
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphene scavenges free radicals to synergistically enhance structural properties in a gamma-irradiated polyethylene composite through enhanced interfacial interactions
Elayaraja
Kolanthai
a,
Suryasarathi
Bose
 a,
K. S.
Bhagyashree
b,
S. V.
Bhat
b,
K.
Asokan
c,
D.
Kanjilal
c and
Kaushik
Chatterjee
*a
a,
K. S.
Bhagyashree
b,
S. V.
Bhat
b,
K.
Asokan
c,
D.
Kanjilal
c and
Kaushik
Chatterjee
*a
aDepartment of Materials Engineering, Indian Institute of Science, Bangalore 560012, India. E-mail: kchatterjee@materials.iisc.ernet.in; Tel: +91-80-22933408
bDepartment of Physics, Indian Institute of Science, Bangalore 560012, India
cInter University Accelerator Centre, Aruna Asaf Ali Marg, New Delhi 110067, India
First published on 28th July 2015
Abstract
A unique strategy for scavenging free radicals in situ on exposure to gamma irradiation in polyethylene (PE) nanocomposites is presented. Blends of ultra-high molecular weight PE and linear low-density PE (PEB) and their nanocomposites with graphene (GPEB) were prepared by melt mixing to develop materials for biomedical implants. The effect of gamma irradiation on the microstructure and mechanical properties was systematically investigated. The neat blend and the nanocomposite were subjected to gamma-ray irradiation in order to improve the interfacial adhesion between PE and graphene sheets. Structural and thermal characterization revealed that irradiation induced crosslinking and increased the crystallinity of the polymer blend. The presence of graphene further enhanced the crystallinity via crosslinks between the polymer matrix and the filler on irradiation. Graphene was found to scavenge free radicals as confirmed by electron paramagnetic resonance spectroscopy. Irradiation of graphene-containing polymer composites resulted in the largest increase in modulus and hardness compared to either irradiation or addition of graphene to PEB alone. This study provides new insight into the role of graphene in polymer matrices during irradiation and suggests that irradiated graphene–polymer composites could emerge as promising materials for use as articulating surfaces in biomedical implants.
1. Introduction
Gamma rays find applications in a wide range of fields such as sterilization of biomedical implants, medical instruments, human blood and food and in diagnostic imaging and treatment of cancer.1,2 In the field of polymer engineering, different kinds of irradiation are frequently used to enhance various properties that are of technological interest such as physicochemical, structural, electrical and thermal properties, etc.3,4 Whereas there is an ample literature on the improvement of mechanical properties in polymeric systems, the effect of gamma irradiation on the mechanical properties of nanoparticle-filled polymer composites is less understood. Blends of ultrahigh-molecular-weight polyethylene (UHMWPE) and linear low-density polyethylene (LLDPE) were chosen as the polymer system in this work to study the effect of graphene and gamma-ray irradiation on the mechanical properties of the polymer.UHMWPE is widely used for preparing acetabular cups and tibial inserts in the human body by virtue of its biocompatibility, low friction coefficient, modulus, toughness and fatigue resistance.5 However, its low wear resistance limits its applications as a desirable implant material owing to the accumulation of wear debris and associated complications such as inflammation, osteolysis, etc.6 Therefore, polymers with superior mechanical properties are in great demand for developing the next generation of prosthetic implants and promise longer lifetime, especially for more active younger patients.7 However, it is particularly difficult to process UHMWPE because of its high molecular weight, high melt viscosity and low solubility in solvents. Blending UHMWPE with a low-viscosity polymer can offer ease of processing by an industrially viable technique like melt mixing.
LLDPE is a linear semicrystalline polyethylene.8 The incorporation of LLDPE with other PEs like low-density PE (LDPE), high-density PE (HDPE) and UHMWPE is expected to improve the mechanical properties of the blend.9 Kyu et al.10 reported that UHMWPE seems to be miscible with LLDPE, HDPE and LDPE in the melt state. Vadhar et al.11 studied the effect of mixing, rheology and mechanical properties of UHMWPE/LLDPE blends. LLDPE is also envisioned to facilitate the homogeneous polymerization of UHMWPE and the formation of a polymer brush on the folded surfaces of the newly developed polyethylene.12
Of late, nanoparticles have been frequently investigated for their role in improving the mechanical properties and wear resistance of UHMWPE/LLDPE blends. Park et al.13 reported that UHMWPE/LLDPE–BaTiO3 nanocomposites exhibited a significant change in mechanical properties. Silicon nitride and alumina nanoparticles in UHMWPE/LLDPE were also reported to improve thermal, dielectric and mechanical properties.14 In recent years, carbonaceous nanoparticles like carbon nanotubes (CNTs)15 and graphene oxide (GO),16etc., are being investigated as reinforcing agents owing to their exceptional mechanical properties and larger surface area. The latter in particular makes these materials effective for load-bearing orthopedic applications.17–19 It has been reported that the incorporation of carbonaceous nanoparticles in UHMWPE led to significant strengthening of composites.20–22 The hardness of GO/UHMWPE composites increased with an increase in GO content in the composites.23 In our previous study, we have studied the effect of rolling on the evolution of the crystallographic texture and mechanical properties of UHMWPE/graphene and UHMWPE/CNT composites.24 Graphene was more effective than CNT in increasing the modulus prior to rolling.
In the recent past, several strategies have been employed to improve the mechanical properties in polymers and polymer-based composites. Irradiation is one such strategy. It is believed to induce scission of the polymer chains and generate free radicals that can lead to the formation of unsaturated crosslinks between adjacent macromolecules.25 In addition, it has been well established that the mechanical properties of UHMWPE vary with the dosage of gamma-ray radiation.26,27 Buchanan et al.28 studied the effect of dosage of radiation on the density and crystallinity of PE. Similar results were also reported in UHMWPE/CNT composites that were irradiated with gamma rays.21 However, the mechanical properties of GO/UHMWPE composites that were irradiated at 90 kGy were shown to be insensitive to irradiation.29 Therefore, it is now well understood that irradiation can improve the structural properties of UHMWPE.26 However, the effect of irradiation on graphene/PE-based composites has not been studied. Hence, we systematically investigated the effects of gamma rays on the mechanical properties of UHMWPE/LLDPE/graphene composites.
In the present study, UHMWPE/LLDPE blends and their composites with graphene were prepared by melt mixing. The structural, thermal, and mechanical properties of non-irradiated and irradiated samples were characterized using X-ray diffraction (XRD), differential scanning calorimetry (DSC), dynamic mechanical analysis (DMA) and microhardness testing. In addition, the effect of gamma-ray irradiation on the blends and composites was studied. Free-radical formation in the irradiated blends and composites was studied by electron paramagnetic resonance (EPR) to determine the role of graphene in scavenging free radicals and synergistically enhancing the mechanical properties of the composites.
2. Experimental
2.1. Materials
LLDPE of melt flow index (MFI) 50 g/10 min and density 0.926 g cc−1 was obtained from Reliance Industries (Reclair M26500). UHMWPE (Mw = 3 × 106 to 6 × 106) was procured from Sigma Aldrich. GO was prepared by a modified Hummers' method using graphite flakes (Superior Company) and reduced graphene oxide (G) via thermal reduction of GO, as previously reported.302.2. Processing of polymer nanocomposites
A neat (70/30 by wt) UHMWPE/LLDPE blend and 1 wt% G were prepared using a conical twin-screw mini-extruder (Haake MiniLab II) at 220 °C with a rotating speed of 60 rpm for 20 min under a nitrogen environment. Rectangular strips of 25 mm length × 6.5 mm width × 1 mm thickness were produced by hot-pressing using a laboratory-scale compression molding machine at 220 °C for 30 min.2.3. Gamma-ray irradiation of polymer samples
Gamma-ray irradiation was performed on the neat blend and graphene-based composites at room temperature at 1 Pa using a 60Co source. Samples were irradiated with two different doses, i.e., 25 and 50 kGy with an average dose rate of 5.5 kGy h−1. Fig. 1 represents schematically the preparation of the composite and subsequent irradiation. The neat UHMWPE/LLDPE blend (PEB), the graphene-based composites (GPEB), and the samples with irradiation dosages of 25 and 50 kGy will be hereafter referred to as PEB, GPEB, PEB25, GPEB25, PEB50, and GPEB50. | ||
| Fig. 1 Schematic of the preparation of the nanocomposite that was subjected to irradiation yielding a crosslinked composite and enhanced interfacial interactions between the polymer chains and the filler in the composite. | ||
2.4. Characterization
 | (1) |
 is the enthalpy of fusion for 100% crystalline UHMWPE (
is the enthalpy of fusion for 100% crystalline UHMWPE (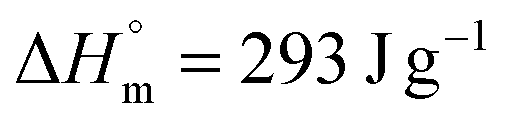 ) and ΔHm is the enthalpy of fusion calculated from the area of the endothermic melting peaks.
) and ΔHm is the enthalpy of fusion calculated from the area of the endothermic melting peaks.
Microindentation experiments were performed using a CSM microhardness tester equipped with a Vickers diamond tip. The maximum load that was used was 100 mN. Loading and unloading were carried out at 200 mN min−1. The dwell time at maximum load was 5 s. Load–displacement data were acquired in real time by a computer and saved for further analysis. At least six independent indentations were performed for analysis. The hardness (H) was determined according to the following equation:31
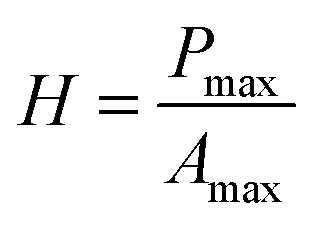 | (2) |
 | (3) |
 | (4) |
 | (5) |
The plasticity index (Ψ) is used to define the relative elastic–plastic behavior of a material that is subjected to external stresses and strains. The plasticity index is calculated using eqn (6):34
 | (6) |
3. Results and discussion
3.1 Characterization of the nanoparticles
The results from XRD, FTIR, and Raman characterization of GO and G are shown in Fig. 2(a–d). Fig. 2a shows the characteristic diffraction peak of GO at 10.8° (d = 8.2 Å) and the diffraction peak at 25.8° (d = 3.45 Å), which confirms the incomplete exfoliation of reduced GO.30 In the FTIR spectrum, the presence of hydroxyl stretching (3440 cm−1), carbonyl stretching (1732 cm−1), sp2-hybridized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1627 cm−1), C–O–C asymmetric stretching (1200–1320 cm−1), C–O stretching of phenol, alcohol or ether (1150–1050 cm−1) and epoxy C–O–C bending motions (850 cm−1) are evident for GO (Fig. 2b). The carbonyl group, along with the phenol, alcohol or ether groups, almost disappeared in the spectrum upon reduction to G. For the latter, the intensity of the hydroxyl stretching peak decreased, whereas the peak at 1577 cm−1 increased, which indicates the partial restoration of sp2 bonds. A broad peak at 1132 cm−1 was observed due to CO vibration, which indicates the reduction of the carboxyl and hydroxyl groups in GO.35,36 G and D bands were observed at 1601 cm−1 and 1359 cm−1 for GO in the Raman spectrum. The peaks slightly shifted to lower (G 1589 cm−1, D 1349 cm−1) wavenumber upon reduction, which is consistent with the literature (Fig. 2c). The SEM image of G shows a typical layered structure (Fig. 2d).
C (1627 cm−1), C–O–C asymmetric stretching (1200–1320 cm−1), C–O stretching of phenol, alcohol or ether (1150–1050 cm−1) and epoxy C–O–C bending motions (850 cm−1) are evident for GO (Fig. 2b). The carbonyl group, along with the phenol, alcohol or ether groups, almost disappeared in the spectrum upon reduction to G. For the latter, the intensity of the hydroxyl stretching peak decreased, whereas the peak at 1577 cm−1 increased, which indicates the partial restoration of sp2 bonds. A broad peak at 1132 cm−1 was observed due to CO vibration, which indicates the reduction of the carboxyl and hydroxyl groups in GO.35,36 G and D bands were observed at 1601 cm−1 and 1359 cm−1 for GO in the Raman spectrum. The peaks slightly shifted to lower (G 1589 cm−1, D 1349 cm−1) wavenumber upon reduction, which is consistent with the literature (Fig. 2c). The SEM image of G shows a typical layered structure (Fig. 2d).
 | ||
| Fig. 2 Characterization of GO and G: (a) XRD patterns, (b) FTIR spectra, (c) Raman spectra and (d) SEM micrograph of G. | ||
3.2 Structural characterization of the nanocomposites
Fig. 3a shows the solid-state 13C NMR spectra of PEB, PEB50 and GPEB50. In the PEB spectrum, the resonance peak at 35.3 ppm and the shoulder at 33.8 ppm correspond to trans–trans methylene and an amorphous phase in the polyethylene blend, respectively.37,38 The PEB50 and GPEB50 spectra also show weak resonance peaks. PEB50 shows a small shoulder at 36.8 ppm, which is plausibly due to additional crystallization in polyethylene upon irradiation. In the case of GPEB50, the peak at 35.3 ppm broadened due to the increase in the number of carbon atoms by the incorporation of graphene in the polymer matrix, which caused this peak to overlap with the shoulder peak. The new resonance peak (indicated by the arrow at 41.2 ppm in Fig. 3b) is related to carbon–carbon crosslinking in polyethylene by irradiation. In addition, there are two more resonance peaks at 15.4 ppm and 27.5 ppm (indicated by arrows) due to methyl end groups and radiation-induced branching in the polymer matrix.38 | ||
| Fig. 3 13C NMR spectra of PEB, PEB50 and GPEB50: (a) normalized-intensity full spectra and (b) magnified spectra. | ||
Fig. 4a shows the normalized IR spectra of non-irradiated and irradiated samples. The IR spectrum of PEB contains four signature bands, namely, strong CH stretching modes at 2911 cm−1 and 2844 cm−1, a polyethylene methylene (CH2) bending mode at 1461 cm−1, and a CH2 rocking mode at 719 cm−1, which are identical to the characteristic features of PE.39,40 In the case of irradiated PEB samples, the additional peak at 965 cm−1 is due to a trans-vinylene group, which indicates crosslinking between the polymer chains induced by irradiation38,41,42 (Fig. 4b).
 | ||
| Fig. 4 FTIR spectra of (a) non-irradiated and gamma ray-irradiated samples of PEB and GPEB and (b) magnified spectra in the 940–990 cm−1 range; (c–f) images of vials showing PEB, PEB 50, GPEB and GPEB50 extracted in hot xylene. | ||
Additional peaks at 1717 cm−1, 1570 cm−1 and 1170 cm−1 are observed in the IR spectra of GPEB, which indicates the presence of G in the blends. The broad peak centered around 1170 cm−1 indicates the C–O stretching and C–O–C vibrations of graphene. A small new trans-vinylene peak is observed at 965 cm−1 for the irradiated composites (GPEB25 and GPEB50), which indicates crosslinking of the polymer chains.
Non-irradiated and irradiated PEB samples were dissolved in hot xylene to determine the effect of irradiation on crosslinking of the polymer chains. Non-irradiated PEB dissolved in hot xylene within a day. In the case of the irradiated sample (PEB50) some insoluble residue remained even after 10 days. This may be attributed to crosslinking between polymer chains upon irradiation (Fig. 4c and d). Non-irradiated and irradiated samples of graphene-based composites were dissolved in hot xylene to extract the graphene particles from the composites (Fig. 4e and f). This enabled us to study the interactions between the polymer matrix and graphene. Whereas the non-irradiated composite was appreciably soluble, as indicated by the visibly suspended graphene (Fig. 4c and d), no suspended graphene was seen in the case of the irradiated composites. This was presumably due to the increased interfacial crosslinking between the graphene and polymer matrix that was induced by irradiation, as depicted schematically in Fig. 1. As discussed further below, the ability of graphene to scavenge free radicals that were generated in the polymer matrix during irradiation presumably induces crosslinking between the filler and the polymer chains for enhanced interfacial interactions.
Fig. 5 shows the EPR spectra of non-irradiated and 50 kGy irradiated samples of PEB and GPEB measured at room temperature four months after irradiation to assess the long-term presence of free radicals, which can dramatically impair the stability and structural properties of the polymer. Both the irradiated samples show a broad peak at ∼3400 G. However, there was essentially no free-radical signal in the two non-irradiated samples. This singlet resonance line in the EPR spectra of PEB50 and GPEB50 was obtained for irradiated polymers only when residual radicals reacted with ambient oxygen. Alternatively, peroxy radicals and alkoxy radicals can react with oxygen to produce oxygen-induced radicals, which may also give rise to a singlet in the spectra. In general, gamma-ray irradiation of polymers generates free radicals but, in this case, the free radicals in the polymer might have been transformed over time into oxygen-centered radicals, similar to the findings reported by Oral et al.,43 where a sharp singlet was observed for UHMWPE at longer time periods (≥4 months). Importantly, the intensity and width of the singlet peak for the irradiated composite considerably decreased compared to the irradiated neat blend. This demonstrates that graphene putatively limited the level of oxidation, as the particles act as radical scavengers in the polymer matrix. An instructive example is the role of MWCNTs in UHMWPE, which act as a radical scavenger, thereby reducing the adverse effects of irradiation on polymer degradation as reported by Martinez-Morlanes et al.44 In a recent study the radical-scavenging activity of graphene-derived particles was observed.45 However, the role of graphene as a free-radical scavenger in polymer matrices has not been reported.
 | ||
| Fig. 5 X-band (mass-normalized) EPR spectra of non-irradiated and 50 kGy irradiated PEB and GPEB. | ||
3.3 Microstructural and thermal characterization of the nanocomposites
Fig. 6a shows the XRD patterns of neat and gamma-ray-irradiated samples. The XRD patterns show diffraction peaks at 21.5°, 23.9° and 36.2°, which correspond to the (110), (200) and (020) planes, respectively, which are the characteristic peaks of the orthorhombic crystal structure of UHMWPE.46 Besides these peaks, the blend also shows a low-intensity broad peak at 19.4°, which is ascribed to the amorphous content in UHMWPE. For LLDPE, diffraction peaks were obtained at the same 2θ positions as in an orthorhombic phase.47 Therefore, the peaks appeared broader but phases can be discerned from the pattern. Vadhar et al.11 reported that a UHMWPE/LLDPE blend that was prepared by melt mixing exhibited broad diffraction peaks in the XRD pattern. On irradiation, the intensity of peaks corresponding to the (110), (200) and (020) planes increased for PEB with increasing radiation dosage. For composites, the addition of 1 wt% G in the blend only served to increase the intensity of the peaks without changing the crystal structure. The percentage crystallinity of non-irradiated and irradiated samples was calculated from XRD data as follows: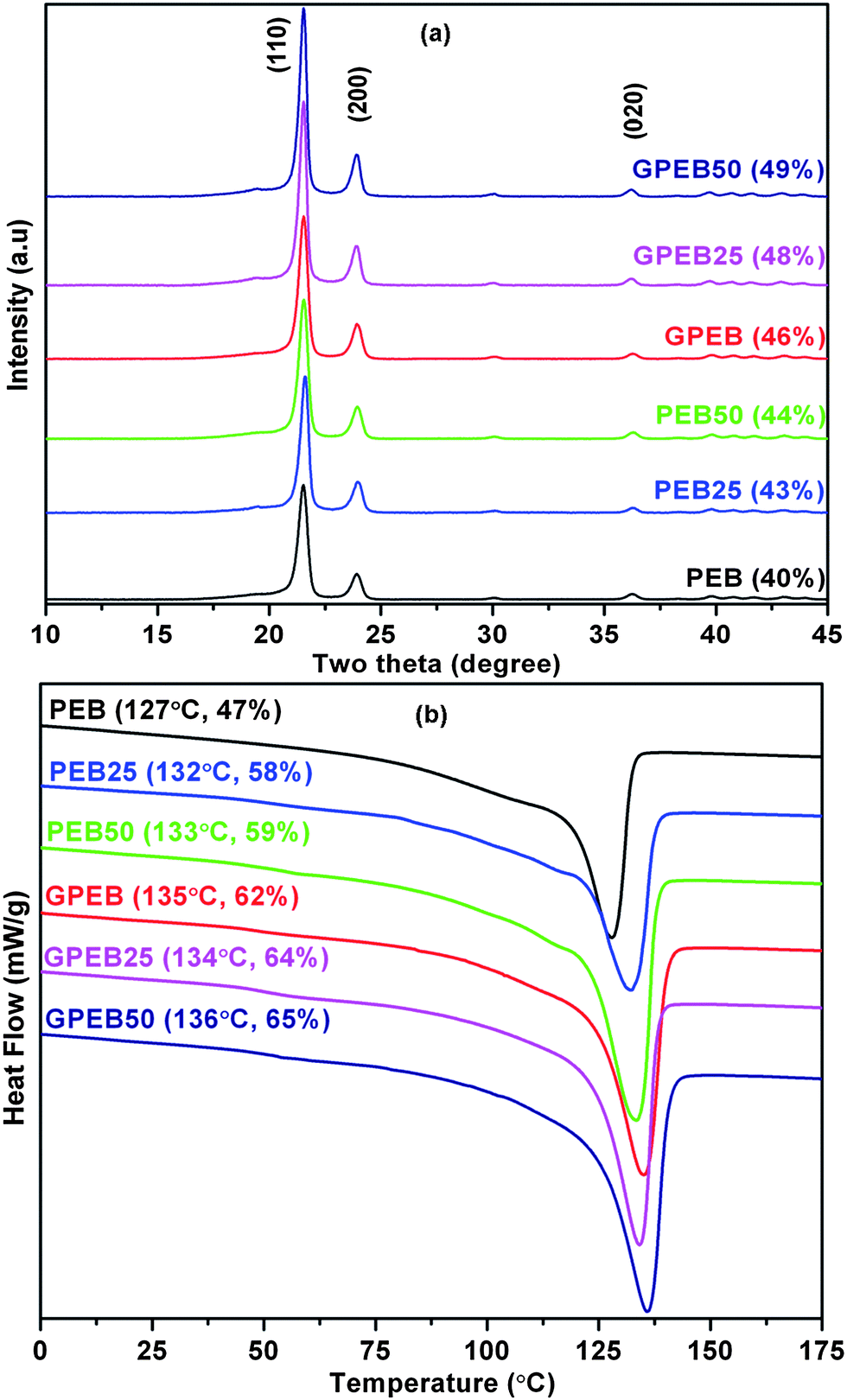 | ||
| Fig. 6 (a) XRD patterns and (b) DSC thermograms of first melting curve of non-irradiated and gamma-ray-irradiated PEB and GPEB. Tm from DSC and % crystallinity from DSC and XRD are indicated for each plot. | ||
% Crystallinity = ((total area of XRD peak-amorphous peak area)/total area) × 100.
The calculated values are listed in Fig. 6a. These results indicate that the degree of crystallinity of the polymer blend is increased by the incorporation of nanoparticles and with an increase in the dosage of radiation. The increase in the crystallinity of irradiated PEB may be attributed to the oxidative reaction of radiation-induced radicals. Irradiation of the blend was performed at room temperature and atmospheric pressure. Atmospheric oxygen, which can diffuse into the amorphous phase of the polyethylene blend, can react with alkyl, allyl and polyenyl radicals to generate highly reactive and short-lived peroxy radicals. These peroxy radicals might trigger chain scission in the amorphous regions of PEB, inducing the formation of additional crystallites. This enables the chains to arrange themselves more easily and pack together, resulting in increased crystallinity of the irradiated blend as has been proposed earlier.48
In composites, the crystallinity of the polymer matrix increased with both the incorporation of graphene and irradiation. Gamma-ray-irradiated composites displayed higher crystallinity as compared to other samples. It has already been established that the incorporation of G into the polymer blend leads to the formation of perfect crystals. This is to state that graphene acts as a heterogeneous nucleation site for polymer crystallization and enhances the kinetics of crystallization.49 Also, gamma irradiation generally leads to chain scission, which is facilitated by oxidative reaction in the amorphous regions. This subsequently leads to the formation of crystallites in the amorphous region due to the generation of free radicals. The free radicals have a high tendency to crosslink with other polymer chains and also with graphene (Fig. 1). The latter scavenges the free radicals, resulting in higher crystallinity. A similar mechanism has been proposed for MWCNTs in irradiated PE composites, where increased crosslinking between the particles and the polymer matrix was observed.21,44
DSC thermograms of non-irradiated and irradiated samples are shown in Fig. 6b. PEB exhibited a single endothermic peak, which indicates co-crystallization between UHMWPE and LLDPE. Similarly to the findings by Vadhar et al.,11 we conclude that melt mixing of 70 wt% UHMWPE and 30 wt% LLDPE at 220 °C led to uniform mixing in the blend. The melting temperature and % crystallinity of non-irradiated and irradiated samples are indicated in Fig. 6b. It is obvious from these results that gamma irradiation increased the melting temperature of PEB because of crosslinking between the polymer chains. The crystallinity of irradiated PEB was found to be higher than that of non-irradiated PEB, which was presumably due to the higher mobility of the new shorter polymer chains that were induced by molecular chain scission during irradiation, as discussed above for the XRD results. A number of studies have observed that irradiation increases the melting temperature and crystallinity in UHMWPE.29,50,51
Fig. 7(a–d) shows SEM micrographs of cryo-fractured surfaces of PEB and the graphene composites before and after irradiation. Two-phase morphology is observed for PEB with a LLDPE phase uniformly dispersed in a UHMWPE matrix, which exhibits fibrillar morphology (Fig. 7a). Irradiation of PEB increased the fibrillar nature (Fig. 7b). Whereas the fractured surface of neat PEB appears smooth, the composite exhibits more pronounced fibrillar morphology (Fig. 7c). Graphene appears well dispersed with no discernable aggregates in the polymer matrix (Fig. 7c). Uniform distribution of the filler and fibrillar morphology are known to enhance the mechanical properties. Irradiation did not lead to the loss of fibrillar morphology in the composites, which is known to yield stronger materials (Fig. 7d).
 | ||
| Fig. 7 SEM micrographs of the cryo-fractured surface of (a) PEB, (b) PEB50, (c) GPEB, (d) GPEB50. | ||
The surface morphology of non-irradiated and irradiated samples was also analyzed (Fig. 8a–f). Whereas PEB and GPEB appear smooth with large uneven features that result from compression molding (Fig. 8a and b), irradiation produced a porous morphology in PEB. The fraction of pores seemed to increase with the radiation dosage (Fig. 8c and e). The composites, on the other hand, acquired a comparatively smoother surface with minimal surface porosity (Fig. 8d and f). This reduction in porosity in the composites can be attributed to the protective effect of graphene against gamma-ray radiation. The ability of graphene to scavenge free radicals appears to have minimized damage to the surface of the polymer matrix.
 | ||
| Fig. 8 SEM micrographs of (a) PEB, (b) GPEB, (c) PEB25, (d) GPEB25, (e) PEB50 and (f) GPEB50 showing surface damage (porosity) from irradiation. | ||
3.4 Mechanical characterization of the nanocomposites
The storage moduli measured at 100 Hz from the tensile dynamic mechanical properties of non-irradiated and irradiated samples are displayed in Fig. 9. The storage modulus of PEB was 532 MPa, which increased to 560 MPa upon 50 kGy exposure (PEB50). This increase in the modulus can be traced back to the increased molecular crosslinking and higher crystallinity in PEB as a result of irradiation. The modulus of PEB25 (563 MPa) was slightly higher than that of PEB50, which was likely due to the large number of pores that had formed on the surface upon higher irradiation (Fig. 8e). It is generally true that lower porosity yields better mechanical properties, as porosity causes local discontinuities in the microstructure. | ||
| Fig. 9 Storage modulus at 100 Hz from DMA measurements of non-irradiated and irradiated PEB and GPEB. | ||
For composites, the storage modulus increased from 532 (PEB) to 599 MPa with the addition of graphene. This increase in modulus was expected on account of the plate-like morphology of graphene. Upon incorporation in the polymer matrix, the large surface area of the graphene phase facilitates stress transfer across the graphene–polymer interface. The presence of such interfaces together with the inherent high hardness of graphene results in an increased modulus. The uniform distribution of graphene in PEB, as indicated by the cryo-fractured morphology (Fig. 7c), and the increased crystallinity of the polymer, as graphene acts as a heteronucleation site, also contribute toward the increase in modulus of the composites. The storage modulus also increased with the radiation dosage, namely to 616 MPa and 630 MPa with dosages of 25 kGy and 50 kGy, respectively. In the case of GPEB the modulus increased monotonically with irradiation unlike in PEB, which was likely because irradiation did not produce pores in the composite. The presence of graphene, which scavenges free radicals, prevented damage to the polymer matrix, minimizing pore formation.
The combination of surface hardness and elastic modulus is often taken to be a qualitative index of wear resistance. Fig. 10a and b shows plots of load versus displacement for non-irradiated and irradiated blends and composites. The elastic modulus, Vickers hardness, contact stiffness and plasticity index are summarized in Table 1. The modulus and surface hardness values of PEB were 1.07 GPa and 0.079 GPa, respectively. The hardness of PEB increased with the radiation dosage, which may be attributed to the increase in crosslinking. The modulus (1.32 GPa) and hardness (0.137 GPa) of GPEB were appreciably higher than those of PEB. The increase in hardness and modulus of the composites can possibly be due to the uniform distribution of graphene in the polymer matrix and the resultant increase in crystallinity, as reported elsewhere.23 Furthermore, irradiation led to a marked increase in the hardness of GPEB (Table 1). The uniform distribution of graphene, increased crystallinity, and irradiation-induced crosslinking are plausible reasons for the increase in hardness and modulus.
 | ||
| Fig. 10 Representative microindentation load–displacement curves of non-irradiated and irradiated (a) PEB and (b) GPEB. | ||
| Sample code | Vickers hardness (GPa) × 10−1 | Elastic modulus (GPa) | Contact stiffness (mN μm−1) | Plasticity index (Ψ) | % Change ina (Ψ) |
|---|---|---|---|---|---|
| a With respect to non-irradiated PEB samples. | |||||
| PEB | 0.79 ± 0.05 | 1.07 ± 0.08 | 51.49 ± 2.35 | 0.43 ± 0.02 | — |
| PEB25 | 0.92 ± 0.06 | 1.25 ± 0.22 | 53.43 ± 1.02 | 0.41 ± 0.17 | 4.7 |
| PEB50 | 1.05 ± 0.06 | 1.28 ± 0.21 | 57.67 ± 2.39 | 0.40 ± 0.12 | 7.0 |
| GPEB | 1.37 ± 0.12 | 1.32 ± 0.15 | 59.73 ± 1.28 | 0.35 ± 0.05 | 19.7 |
| GPEB25 | 1.40 ± 0.09 | 1.48 ± 0.07 | 61.58 ± 2.37 | 0.31 ± 0.04 | 28.0 |
| GPEB50 | 1.55 ± 0.16 | 1.58 ± 0.32 | 65.53 ± 1.25 | 0.25 ± 0.08 | 47.9 |
The contact stiffness was calculated from the slope of the unloading portion of the load–displacement curve. The stiffness of the polymer increased, which was due both to the incorporation of graphene and subsequent irradiation (Table 1). The plasticity index is of particular interest and sheds more light on the mechanical properties of polymer-based materials. It is used to characterize the self-healing ability of a material. As shown in Table 1, the incorporation of graphene and irradiation caused a significant decrease in the plasticity index of the materials. According to Archard's prediction,52 increased hardness can lower the friction between polymers and metals via minimization of the plastic contact area. With the observed marked increase in hardness in irradiated GPEB, these composites can be further investigated as candidate materials for use as articulating surfaces in biomedical implants.
Carbonaceous particles are believed to exhibit high electron donor–acceptor capacity due to the presence of a network of conjugated double bonds.29 Similarly to graphite, fullerenes and CNTs, which are known for the ease with which they react with free radicals,53 graphene is also reported to exhibit this characteristic.45,54 It has previously been reported that the carbon lattice in the graphene structure was strongly affected by gamma-ray irradiation, with variations in the oxygen levels being negligible.29,55 It was proposed that graphene fillers in polymeric composites could act as radical scavengers and crosslinking agents upon gamma-ray irradiation. Goncalves et al.56 reported on the role of GO as a scavenger of radicals that were generated during a polymerization reaction. We hereby propose that irradiation of the graphene-based polyethylene composite enhanced the mechanical properties of the polymer matrix as a result of the following effects: (a) the presence of a well-dispersed hard filler with a large surface area that facilitates stress transfer; (b) the strengthening of the polymer matrix due to the crosslinking of the chains induced by irradiation; (c) the increased crystallinity of the polymer matrix resulting from the presence of a filler, which acts as a heteronucleation site, and irradiation, which induces chain scission and thereby local reorganization of the chains; and (d) the enhanced interfacial interactions between the polymer chains and the filler, owing to the ability of graphene to scavenge free radicals generated as a result of irradiation, which will facilitate better stress transfer to the filler. It is also important to note an additional benefit: the incorporation of graphene can offset the adverse effects of gamma irradiation by minimizing oxidation of the polymer that is induced by free radicals due to its free-radical scavenging mechanism. EPR results are in good agreement with these conclusions. Therefore, the incorporation of graphene in irradiated PEB is observed to lead to superior bulk and surface mechanical properties. Hence, gamma-ray-irradiated graphene composites can be considered as candidate materials with improved wear resistance.
4. Conclusion
UHMWPE/LLDPE blends and their composites with graphene were prepared by melt mixing and thermo-compression and subsequently subjected to 25 and 50 kGy gamma irradiation in ambient conditions. Irradiation induced crosslinking in the polymer matrix and enhanced the crystallinity in the blend. EPR analysis indicated a reduced concentration of radicals, which suggests that graphene acts as a radical scavenger in the irradiated composites, which can minimize deterioration of the polymer. This in turn led to the grafting of polymer chains on the filler, yielding enhanced interactions at the polymer–graphene interface. The incorporation of graphene thus synergistically increased the modulus and hardness of the composite that was subjected to irradiation, yielding materials that may be better suited as strong and wear-resistant materials for use in biomedical implants. These findings should help guide the design of new materials as articulating surfaces in biomedical implants.Acknowledgements
This study was funded by the Department of Science and Technology (DST), India. E.K. gratefully acknowledges the Department of Biotechnology, India for the Postdoctoral Research Associate Fellowship. K.C. acknowledges the Ramanujan fellowship from DST.References
- G. Calais, M. Alfonsi, E. Bardet, C. Sire, T. Germain, P. Bergerot, B. Rhein, J. Tortochaux, P. Oudinot and P. Bertrand, J. Natl. Cancer Inst., 1999, 91, 2081 CrossRef CAS PubMed.
- K. A. da Silva Aquino, Sterilization by gamma irradiation, Gamma Radiation, InTech, 2012 Search PubMed.
- A. Sionkowska, Prog. Polym. Sci., 2011, 36, 1254 CrossRef CAS PubMed.
- T. J. Singh, Ganeshsanjeev, K. Siddappa and S. V. Bhat, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 1299 CrossRef CAS PubMed.
- E. Gomez-Barrena, J.-A. Puertolas, L. Munuera and Y. T. Konttinen, Acta Orthop., 2008, 79, 832 CrossRef PubMed.
- M. Slouf, S. Eklova, J. Kumstatova, S. Berger, H. Synkova, A. Sosna, D. Pokorny, M. Spundova and G. Entlicher, Wear, 2007, 262, 1171 CrossRef CAS PubMed.
- S. Kearns, B. Jamal, C. Rorabeck and R. Bourne, Clin. Orthop., 2006, 453, 103 CrossRef CAS PubMed.
- S. L. Sakellarides and A. J. McHugh, Polym. Eng. Sci., 1985, 25, 1179 CAS.
- A. K. Gupta, S. K. Rana and B. L. Deopura, J. Appl. Polym. Sci., 1992, 44, 719 CrossRef CAS PubMed.
- T. Kyu and P. Vadhar, J. Appl. Polym. Sci., 1986, 32, 5575 CrossRef CAS PubMed.
- P. Vadhar and T. Kyu, Polym. Eng. Sci., 1987, 27, 202 CAS.
- S. Ronca, G. Forte, A. Ailianou, J. A. Kornfield and S. Rastogi, ACS Macro Lett., 2012, 1, 1116 CrossRef CAS.
- H. S. Park, J. H. Lee, J.-D. Nam, S. J. Seo, Y. K. Lee, Y. S. Oh and H.-C. Jung, Macromol. Res., 2006, 14, 430 CrossRef CAS.
- W. Zhou, C. Wang, T. Ai, K. Wu, F. Zhao and H. Gu, Composites, Part A, 2009, 40, 830 CrossRef PubMed.
- S. Bose, R. A. Khare and P. Moldenaers, Polymer, 2010, 51, 975 CrossRef CAS PubMed.
- P. K. Mural, M. Sharma, G. Madras and S. Bose, RSC Adv., 2015, 5, 32078–32087 RSC.
- S. Kumar, S. Bose and K. Chatterjee, RSC Adv., 2014, 4, 19086 RSC.
- S. Kumar and K. Chatterjee, Nanoscale, 2015, 7, 2023 RSC.
- S. Kumar, S. Raj, E. Kolanthai, A. K. Sood, S. Sampath and K. Chatterjee, ACS Appl. Mater. Interfaces, 2015, 7, 3237 CAS.
- P.-G. Ren, Y.-Y. Di, Q. Zhang, L. Li, H. Pang and Z.-M. Li, Macromol. Mater. Eng., 2012, 297, 437 CrossRef CAS PubMed.
- M. Martinez-Morlanes, P. Castell, V. Martinez-Nogues, M. T. Martinez, P. Alonso and J. Puertolas, Compos. Sci. Technol., 2011, 71, 282 CrossRef CAS PubMed.
- M. Sturzel, F. Kempe, Y. Thomann, S. Mark, M. Enders and R. Mülhaupt, Macromolecules, 2012, 45, 6878 CrossRef.
- Z. Tai, Y. Chen, Y. An, X. Yan and Q. Xue, Tribol. Lett., 2012, 46, 55 CrossRef CAS.
- E. Kolanthai, R. Kalsar, S. Bose, S. Suwas and K. Chatterjee, Phys. Chem. Chem. Phys., 2014, 16, 23108 RSC.
- G. Lewis, Biomaterials, 2001, 22, 371 CrossRef CAS.
- S. M. Kurtz, O. K. Muratoglu, M. Evans and A. A. Edidin, Biomaterials, 1999, 20, 1659 CrossRef CAS.
- E. Oral and O. K. Muratoglu, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 265, 18 CrossRef CAS PubMed.
- F. Buchanan, J. White, B. Sim and S. Downes, J. Mater. Sci.: Mater. Med., 2001, 12, 29 CrossRef CAS.
- J. Puertolas and S. Kurtz, J. Mech. Behav. Biomed. Mater., 2014, 39, 129 CrossRef CAS PubMed.
- P. Xavier, K. Sharma, K. Elayaraja, K. Vasu, A. Sood and S. Bose, RSC Adv., 2014, 4, 12376 RSC.
- W. C. Oliver and G. M. Pharr, J. Mater. Res., 1992, 7, 1564 CrossRef CAS.
- A. R. Franco Jr, G. Pintaúde, A. Sinatora, C. E. Pinedo and A. P. Tschiptschin, Mater. Res., 2004, 7, 483 CrossRef.
- D. Bartel, J. Rawlinson, A. Burstein, C. Ranawat and W. Flynn Jr, Clin. Orthop., 1995, 317, 76 Search PubMed.
- R. Rohini and S. Bose, Phys. Chem. Chem. Phys., 2015, 17, 7907 RSC.
- N. Wu, X. She, D. Yang, X. Wu, F. Su and Y. Chen, J. Mater. Chem., 2012, 22, 17254 RSC.
- J. Oh, J.-H. Lee, J. C. Koo, H. R. Choi, Y. Lee, T. Kim, N. D. Luong and J.-D. Nam, J. Mater. Chem., 2010, 20, 9200 RSC.
- R. Kitamaru, F. Horii and K. Murayama, Macromolecules, 1986, 19, 636 CrossRef CAS.
- A. L. Cholli, W. M. Ritchey and J. L. Koenig, Appl. Spectrosc., 1987, 41, 1418 CrossRef CAS.
- S. Krimm, C. Liang and G. Sutherland, J. Chem. Phys., 1956, 25, 549 CrossRef CAS PubMed.
- S. A. Umapathi, P. Komaragounder, S. ShailadraKumar and N. S. Kumar, J. Biorem. Biodegrad., 2012, 3, 1000142 Search PubMed 1.
- D. C. Waterman and M. Dole, J. Phys. Chem., 1970, 74, 1913 CrossRef CAS.
- O. K. Muratoglu, C. R. Bragdon, D. O. O'Connor, M. Jasty, W. H. Harris, R. Gul and F. McGarry, Biomaterials, 1999, 20, 1463 CrossRef CAS.
- E. Oral, S. L. Rowell and O. K. Muratoglu, Biomaterials, 2006, 27, 5580 CrossRef CAS PubMed.
- M. Martinez-Morlanes, P. Castell, P. Alonso, M. T. Martinez and J. Puértolas, Carbon, 2012, 50, 2442 CrossRef CAS PubMed.
- Y. Qiu, Z. Wang, A. C. E. Owens, I. Kulaots, Y. Chen, A. B. Kane and R. H. Hurt, Nanoscale, 2014, 6, 11744 RSC.
- H. Kiho, A. Peterlin and P. H. Geil, J. Appl. Phys., 1964, 35, 1599 CrossRef CAS PubMed.
- K. A. Moly, H. J. Radusch, R. Androsh, S. S. Bhagawan and S. Thomas, Eur. Polym. J., 2005, 41, 1410 CrossRef CAS PubMed.
- M. Shafiq, M. S. Mehmood and T. Yasin, Mater. Chem. Phys., 2013, 143, 425 CrossRef CAS PubMed.
- J. Liang, Y. Huang, L. Zhang, Y. Wang, Y. Ma, T. Guo and Y. Chen, Adv. Funct. Mater., 2009, 19, 2297 CrossRef CAS PubMed.
- M. Goldman, R. Gronsky, R. Ranganathan and L. Pruitt, Polymer, 1996, 37, 2909 CrossRef CAS.
- S. M. Lee, H.-J. Jeon, S. W. Choi, H. H. Song, Y. C. Nho and K. Cho, Macromol. Res., 2006, 14, 640 CrossRef CAS.
- S. Ge, S. Wang and X. Huang, Wear, 2009, 267, 770 CrossRef CAS PubMed.
- A. Galano, Nanoscale, 2010, 2, 373 RSC.
- P. A. Denis, J. Phys. Chem. C, 2009, 113, 5612 CAS.
- A. Anson-Casaos, J. A. Puertolas, F. J. Pascual, J. Hernandez-Ferrer, P. Castell, A. M. Benito, W. K. Maser and M. T. Martinez, Appl. Surf. Sci., 2014, 301, 264 CrossRef CAS PubMed.
- G. Goncalves, S. M. A. Cruz, A. Ramalho, J. Gracio and P. A. A. P. Marques, Nanoscale, 2012, 4, 2937 RSC.
| This journal is © the Owner Societies 2015 |