
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of water on the binding of glycosaminoglycan saccharides to hydroxyapatite surfaces: a molecular dynamics study†
Sergio E.
Ruiz Hernandez
*ab,
Ian
Streeter
c and
Nora H.
de Leeuw
ab
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
bSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: RuizHernandezS@cardiff.ac.uk
cEMBL-European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, CB10 1SD, UK
First published on 31st July 2015
Abstract
Classical molecular dynamics (MD) simulations have been employed to study the interaction of the saccharides glucuronic acid (GlcA) and N-acetylgalactosamine (GalNAc) with the (0001) and (01![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) surfaces of the mineral hydroxyapatite (HAP). GlcA and GalNAc are the two constituent monosaccharides of the glycosaminoglycan chondroitin sulfate, which is commonly found in bone and cartilage and has been implicated in the modulation of the hydroxyapatite biomineralization process. MD simulations of the mineral surfaces and the saccharides in the presence of solvent water allowed the calculation of the adsorption energies of the saccharides on the HAP surfaces. The calculations show that GalNAc interacts with HAP principally through the sulfate and the carbonyl of acetyl amine groups, whereas the GlcA interacts primarily through the carboxylate functional groups. The mode and strength of the interaction depends on the orientation of the saccharide with respect to the surface and the level of disruption of the layer of water competing with the saccharide for adsorption sites on the HAP surface, suggesting that chondroitin 4-sulfate binds to the layer of solvent water rather than to HAP.
0) surfaces of the mineral hydroxyapatite (HAP). GlcA and GalNAc are the two constituent monosaccharides of the glycosaminoglycan chondroitin sulfate, which is commonly found in bone and cartilage and has been implicated in the modulation of the hydroxyapatite biomineralization process. MD simulations of the mineral surfaces and the saccharides in the presence of solvent water allowed the calculation of the adsorption energies of the saccharides on the HAP surfaces. The calculations show that GalNAc interacts with HAP principally through the sulfate and the carbonyl of acetyl amine groups, whereas the GlcA interacts primarily through the carboxylate functional groups. The mode and strength of the interaction depends on the orientation of the saccharide with respect to the surface and the level of disruption of the layer of water competing with the saccharide for adsorption sites on the HAP surface, suggesting that chondroitin 4-sulfate binds to the layer of solvent water rather than to HAP.
1 Introduction
Mineralised tissues such as bone and tooth enamel have a complex composite micro-architecture, in which an underlying matrix of fibrous organic material becomes strengthened by the deposition of inorganic apatite crystals. This biological apatite is similar in stoichiometry and structure to hydroxyapatite (HAP),1 Ca10(PO4)6(OH)2, but with crystal defects such as substitutions by carbonate, water and various metal cations.2 The microscopic structures of these mineralised tissues are ultimately regulated by cells such as osteoblasts, ameloblasts and odontoblasts (in bone, tooth enamel and dentine, respectively), which are able to control the growth of apatite crystals to a specific size, shape, orientation, chemical stoichiometry, and spatial distribution. It is remarkable that cells achieve this level of control when they can only manipulate the apatite via an indirect approach: by secreting various biomolecules into the surrounding tissue they create chemical conditions that are favourable for the spontaneous extracellular formation of the appropriate apatite structures.Glycosaminoglycans (GAGs) are polysaccharide molecules that are abundant in mineralised tissues, and are known to interact strongly with apatite crystals.3–5 A GAG molecule is an unbranched polymer chain of two different types of monosaccharides linked together in alternation.6 The most prevalent GAG found in mineralised tissues is chondroitin sulfate, shown in Fig. 1(a), in which the alternating monosaccharides are glucuronic acid (GlcA) and N-acetylgalactosamine (GalNAc) shown in Fig. 1(b) and (c) respectively. This GAG can be sulfated to some extent on any of its hydroxy groups, and the most commonly sulfated position is the 4-hydroxy group of the GalNAc monosaccharide. GAGs, including chondroitin sulfate, are usually found as part of a larger macromolecular complex known as a proteoglycan, in which multiple GAG molecules are covalently attached to a central protein, and each GAG chain typically comprises 40–120 monosaccharide units.7
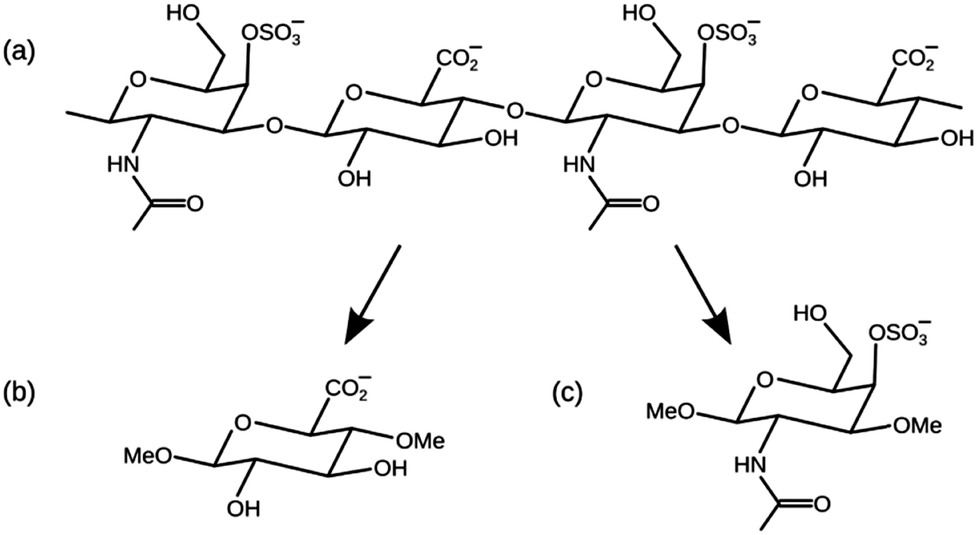 | ||
| Fig. 1 (a) A short fragment of chondroitin-4-sulfate. (b) Glucuronic acid (GlcA) methylated in the 1- and 4-hydroxy positions. (c) N-Acetylgalactosamine-4-sulfate (GalNAc-4S) methylated in the 1- and 3-hydroxy positions. | ||
There are a number of ways in which the biomolecules secreted by cells can influence the mechanism of tissue mineralisation: either by providing a template for crystal nucleation, by adsorbing onto crystal surfaces and thus inhibiting crystal growth, or by sequestering dissolved mineral ions.8 A plethora of biomolecules have been implicated as either promoters or inhibitors of tissue mineralisation, including GAG-containing proteoglycans9 and the collagen,10 alkaline phosphatase11 and SIBLING proteins.12 It is likely, however, that mineralisation is controlled by many different organic molecules simultaneously, with exquisite control of the process effected by the cell's use of system feedback loops.
It has been found that under in vitro conditions of limited calcium availability, GAGs, including chondroitin sulfate, and their proteoglycans inhibit the deposition of hydroxyapatite, perhaps because the adsorption of GAG molecules on the mineral surfaces is an obstacle to the mechanism of crystal growth.3–5 The role of proteoglycans in the mechanism of apatite formation in vivo is more difficult to interpret, because proteoglycans and GAGs have multiple functions in physiological tissues, and interact with many other biomolecules and with cells.8,9,13–15 Studies in vivo and in cell culture have often produced conflicting results, with proteoglycans observed either to promote or to inhibit apatite formation.16 Evidence for the importance of the GAG–apatite interface in vivo has come from solid-state NMR experiments of bones,17,18 teeth,19 plaque20 and kidney stones:21 in all cases 31P–13C coupling showed that the apatite mineral was predominantly in contact with GAG molecules rather than with any other organic component.
In this paper we have used computational methods to investigate the interfacial interactions between GAGs and HAP. We present molecular dynamics (MD) simulations of chondroitin sulfate saccharides adsorbed onto a slab of HAP in the presence of interfacial water. Adsorption structures have been calculated for the (0001) surface of HAP, which is the thermodynamically most stable surface, and the (010) surface, which is the dominant plane of apatite nanocrystals in a biological environment.22 We have simulated the adsorption of the monosaccharides GlcA and GalNAc 4-sulfate (GalNAc-4S) independently, rather than longer chondroitin sulfate chains, because this reduces the number of possible HAP–saccharide interactions in each simulation, and simplifies the interpretation of the results. The simulated monosaccharides were methylated in two different positions, as shown in Fig. 1, in order to the reproduce the effect of their position within a long polymer GAG chain.
Classical MD simulations have been used previously to investigate the adsorption of small organic molecules at an apatite–water interface.23,24 For these molecules it was reported that the principal adsorption interactions were the electrostatic attractions between surface calcium ions and the adsorbate oxygen and nitrogen atoms, and that an important secondary interaction was hydrogen-bonding from adsorbate polar hydrogen atoms to the surface phosphate oxygen atoms. Recently we have used DFT calculations to predict the adsorption structures of GlcA and GalNAc (not sulfated) on HAP in vacuo.25 It was found that GalNAc interacts with HAP principally through its hydroxy and acetyl amine functional groups, and that GlcA interacts principally through its hydroxy and carboxylate functional groups. However, the inclusion of interfacial water in the present study will give us a better understanding of adsorption processes under more realistic physiological conditions, rather than in vacuo.
2 Methods
The adsorption of GAG saccharides on HAP surfaces was modelled using interatomic potential-based molecular dynamics simulations in order to calculate the structures and energies of these systems. MD trajectories were calculated using the DL_POLY_2 code,26 with an integration algorithm based on the Verlet leap-frog scheme.27 The simulations were performed at constant volume (using cell parameters obtained from constant pressure calculations in the bulk) and constant temperature (310 K) using the Nosé–Hoover thermostat28,29 with 0.5 ps for the thermostat and barostat relaxation times.Following conventional MD simulation procedures, the system was described by a simulation cell with three-dimensional periodic boundary conditions, containing a slab of HAP with a surface on either side. The thickness of the slab was greater than 20 Å for each surface studied, and the slab was separated by a gap of approximately 53 Å from its images in the next cell. Under these conditions the system was run for 1200 ps with 400 ps of equilibration. Then the gap between the HAP slabs was filled with water molecules, leading to a simulation cell containing approximately 3000 atoms in total. Now the simulation times were increased to 1600 ps with 400 ps of equilibration. The Shake algorithm was also employed to constrain intramolecular hydrogen-bonds.
2.1 Interatomic potential model
A combination of interatomic potential models was employed to describe the interactions of this multi-component system. We have used the Glycam06 forcefield for the saccharide molecules and for non-bonded saccharide–water interactions. The Glycam06 forcefield is an established parameter set optimised specifically for carbohydrate molecules,30 in which bond lengths and angles are described by harmonic potentials, dihedral angles by sinusoidal potentials, and non-bonded interactions by Lennard-Jones potentials. Intramolecular 1–4 (and greater) electrostatic and Van der Waals interactions are non-zero and are unscaled. The water molecules were described by the TIP3P model, to ensure compatibility with the parameterisation of the saccharide in the Glycam06 forcefield, which includes TIP3P water.The mineral slab was described by the interatomic potential model developed by de Leeuw for modelling apatite crystals.31 In this forcefield, phosphate and hydroxy group bonds are parameterised as the sum of a Morse potential and a coulombic potential, phosphate bond angles by a harmonic potential, and non-bonded interactions by Buckingham potentials (see Table S1, ESI†). This forcefield makes use of a shell model,32 in which each oxygen anion in the phosphate and hydroxy groups consists of both a core and a massless shell connected by a spring, in order to model the atom's electronic polarisability. The forcefield has been validated by comparison with DFT calculations,31 and produces consistent defect and dissolution energies.33–35
HAP–water interactions and HAP–GAG interactions were parameterised using non-bonded potentials derived in previous MD studies,23,24 which are compatible with the current HAP shell model but do not consider pH effects (see Section 2.2). Specifically, the interfacial interactions were described by Buckingham potentials and Lennard-Jones potentials, and the repulsive portion of the potential was scaled for consistency with the charges on the adsorbate atoms, using a method originally described by Schröder et al.,36 and subsequently used in a number of surface adsorption studies.23,24,31,37,38
2.2 Surface structures
HAP (Fig. 2) is found experimentally to have a P63/m space group, for which the hexagonal unit cell parameters are a = b = 9.432 Å, c = 6.881 Å, α = β = 90° and γ = 120°.1 The mineral contains tetrahedral phosphate groups, hydroxy groups, and calcium cations. There are two different Ca2+ sites in the HAP crystals: ‘columnar’ Ca2+ ions (CaI), which are arranged in single-ion columns parallel to the c axis and are bound by phosphate tetrahedra, and ‘axial’ Ca2+ ions (CaII), which form hexagonal columns, at the centre of which are the hydroxy groups.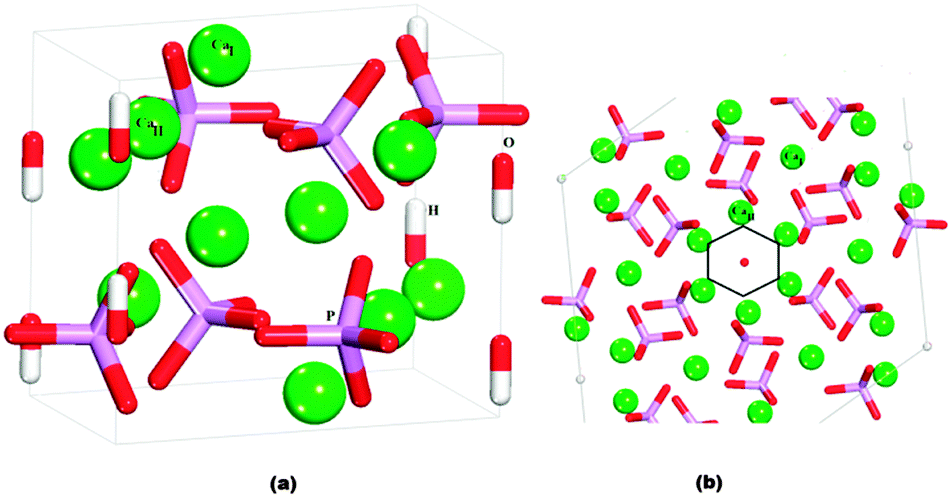 | ||
| Fig. 2 (a) Hydroxyapatite unit cell. (b) View onto (0001) surface, showing the hexagonal channels: Ca green, P purple, O red, H white. CaI atoms are columnar calcium and CaII atoms are axial calcium. | ||
The crystallographic structure of HAP contains columns of hydroxy groups in the hexagonal channels that are oriented parallel to the c axis. The OH groups are oriented either hydrogen "up" or hydrogen "down" in the channels, where all "up" and "down" OH− positions have occupancy of 0.5.1 In an MD simulation the occupancy of a site is necessarily restricted to either unity or zero, so in our simulated slabs of HAP the hydroxy groups were placed in only half of the possible positions, such that within each column they all pointed in the same direction. The unit cell was then doubled in the b direction, so that neighbouring columns alternated in the orientation of the hydroxy groups. Previous DFT calculations have identified this arrangement of hydroxy groups as the lowest energy configuration of bulk HAP,39 and it is similar to the synthetic monoclinic HAP material, where all hydroxy positions are fully ordered in a similar configuration as modelled here.
In calcium deficient environments (i.e. bones, teeth), the apatite obtained is nonstoichiometric, whereas the surface phosphate ions are usually protonated,40–45 with the protonation rate of the ions typically ranging from PO3–OH at pH ∼10.5 to PO2–(OH)2 for pH ∼5.5.46
We have used three different HAP slabs for the MD simulations of the adsorption of GAGs, which are shown in Fig. 3. We have considered the low index (0001) and (010) crystal surfaces, which are both present in the experimental morphology of HAP, where for the (010) surface we have considered two different possible terminating planes.
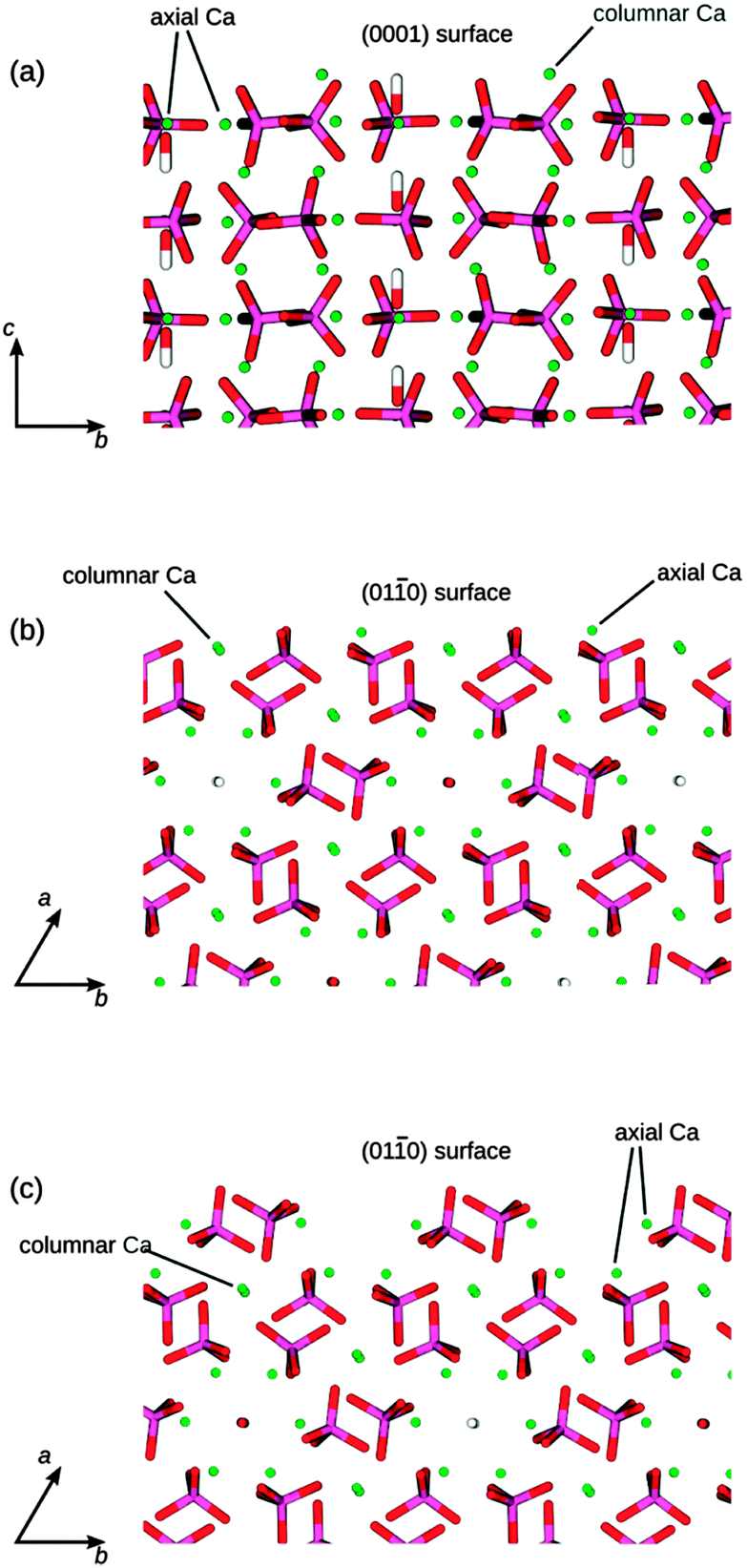 | ||
| Fig. 3 The three different HAP surfaces shown at the top of each side view image: (a) (0001) surface, (b) flat (010) surface, cutting through the columns of Ca atoms, (c) crenellated (010) surface, cutting through the hydroxy channels. P is purple, O is red, Ca is green, and H is white. | ||
The (010) termination shown in Fig. 3b contains columnar Ca atoms in the surface, with the hydroxy channels remaining intact below the surface, whereas the terminating plane in Fig. 3c cuts through the hydroxy channels and leaves the columnar Ca atoms below the surface. The surface in Fig. 3b has been identified previously as the most stable (010) termination, using computational methods,47 but the surface in Fig. 3c has been identified as the principal (010) surface of fluorapatite using high-resolution specular X-ray reflectivity data.48 We note that both of these possible terminations are likely to be present transiently as a crystal grows in the (010) direction.
These three stoichiometric surface structures allow us to compare our results with previous theoretical results23,24,38,49 while also keeping the Ca/PO4 ratio, which is important to investigate experimental findings regarding the high Ca2+ binding capacity of chondroitin 4-sulfate.50
2.3 Adsorption calculations
The monosaccharides GlcA and GalNAc-4S were placed near the HAP slabs in a number of different positions and orientations with respect to the surface. We added one saccharide molecule per surface simulation cell, which was large enough to avoid interactions between it and the saccharide molecules in neighbouring repeat cells.For each monosaccharide and for each HAP slab, we also simulated the system where the saccharide was fully solvated, i.e. the saccharide was situated in the solution midway between the upper and lower surfaces of two slabs from neighbouring repeat cells. The adsorption energy of a saccharide on a particular surface, Eads, is given by:
| Eads = Esys,ads − Esys,solv | (1) |
The hydration energy of the surfaces is given by:
| Ehyd = (Esurf,solv − Esurf + nEH2O)/A | (2) |
3 Results and discussion
In order to obtain a plausible initial orientation of the monosaccharides over the three HAP surfaces, while reducing the number of possibilities and consequently the computational cost of the more realistic calculations in aqueous environment, we have first carried out simulations of both surface and GAG systems in a vacuum. The final configurations of the GAGs at the surfaces in the absence of solvent were used as starting points for a more complex scenario in the presence of water.3.1 Surfaces in vacuum
In the absence of solvent the three surfaces evolve in various ways. The first and, to a lesser extent, the second layers of atoms in the surface slabs readjust their positions to re-balance the forces, which have become unbalanced upon the creation of the surfaces from the bulk material. We observe rotations of the phosphate groups and displacements of the topmost calcium ions to regain the maximum possible coordination. The P–O bond distances remain ∼1.55 Å with the Ca–O distances ranging from 2.15 to 2.88 Å. The (0001) surface is the only surface with hydroxy groups and both types of Ca2+ in the first layer of the slab (Fig. 3a). The columnar calcium ions (CaI) form one extra bond, to a total of six bonds (out of nine in the bulk) with a phosphate oxygen (OPO3), while the axial calcium ions (CaII) also form an additional bond with the oxygen of the hydroxy group, which has moved closer to the cations. In this termination of the surface we do not observe the OH groups moving more into the bulk of the material, as was reported after relaxation for a different termination.24 The flat (010) surface (Fig. 3b) also has the two types of calcium in the top layer of the slab and they both form an additional bond with the phosphate oxygen. This surface has more calcium in the top layer than the (0001), and therefore shows the greatest rotations of the phosphate groups to enhance their coordination. We also observe that some of the columnar calcium ions move into the bulk to retain the coordination with the phosphate group that has rotated to coordinate to CaII. In the crenellated (010) surface the phosphate groups experience little rotation to form two extra bonds between their oxygen and the axial calcium ions in the trench of the surface.
3.2 Monosaccharides in vacuum
The glycosaminoglycans are polar molecules and highly soluble. We have first simulated the two monosaccharides in the absence of solvent to analyse the conformational changes that might occur once the molecule interacts with the surfaces. Under these conditions we observe that the methylated N-acetylgalactosamine-4-sulfate (GalNAc-4S) remains in the chair conformation, with the acetyl-amine group in an equatorial position and both methoxy groups in equatorial positions. These methoxy groups represent the oxygen-linked attachments to the next saccharides in the GAG polymer (see Fig. 1), whereas the sulfate group is in an axial position forming intramolecular hydrogen bonds between its O and the H of the hydroxy functional group. Methylated glucuronic acid (GlcA), is a smaller molecule and without solvent the conformation of the molecule is more variable than GalNAc-4S. The methoxy and hydroxy groups alternate between equatorial and axial positions, whereas the carboxylate also moves between the equatorial to a pseudo-axial position to form hydrogen-bonds between its oxygen and the hydrogen of hydroxy group.3.3 Adsorption of GlcA on the surfaces in vacuum
The saccharide was placed onto the three surfaces in twelve different initial orientations following chemical intuition. Under these conditions the GlcA molecule often changed its conformation in order to direct all of its functional groups towards the surfaces, even though there is an energy penalty (173 kJ mol−1) associated with this change in conformation, which is, however, outweighed by the energetically favourable interactions with the surfaces. In all positions, the carboxylate group is directed towards the surface, with examples of the interactions on the (010) surfaces shown in Fig. 4a and b. Fig. 4 shows that sometimes the carboxylic oxygen is bridging two axial Ca2+ or one columnar and one axial Ca2+ on the flat (010) surface (Fig. 4a). The interactions of the carboxylate groups with the Ca2+ of HAP has been reported previously in the literature,51–53 It binds more strongly to the columnar calcium ions than to the axial calcium ions in the surface, because the columnar calcium ions are more closely spaced in the plane (3.48 Å), and the carboxylate can hence bridge more easily between two columnar calcium ions. For the axial calcium atoms (6.95 Å spacing in the surface) the carboxylate can only bind to one Ca2+ at a time, which is less energetically favourable. On the (0001) the molecule lies flat on the surface in all cases, reaching the minimum energy value when the molecule adsorbs as shown in Fig. 4c.
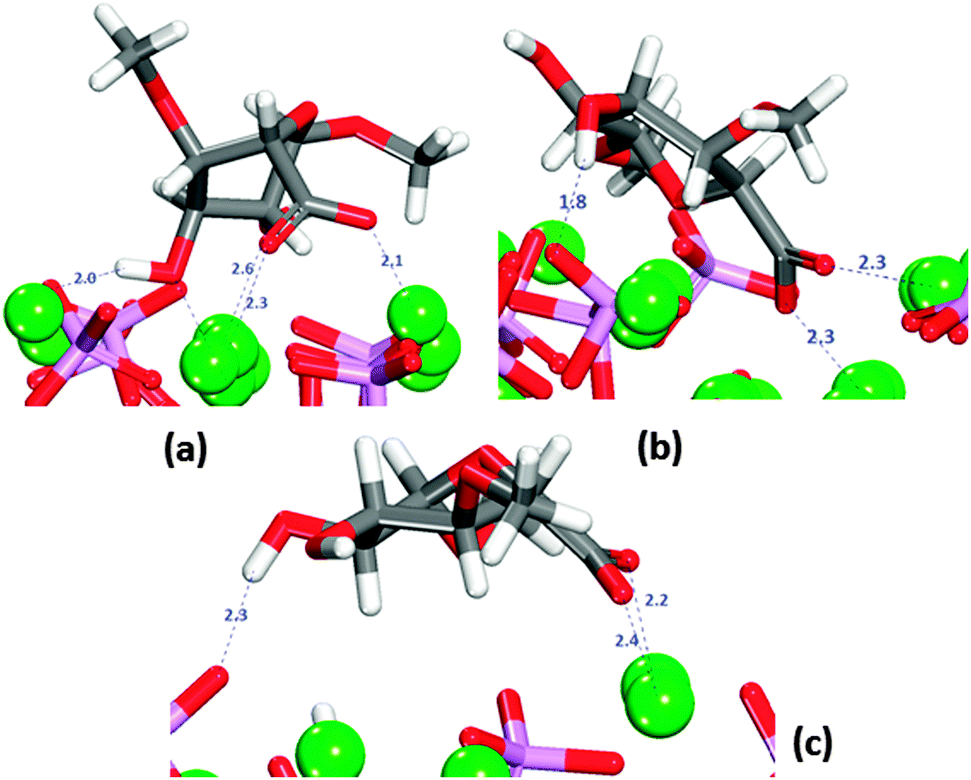 | ||
| Fig. 4 Most stable adsorptions of GlcA on HAP surfaces in the absence of solvent. (a) flat (010). (b) Crenellated (010) (c) (0001). Oxygen in red, calcium in green, phosphate in purple, hydrogen in white. | ||
3.4 Adsorption of GalNAc-4S on the HAP surfaces in vacuum
In the same way as GlcA, the saccharide was placed in twelve different initial orientations on the three surfaces of interest. In all final configurations, the sulfate group is oriented towards the surface (Fig. 5).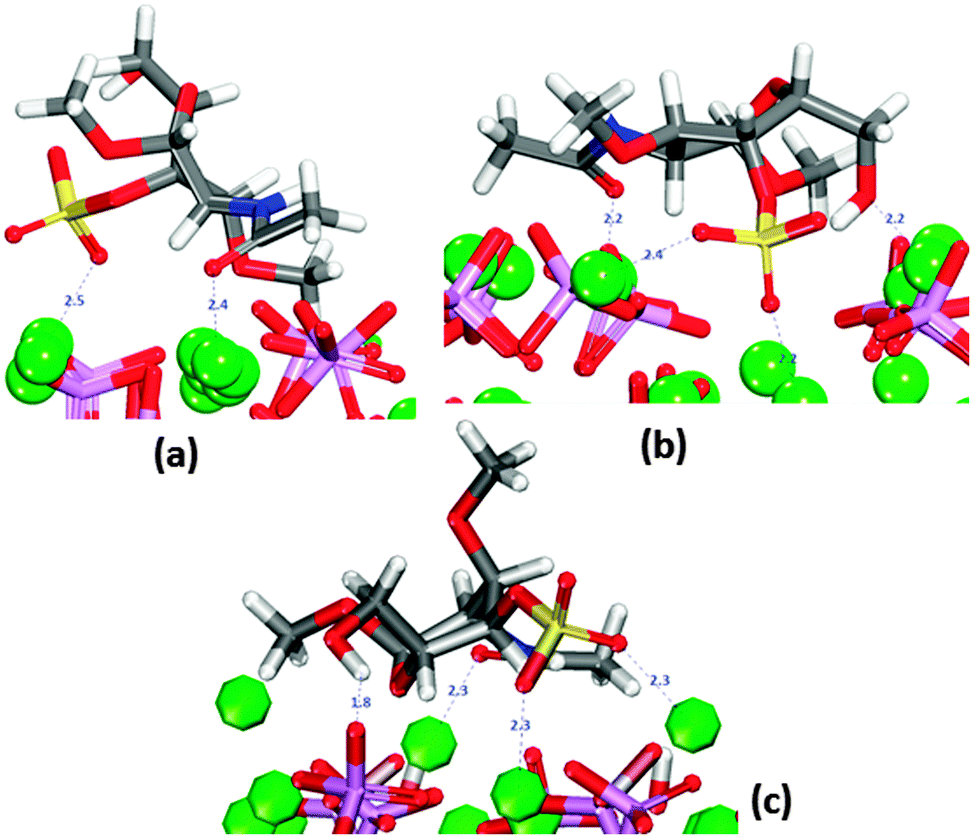 | ||
| Fig. 5 Most stable adsorptions of GalNAc-4S on HAP surfaces in the absence of solvent. (a) Flat (010). (b) crenellated (010) (c) (0001). Oxygen in red, calcium in green, phosphate in purple, hydrogen in white. sulfur in yellow. | ||
On the (010) surface it can form bridges between two calcium ions, all at distances similar to those shown in Fig. 5b. The oxygen of the carbonyl group is also often oriented towards surface calcium (Fig. 5a and b).
In the case of the crenellated (010) plane, the sulfate group is initially located in the “trench” where the hydroxy group is also interacting with the surface. In all configurations the molecule lies flat on the surface. On the (0001) surface, the hydroxy group is closest to the surface (Fig. 5c) and the sulfate is only coordinating more than one calcium ion in two configurations, whereas the molecule lies flat on the surface in four of the configurations.
3.5 Monosaccharides in water
In aqueous environment the GalNAc-4S stays in the low energy chair conformation throughout the MD simulation (Fig. S4a, ESI†) as described in the absence of solvent. However, there are fewer intramolecular hydrogen-bonds between the H of the hydroxy functional group and the O of the sulfate, with all the polar functional groups generally interacting with the surrounding water molecules. The sulfate group is negatively charged and it polarises water (more than the other functional groups), at any time forming multiple water–sulfate hydrogen bonds. There are always typically 2–3 water molecules within 2.5 Å of the sulfate oxygen, but the other polar functional groups are also observed to form hydrogen bonds to water.GlcA remains in the low-energy chair conformation (Fig. S4b, ESI†), in the presence of solvent. The carboxylate hydroxy and methoxy groups are all in equatorial positions. Intramolecular non-bonded interactions are absent: the polar functional groups generally interact with the surrounding water molecules, rather than with each other. The carboxylate group is negatively charged, and it therefore attracts the water molecule's hydrogen atoms. There are always typically 1–2 water molecules within 2.5 Å of the carboxylate oxygen and all other polar functional groups are also observed to form hydrogen bonds to water.
3.6 Hydrated HAP surfaces
We first investigated the flat (010) surface. A plot of the distances between the oxygen atoms of the water molecules and the surface plane, considered as the topmost calcium atoms, is shown as open circles in Fig. 6a. During the MD simulations, a group of water molecules forms a monolayer on the surface (Fig. 6c), primarily interacting via their oxygen atoms with the surface calcium ions at an average distance of 2.52 Å. Hydrogen bonds are also formed between the oxygen atoms of the phosphate groups and the hydrogens of the water molecules, but few of them are at distances of less than ∼2 Å. The water molecules do not form a regular adsorption pattern on the surface and a frequent exchange with molecules in the bulk solution is observed, which we can express as an exchange frequency per surface Ca ion. Here ∼11 times water molecules coordinated to Ca2+ exchange with bulk water during 1.2 ns of data collection time, i.e. a frequency of ∼9.2 s−9. The hydration energy of the surface is −0.73 J m−2. As already mentioned the (010) surface with a crenellated termination (Fig. 3c) has been identified as the principal (010) surface of fluorapatite using high-resolution specular X-ray reflectivity data.48 This surface termination cuts through the channels of hydroxy groups. The surface contains trenches and ridges which run parallel to the c direction; in this case (open triangles in Fig. 6a) the water molecules form a more regular arrangement on the surface (Fig. 6d). In particular, the surface trenches are filled with columns of water molecules with a specific orientation, in agreement with X-ray reflectivity data,48,54 with an average distance from calcium to oxygen of the water of 2.57 Å. In this surface the water molecules in the monolayer prefer to interact more with the surface than with each other forming more hydrogen bonds between the water and the phosphate ions. The frequency of exchanges of water molecules in the monolayer with the bulk is 26.7 s−9, but the water molecules in the trench have an exchange frequency of less than 1 s−9. The hydration energy of the surface is −0.81 J m−2, i.e. the average water–HAP interactions are stronger in the crenellated termination than on the flat (010) termination and the water appears to be more ordered on this termination, where order here is defined as the geometrical arrangement of the water molecules close to the surface.
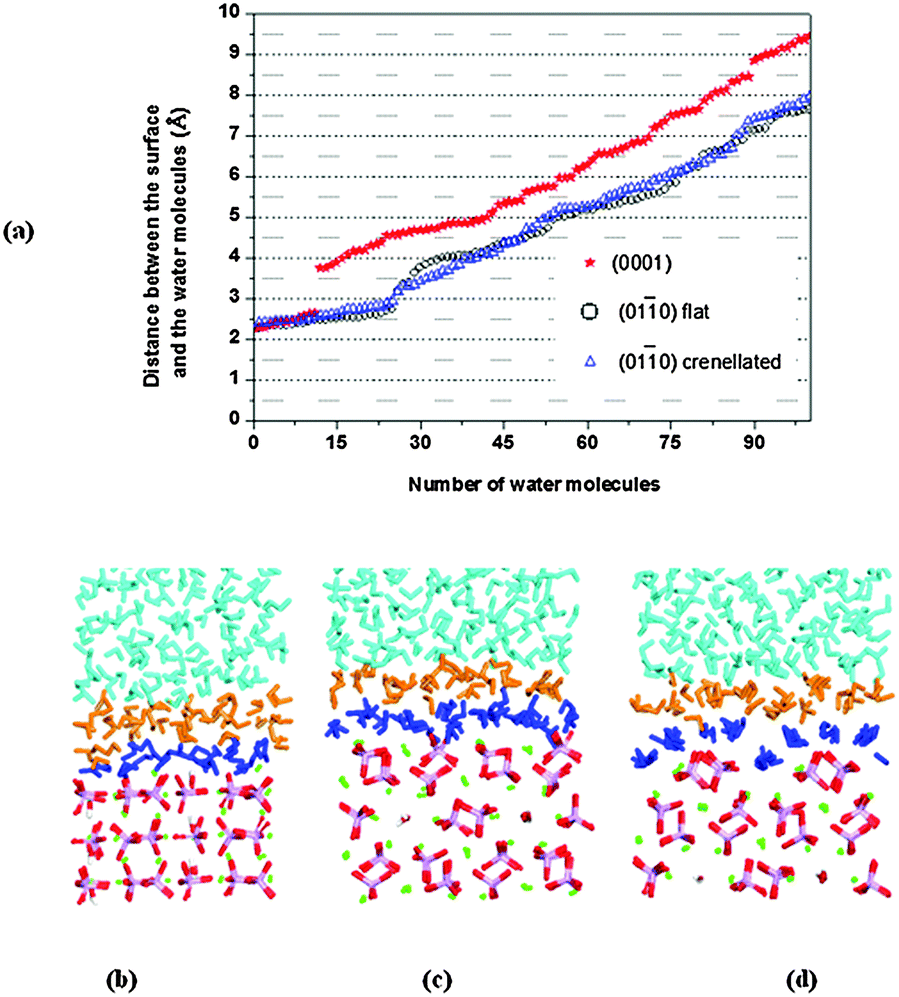 | ||
Fig. 6 (a) Diagram showing Ca2+–Owater distances for all water molecules.  (0001) ○ flat (010) (0001) ○ flat (010)  crenellated (010) (b) the water molecules close to (0001), (c) the water molecules close to flat (010). (d) The water molecules close to crenellated (010), note the water molecules in the trench. P is purple, O is red, Ca is green, and H is white. First layer of water in dark blue. crenellated (010) (b) the water molecules close to (0001), (c) the water molecules close to flat (010). (d) The water molecules close to crenellated (010), note the water molecules in the trench. P is purple, O is red, Ca is green, and H is white. First layer of water in dark blue. | ||
The (0001) surface is the thermodynamically most stable surface in both fluorapatite38 and HAP,23,25 with columns of calcium atoms and hydroxy groups running perpendicular to the surface. The water molecules interact with surface calcium ions via their oxygen atoms at distances from 2.28 Å to 2.61 Å, in line with the 2.28 Å reported by the experimental work by Pareek et al.54 and the 2.29 Å and 2.44 Å obtained by DFT calculations by Corno et al.55 The water molecules (red stars in Fig. 5a) also interact with surface hydroxy and phosphate oxygen atoms via their hydrogen atoms, with almost all the phosphate ions involved in the interactions at distances of less than 2 Å, in a good agreement with the 1.8 Å found by Pareek et al.54 and the 1.5 Å by Corno et al.55 The water molecules closest to the surface do not form a regular adsorption pattern on the surface (Fig. 6b), and they are barely exchanging with the bulk solution (∼1.7 s−9). This surface has a smaller surface area than the (010) and the hydration energy is −1.36 J m−2, indicating that the water is more strongly attracted to this surface than to either termination of the (010) surface, which agrees with the much lower frequency of water exchange. The gap (∼1.5 Å in Fig. 6a) between the first and second layers of water molecules on the (0001) surface is due to the stronger interactions with surface ions and also the existence of an extensive network of hydrogen-bonded interactions between the water molecules within the layer nearest the surface, at the expense of interactions with the water molecules in the second layer. Although a more stable surface may be expected to be less reactive towards water, when the geometry and composition of the surface allows strong interactions between water and surface, in addition to extensive hydrogen-bonding between the molecules within the water layer, the formation of a regular water layer is often found to be an especially favourable process. A similar situation was observed in previous work on the α-quartz (0001) surface,56 but in that case the gap was even more pronounced (∼2.5 Å) due the highly ordered nature of the first monolayer of water.
3.7 Adsorption of the GlcA on the hydrated surfaces
The saccharide was placed onto the surfaces in five different orientations (labelled from 1 to 5 in Table 1), corresponding to the fifth configurations with lowest energy values obtained for the adsorption simulations in vacuum.| GlcA | |||
|---|---|---|---|
| Configuration | E ads/kJ mol−1 | ||
| (0001) | Flat (010) |
Crenellated (010) |
|
| 1 | 37.6 | 23.5 | 22.7 |
| 2 | −34.7 | 12.2 | 24.9 |
| 3 | −55.7 | 30.6 | 13.9 |
| 4 | 52.2 | 30.2 | 28.7 |
| 5 | 17.7 | 64.9 | 61.0 |
Table 1 shows that GlcA adsorbs favourably onto the (0001) surface for two configurations (2 and 3). The adsorption energies on the (010) surfaces are positive for all configurations, suggesting unfavourable adsorption of the saccharide on to these surfaces. Although the values of adsorption energy are less positive in the case of the flat terminated (010) surface than for the crenellated termination and for the (0001) surface. In all cases the GAG and the solvent compete for the adsorption sites on the surfaces and generally the saccharides interact with the mineral surfaces via a monolayer of interfacial water, but the few exceptions to this general behaviour, corresponding to the configurations with the least positive or negative adsorption energies in Table 1 are discussed in the sections below.
![[1 with combining macron]](https://www.rsc.org/images/entities/b_char_0031_0304.gif) 0) hydrated surface.
Table 1 shows that for all configurations the adsorption energy of GlcA onto the flat (010) surface in the presence of water is unfavourable. All configurations considered show interactions by oxygen of the carboxylate to calcium of the surface (Fig. S7, ESI†), but all the initial bridges and hydroxy interactions are no longer present because GlcA does not lie as flat on the surface as it does in the absence of solvent, with only the carboxylate and occasionally the hydroxy groups being located at the level of the first layer of water. In the system with the least positive adsorption energy, the strongest interactions occur between the carboxylate oxygen atoms and axial calcium ions (Fig. 7) while the hydroxy groups form hydrogen-bonds.
0) hydrated surface.
Table 1 shows that for all configurations the adsorption energy of GlcA onto the flat (010) surface in the presence of water is unfavourable. All configurations considered show interactions by oxygen of the carboxylate to calcium of the surface (Fig. S7, ESI†), but all the initial bridges and hydroxy interactions are no longer present because GlcA does not lie as flat on the surface as it does in the absence of solvent, with only the carboxylate and occasionally the hydroxy groups being located at the level of the first layer of water. In the system with the least positive adsorption energy, the strongest interactions occur between the carboxylate oxygen atoms and axial calcium ions (Fig. 7) while the hydroxy groups form hydrogen-bonds.
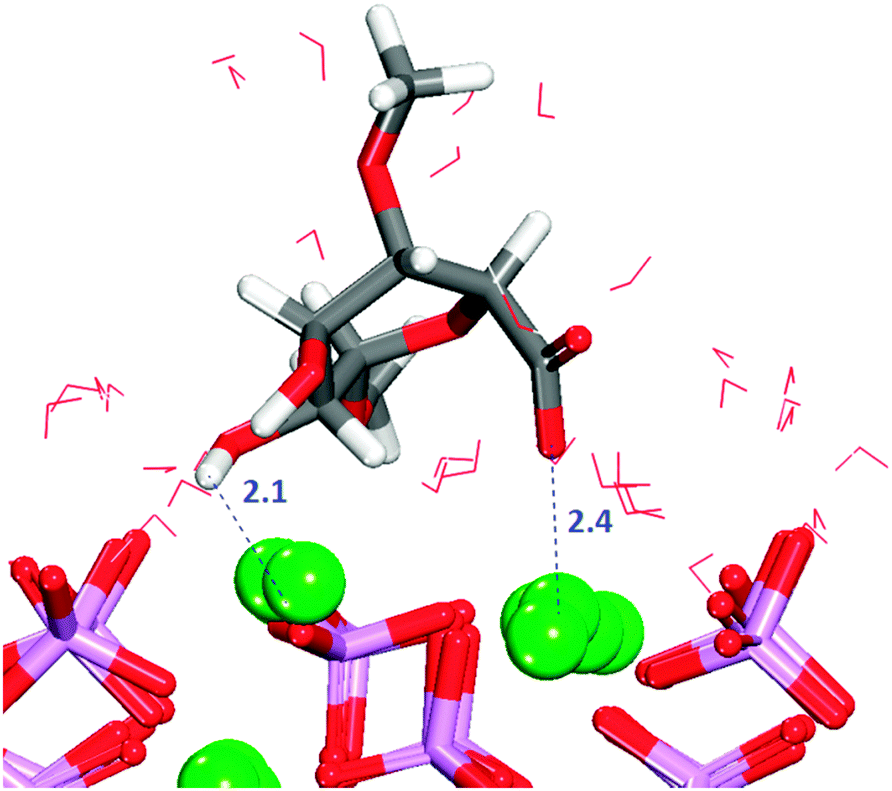 | ||
| Fig. 7 Configuration 2 for GlcA on the flat (010) surface. The image shows the interaction of carboxylate with Ca2+ and the hydrogen-bonds formed with the surface, water molecules not shown. Oxygen in red, calcium in green, phosphate in purple, hydrogen in white. | ||
The GAG has undergone a conformational change and the carboxylate group is now in the axial position, not in the most energetically favoured equatorial position. The GlcA forms hydrogen-bonds with the surface in all five configurations studied (Fig. S8, ESI†), but these interactions by themselves are not enough to stabilize the adsorption of the saccharide.
A combination of a strong and long-lasting interaction through the carboxylate group, the formation of hydrogen-bonds and the relative position of the molecule with respect to the surface plane (discussed further in Section 3.9) are the main factors contributing to the relatively small positive energy of configuration (2).
0) hydrated surface.
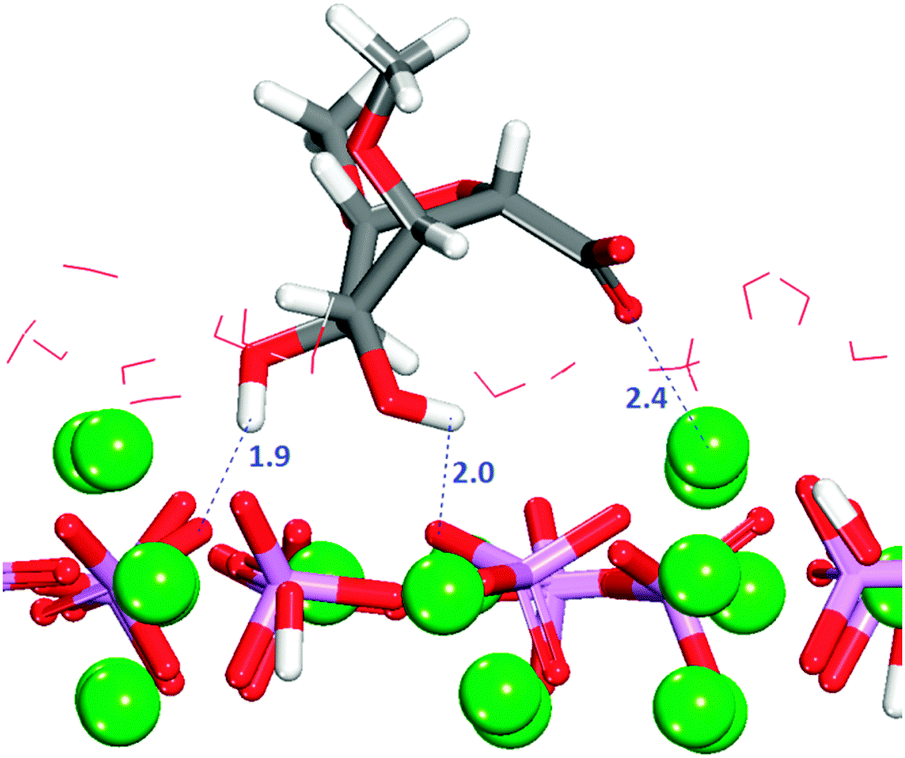
The crenellated (010) surface has a higher hydration energy than the flat surface, indicating greater surface reactivity, but all GlcA adsorptions are again energetically unfavourable (Table 1). Similar to the flat termination, in all configurations interactions occur between the oxygen of the carboxylate and the calcium in the surface, with the carboxylate group piercing the first hydration layer and sharing the trench with water molecules. In two configurations, the adsorbate is lying flat on the surface at the level of the water molecules in the first layer of hydration. Only in three configurations are hydrogen-bonds between hydroxy groups and phosphates also observed (Fig. S9, ESI†). The energetically most favourable configuration (3) is obtained when the organic molecule is not very deep in the surface trench, as indicated by the first peak shifted along the r axis of the RDF Ca2+–Ocarboxylate plot (Fig. S10, ESI†), but with the carboxylate still able to form a bridge between two calcium atoms in the bottom of the trench (Fig. 8).
 | ||
| Fig. 8 Configuration 3 for GlcA on the crenellated (010) surface. The image shows the GlcA in the trench of the surface with its carboxylate groups bridging two columnar Ca2+ ions. Oxygen in red, calcium in green, phosphate in purple, hydrogen in white. | ||
It is noted that configuration 3 does not form hydrogen-bonds to the surface as found for the most strongly bound configuration on the flat (010) surface. GlcA in configuration 3 is adsorbed on the crenellated surface, with the molecule pinned to the surface in a vertical position in which the hydroxy groups are more exposed to the solution, hence interacting more with the water molecules, which also stabilises this configuration.
When the organic molecule is lying along the trench in the surface, all functional groups are able to interact strongly with the surface atoms. The interactions between the oxygen atoms of the saccharide functional groups and the calcium ions help to lower some of the positive adsorption energies compared to the flat (010) surface.
 | ||
| Fig. 9 Configuration 3 for GlcA on the (0001) surface. The image shows the interaction of carboxylate with Ca2+ and multiple hydrogen bonds. Oxygen in red, calcium in green, phosphate in purple, hydrogen in white. | ||
The intensity of the RDF peak (Fig. S11, ESI†) is less than for the (010) surfaces, indicating a weaker interaction, because the oxalate group is not able to form bridges with the calcium ions. For all the configurations in which GlcA remains in the first layer of adsorption, it forms hydrogen-bonds with the surface.
As occurs on the (010) surfaces, the combination of the various interactions lowers the positive adsorption energies, reaching negative values with a change in conformation of the organic molecule. However, when the GAG remains in its most stable conformation in solution the adsorption is even weaker than on the (010) surfaces, for two reasons: The interactions between surface and carboxylate are not particularly strong, because it cannot interact with more than one calcium ion at a time, and the relative position of the organic molecule with respect to the surface in certain cases removes the solvent from the monolayer, as is discussed in more detail in Section 3.9.
3.8. Adsorption of GalNAc-4S on the hydrated surfaces
Similarly to GlcA, the GalNAc-4S molecule was placed onto the surfaces in five different orientations (labelled from 1 to 5 in Table 2), corresponding to the fifth configurations with lowest energy values obtained for the adsorption simulations in vacuum.| GalNAc-4S | |||
|---|---|---|---|
| Configuration | E ads/kJ mol−1 | ||
| (0001) | Flat (010) |
Crenellated (010) |
|
| 1 | 113.4 | — | 59.2 |
| 2 | — | 4.8 | 85.0 |
| 3 | 41.8 | 154.9 | 142.7 |
| 4 | 50.7 | 27.1 | 75.8 |
| 5 | — | 60.0 | 84.2 |
The values in Table 2 indicate the adsorption of the saccharide on to the three surfaces covered in this study is thermodynamically unfavourable. For some of the configurations analysed, the molecule experiments complete desorption from the material. The adsorption energies are generally higher than those obtained for GlcA. The GalNac-4S also prefers to interact with the material through the monolayer of water and as it occurs the case of GlcA the least positive values of adsorption energies are for the (010) surface with the flat termination.
In the sections below we will discuss the geometries leading to the least unfavourable adsorptions on each surface.
0) surface.
The GalNAc-4S is sulfated in position 4, Fig. 1c. This sulfate group has three prominent negatively-charged oxygen atoms that can interact with HAP calcium ions, and it has more volume than the carboxylate present in GlcA. This functional group also attracts more water than the carboxylate.
In configuration 1 (Table 2), the organic molecule desorbs from the surface. The other configurations exhibit interactions between the sulfate and the surface, where in all cases the sulfate, and in one case the hydroxy group, are the only groups of the GAG within the first adsorption layer. The configuration with the least positive adsorption energy (Fig. 10) is the only configuration forming hydrogen-bonds with the surface (Fig. S12, ESI†), which as we described in Section 3.7.1 for GlcA is a determinant factor to increase the strength of binding. Configuration 2 in Fig. 10 is showing a very similar adsorption to the one obtained for GlcA (Fig. 7). As no other functional group of GalNAC-4S is interacting with the surface, the stabilization of the adsorption of GalNAC-4S compared with the GlcA is due to the nature of the sulfate, the adsorption position of the molecule and the more frequent occurrence of hydrogen-bonds, between the hydroxy group of the organic molecule and the phosphate ions, shown in the RDF plot in Fig. S12 (ESI†). The intensity of the first peak is higher than the one obtained for a similar configuration of adsorbed GlcA (Fig. S8, ESI†), even though GalNAc-4S only has one hydroxy group while there are two in GlcA. However, we suggest that hydrogen-bonding is more transient, breaking and re-forming frequently during the simulation.
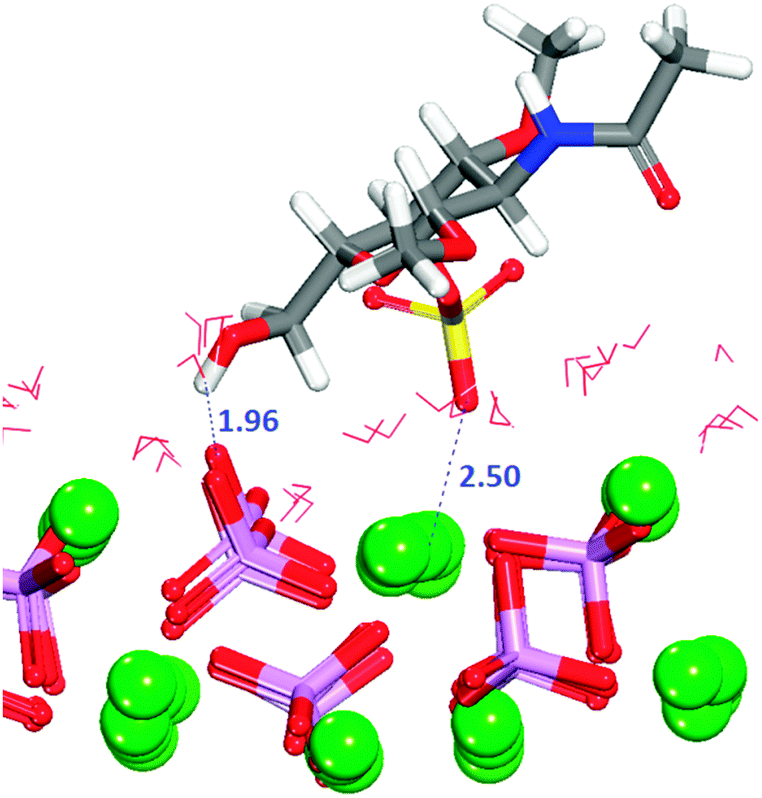 | ||
| Fig. 10 Configuration 2 for GalNAc-4S on the flat (010) surface. The image shows the interaction of sulfate with Ca2+ and a hydrogen-bond. Oxygen in red, calcium in green, phosphate in purple, sulfur in yellow, hydrogen in white, nitrogen in blue. | ||
0) hydrated surface.
During the simulation, the sulfate group of the saccharide inside the trench gradually moves upwards. In three configurations (2, 3 and 4) the GAG as a whole is lying flat on the surface, at the level of the first layer of hydration. In the other two configurations (1 and 5), only the sulfate and in one case the carbonyl groups are level with the first water layer or sharing the trench with water molecules. The adsorption energy is positive for all the five configurations (Table 2), with the weakest bindings seen in any of the systems analyzed in this study, indicating that the adsorption of GalNAc-4S on the (010) surface is thermodynamically highly unfavourable.
Configuration 1 (Fig. 11) exhibits the strongest interaction between surface and adsorbate, with the sulfate group as the only functional group inside the trench, and an additional interaction between the carbonyl oxygen and the calcium ion of the surface. With the main part of the molecule outside the trench, no formation of hydrogen-bonds with the surface is observed (Fig. S13, ESI†).
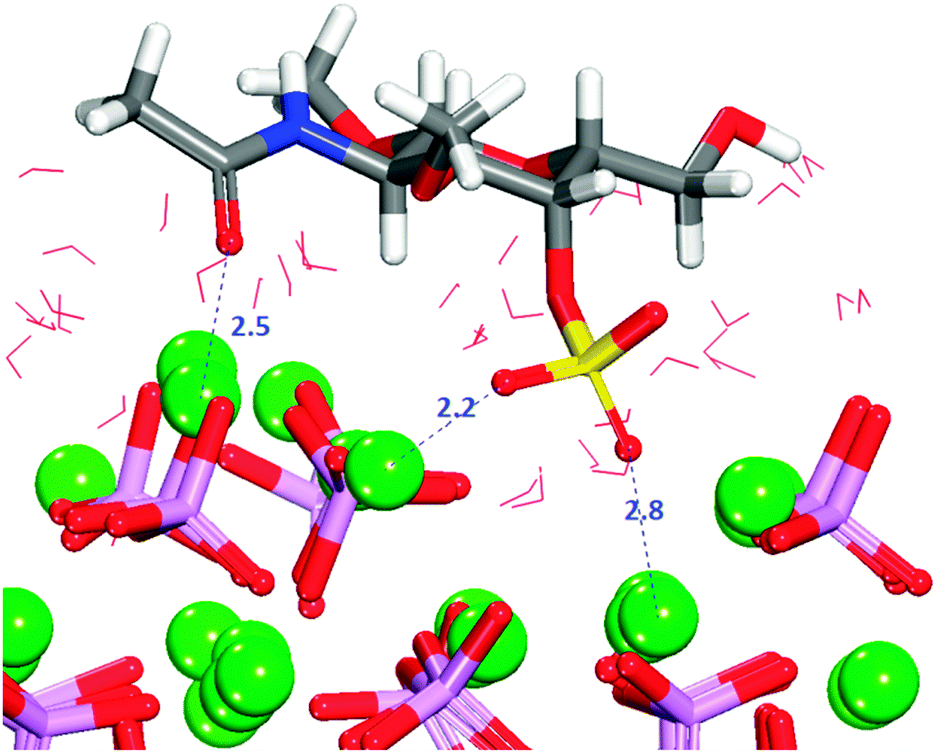 | ||
| Fig. 11 Configuration 1 for GalNAc-4S on the crenellated (010) surface. The molecule is lying flat on the surface blocking access of the solvent. Oxygen in red, calcium in green, phosphate in purple, sulfur in yellow, hydrogen in white. Nitrogen in blue. | ||
As the GalNAc-4S molecule has a single hydroxy group, the presence of peaks in Fig. S13 (ESI†), also supports the fact that in the other configurations the organic molecule resides further into the trench of the surface, to allow the hydroxy group to interact with surface phosphate oxygens. However, here the several interactions occurring at the same time do not produce the same effects observed for GlcA on this surface.
The presence of the sulfate and the acetylamine groups in the molecule makes it a bigger system compared to GlcA, and the diffusion of the GalNAc-4S in the trench causes more rotations of the phosphate groups, and hence less coordination between Ca2+–Ophosphate.
The distance between calcium ions is decreased in comparison with the pure surface, causing more strain in the surface and further instabilities. A combination of these effects due to the presence of the molecule, and the loss of interactions with the solvent (discussed in Section 3.9), causes the relative instabilities of these systems.
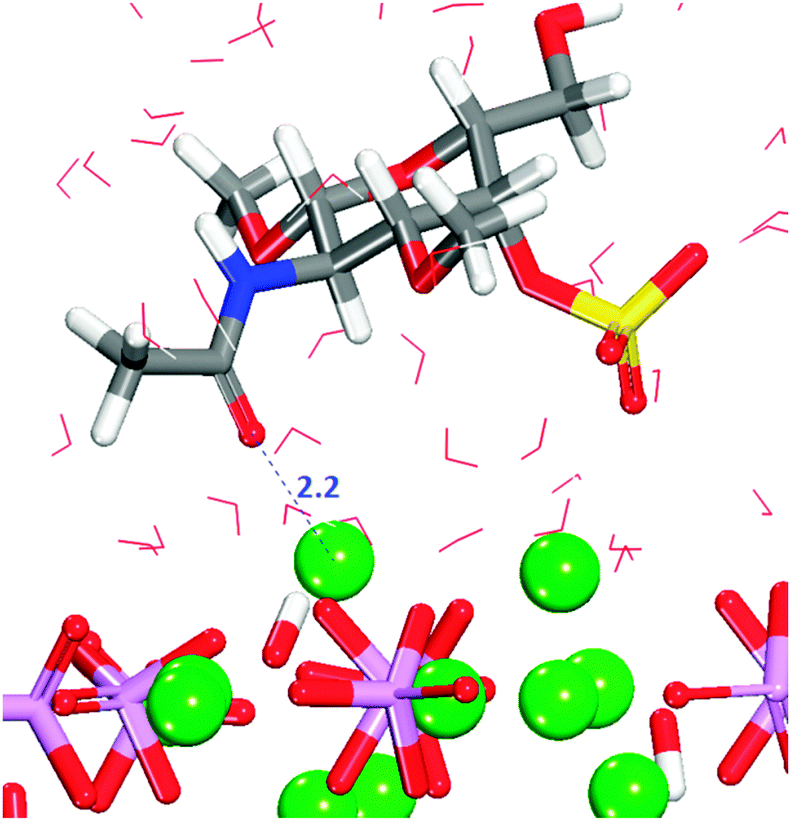 | ||
| Fig. 12 Configuration 3 for GalNAc-4S on the (0001) surface. The main part of the molecule is away from the surface. Oxygen in red, calcium in green, phosphate groups are pink tetrahedra, sulfur in yellow, hydrogen in white. | ||
Configuration 3 is another example of the importance of the position of the adsorbate on the surface and the formation of hydrogen-bonds between the organic and the surface, but the interactions between the surface and the sulfate are transient during the entire simulation time.
3.9 The effect of the aqueous environment
Fig. 4 and 5, show the most favourable adsorption configurations of the two saccharides (GlcA and GalNAC-4S) in the absence of solvent. The results of the MD simulations are in a good agreement with previous DFT calculations on GlcA,25 showing the same interactions and very similar interaction distances and distortions in the GAG structure. The same study analysed a non-sulfonated GalNAc, so our results are not directly comparable, but any trends are similar. The discrepancies between the adsorption energies of the biomolecules obtained with DFT in vacuum and those reported in this work are due to the high stability of the GAGs in water, as well as the strong affinity of the hydroxyapatite surfaces for water. Both factors contribute to making the adsorption of the GAGs onto the HAP surfaces less favourable.A common issue affecting the stabilization of both saccharides at the three hydroxyapatite surfaces is the interaction of the organic molecule with the solvent molecules near the surface, which are identified in Section 3.6 and represented in dark blue in Fig. 6b–d.
A similar situation was reported in previous studies of the behavior of water at quartz surfaces, when a desorbed Si(OH)4 molecule remained near the quartz/water interface56 in a process calculated to be endothermic. During adsorption on the flat (010) and (0001) surfaces both saccharides generally displace ∼10% of the water molecules closest to the surface, which value rises (up to 20% for the (0001) surface), if the molecule lies flat on the surface. In the case of the crenellated (010) surface, both GAGs affect significantly the order of the water adsorbed in the bottom of the trench. The displacement of the water molecules in the first hydration layer prevents the formation of hydrogen-bonds between the water and the phosphate, and there are also fewer interactions between Ca2+ and oxygen of the water, hence the system becomes less stable than the hydrated surface in the absence of GAGs. The strongest binding of GAGs is obtained for the systems in which the first hydration layer is less affected by the adsorbate, confirming that the interactions between calcium ions and water are very important, as was also shown in a previous study.24
To overcome the effect of the loss of water at the surface, the saccharide has to mimic the water interaction in the same area, in a similar way as described in Section 3.7.3 for configuration 3 of GlcA on the (0001) surface (Fig. 8), where less than the typical amount of water is displaced by the adsorbate and where interactions between the oxygen of the hydroxy group and the surface calcium ions replace the Ca2+–Ow interaction of the lost water (see Fig. S14 and S15 in the ESI†). The hydroxy groups of the saccharide also coordinate to the remaining water molecules closest to the surface. As the (0001) surface is also the surface with the largest value for its hydration energy, and is the one where the water monolayer is most pronounced (Fig. 5a), the competition between GAG and water for the adsorption sites on the surface will have the highest impact on GAG adsorption energies. The crenellated (010) surface, where the water molecules are highly ordered (Fig. 6d), which ordering is broken after adsorption, will be the second surface most affected by the competition mentioned above.
Recent simulations of the adsorption of peptides considering pH effects46 report considerable changes of adsorption sites and geometries as a result of change in the pH of the solvent, leading to a change in adsorption energies with more negative values at lower pH. In view of those findings and suggestions by Posner and Park40,48 regarding the influence of phosphate protonation on the structure and binding strength of the water monolayer, we can speculate that at the typical pH of ∼6–7.4 in the kidneys,57 the changes in the structure of the water monolayer could lessen the competition between the organic and the water in certain adsorption sites, thereby increasing the binding probabilities for the GAGs. Some proteoglycans for example, have shown an increase in affinity at low pH.50
4. Conclusions
We have performed a series of molecular dynamics simulations to investigate the adsorption of two monosaccharides of a common glycosaminoglycan at three major surfaces of hydroxyapatite in aqueous environment. The results have shown that the solvent is competing with the monosaccharides for certain adsorption sites at the surfaces, but that the water molecules also solvate the organic molecules. The results are in a good agreement with previous simulations of HAP–citric acid interactions23 and experimental solid state NMR findings identifying the presence of a layer of water between the GAG and the surface of HAP.58 In other studies of interactions of hydroxyapatite with amino acids, such as HAP–glycine,59 the water also competes with the biomolecule for adsorption sites. Our results also suggest that when the surface-bound water molecules form a regular arrangement, as in the case of the crenellated (010) surface, the adsorption of GalNAc-4S is thermodynamically unlikely.
Previous experimental work established that in general, there is a direct relationship between the ability of a macromolecule to bind to HAP and its ability to block crystal growth60 where the interaction occurs mainly through surface calcium ions,61 demonstrating that the calcium binding capacity of GAGs is a significant factor in their ability to inhibit HAP growth.50 In the present study we have shown that only those monosaccharide functional groups able to bind to Ca2+ and causing less perturbation of the surface hydration layer are viable for adsorption. Sulfate and carboxylate groups are the desirable functional groups for adsorption to take place, with the sulfate group showing the greater affinity for calcium, also in agreement with experimental findings demonstrating the importance of the sulfate group for the adsorption capacities of chondroitin 4-sulfate.62
The values of the adsorption energies obtained in this work support experimental reports, which considered that proteoglycans and GAGs with more sulfate groups are better adsorbates to HAP than chondroitin 4-sulfate.3,5 The present work suggests that further stabilization of the surface–saccharide complex compared to the purely hydrated surface is due to hydroxy groups in the organic molecule replacing the missing water–water and water−surface hydrogen-bonds, while other stronger interactions also take place, e.g. between the carboxylate and sulfate functional groups and the surface.
The GlcA displaces fewer surface water molecules and maintains multiple surface−organic interactions for longer periods of time on the (0001) than on the flat (010) surface, leading to stronger binding on the (0001), but it is also possible that the kinetic barrier to desorption is too large for this to be observed on the timescale of the simulations. The interactions of the saccharide with the surfaces are within the energy penalty of a conformational change in the adsorbate, while the GalNAc-4S is not able to sustain long lasting interactions with (0001), but its adsorptions onto the flat (010) surface show very small positive values.
However if the saccharides are forming chondroitin 4-sulfate, the conformational changes observed in the monomers, specifically those stabilizing the interactions with (0001), are energetically forbidden by the presence of the glycosidic linkage and the length of the chain. Our simulations suggest that amino acids and GAGs behave differently when adsorbing onto HAP surfaces in the presence of solvent. The amino acids or peptides manage to displace the water and bind favourably to the surfaces,24,53 whereas the trajectories and values of the adsorption energies obtained here for the GAGs support previous experimental findings which show that disaccharides present in proteoglycans do not bind to HAP for long enough to affect crystal growth.63 If water is present, chondroitin sulfate binds to the layer of water rather than to HAP.58 Our results also support suggestions from experiments at pH > 10.5, that GAGs act as templates for HAP formation, instead of moderators of crystal growth by binding to the surfaces.58 The same experimental work showed that when HAP was formed in situ at pH ∼7−8 in the presence of the GAG, the polysaccharide was more closely attached to the surface, confirming the importance of the pH and therefore the inclusion of both solvent and surface protonation in future simulations to obtain more accurate adsorption energies.
Acknowledgements
We acknowledge the UK Medical Research Council and the UK Engineering and Physical Sciences Research Council for funding. Sergio E. Ruiz Hernandez is grateful to UCL for an Overseas Research Scholarship. Via our membership of the UK's HPC Materials Chemistry Consortium, funded by EPSRC (EP/L000202), this work made use of the facilities of HECToR and ARCHER, the UK's national high-performance computing service, which is funded by the Office of Science and Technology through EPSRC's High End Computing Programme.References
- M. I. Kay, R. A. Young and A. S. Posner, Nature, 1964, 204, 1050–1052 CrossRef CAS PubMed.
- R. A. Young, Clin. Orthop. Relat. Res., 1975, 249–262 CAS.
- S. G. Rees, R. P. Shellis and G. Embery, Biochem. Biophys. Res. Commun., 2002, 292, 727–733 CrossRef CAS PubMed.
- R. Hall, G. Embery, R. Waddington and A. Gilmour, Calcif. Tissue Int., 1995, 56, 236–239 CrossRef CAS.
- J. Okazaki, G. Embery, R. C. Hall, D. T. H. Wassell, R. J. Waddington and A. Kamada, Biomaterials, 1999, 20, 309–314 CrossRef CAS.
- L. Kjellen and U. Lindahl, Annu. Rev. Biochem., 1991, 60, 443–475 CrossRef CAS PubMed.
- M. Ly, T. N. Laremore and R. J. Linhardt, OMICS, 2010, 14, 389–399 CrossRef CAS PubMed.
- A. L. Boskey, Clin. Orthop. Relat. Res., 1992, 244–274 CAS.
- G. K. Hunter, Clin. Orthop. Relat. Res., 1991, 256–280 CAS.
- F. Nudelman, K. Pieterse, A. George, P. H. H. Bomans, H. Friedrich, L. J. Brylka, P. A. J. Hilbers, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1004–1009 CrossRef CAS PubMed.
- H. I. Roach, Histochem. J., 1999, 31, 53–61 CrossRef CAS.
- A. George and A. Veis, Chem. Rev., 2008, 108, 4670–4693 CrossRef CAS PubMed.
- M. Goldberg, D. Septier, O. Rapoport, R. V. Iozzo, M. F. Young and L. G. Ameye, Calcif. Tissue Int., 2005, 77, 297–310 CrossRef CAS PubMed.
- F. Lamoureux, M. Baud'huin, L. Duplomb, D. Heymann and F. Redini, BioEssays, 2007, 29, 758–771 CrossRef CAS PubMed.
- W. Schneiders, A. Reinstorf, A. Biewener, A. Serra, R. Grass, M. Kinscher, J. Heineck, S. Rehberg, H. Zwipp and S. Rammelt, J. Orthop. Res., 2009, 27, 15–21 CrossRef CAS PubMed.
- A. L. Boskey, Connect. Tissue Res., 1996, 35, 357–363 CrossRef CAS.
- E. R. Wise, S. Maltsev, M. E. Davies, M. J. Duer, C. Jaeger, N. Loveridge, R. C. Murray and D. G. Reid, Chem. Mater., 2007, 19, 5055–5057 CrossRef CAS.
- M. J. Duer, T. Friscic, R. C. Murray, D. G. Reid and E. R. Wise, Biophys. J., 2009, 96, 3372–3378 CrossRef CAS PubMed.
- D. G. Reid, M. J. Duer, R. C. Murray and E. R. Wise, Chem. Mater., 2008, 20, 3549–3550 CrossRef CAS.
- M. J. Duer, T. Friscic, D. Proudfoot, D. G. Reid, M. Schoppet, C. M. Shanahan, J. N. Skepper and E. R. Wise, Arterioscler., Thromb., Vasc. Biol., 2008, 28, U2030–U2208 CrossRef PubMed.
- D. G. Reid, G. J. Jackson, M. J. Duer and A. L. Rodgers, J. Urol., 2011, 185, 725–730 CrossRef CAS PubMed.
- J. Moradianoldak, S. Weiner, L. Addadi, W. J. Landis and W. Traub, Connect. Tissue Res., 1991, 25, 219–228 CrossRef CAS.
- N. H. de Leeuw and J. A. L. Rabone, CrystEngComm, 2007, 9, 1178–1186 RSC.
- N. Almora-Barrios and N. H. de Leeuw, CrystEngComm, 2010, 12, 960–967 RSC.
- I. Streeter and N. H. de Leeuw, Proc. R. Soc. A, 2011, 467, 2084–2101 CrossRef CAS PubMed.
- W. Smith and T. R. Forester, J. Mol. Graphics, 1996, 14, 136–141 CrossRef CAS.
- L. Verlet, Phys. Rev., 1967, 159, 98–103 CrossRef CAS.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef PubMed.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef.
- K. N. Kirschner, A. B. Yongye, S. M. Tschampel, J. Gonzalez-Outeirino, C. R. Daniels, B. L. Foley and R. J. Woods, J. Comput. Chem., 2008, 29, 622–655 CrossRef CAS PubMed.
- N. H. de Leeuw, Phys. Chem. Chem. Phys., 2004, 6, 1860–1866 RSC.
- B. G. Dick and A. W. Overhauser, Phys. Rev., 1958, 112, 90–103 CrossRef.
- N. H. de Leeuw, J. Phys. Chem. B, 2004, 108, 1809–1811 CrossRef CAS.
- S. Peroos, Z. M. Du and N. H. de Leeuw, Biomaterials, 2006, 27, 2150–2161 CrossRef CAS PubMed.
- N. H. de Leeuw, J. Mater. Chem., 2010, 20, 5376–5389 RSC.
- K. P. Schröder, J. Sauer, M. Leslie, C. R. A. Catlow and J. M. Thomas, Chem. Phys. Lett., 1992, 188, 320–325 CrossRef.
- N. H. de Leeuw and T. G. Cooper, J. Mater. Chem., 2003, 13, 93–101 RSC.
- D. Mkhonto, P. E. Ngoepe, T. G. Cooper and N. H. de Leeuw, Phys. Chem. Miner., 2006, 33, 314–331 CrossRef CAS.
- N. H. de Leeuw, Chem. Commun., 2001, 1646–1647, 10.1039/B104850n.
- A. S. Posner, J. Biomed. Mater. Res., 1985, 19, 241–250 CrossRef CAS PubMed.
- T. Ishikawa, M. Wakamura and S. Kondo, Langmuir, 1989, 5, 140–144 CrossRef CAS.
- H. Tanaka, M. Chikazawa, K. Kandori and T. Ishikawa, Phys. Chem. Chem. Phys., 2000, 2, 2647–2650 RSC.
- E. Jimenez-Izal, F. Chiatti, M. Corno, A. Rimola and P. Ugliengo, J. Phys. Chem. C, 2012, 116, 14561–14567 CAS.
- R. Astala and M. J. Stott, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 075427 CrossRef.
- F. Chiatti, M. Corno, Y. Sakhno, G. Martra and P. Ugliengo, J. Phys. Chem. C, 2013, 117, 25526–25534 CAS.
- H. Heinz, J. Phys.: Condens. Matter, 2014, 26, 244105–244112 CrossRef PubMed.
- D. Mkhonto and N. H. de Leeuw, J. Mater. Chem., 2002, 12, 2633–2642 RSC.
- C. Y. Park, P. Fenter, Z. Zhang, L. W. Cheng and N. C. Sturchio, Am. Mineral., 2004, 89, 1647–1654 CAS.
- N. Almora-Barrios and N. H. de Leeuw, Geochim. Cosmochim. Acta, 2009, 73, A31 Search PubMed.
- G. Embery, S. Rees, R. Hall, K. Rose, R. Waddington and P. Shellis, Eur. J. Oral Sci., 1998, 106, 267–273 CrossRef CAS PubMed.
- A. Rimola, M. Corno, C. M. Zicovich-Wilson and P. Ugliengo, J. Am. Chem. Soc., 2008, 130, 16181–16183 CrossRef CAS PubMed.
- P. Canepa, F. Chiatti, M. Corno, Y. Sakhno, G. Martra and P. Ugliengo, Phys. Chem. Chem. Phys., 2011, 13, 1099–1111 RSC.
- A. Rimola, M. Corno, C. M. Zicovich-Wilson and P. Ugliengo, Phys. Chem. Chem. Phys., 2009, 11, 9005–9007 RSC.
- A. Pareek, X. Torrelles, J. Rius, U. Magdans and H. Gies, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 035148 Search PubMed.
- M. Corno, C. Busco, V. Bolis, S. Tosoni and P. Ugliengo, Langmuir, 2009, 25, 2188–2198 CrossRef CAS PubMed.
- Z. M. Du and N. H. de Leeuw, Dalton Trans., 2006, 2623–2634, 10.1039/B516258k.
- R. A. Rhoades and D. R. Bell, Medical Physiology: Principles for Clinical Medicine, Wolters Kluwer Health, 2012 Search PubMed.
- S. M. Best, M. J. Duer, D. G. Reid, E. R. Wise and S. Zou, Magn. Reson. Chem., 2008, 46, 323–329 CrossRef CAS PubMed.
- M. Corno, A. Rimola, V. Bolis and P. Ugliengo, Phys. Chem. Chem. Phys., 2010, 12, 6309–6329 RSC.
- T. Aoba, M. Fukae, T. Tanabe, M. Shimizu and E. C. Moreno, Calcif. Tissue Int., 1987, 41, 281–289 CrossRef CAS.
- H. Tanaka, T. Arai, K. Miyajima, S. Shimabayashi and M. Nakagaki, Colloids Surf., 1989, 37, 357–368 CrossRef.
- C. C. Chen and A. L. Boskey, Calcif. Tissue Int., 1985, 37, 395–400 CrossRef CAS.
- E. D. Eanes and A. W. Hailer, Calcif. Tissue Int., 1994, 55, 176–179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Potential parameters used in this work. Adsorption positions of GlcA and GalNAc-4s on to the dry surfaces. RDF graphs. See DOI: 10.1039/c5cp02630j |
| This journal is © the Owner Societies 2015 |