
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure determination of trans-cinnamaldehyde by broadband microwave spectroscopy†
Sabrina
Zinn
abc,
Thomas
Betz
abc,
Chris
Medcraft
ac and
Melanie
Schnell
*abc
aMax Planck Institute for the Structure and Dynamics of Matter, Luruper Chaussee 149, D-22761 Hamburg, Germany. E-mail: melanie.schnell@mpsd.mpg.de
bThe Hamburg Centre for Ultrafast Imaging, Luruper Chaussee 149, D-22761 Hamburg, Germany
cCenter for Free-Electron Laser Science, Notkestrasse 85, D-22607 Hamburg, Germany
First published on 15th May 2015
Abstract
The rotational spectrum of trans-cinnamaldehyde ((E)-3-phenyl-2-propenal, C9H8O) was recorded by chirped-pulse Fourier transform microwave spectroscopy in the frequency range of 2–8.5 GHz. The odourant molecule is the essential component of cinnamon oil and causes the characteristic smell. The rotational signatures of two conformers were observed: s-trans–trans- and s-cis–trans-cinnamaldehyde. The rotational spectra of s-trans–trans-cinnamaldehyde and all of its 13C-monosubstituted species in natural abundance were assigned and the corresponding carbon backbone structure was determined. The second conformer s-cis–trans-cinnamaldehyde is about 9 kJ mol−1 higher in energy and could also be identified in the spectrum.
1. Introduction
The odourant molecule trans-cinnamaldehyde is the major component of cinnamon oil1 and is responsible for its characteristic smell. The molecule consists of a phenyl ring with an unsaturated aldehyde attached to it. The structure of the molecule is planar due to the conjugation of the π-electron system, as seen in Fig. 1. This conjugation causes a change in the typical carbon–carbon bond lengths, which is an interesting effect to study. Since both the structure and the structural flexibility of an odourant molecule are important for molecular recognition by olfactory receptors, a precise experimentally obtained molecular structure can be useful for further studying the structure–odour relationship.2 The importance of studying odourant molecules was already shown before,3,4 and it is also demonstrated that olfaction is very complicated to describe because there are many structures and a vast number of odours. One major aim in that research field is to identify the common structural features that cause the same odour perception. However, as in many molecular recognition processes, conformational flexibility is known to play a role. A better understanding of this as well as the identification of such common structural features will be an important step towards predicting the odour of a respective molecule.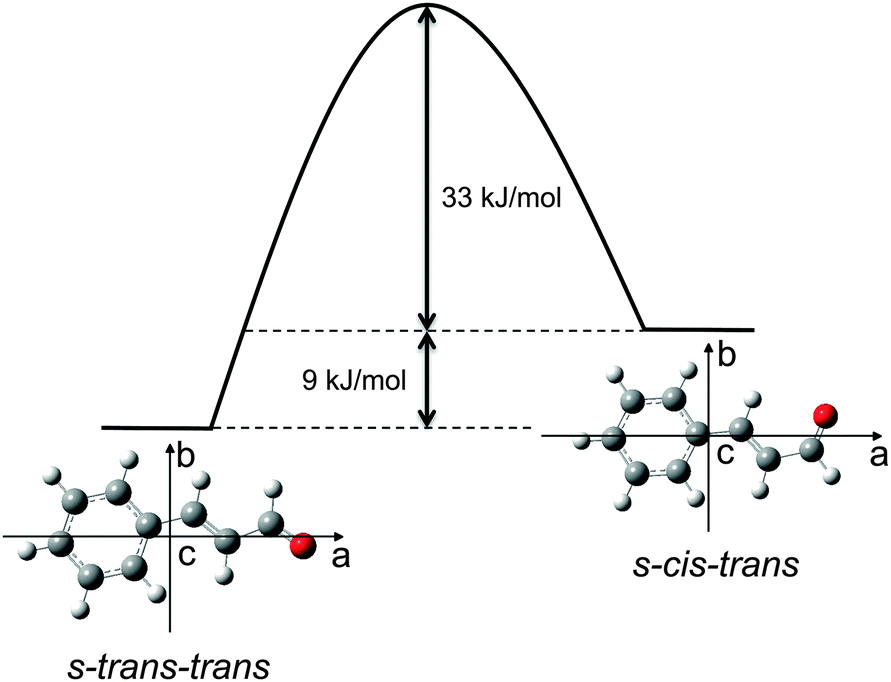 | ||
| Fig. 1 Scheme of the molecular structure of s-trans–trans-cinnamaldehyde ((E)-3-phenyl-2-propenal) and s-cis–trans-cinnamaldehyde in the coordinate system of the molecular moments of inertia and the calculated energy difference and isomerisation barrier of the two conformers, using the MP2 level of theory and the 6-311++G(2d,2p) basis set. | ||
Cinnamaldehyde was recently found to activate the transient receptor potential ion channel TRPA1, which mediates thermosensation, and thus cinnamaldehyde can be a potential molecular model for studying why noxious cold can be perceived as a burning pain.5 For this, the molecular recognition process, especially the conformational flexibility, of the molecule is of interest.
In a photochemical reaction with UV-light trans-cinnamaldehyde isomerises to cis-cinnamaldehyde.6 Biochemical reactions with this motif can, for example, also be found in the retina of the (human) eye. Recently, these light-triggered dynamics were studied by femtosecond crystallography on minicrystals of photoactive yellow protein (PYP).7 The chromophore of this protein reveals structural similarities with trans-cinnamaldehyde, which can be seen as a model system for this photoactive protein in gas-phase studies, another example for the physiological relevance of a detailed structure determination of trans-cinnamaldehyde.
Four possible stereoisomers of cinnamaldehyde can exist: two diastereomers, differing in the cis- or trans-orientation of the carbon–carbon double bond of the side chain. Each of these diastereomers can consist of two conformers, differing by the rotation around the single bond in between the two double bonds. Here we use broadband rotational spectroscopy to study the two conformers of the trans-diastereomer, s-trans–trans-cinnamaldehyde and the higher energy conformer s-cis–trans-cinnamaldehyde that are depicted in Fig. 1.
High-resolution microwave spectroscopy combined with supersonic expansion is an ideal tool to study the structure of molecules in the gas phase, since the rotational constants obtained from the spectrum are based on the moments of inertia of the molecule. From rotational parameters of the parent molecule and the isotopically substituted species precise information on the molecular structure can be obtained. The slightly different mass of the singly substituted molecules leads to a change in the molecular moment of inertia. From this change the respective atom positions in the molecule with respect to its center of mass can be determined using, for example, Kraitchman's equations (substitution structure, rs).8 To obtain a structure that is closer to the theoretical equilibrium structure compared to the rs-structure and thus supports a comparison between the experiment and quantum chemical structural parameters, a mass dependent structure (rm-structure) can also be calculated. This method corrects for the isotope-dependent rovibrational contribution to the moments of inertia of the molecule.11
This study extends the earlier work on trans-cinnamaldehyde using low-resolution microwave spectroscopy by Steinmetz et al.,9 in which only one rotational constant could be determined due to low spectral resolution. Here, we report complete sets of rotational constants for both low-energy conformers of trans-cinnamaldehyde. We were also able to obtain the rotational constants for all the mono-substituted 13C-isotopologues of the energetically lower conformer in natural abundance and hence determine its carbon backbone structure. The obtained structure is compared to a recent gas-electron diffraction (GED) study.10 Such a comparison is also interesting from a conceptional point of view. Typically, gas-electron diffraction studies are performed at the elevated temperatures necessary to generate sufficient vapour pressure of the respective molecules. As a consequence, the molecules can be vibrationally excited and several conformers can be populated, which complicates the analysis. The suitability of the analysis of the experimental data often strongly relies on the quality of the quantum-chemical calculations. A direct comparison between microwave data and GED data will thus be helpful to evaluate the quality of the GED data in the particular case of trans-cinnamaldehyde, for which two conformers are present.
2. Experimental details
The spectra presented in this work were recorded using broadband microwave spectroscopy employing the chirped-pulse technique (CP-FTMW).12 Our spectrometer covers a frequency range from 2 to 8.5 GHz. The detailed description of the experimental setup can be found in ref. 13 and 14.A sample of trans-cinnamaldehyde was purchased from Sigma-Aldrich (≥95% purity) and used without further purification. The sample is a liquid at room temperature and has a reported boiling point of 251 °C. It has a strong total dipole moment of 5.1 D with the two dipole moment components of 4.9 D for μa and −1.5 D for μb. For the measurements, the sample was heated to 100 °C in a reservoir. Neon was used as a carrier gas with an absolute backing pressure of 1.8 bar. The gas mixture was supersonically expanded into the vacuum chamber via a pulsed nozzle. After this, the molecules were excited by a 1 μs long microwave pulse, which was linearly chirped in frequency. Fifty microseconds of the resulting free induction decay (FID) were recorded, this results in a spectral resolution of 20 kHz. In total, 1.2 million FIDs were recorded and averaged. The rotational spectrum was obtained by Fourier transforming the experimental data. A Kaiser–Bessel window function was applied to the spectrum to identify weak transitions that otherwise, in some cases, would overlap with stronger transitions.
Predictions of the rotational constants for different possible conformers of cinnamaldehyde were obtained from quantum chemical calculations. Geometry optimisations were performed using the density-functional theory (DFT) methods M06-2X and B3LYP as well as the MP2 level of theory, all with a 6-311++G(2d,2p) basis set as implemented in the program package Gaussian. The results are listed in Tables 1 and 2.
| Parameter | Experiment | B3LYP | M06-2X | MP2 | Steinmetz et al.9 |
|---|---|---|---|---|---|
| a Δ = Ic − Ia − Ib; u is the atomic mass unit. | |||||
| A [MHz] | 4866.3795(13) | 4915.85 | 4926.28 | 4884.51 | |
| B [MHz] | 579.05960(17) | 578.73 | 582.94 | 579.03 | |
| C [MHz] | 517.81608(16) | 517.78 | 521.26 | 517.67 | |
| B + C [MHz] | 1096.87568(33) | 1096.51 | 1104.20 | 1096.70 | 1106.3(1) |
| D K [kHz] | 1.72(12) | 1.2 | |||
| D JK [kHz] | 0.076(10) | 0.08 | |||
| D J [kHz] | 0.01005(82) | 0.008 | |||
| d 1 [kHz] | 0.130(32) | 0.105 | |||
| d 2 [kHz] | 0.00123(20) | 0.00113 | |||
| |μa| [D] | 4.73 | 4.31 | 4.90 | ||
| |μb| [D] | 1.17 | 1.21 | 1.53 | ||
| |μc| [D] | 0 | 0 | 0 | ||
| Δ [μÅ2]a | −0.628 | ||||
| J max | 17 | ||||
| Assigned lines | 93 | ||||
| Error [kHz] | 6.5 | ||||
| Parameter | Experiment | B3LYP | M06-2X | MP2 | Steinmetz et al.9 |
|---|---|---|---|---|---|
| a Δ = Ic − Ia − Ib; u is the atomic mass unit. | |||||
| A [MHz] | 4494.1094(21) | 4526.23 | 4534.55 | 4495.56 | |
| B [MHz] | 626.04519(27) | 625.32 | 631.43 | 626.84 | |
| C [MHz] | 549.97612(24) | 549.42 | 554.25 | 550.13 | |
| B + C [MHz] | 1176.02131(26) | 1174.74 | 1185.68 | 1176.96 | 1179.1(1) |
| |μa| [D] | 3.58 | 3.08 | 3.50 | ||
| |μb| [D] | 1.85 | 1.89 | 2.17 | ||
| |μc| [D] | 0 | 0 | 0 | ||
| D K [kHz] | 0.40(17) | ||||
| D J [kHz] | 0.0058(29) | ||||
| Δ [μÅ2]a | −0.801 | ||||
| J max | 10 | ||||
| Assigned lines | 33 | ||||
| Error [kHz] | 5.2 | ||||
The spectra were fitted using an asymmetric rotor Hamiltonian (Watson S-reduction in Ir representation15) as implemented in the PGOPHER programme suite.16
3. Results and discussion
The measured spectrum and the corresponding fits for both conformers are shown in Fig. 2. The experimental spectrum is dominated by strong a-type transitions, as predicted from the dipole moments. Together with weaker b-type transitions they could be assigned to s-trans–trans-cinnamaldehyde by comparison with the rotational parameters obtained from ab initio calculations and the results of the previous low-resolution microwave study.9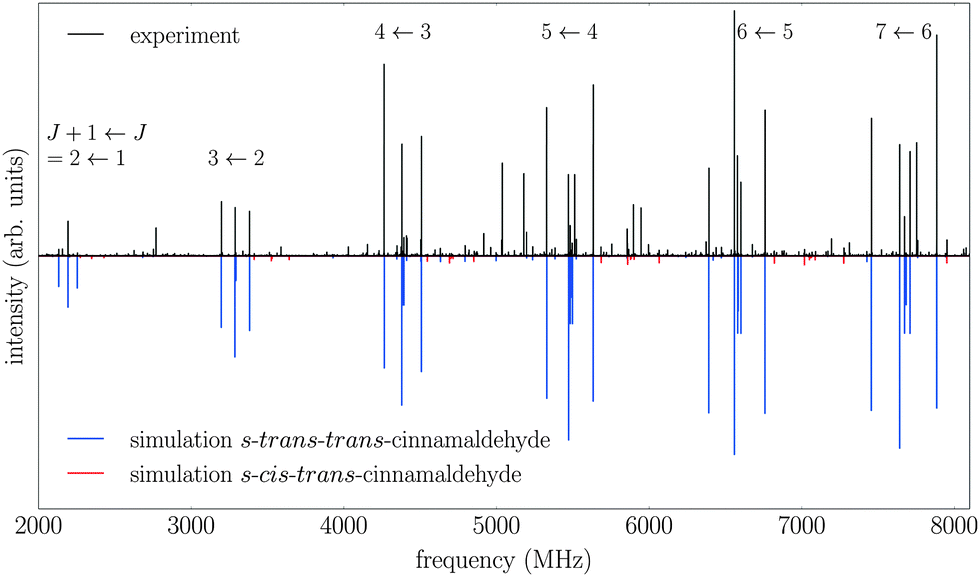 | ||
| Fig. 2 Rotational spectrum of s-trans–trans- (blue trace) and s-cis–trans-cinnamaldehyde (red trace). The upper trace shows the experimental data and the lower trace represents the result of fitting the asymmetric rotor Hamiltonians of both conformers to the spectrum. Additional transitions seen in the spectrum may arise from complexes of water and cinnamaldehyde but are not assigned yet. | ||
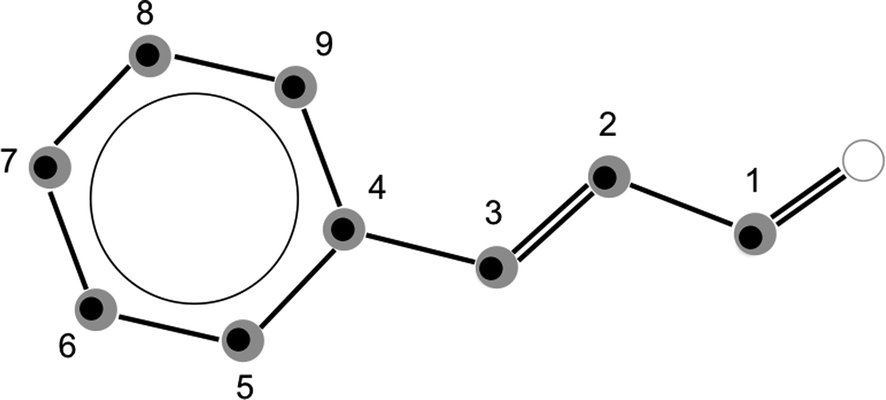
In addition, the spectrum of the second conformer, s-cis–trans-cinnamaldehyde, could be assigned, which is about 40 times weaker. A zoom-in view of the measured spectrum, illustrating the spectral features of s-cis–trans-cinnamaldehyde, is depicted in Fig. 3. The two conformers differ in the orientation of the terminal aldehyde group with respect to the C–C double bond of the alkenyl side chain, as depicted in Fig. 1. The most abundant conformer s-trans–trans-cinnamaldehyde is calculated (MP2/6-311++G(2d,2p)) to be about 9 kJ mol−1 lower in energy than s-cis–trans-cinnamaldehyde. They are separated by an isomerisation barrier of 33 kJ mol−1 (see Fig. 1), in accordance with the significant intensity difference. The higher energy of the cis-conformer seems to be caused by steric repulsion of the oxygen and the C3 carbon atom and the two double bonds (C2–C3 and C1–O). The atom labels are given in Fig. 4.
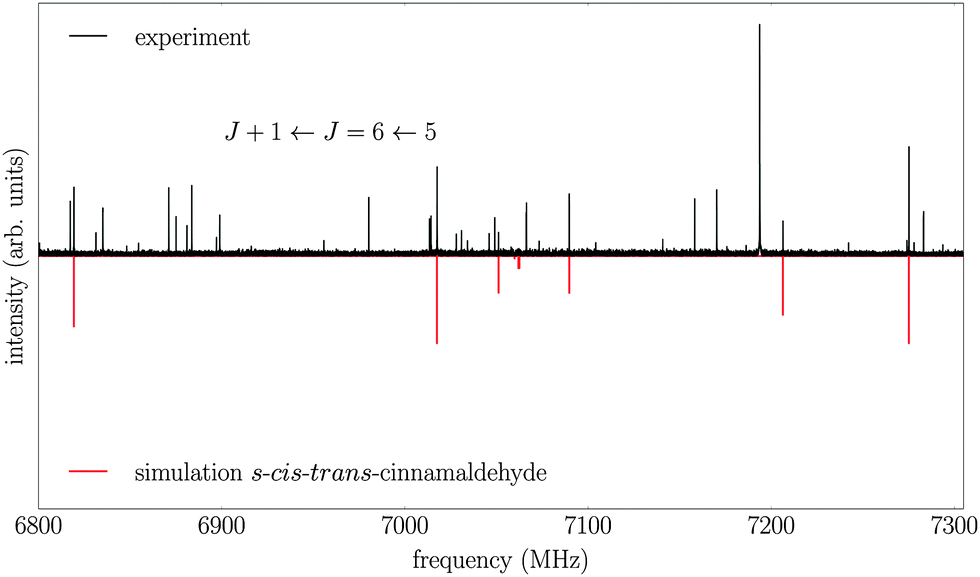 | ||
| Fig. 3 Part of the broadband rotational spectrum (intensity scaled by a factor of 20 compared to Fig. 2), showing the J + 1 ← J = 6 ← 5 transition of the higher-energy conformer, s-cis–trans-cinnamaldehyde (red trace). The upper trace shows the experimental data and the lower trace represents the result of fitting an asymmetric rotor Hamiltonian to it. Additional transitions seen in the spectrum may arise from complexes of water and cinnamaldehyde but are not assigned yet. | ||
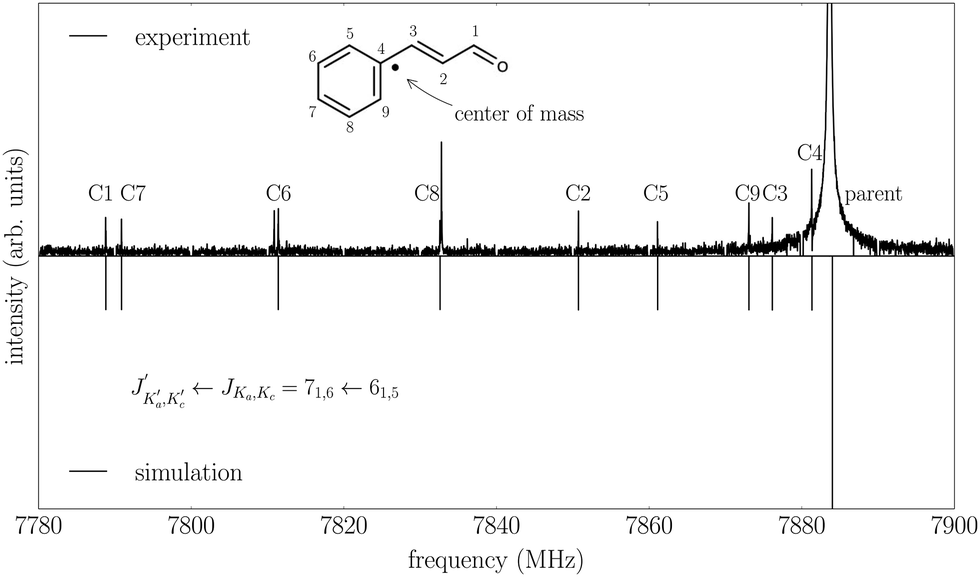 | ||
Fig. 4 Satellite lines arising from all singly substituted 13C isotopologues of s-trans–trans-cinnamaldehyde and the parent line of the 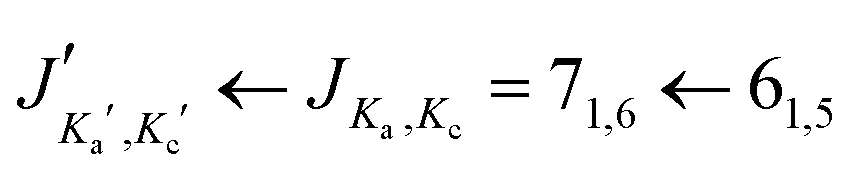 transition. Note that the line shift from the parent transition increases with the distance of the substituted carbon atom to the center of mass of the molecule. transition. Note that the line shift from the parent transition increases with the distance of the substituted carbon atom to the center of mass of the molecule. | ||
The obtained energies are very similar to those found for acrolein (H2CCHCOH), which is the simplest member in the group of α,β-unsaturated aldehydes. Using microwave spectroscopy,17 the structures of both the s-cis- and s-trans-conformers of acrolein were obtained, with a calculated energy difference of 8.5 kJ mol−1 and a calculated barrier height of 29.3 kJ mol−1 (CCSD(T)/CBS+CV).18 This similarity might hint at the dominance of the local electronic environment in the side chain of trans-cinnamaldehyde.
The structural isomer cis-cinnamaldehyde, which is the product of a photochemical reaction of trans-cinnamaldehyde, was not observed. Furthermore, no spectroscopic indications for internal dynamics, such as rotation around single bonds, were found.
In the following, the results of the individual conformers are discussed in more detail.
s-trans–trans-Cinnamaldehyde
The experimentally determined spectroscopic constants for s-trans–trans-cinnamaldehyde and a comparison to calculated parameters are given in Table 1. In total, 93 rotational transitions (52 a- and 41 b-type transitions) could be assigned with an average error of the fit of 6.5 kHz. Anharmonic calculations were carried out using the B3LYP hybrid functional using the 6-311++G(2d,2p) basis set. The fitted distortion constants compare well with the ones obtained from these calculations. Comparing the rotational constants, the MP2 level of theory agrees best with the experimental values, with deviations of 0.5% and less.In earlier studies of trans-cinnamaldehyde, where the molecule was regarded as a near symmetric top molecule, only the sum of B + C could be determined.9
The inertial defect Δ of the molecule, calculated from the obtained rotational constants, is rather small. Its value is a scale for the deviation of the molecular structure from planarity (Δ = 0 for planar molecules) and will be discussed in more detail below.
s-cis–trans-Cinnamaldehyde
A closer look into the broadband spectrum reveals rotational transitions of the higher-energy conformer s-cis–trans-cinnamaldehyde (Fig. 3). In total, 33 transitions (19 a-type and 14 b-type) were assigned to this conformer and fit to an asymmetric rotor Hamiltonian with an error of the fit of 5.2 kHz. The experimentally determined constants and calculated values can be found in Table 2. Again, the MP2 level of theory gives the best agreement with the experimentally determined rotational constants. The results from the earlier low-resolution study9 are also in good agreement.Due to the lower intensity only transitions up to J = 10 could be assigned in the spectrum, this leads to less well-defined distortion constants for this conformer. The calculated inertial defect is small and of the same order of magnitude as the s-trans–trans-conformer.
13C-isotopologues of s-trans–trans-cinnamaldehyde
For the s-trans–trans-conformer, we could also assign rotational transitions arising from all singly substituted 13C-isotopologues. The assignments of these satellite lines for the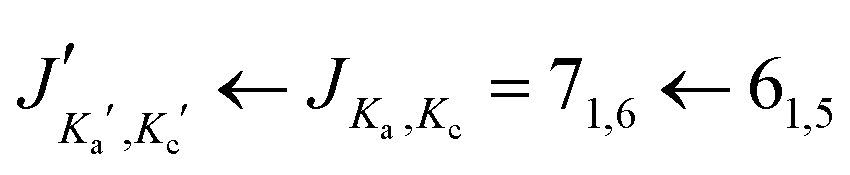 transition are depicted in Fig. 4.
transition are depicted in Fig. 4.
For each 13C isotopologue, between 12 and 22 rotational transitions could be assigned with an error for the fits between 3 kHz and 9 kHz. From this overall set of 30 rotational constants, the carbon atom positions, and thus the bond lengths and angles, were calculated using Kraitchman's equations for planar molecules (rs-structure), as implemented in the KRA program.19 The rotational constants of these fits and the calculated carbon atom positions are given in the ESI.†
The calculated inertial defect, Δ = Ic − Ia − Ib, of the experimentally determined structure has a non-zero value of −0.63 μÅ2, due to low-lying out-of-plane vibrations,20 for example, at around 60 cm−1 (about the dihedral angle C5–C4–C3–C2, atom labels are given in Fig. 4), contributing to the zero point motion of the molecule in the ground vibrational state. This inertial defect is neglected in Kraitchman's equations for planar molecules. Therefore, we carried out a mass-dependent structural fit (r(1)m-structure), which is typically closer to the equilibrium geometry of the molecule as it takes the inertial defect into account.11 For this, a least-square fit of the structure to the rotational constants was performed as, for example, implemented in the STRFIT-program.19 Additionally, an adjustable constant was fit, which accounts for the isotope-dependent rovibrational contribution to the moments of inertia of the molecule.11 Details of this fit are given in the ESI.† A least-square fit of the structure without additional fit parameters (r0-structure) led to a poorly defined structure and will not be discussed further.
The comparison of the structural parameters of the carbon backbone of s-trans–trans-cinnamaldehyde, obtained from these different approaches, is given in Tables 3 and 4.
| Bond lengths [Å] | MW rs | MW r(1)m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
MP2 re | GED rg10 | Acrolein17 |
|---|---|---|---|---|---|
| a The r(1)m-fit was performed using one additional adjustable parameter: cc = −0.0218(13). | |||||
| C1–C2 | 1.4745(18) | 1.456(34) | 1.465 | 1.473(8) | 1.468(4) |
| C2–C3 | 1.3568(26) | 1.341(65) | 1.349 | 1.348(1) | 1.340(4) |
| C3–C4 | 1.4270(7) | 1.464(62) | 1.460 | 1.470(8) | |
| C4–C5 | 1.4254(11) | 1.403(74) | 1.404 | 1.406(1) | |
| C5–C6 | 1.3980(5) | 1.410(24) | 1.394 | 1.392(1) | |
| C6–C7 | 1.3824(24) | 1.390(54) | 1.395 | 1.395(1) | |
| C7–C8 | 1.4081(18) | 1.399(41) | 1.398 | 1.398(1) | |
| C8–C9 | 1.3935(4) | 1.362(32) | 1.391 | 1.390(1) | |
| C9–C4 | 1.4003(13) | 1.414(89) | 1.405 | 1.408(1) | |
| Bond angles [°] | MW rs | MW r(1)ma |
MP2 re | GED rg10 | Acrolein17 |
|---|---|---|---|---|---|
| a The r(1)m-fit was performed using one additional adjustable parameter: cc = −0.0218(13). | |||||
| C1–C2–C3 | 119.71(29) | 121.3(59) | 119.7 | 115.3(27) | 120.4 |
| C2–C3–C4 | 126.55(16) | 127.1(53) | 127.3 | 128.3(26) | |
| C3–C4–C9 | 124.32(8) | 124.5(54) | 122.6 | 122.0(26) | |
| C4–C5–C6 | 121.14(7) | 120.5(34) | 120.9 | 121.4(3) | |
| C5–C6–C7 | 120.17(9) | 120.1(5) | 119.9 | 118.3 | |
| C6–C7–C8 | 119.81(4) | 119.8(13) | 119.7 | 122.1 | |
| C7–C8–C9 | 120.00(7) | 119.5(23) | 120.4 | 118.7 | |
| C8–C9–C4 | 121.54(4) | 123.1(30) | 120.5 | 121.0(3) | |
| C9–C4–C5 | 117.35(4) | 116.9(35) | 118.6 | 118.6(3) | |
The effect of the conjugated π-electron system becomes obvious by evaluating the individual C–C bond lengths (Table 3). In a conjugated system, the C–C single bonds are shorter compared to non-conjugated C–C single bonds (typically 1.53 Å), while the C–C double bonds are longer compared to non-conjugated C–C double bonds (typically 1.34 Å), which can be seen in both reported structures.
For this planar molecule, the results obtained from the different methods are close, because the rather small inertial defect of the molecules does not lead to large deviations. Nearly all determined structural parameters are in good agreement with each other and the ab initio values. This is also displayed in Fig. 5, which is a comparison between the calculated and experimentally determined carbon backbone structure. It is therefore appropriate to compare the calculated structural parameters (MP2/6-311++G(2d,2p)) for s-trans–trans-cinnamaldehyde with the ones for the higher energy conformer s-cis–trans-cinnamaldehyde. The steric repulsion between the carbon atom C3 and the oxygen atom as well as between the two π-electron clouds of the double bonds might cause the energy difference between the two conformers. Its occurrence is confirmed by an increase of the C1–C2–C3 angle and an elongation of the C1–C2 single bond in the s-cis–trans-conformer compared to the s-trans–trans-conformer. The same behavior can also be found in the experimentally determined values of s-cis- and s-trans-acrolein,17 which underlines again the similarity of the electronic environment in the side chain of trans-cinnamaldehyde and acrolein. The comparison of these values can be found in the ESI.†
 | ||
| Fig. 5 Comparison of the calculated (grey) and the experimentally determined (black) carbon backbone of s-trans–trans-cinnamaldehyde. The calculated oxygen position (white) is given as well. The calculated atom positions are obtained from MP2/6-311++G(2d,2p) calculations, while the experimentally determined values originate from the substitution method (rs-structure). | ||
The structural parameters obtained in the gas-electron diffraction study10 performed at 165 °C differ from the ab initio calculations and from the ones determined in this work (Tables 3 and 4) for the values of the side chain. In that study the authors could only identify the s-trans–trans-conformer to be the most dominant species in their 165 °C sample and could not unambiguously identify the presence of the second conformer, s-cis–trans-cinnamaldehyde. Since gas-electron diffraction structures are an average over all populated rovibrational states, the elevated temperature of this study is especially problematic as only a very small fraction of the molecules will be in the ground vibrational state. Therefore the structure obtained is not representative of the ground vibrational state or the equilibrium structure. This might explain the deviation of the bond angles of the side chain in the gas-electron diffraction structure. Furthermore, the analysis of the experimental data also depends on fixing the molecular parameters to calculated values and can be complicated by the presence of more than one conformer. For example, in the GED-study on trans-cinnamaldehyde reported in ref. 10 structural constraints based on the theoretical molecular structure were introduced, which might influence the resulting structure.
4. Conclusions
Two conformers of the odourant molecule trans-cinnamaldehyde were investigated using broadband microwave spectroscopy in the frequency range of 2–8.5 GHz. The rotational transitions of the s-trans–trans-conformer are about 40 times stronger than the transitions of the higher energy s-cis–trans-conformer. The high sensitivity of the spectrometer allowed us to record the rotational spectra of all 13C-singly substituted species of s-trans–trans-cinnamaldehyde and thereby determine the positions of all carbon atoms of this conformer. Different methods were used to obtain the structure from the measured values giving comparable results, within the given errors. A comparison with a recent gas-electron-diffraction study could be performed and the structural similarities between trans-cinnamaldehyde and acrolein were discussed. The similarities with acrolein showed that the phenyl ring has little influence on the structure of the side chain in trans-cinnamaldehyde.Acknowledgements
The authors thank Zbigniew Kisiel for help with the STRFIT program and David Schmitz for scientific discussions. This work has been supported by the excellence cluster ‘The Hamburg Centre for Ultrafast Imaging – Structure, Dynamics and Control of Matter at the Atomic Scale’ of the Deutsche Forschungsgemeinschaft. M.S. acknowledges financial support from the Fonds der Chemischen Industrie via a Dozentenstipendium.References
-
R. Scharnow, Codiertes Handbuch der Güter des Seetransports, 1986 Search PubMed
.
- J. C. Brookes, A. P. Horsfield and A. M Stoneham, Odour character differences for enantiomers correlate with molecular flexibility, J. R. Soc., Interface, 2009, 6, 75–86 CrossRef CAS PubMed
- H. Mouhib, W. Stahl, M. Lüthy, M. Büchel and P. Kraft, Cassis odor through microwave eyes: olfactory properties and gas-phase structures of all the cassyrane stereoisomers and its dihydro derivatives, Angew. Chem., Int. Ed., 2011, 50, 5576–5580 CrossRef CAS PubMed
- H. Mouhib and W. Stahl, Conformational analysis of green apple flavour: the gas-phase structure of ethyl valerate validated by microwave spectroscopy, ChemPhysChem, 2012, 13, 1297–1301 CrossRef CAS PubMed
- M. Bandell, G. M. Story, S. W. Hwang, V. Viswanath, S. R. Eid, M. J. Petrus, T. J. Earley and A. Patapoutian, Noxious cold ion channel trpa1 is activated by pungent compounds and bradykinin, Neuron, 2004, 41, 849–857 CrossRef CAS PubMed
- J. Dennis and T. Y. Shibamoto, Photochemical Products of Trans-Cinnamic Alcohol: Possible Formation of Skin Irritants and Allergens, J. Toxicol., Cutaneous Ocul. Toxicol., 1990, 9(2), 149–157 CrossRef
- J. Tenboer, S. Basu, N. Zatsepin, K. Pande, D. Milathianaki, M. Frank, M. Hunter, S. Boutet, G. J. Williams, J. E. Koglin, D. Oberthuer, M. Heymann, C. Kupitz, C. Conrad, J. Coe, S. Roy-Chowdhury, U. Weierstall, D. James, D. Wang, T. Grant, A. Barty, O. Yefanov, J. Scales, C. Gati, C. Seuring, V. Srajer, R. Henning, P. Schwander, R. Fromme, A. Ourmazd, K. Moffat, J. J. Van Thor, J. C. H. Spence, P. Fromme, H. N. Chapman and M. Schmidt, Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein, Science, 2014, 346(6214), 1242–1246 CrossRef CAS PubMed
- J. Kraitchman, Determination of Molecular Structure from Microwave Spectroscopic Data, Am. J. Phys., 1953, 21(1), 17 CrossRef CAS
- W. E. Steinmetz, The Application of Low-Resolution Microwave Spectroscopy to Conformational Analysis, J. Am. Chem. Soc., 1973,(96), 685–692 Search PubMed
- T. Egawa, R. Matsumoto, D. Yamamoto and H. Takeuchi, Molecular structure of trans-cinnamaldehyde as determined by gas electron diffraction aided by DFT calculations, J. Mol. Struct., 2008, 892, 158–162 CrossRef CAS
- J. K. G. Watson, A. Roytburg and W. Ulrich, Least-Squares Mass-Dependence Molecular Structures, J. Mol. Spectrosc., 1999, 196, 102–119 CrossRef CAS PubMed
- G. G. Brown, B. C. Dian, K. O. Douglass, S. M. Geyer, S. T. Shipman and B. H. Pate, A broadband Fourier transform microwave spectrometer based on chirped pulse excitation, Rev. Sci. Instrum., 2008, 79, 053103 CrossRef PubMed
- T. Betz, S. Zinn, J. B. Graneek and M. Schnell, Nuclear quadrupole coupling constants of two chemically distinct nitrogen atoms in 4-aminobenzonitrile, J. Phys. Chem. A, 2014, 118(28), 5164–5169 CrossRef CAS PubMed
- D. Schmitz, V. Alvin Shubert, T. Betz and M. Schnell, Multi-resonance effects within a single chirp in broadband rotational spectroscopy: the rapid adiabatic passage regime for benzonitrile, J. Mol. Spectrosc., 2012, 280, 77–84 CrossRef CAS
-
J. K. G. Watson, Vibrational Spectra and Structure, Elsevier, 1977 Search PubMed
-
C. M. Western, PGOPHER, a Program for Simulating Rotational Structure, University of Bristol, http://pgopher.chm.bris.ac.uk Search PubMed
- C. E. Blom, G. Grassi and A. Bauder, Molecular structure of s-cis- and s-trans-acrolein determined by microwave spectroscopy, J. Am. Chem. Soc., 1984, 106, 7427–7431 CrossRef CAS
- C. Puzzarini, E. Penocchio, M. Biczysko and V. Barone, Molecular structure and spectroscopic signatures of acrolein: theory meets experiment, J. Phys. Chem. A, 2014, 118, 6648–6656 CrossRef CAS PubMed
-
Z. Kisiel, PROSPE. Programs for Rotational Spectroscopy, http://info.ifpan.edu.pl/%E2%88%BCkisiel/prospe.htm Search PubMed
- T. Oka, On negative inertial defect, J. Mol. Struct., 1995, 352/353(95), 225–233 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp02582f |
| This journal is © the Owner Societies 2015 |