinal in C1C2. The green bar indicates the shift with respect to vacuum, resulting from the remaining protein backbone. | ||
Conclusions
Static aspects and reaction path of the retinal photoconversion in C1C2 have been investigated with QM/MM methods and were compared to experimental findings. A crucial point in the generation of valid data is the choice of the quantum mechanical treatment to describe the chromophore in its electrostatic environment. Geometry optimisations of the chromophore in the protein binding pocket with HF, DFT and CASSCF methods lead to a slightly different orientation of the NH+ Schiff base moiety with respect to the crystal structure. In all models, this fragment moves towards the suggested primary counter-ion D292 and forms a strong hydrogen bond with the latter, an effect which might be balanced by the initial configuration of the crystal structure and the restrictions applied in our simulations. We could however show, that relaxation of the binding pocket does not destabilise this configuration, and we were able to obtain a stable structure with hydrogen bonding to E162 through a different optimisation strategy (see ESI†). The latter shows a similar absorption value and state composition, such that, from a computational point of view, both initial configurations appear plausible.The optimisation with DFT and CASSCF methods furthermore leads to a strong difference in bond length alternation of the chromophore. Similar to observations made by Larsson and coworkers for retinal in vacuo,41 DFT tends to overestimate the conjugation along the chain, while CASSCF yields a more polyenic structure with larger BLA. The level of conjugation however strongly affects the maximum absorption of the chromophore.42 In any case, for an adequate description of excitation energies, especially with charged residues in the vicinity of the chromophore, a multi-state multi-reference treatment is inevitable. The best agreement with the experimental 470 nm value was obtained by using an MS-CASPT2 strategy on top of the CASSCF(12,12)//Amber optimised chromophore, employing simultaneous optimisation of both counter-ion positions (479 nm). Single state CASPT2 largely overestimates the influence of the nearby charged residues and consequently yields too high excitation energies (see ESI†). DFT-MRCI yields reasonable results with the CASSCF geometry (452 nm), but underestimates the absorption with a DFT-optimised structure (498 nm). The missing polarisation contribution in our study may slightly alter these values, a lowering of the excitation energy by ca. 5 kcal mol−1 has e.g. been reported in the study of Sneskov et al. due to polarisation effects. Consideration of excitonic coupling effects and charge transfer, as e.g. applied in the works of Hasegawa et al.43 and Wanko et al.,44 may further improve the obtained values. Recently, it has been reported that MS-CASPT2 may overestimate the state energy splitting near conical intersections.45 Application of more elaborate XMS-CASPT2 or XMCQDPT2 strategies may thus lead to more accurate PESs near the S1/S0 state crossing in the studied system.
The alternative protonation of the E162 residue yields in all computed models a too red-shifted absorption value. From this point of view, it does not significantly improve the initially suggested two-counter-ion model, suggesting that E162 should be deprotonated. A final conclusion on this issue can however not be drawn solely based on the comparison of the absorption values in our models.
The first excited state is associated with 1Bu-like HOMO–LUMO excitation with large oscillator strength. The higher S2 and S3 states also exhibit small but non-negligible contributions in the corresponding part of the absorption spectrum. In MS-CASPT2, the second excited state is similar to the polyenic 2Ag state, with significant contribution of HOMO → LUMO double and HOMO−1 → LUMO single excitations. Higher excited states are of strongly mixed character.
At the Franck–Condon (FC) point, the S1 and S2 states are well separated. This situation changes dramatically after bond length relaxation in S1. The energy difference drops to 7.3 kcal mol−1, thus, further interaction of these states in the photoconversion dynamics cannot be consequently excluded. Bond relaxation in S1 leads to equalisation of the bond lengths along the conjugated chain, very much like in the 1Bu state of symmetric carotenoids. This is a consequence of the nearby charges, which hinder the relocation of the positive charge at the Schiff base terminus upon excitation, thus resembling the situation in the neutral symmetric polyenes where no large charge migration takes place. As a consequence, the retinal chromophore has a more “polyenic” character in C1C2.
Different from reaction pathways in bR,21 Rh14 and anabaena sensory Rh,22 which are strictly unidirectional, the computed CD-MS-CASPT2//CASSCF profile along the C13C14 torsion suggests a bidirectional pathway in the conversion of C1C2. The negative torsion path in C1C2 has a small barrier of ca. 3 kcal mol−1 which, however, can easily be overcome with thermal excess energy (i.e. vibrational energy) after FC excitation. The location of the barrier may switch to the other side of the torsion potential, when retinal establishes a salt bridge with D292. With these properties, the reaction dynamics of retinal in C1C2 may share more similarities to the photoprocess observed in iso-rhodopsin as in bR.46 Reaction rates, the distribution in the proposed reaction channels and resulting photoproducts can, however, only be obtained through molecular dynamics computations, which due to the state order problems at the CASSCF level with electrostatic embedding will form a major challenge.
In the 13-cis isomer, which was generated from the computed positive torsion path, the hydrogen bonding to the assumed primary proton acceptor D292 is released, and the Schiff base proton unexpectedly points toward a neighboring serine. During the ultrafast photoisomerisation, the protein environment will not participate, therefore we performed the optimisation within a frozen environment. Proton release however is reported to take place within 700 ns, which is enough time for the protein to reorient and offer a new partner for deprotonation of the Schiff base.
Computational C1C2 resonance Raman spectra indicate significant differences for all-trans and 13-cis chromophores. The computed all-trans retinal spectrum reproduces location and intensity of distinct bands in ChR2, indicating that, despite their differences in protein sequence, the steric and electrostatic environments which influence the excited state properties of retinal are not too different in C1C2 and ChR2.
Finally, electrostatic screening of the nearby residues reveals 3 sites with major influence on the retinal absorption: the two counter-ions E162 and D292 are mainly responsible for the strong blue shift, while a lysine K132 positioned close to the Schiff base terminus induces a major red shift. The contribution of other residues is only minor.
Acknowledgements
We thank Prof. Christel Marian (University of Düsseldorf) and Prof. Marco Garavelli (ENS Lyon) for valuable discussions. Financial support was provided through “Strategischer Forschungsfonds” of the Heinrich-Heine-University Düsseldorf, project no. F 2013 – 442-9. Computational support and infrastructure was provided by the “Centre for Information and Media Technology” (ZIM) at the University of Düsseldorf (Germany).Notes and references
- H. Harz and P. Hegemann, Nature, 1991, 351, 489 CrossRef CAS PubMed.
- S. Ehlenbeck, G. Gradmann, F.-J. Braun and P. Hegemann, Biophys. J., 2001, 82, 740 CrossRef.
- G. Nagel, T. Szellas, W. Huhn, S. Kateriya, N. Adeishvili, P. Berthold, D. Ollig, P. Hegemann and E. Bamberg, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13940 CrossRef CAS PubMed.
- G. Nagel, D. Ollig, M. Fuhrmann, S. Kateriya, S. Musti, E. Bamberg and P. Hegemann, Science, 2002, 296, 2395 CrossRef CAS PubMed.
- R. R. Birge, Annu. Rev. Phys. Chem., 1990, 41, 683 CrossRef CAS PubMed.
- E. S. Boyden, F. Zhang, E. Bamberg, G. Nagel and K. Deisseroth, Nat. Neurosci., 2005, 8, 1263 CrossRef CAS PubMed.
- X. Li, et al. , Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17816 CrossRef CAS PubMed.
- A. Bi, J. Cui, Y.-P. Ma, E. Olshevskaya, M. Pu, A. M. Dizhoor and Z.-H. Pan, Neuron, 2006, 50, 23 CrossRef CAS PubMed.
- M.-K. Verhoefen, C. Bamann, R. Blöcher, U. Förster, E. Bamberg and J. Wachtveitl, ChemPhysChem, 2010, 11, 3113 CrossRef CAS PubMed.
- P. Hegemann, W. Gärtner and W. Uhl, Biophys. J., 1991, 60, 1477 CrossRef CAS.
- H. E. Kato, F. Zhang, O. Yizhar, C. Ramakrishnan, T. Nishizawa, K. Hirata and J. Ito, et al. , Nature, 2012, 482, 369 CrossRef CAS PubMed.
- M. Nack, I. Radu, C. Bamann, E. Bamberg and J. Heberle, FEBS Lett., 2009, 583, 3676 CrossRef CAS PubMed.
- N. Ferré and M. Olivucci, J. Am. Chem. Soc., 2008, 125, 6868–6869 CrossRef PubMed.
- (a) V. Bonačić-Koutecký, K. Schöffel and J. Michl, Theor. Chim. Acta, 1987, 72, 459–474 CrossRef; (b) M. Garavelli, F. Bernardi, M. Olivucci, T. Vreven, S. Klein, P. Celani and M. A. Robb, Faraday Discuss., 1998, 110, 51–70 RSC; (c) G. Tomasello, G. Olaso-González, P. Altoè, M. Stenta, L. Serrano-Andrés, M. Merchán, G. Orlandi, A. Bottoni and M. Garavelli, J. Am. Chem. Soc., 2009, 131, 5172–5186 CrossRef CAS PubMed.
- (a) P. Tavan and K. Schulten, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 36, 4337–4358 CrossRef CAS; (b) S. Knecht, C. M. Marian, J. Kongsted and B. Menucci, J. Phys. Chem. B, 2013, 117, 13808 CrossRef CAS PubMed; (c) C. M. Marian and N. Gilka, J. Chem. Theory Comput., 2008, 4, 1501 CrossRef CAS.
- K. Welke, H. C. Watanabe, T. Wolter, M. Gaus and M. Elstner, Phys. Chem. Chem. Phys., 2013, 15, 6651 RSC.
- M. Kamiya, H. E. Kato, R. Ishitani, O. Nureki and S. Hayashi, Chem. Phys. Lett., 2013, 556, 266 CrossRef CAS PubMed.
- K. Sneskov, J. M. H. Olsen, T. Schwabe, C. Hättig, O. Christiansen and J. Kongsted, Phys. Chem. Chem. Phys., 2013, 15, 7567 RSC.
- D. Polli, I. Rivalta, A. Nenov, O. Weingart, M. Garavelli and G. Cerullo, Photochem. Photobiol. Sci., 2015, 14, 213 CAS.
- I. Schapiro and S. Ruhman, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 589–597 CrossRef CAS PubMed.
- A. Strambi, B. Durbeej, N. Ferré and M. Olivucci, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21322–21326 CrossRef CAS PubMed.
- P. Altoè, A. Cembran, M. Olivucci and M. Garavelli, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20172–20177 CrossRef PubMed.
- A. Warshel, Nature, 1976, 260, 679–683 CrossRef CAS PubMed.
- P. Altoé, M. Stenta, A. Bottoni and M. Garavelli, Theor. Chem. Acc., 2007, 118, 219 CrossRef.
- D. A. Case, T. A. Darden, T. E. Cheatham, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Goetz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M. J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, Amber 12, 2012 Search PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950 CAS.
- M. H. M. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and A. Wolf, Molpro 2010, 2010 Search PubMed.
- S. Grimme and M. Waletzke, J. Chem. Phys., 1999, 111, 5645 CrossRef CAS PubMed.
- G. Karlström, R. Lindh, P.-Å. Malmqvist, B. O. Roos, U. Ryde, V. Veryazov, P.-O. Widmark, M. Cossi, B. Schimmelpfennig, P. Neogrády and L. Seijo, MOLCAS: a program package for computational chemistry, Comput. Mater. Sci., 2003, 28, 222 CrossRef.
- TURBOMOLE V6.3 2010, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- A. ten Wolde, H. J. C. Jacobs, F. Langkilde, K. Bajdor, R. Wilbrandt, F. Negri, F. Zerbetto and G. Orlandi, J. Phys. Chem., 1994, 98, 9437 CrossRef CAS.
- M. Garavelli, F. Negri and M. Olivucci, J. Am. Chem. Soc., 1999, 121, 1023 CrossRef CAS.
- S. Ito, H. E. Kato, R. Taniguchi, T. Iwata, O. Nureki and H. Kandori, J. Am. Chem. Soc., 2014, 136, 3475 CrossRef CAS PubMed.
- J. Kuhne, K. Eisenhauer, E. Ritter, P. Hegemann, K. Gerwert and F. Bartl, Angew. Chem., Int. Ed., 2015, 54, 4953–4957 CrossRef CAS PubMed.
- E. N. Laricheva, S. Gozem, S. Rinaldi, F. Melaccio, A. Valentini and M. Olivucci, J. Chem. Theory Comput., 2012, 8(8), 2559–2563 CrossRef CAS.
- K. C. Hasson, F. Gai and P. A. Anfinrud, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 15124 CrossRef CAS.
- D. Polli, P. Altoè, O. Weingart, K. M. Spillane, C. Manzoni, D. Brida, G. Tomasello, G. Orlandi, P. Kukura, R. A. Mathies, M. Garavelli and G. Cerullo, Nature, 2010, 467, 440–443 CrossRef CAS PubMed.
- R. Liu, Acc. Chem. Res., 2001, 34, 555 CrossRef CAS PubMed.
- A. Inaguma, H. Tsukamato, H. E. Kato, T. Kimura, T. Ishizuka, S. Oisho, H. Yawo, O. Nureki and Y. Furutani, J. Biol. Chem., 2015, 290, 11623–11634 CrossRef CAS PubMed.
- F. Blomgren and S. Larsson, J. Comput. Chem., 2005, 26, 738–742 CrossRef CAS PubMed.
- P. D. Patel and A. E. Masunov, J. Phys. Chem. A, 2009, 113, 8409 CrossRef CAS PubMed.
- J. Hasegawa, Chem. Phys. Lett., 2013, 571, 77–81 CrossRef CAS PubMed.
- M. Wanko, M. Hoffmann, T. Frauenheim and M. Elstner, J. Phys. Chem. B, 2008, 112, 11462–11467 CrossRef CAS PubMed.
- S. Gozem, M. Huntress, I. Schapiro, R. Lindh, A. Granovsky, C. Angeli and M. Olivucci, J. Chem. Theory Comput., 2012, 8, 4069–4080 CrossRef CAS.
- D. Polli, O. Weingart, D. Brida, E. Poli, M. Maiuri, K. M. Spillane, A. Bottoni, P. Kukura, R. A. Mathies, G. Cerullo and M. Garavelli, Angew. Chem., Int. Ed., 2014, 53, 2504 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Excitation energies, detailed resonance Raman data and Cartesian coordinates. See DOI: 10.1039/c5cp02650d |
| This journal is © the Owner Societies 2015 |