
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Li+ intercalation in isostructural Li2VO3 and Li2VO2F with O2− and mixed O2−/F− anions†
Ruiyong
Chen
*ab,
Shuhua
Ren
b,
Murat
Yavuz
acd,
Alexander A.
Guda
e,
Viktor
Shapovalov
e,
Raiker
Witter
bf,
Maximilian
Fichtner
ab and
Horst
Hahn
abg
aHelmholtz Institute Ulm for Electrochemical Energy Storage, 89081 Ulm, Germany. E-mail: ruiyong.chen@kit.edu; hc18chen@gmail.com
bInstitute of Nanotechnology, Karlsruhe Institute of Technology, P.O. Box 3640, 76021 Karlsruhe, Germany
cInstitute for Applied Materials, Karlsruhe Institute of Technology, P.O. Box 3640, 76021 Karlsruhe, Germany
dMaterials Science, Technische Universität Darmstadt, 64287 Darmstadt, Germany
eInternational Research Center–Smart Materials, Southern Federal University, 344090 Rostov-on-Don, Russia
fTechnomedicum, Tallinn University of Technology, 19086 Tallinn, Estonia
gJoint Research Laboratory Nanomaterials, Institute of Materials Science, Technische Universität Darmstadt, 64287 Darmstadt, Germany
First published on 8th June 2015
Abstract
Mixed-anion materials for Li-ion batteries have been attracting attention in view of their tunable electrochemical properties. Herein, we compare two isostructural (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) model intercalation materials Li2VO3 and Li2VO2F with O2− and mixed O2−/F− anions, respectively. Synchrotron X-ray diffraction and pair distribution function data confirm large structural similarity over long-range and at the atomic scale for these materials. However, they show distinct electrochemical properties and kinetic behaviour arising from the different anion environments and the consequent difference in cationic electrostatic repulsion. In comparison with Li2VO3 with an active V4+/5+ redox reaction, the material Li2VO2F with oxofluoro anions and the partial activity of V3+/5+ redox reaction favor higher theoretical capacity (460 mA h g−1vs. 230 mA h g−1), higher voltage (2.5 V vs. 2.2 V), lower polarization (0.1 V vs. 0.3 V) and faster Li+ chemical diffusion (∼10−9 cm2 s−1vs. ∼10−11 cm2 s−1). This work not only provides insights into the understanding of anion chemistry, but also suggests the rational design of new mixed-anion battery materials.
m) model intercalation materials Li2VO3 and Li2VO2F with O2− and mixed O2−/F− anions, respectively. Synchrotron X-ray diffraction and pair distribution function data confirm large structural similarity over long-range and at the atomic scale for these materials. However, they show distinct electrochemical properties and kinetic behaviour arising from the different anion environments and the consequent difference in cationic electrostatic repulsion. In comparison with Li2VO3 with an active V4+/5+ redox reaction, the material Li2VO2F with oxofluoro anions and the partial activity of V3+/5+ redox reaction favor higher theoretical capacity (460 mA h g−1vs. 230 mA h g−1), higher voltage (2.5 V vs. 2.2 V), lower polarization (0.1 V vs. 0.3 V) and faster Li+ chemical diffusion (∼10−9 cm2 s−1vs. ∼10−11 cm2 s−1). This work not only provides insights into the understanding of anion chemistry, but also suggests the rational design of new mixed-anion battery materials.
1. Introduction
Rechargeable Li-ion batteries are among the most desired choices for future electrochemical energy storage.1,2 The performance of Li-ion batteries is governed by the crystal structure and the active element(s) of the electrode materials. Understanding the role of anions, which acts as pillars for the crystal framework, is of importance for advancing the performance of electrode materials. Transition metal oxides (MOx)3 and lithium transition metal oxides (Li–M–O)4–7 with O2− anions have demonstrated pronounced variation as the materials to be used or considered. The metal–ligand bonding character (ionic/covalent) and the bond strength are crucial factors to determine the redox potentials8 and the Li+ storage mechanism (intercalation/conversion).9 The incorporation of guest anions of high electronegativity into the crystal lattice is found to be an effective strategy to enhance the electrode performance.10,11 Mixed-anion materials have become a new avenue for developing promising candidates with versatile crystal chemistry and physicochemical properties.12–15 However, the parent crystal symmetry is often altered by introducing guest anions, making it difficult to identify the individual function of anions governing the performance. Anion partitioning is often observed in mixed-anion materials upon lithiation (such as FeOF, with the formation of separate oxide and fluoride phases) in the case of large mismatch in the bonding between metal and different anion ligands.13,16 This leads to a conversion reaction at low voltage plateaus and the initial active phase can hardly be recovered.16 In contrast, early transition metal compounds with strong metal-ligand bonding (such as vanadium-based oxides and vanadates) are favourable for Li+ intercalation reaction without metal–oxygen bond cleavage.17 So far, candidate compounds of vanadium-based mixed anion materials for Li-ion batteries are extremely rare. We have previously reported a new oxyfluoride intercalation compound Li2VO2F,18 which has a high theoretical capacity of about 462 mA h g−1 and a stable crystal structure for more than one-electron reactions. It outperforms any known vanadium oxide and vanadate with respect to reversible capacity and structural stability.17 The realization of high capacity intercalation compounds based on mixed-anion oxyfluoride is of great importance. Identifying the fundamental aspects related to a number of material properties requires detailed knowledge of how the intermixed O2−/F− anion environment affects the structural features and the kinetic quantities.The Li+ storage hosts based on oxides with disordered rocksalt (DRS) structure (space group Fmm, No. 225) have received renewed interest because of their capability to extend the reversible capacity compared to classic cathode materials, stable phase for Li+ intercalation storage and a low lattice volume change for Li+ uptake/removal.19–24 To date, several such cation-DRS materials with a typical formula of Li1.211Mo0.467Cr0.3O2,19 Li1.3Nb0.3Mn0.4O2,20 Li1.2Ni0.4Ti0.4O221 and Li2MTiO422–24 have been reported, as summarized in Fig. 1. These Li-rich materials show an average operating voltage of about 2.2–3.1 V. All Li+ and metal cations are distributed randomly at the crystallographically equivalent site (4a Wyckoff site) in the DRS structure. The anionic sublattice (4b Wyckoff site) is exclusively occupied by close-packed oxygen. Note that the stoichiometric LiMO2 with DRS showed negligible Li+ storage activity25 due to the high energy barrier for Li+ diffusion.19,26 The structure-Li+ mobility relationships for such a class of materials have recently been clarified by Ceder et al.26 The Li-rich composition creates macroscopic percolation pathways spanning the entire DRS structure for Li+ diffusion. However, experimental observations elucidating the dependence of Li+ diffusion on the anion environment in isostructural compounds are still lacking. Compared to reported cation-DRS materials with only mono O2− anions,19–24 the material with mixed anions provides a wide variety to develop new disordered electrode materials with enhanced performance.
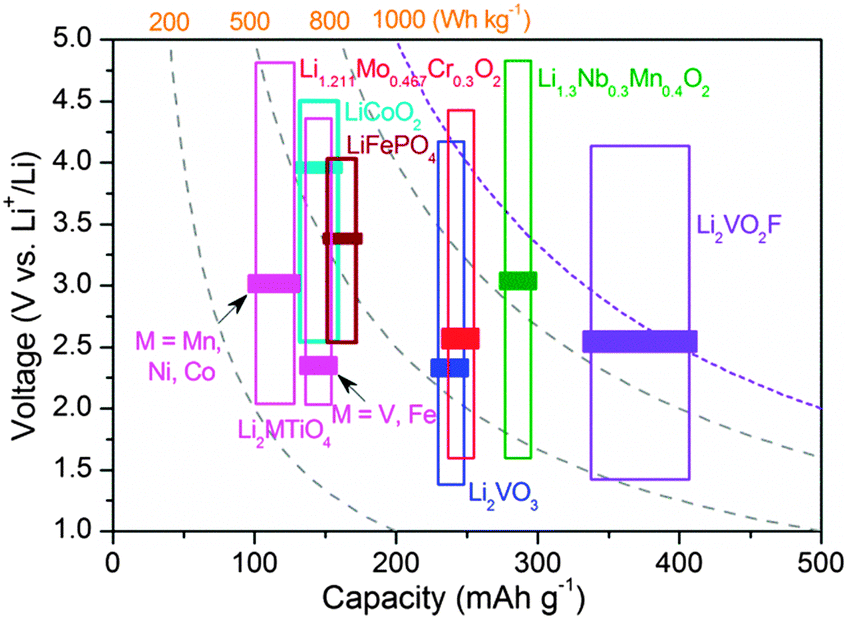 | ||
| Fig. 1 Typical operating voltage/capacity range/energy density plots for various reported DRS (space group Fmm) cathode materials of Li-ion batteries.18–24 The bars indicate the average voltages. LiFePO4 and LiCoO2 are also shown for comparison. | ||
To access the importance of the anion chemistry and to correlate the chemical features of materials to their electrochemical properties, we herein perform a direct experimental comparison between two isostructural model intercalation compounds Li2VO3 and Li2VO2F with oxo and oxofluoro anions, respectively. We show how F− anion incorporation can affect positively the key electrochemical properties in terms of capacity, voltage, hysteresis, charge transfer resistance and Li+ transport, without alternating the DRS crystal structure. These insights into anion chemistry in disordered materials are suggestive for tuning the material properties and therefore open up new possibilities for energy storage materials based on the DRS structure with a rational design of the anionic sublattice.
2. Experimental
2.1. Synthesis
All materials were synthesized by a ball-milling route (Retsch PM100, WC jar and balls) in an argon atmosphere. For Li2VO2F, the precursors of Li2O, LiF and V2O3 were used (450 rpm, 40 h). For Li2VO3, the precursors of Li2O and V2O4 were used (400 rpm, 14 h). The starting materials were mixed with intended stoichiometric amounts together with an excess of 10 wt% Li2O to compensate for lithium loss during synthesis.2.2. Electrochemical studies
For electrochemical performance tests, the as-obtained materials were ball-milled with Super P carbon (20 wt%) at 300 rpm for 10 h. A slurry was fabricated by mixing the active powder (72 wt%)/Super P (18 wt%) composite and 10 wt% poly(vinylidene fluoride-hexafluoropropylene) (Solef 5130) in dimethylformamide (DMF). The slurry was coated on a stainless steel current collector and dried under vacuum at 90 °C. The loading of the active material on the current collector was about 3 mg cm−2. The electrochemical charge–discharge performance was evaluated with lithium as the anode and LiPF6 (1 M in ethylene carbonate and dimethyl carbonate, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio) as electrolyte using a Swagelok cell in the same voltage range of 1.3–4.1 V at 25 °C. For ex situ structural characterization, samples were collected after disassembling the cell and washed with dimethyl carbonate. Electrochemical impedance spectroscopy (EIS) measurements were performed at 25 °C using a Bio-Logic instrument in the frequency range of 100 kHz–1 mHz with an AC voltage of 5 mV. Prior to EIS measurements, the cathode materials were cycled to the requested voltages and then held for 2 h to reach a quasi-equilibrium state.
:1 volume ratio) as electrolyte using a Swagelok cell in the same voltage range of 1.3–4.1 V at 25 °C. For ex situ structural characterization, samples were collected after disassembling the cell and washed with dimethyl carbonate. Electrochemical impedance spectroscopy (EIS) measurements were performed at 25 °C using a Bio-Logic instrument in the frequency range of 100 kHz–1 mHz with an AC voltage of 5 mV. Prior to EIS measurements, the cathode materials were cycled to the requested voltages and then held for 2 h to reach a quasi-equilibrium state.
2.3. Synchrotron XRD and PDF analysis
Synchrotron X-ray diffraction (XRD) data were collected at beamline BL04, MSPD (λ = 0.41343 Å) at the ALBA Synchrotron Light Source, Spain. The powders were loaded into 0.7 mm glass capillaries in an argon-filled glovebox. The high energy X-rays were used to access the high magnitude of scattering vectors of Qmax = 26 Å−1 (Q = 4π sin θ/λ) for the success of atomic pair distribution function (PDF) analysis. The scattered radiation was collected using a Mythen II detector. The diffraction patterns were refined using the Rietveld method using the FullProf program.27 The PDF, G(r), provides the probability of finding atomic pairs separated by the real space distance (r) in a sample, weighted by the concentration and scattering power of the contributing atoms. Raw X-ray total scattering data were processed using the program Fit2D28 and then Fourier transformed to yield the PDF, G(r), using PDFgetX3.29 PDFFIT30 was used to refine the structural models.2.4. V K-Edge XANES
The vanadium K-edge X-ray absorption near edge structure (XANES) spectra was recorded using an in-house Rigaku R-XAS spectrometer in transmission mode at room temperature with a crystal monochromator Ge(311) and an energy resolution of 0.7 eV, at the Southern Federal University, Russia. Pellets were prepared in a glovebox and sealed in an X-ray transparent plastic bag in an inert atmosphere. An argon-filled ionization chamber was used to detect the incident intensity on the sample and a scintillation counter was used for the detection of transmitted intensity. The goniometer section of spectrometer was filled with helium buffer gas to avoid the strong air absorption of X-rays. Ten spectra were acquired and averaged for each sample.2.5. Raman, 7Li MAS NMR and EPR
Raman spectra were collected using a He–Ne laser at 632.8 nm with a laser power of about 1 mW. An incident laser spot (2 μm) was focused on the powder samples sealed in glass capillaries, under the assistance of a microscope objective. Spectra were recorded at several different sample positions. Solid state 7Li magic angle spinning (MAS, with a spinning speed of 40 kHz) nuclear magnetic resonance (NMR) experiments were performed on a Bruker Advance spectrometer using 1.8 mm MAS probe at room temperature (90°/180° pulses of 2/4 μs). Spectra were referenced to a 1 M LiCl solution at 0 ppm. Electron paramagnetic resonance (EPR) spectra were recorded at room temperature with powders sealed in a 4 mm quartz tube.3. Results and discussion
3.1. X-ray diffraction
Nanoscale isostructural Li2VO2F and Li2VO3 were synthesized by a high energy ball-milling technique using lithium oxide/fluoride and vanadium oxide as precursors in an inert atmosphere. The total scattering data were collected with synchrotron X-ray diffraction and subsequently converted to d spacing values (Fig. 2a). The XRD pattern for Li2VO3 resembles that for Li2VO2F, suggesting long-range structural similarities. Rietveld refinement confirms that both materials crystallize in the same cubic Fmm symmetry with a DRS structure (Fig. S1†). Li and V cations (2:1) intermix evenly at the 4a Wyckoff sites, whereas the anion sublattice is occupied by O2− for Li2VO3 or mixed O2−/F− for Li2VO2F. The interplanar spacing d200 is directly related to the nearest cation–anion distance (a/2) in the cubic symmetry, which shows quite close values for Li2VO3 and Li2VO2F. The lattice constant was further refined to be a = 4.1178 Å for Li2VO2F and a = 4.1043 Å for Li2VO3, respectively.
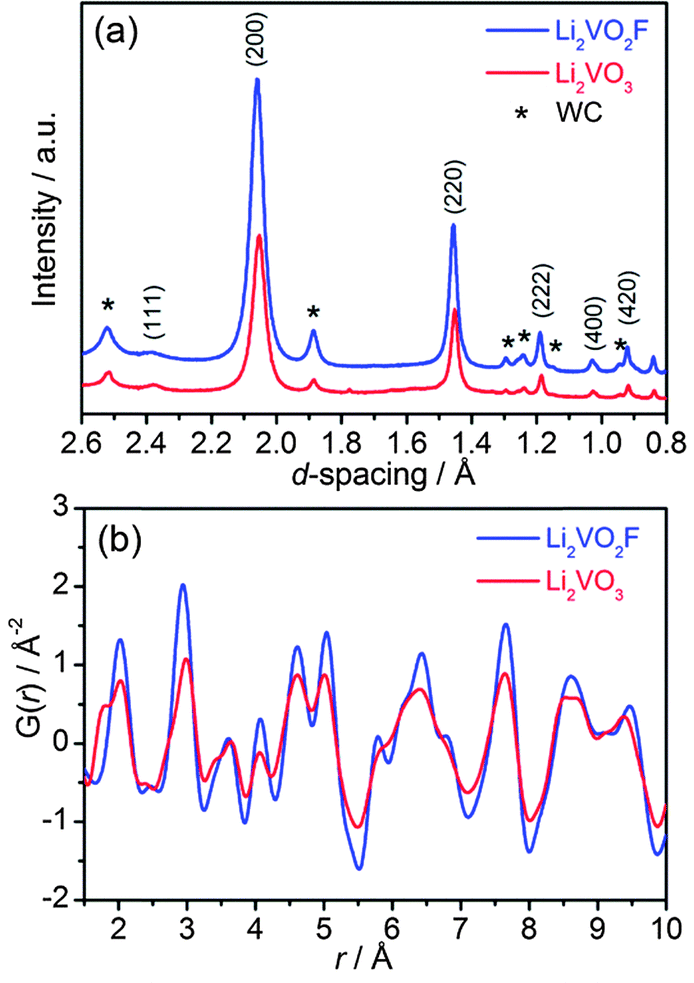 | ||
| Fig. 2 (a) Synchrotron XRD patterns (λ = 0.41343 Å), and (b) atomic PDF profiles in the r-range from 1.5 to 10 Å for Li2VO2F and Li2VO3. | ||
Note that the Rietveld technique is not able to account for the local distortion at the atomic scale. Hence, the atomic PDF analysis of the diffraction data, which considers both Bragg and diffuse scatterings, was performed to probe the short-range and intermediate-range structural information independent of the long-range order.
3.2. Atomic PDF analysis
The atomic PDF, G(r), decays to zero at r ≈ 100 Å for Li2VO2F and Li2VO3 (Fig. S2†), indicating a crystallite size of about 10 nm. For r-range from 1.5 to 10 Å, the experimental PDF profiles for all samples show large similarities (Fig. 2b). For Li2VO2F, the PDF profile can be well fitted using the average Rietveld structure model (Fig. 3a). Moreover, the local structural imperfections can be visualized directly from the PDF fitting. The local distortions are evidenced by the deviations of the first two PDF peaks (d4a–4b, d4a–4a or d4b–4b) from the expected interatomic distances of a/2 and a/√2 (indicated with arrows in Fig. 3a) in the lattice with atoms constrained to particular positions. Such distortions reveal the difference in local bond lengths in the octahedral units. By further refining the atomic positions of F and the isotropic atomic displacement parameter (Uiso,F = 0.028 Å2), one can obtain an improvement in the reliability factor (Rwp) from 19% to 6.8%.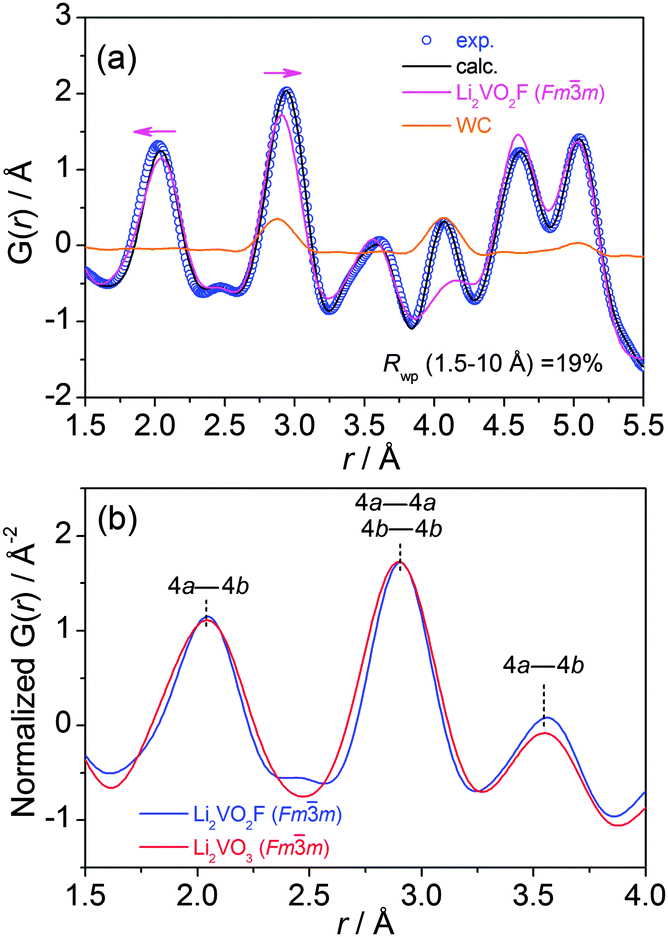 | ||
| Fig. 3 (a) Experimental and calculated (using Rietveld average structure model) atomic PDF profile for Li2VO2F, Uiso (Å2) parameters for Li, V, O and F are 0.0024, 0.0122, 0.0128 and 0.1721, respectively. (b) Modelled PDF profiles for two Fmm phases with oxo and oxofluoro anion ligands in the r-range of 1.5–4 Å. | ||
For Li2VO3, a shoulder PDF peak located at a shorter bond length (1.74 Å) was observed (Fig. 2b). The PDF profile for Li2VO3 cannot be successfully fitted using crystallographic structure models derived from Rietveld refinement by considering only a single Fmm phase. Attempts to refine the structure using an assumed starting model with a NaCl superstructure (LiVO2-like phase, Fdm), as previously discussed by Pralong31 and Chieh,32 do not yield a good fit. Instead, PDF refinement by introducing a monoclinic phase (LiVO3, C2/c) yields an improved fit (Rwp = 16%) (Fig. S3†). This suggests that LiVO3 exists as an amorphous component in the as-milled material, which is undetectable by synchrotron XRD. This shoulder at 1.74 Å can be assigned to the V–O bonds of the tetrahedral VO4 structural units in the monoclinic LiVO3 (C2/c) amorphous phase.33 A quantification analysis from the PDF data suggests a phase fraction of 25% for the amorphous component.
The presence of the amorphous LiVO3 was further checked by Raman spectroscopy. The Raman spectrum for the disordered Li2VO2F (Oh5 symmetry) is nearly featureless (Fig. S4†), which arises from the random distribution of cations/anions and the fluctuation in the cation–anion bond distances. In contrast, several Raman bands at 818, 787 and 369 cm−1 were observed for the as-milled Li2VO3 (Fig. S4†). These Raman features bear a likeness to that of a high-pressure phase of amorphous LiVO3 with structural deformation.34
The PDF corresponding to the Fmm phase with oxo and oxofluoro anions was extracted from the raw PDF multi-phase data and plotted together with intensities normalized arbitrarily, keeping the first two peaks superimposed (Fig. S5†). It can be seen that Li2VO2F has slightly larger lattice dimension compared to Li2VO3. In Fig. 3b, PDF profiles in short range (1.5–4 Å) are shown to highlight the nearest bonding character at approximately a unit lattice dimension. The three PDF peaks correspond to the bond lengths of the nearest cation–anion (4a–4b), the nearest cation–cation (4a–4a) or anion–anion (4b–4b), and the second nearest cation–anion (4a–4b) in the cubic Fmm structure. The V(Li)–O(F) bond distances are centered around the same values. The relative intensities of the correlations are nearly identical. In addition, the width of the PDF peak reflects the distribution of atomic distances and the correlation of atomic thermal motions. The oxyfluoride shows narrower PDF peaks than the oxide. It is deduced that the oxofluoro ligands do not cause a significant increase in the degree of distortion compared to the oxo counterpart.
3.3. 7Li NMR and EPR
7Li MAS NMR measurements were performed for the as-milled Li2VO3 in order to probe the local Li environments. A broad 7Li resonance line (with a linewidth of 58 ppm) centred at −24.6 ppm was observed (Fig. S6†), together with a sharp signal at ∼0 ppm (Li-containing species from diamagnetic impurities). Compared to Li2VO2F (−9.1 ppm),18 the more negatively shifted 7Li isotropic line for Li2VO3 indicates the presence of Li+–O–V4+ interactions. The random cationic distribution in the DRS phase is responsible for the large distribution of chemical shifts and the broad 7Li signal,35 as also observed for Li2VO2F.18 Li species in the amorphous LiVO3 cannot be resolved. EPR was applied to probe the presence of V4+ in these samples, which can supplement NMR. Note that V5+ is diamagnetic and cannot exhibit EPR, whereas V3+ is paramagnetic and its EPR is rarely observed under ambient experimental conditions.36 Li2VO3 shows a reasonably intense EPR signal (Fig. S6†), compared to Li2VO2F (with trace amounts of V4+ due to a non-stoichiometric composition).183.4. V K-Edge XANES
Further spectroscopic characterization using element-specific and oxidation state-sensitive techniques were performed to discriminate the electronic structures of V for both materials. The normalized V K-edge XANES spectra together with the first derivative spectra for the as-milled Li2VO2F and Li2VO3 are shown in Fig. 4. For comparison, the XANES spectra for several reference materials V2O3 (V3+), V2O5 (V5+) and VOF3 (V5+) are also shown. Similar to V2O3, Li2VO2F has a weak pre-edge absorption located at about 5.468 keV. In contrast, a more prominent pre-edge peak located at a higher energy of 5.469 keV was observed for Li2VO3. Pre-edge peaks can be attributed to quadrupole 1s–3d transitions or dipole transitions to the 3d states hybridized with p-states. The high intensity of the pre-edge feature in Li2VO3 indicates noncentrosymmetric coordination, as is found in several vanadium oxides.37 Both Li2VO2F and Li2VO3 show strong whiteline peaks, compared to the reference materials. The main absorption edge (dipole-allowed 1s–4p transition) for Li2VO3 is located at higher energy (an energy shift by ∼2 eV) than that for Li2VO2F, indicating a higher average oxidation state of V. In addition, the main absorption edge shapes for Li2VO2F and Li2VO3 are very similar with shoulder peaks at 5.475 keV (1s–4p shakedown transition). Such absorption features can be better seen in the derivative spectra. The local coordination symmetries of V absorbers in Li2VO2F and Li2VO3 will be further studied by extended X-ray absorption fine structure using synchrotron radiation in future.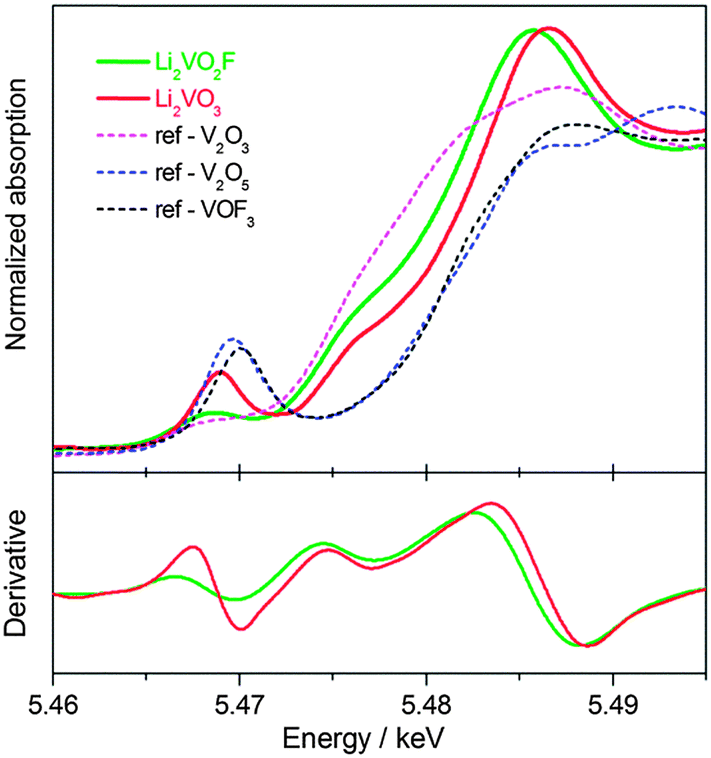 | ||
| Fig. 4 V K-edge XANES spectra and the derivative plots for Li2VO2F and Li2VO3. | ||
3.5. Galvanostatic electrochemical cycling
The above-mentioned analysis confirms the structural similarity between the Li2VO3 and Li2VO2F, which makes these materials ideally suitable for a comparative study of the anion chemistry in electrochemical intercalation reactions. For Li2VO3, only the V4+/V5+ redox couple is active for lithium storage. In contrast, Li2VO2F has a high theoretical capacity of 462 mA h g−1, assuming a two-electron V3+/V5+ oxidation reaction. The charge–discharge performance for Li2VO3 and Li2VO2F was compared between 4.1 and 1.3 V at 25 °C and at a rate of C/20 based on their theoretical capacities. The distinct voltage profiles for the first two cycles are shown in Fig. 5a. The charge–discharge capacity was calculated based on the mass of corresponding active materials. Li2VO2F delivers a capacity of 330 mA h g−1 (exchange of about 1.4Li+) at 25 °C. Up to 1.8Li+ exchange has been previously observed at elevated temperature and at low current rate.18 For Li2VO3, a capacity of about 295 mA h g−1 was observed, indicating a possible large contribution from amorphous LiVO3.38 Above 3.7 V, the charge curve for Li2VO3 shows a fast increase in voltage compared to that for Li2VO2F. Note that a full capacity for a Fdm Li2−xVO3 and a C2/c LiVO3 has been reached when cycling between 3.5 and 1 V.31,38 Thus, the overcharge of Li2VO3 to 4.1 V may trigger the electrolyte decomposition. The second charge curves for both materials are roughly overlapped. However, the discharge curves shift down by about 0.3 V for Li2VO3 compared to that for Li2VO2F. The onset discharge voltage is about 3.5 V for Li2VO3, which is lower than that of about 4 V for Li2VO2F. The average discharge voltage is about 2.2 V for Li2VO3 and about 2.5 V for Li2VO2F. The higher lithiation voltage for the oxyfluoride is believed to be related to the presence of the F ligand with high electronegativity. The sloping profiles suggest a single-phase intercalation mechanism for both materials.
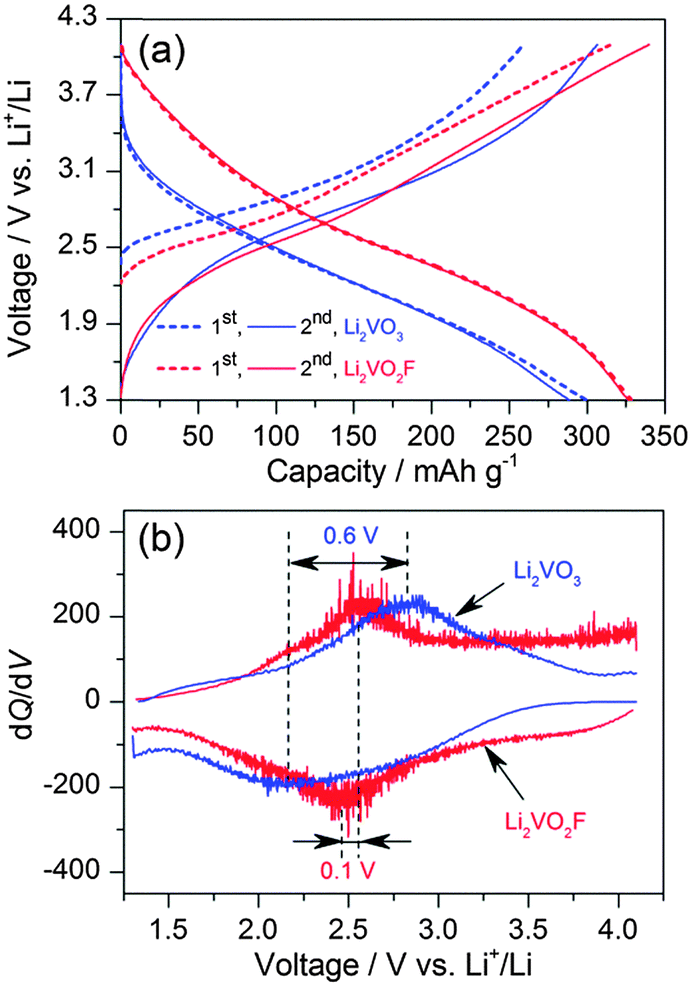 | ||
| Fig. 5 (a) Voltage profiles for Li2VO3 and Li2VO2F measured between 1.3 and 4.1 V. (b) dQ/dV plots from the second cycles. | ||
The dQ/dV curves in Fig. 5b show the difference in the centred redox voltages, which is about 0.6 V for Li2VO3 in comparison with only 0.1 V for Li2VO2F. Bearing in mind the above observations, it is remarkable that Li2VO2F allows about 1.4Li+ intercalation reaction (a dominate V4.4+/V3+ reduction) centred at ∼2.5 V with oxofluoro ligands, in contrast to the 1.0Li+ intercalation process (V5+/V4+ reduction) at ∼2.2 V for Li2VO3 with oxo ligands. Low onset discharge voltage (∼3.5 V), low average discharge voltage (∼2.3 V) and large voltage hysteresis (∼0.7 V) were also found in other vanadium-based DRS materials with oxo ligands.24 These observations suggest that the ligand anions play a predominate role in the electrochemical properties in terms of energy density and energy efficiency.
3.6. EIS Studies
EIS spectra were recorded to extract the characteristic kinetic parameters such as the charge transfer resistance (Rct) and the Li+ chemical diffusion coefficients (DLi+) for Li2VO3 and Li2VO2F. All the Nyquist plots consist of two depressed semicircles at high and middle frequency regions and an inclined straight line at the low frequency region (Fig. 6). The Rct, which defines the transport rate of charge carriers across the electrode/electrolyte interface, is estimated from the EIS fitting (χ2 ranging from 10−4 to 10−6) of the high and middle frequency semicircle regions (3 Hz–10 kHz for Li2VO3, and 10 Hz–10 kHz for Li2VO2F) using a proposed equivalent circuit by Levi et al.,39 consisting of the solution resistance and two parallel RQ elements in series.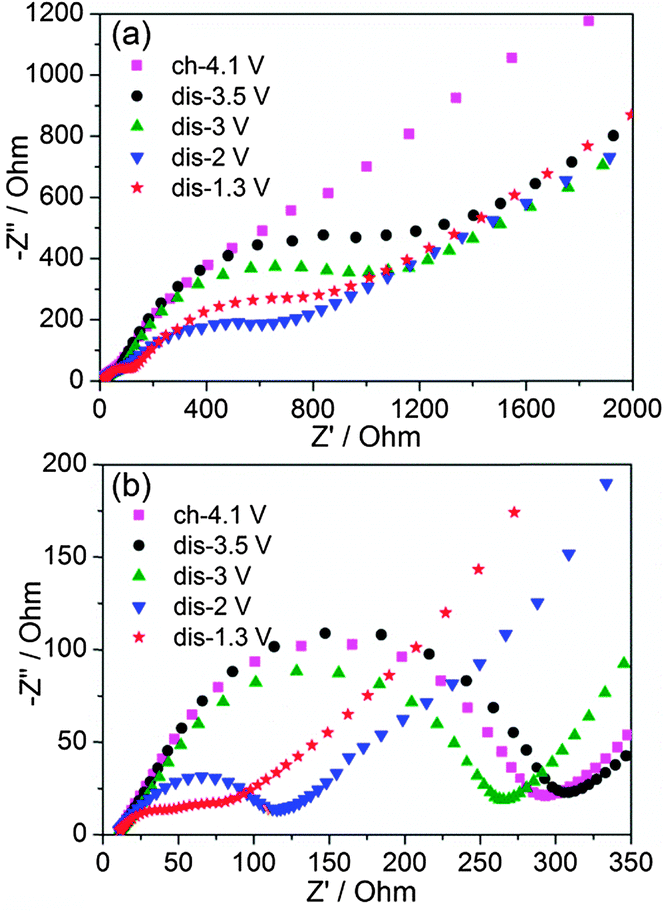 | ||
| Fig. 6 Nyquist plots of (a) Li2VO3 and (b) Li2VO2F at various voltages. | ||
Fig. 7a shows the variation of Rct at different voltages. Li2VO2F showed lower Rct compared to Li2VO3, indicating fast kinetics of the electrode reaction. This provides physical interpretation of the electrochemical polarization behaviour for both materials. In addition, for Li2VO2F, Rct showed a relatively small variation with voltage (i.e., Li content), compared to that for Li2VO3. These observations suggest that the microscopic Li+ diffusion behaviour can be largely different due to the difference in atomic configuration of the rocksalt structural units although the dilithium composition in DRS assures a macroscopic percolating network for Li+ migration.26
3.7. Li+ Diffusivity
The diffusion-controlled Warburg impedance and its dependence on lithium content for Li2VO3 and Li2VO2F were studied by analysing the low frequency complex impedance regions. The DLi+ at different voltages were calculated according to the equation: DLi+ = 1/2(Vm/SFσ)2(dE/dx)2,40 where Vm is the molar volume, S is the contact area between electrolyte and sample, F is the Faraday constant, σ is the Warburg coefficient and dE/dx is the slope of open-circuit voltage versus Li+ concentration. The Warburg coefficient (σ) can be determined from the slope of the real resistance (Z′) versus the inverse square root of the angular frequency (ω−0.5) of the low-frequency impedance data (Fig. S7†). A linear behaviour was observed for frequency between 10 and 1 mHz. DLi+ as a function of lithium content for the materials is shown in Fig. 7b. The experimentally estimated DLi+ is about 10−9 cm2 s−1 for Li2VO2F, which is about two orders of magnitude higher than that for Li2VO3 (10−11 cm2 s−1). Considering that a high DLi+ (∼10−9 cm2 s−1) has been reported for a C2/c LiVO3 for lithium intercalation,38 the presence of the amorphous LiVO3 in the material should not affect largely the apparent EIS behaviour of Li2VO3. Hence, these results indicate that the Li+ migration is relatively facile for Li2VO2F. These transport properties correlate directly to their distinct kinetic behaviour.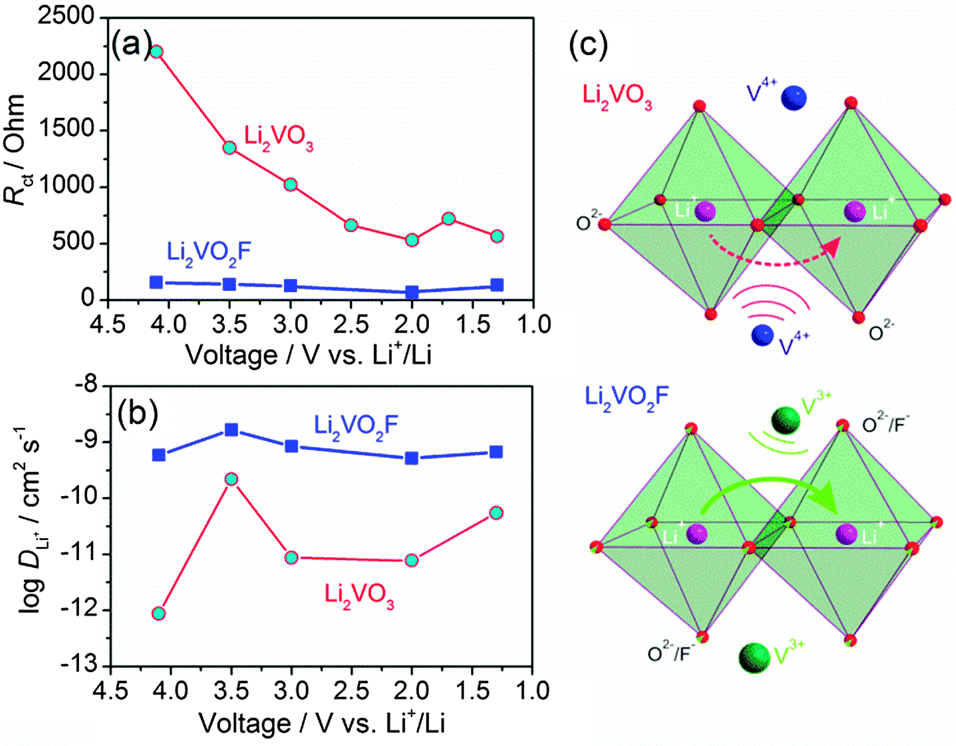 | ||
| Fig. 7 Variation of (a) Rct and (b) DLi+ at different voltages for Li2VO3 and Li2VO2F. (c) Schematic view of the Li+ migration through the o–t–o path indicated by curved arrows, which is subjected to the electrostatic repulsion from neighbouring vanadium cations in the lattice. Note that Li (purple spheres) and V (blue/green spheres) have a statistical distribution of 2:1 at the cationic sites. Anions O2− and O2−/F− are represented by small red and red/green spheres, respectively. | ||
It is assumed that an unimpeded Li+ diffusion pathway is maintained for Li2−xVO2F with the variation of x upon cycling, arising from the free Li+ transport manner in a DRS structure. This is in contrast to the increase in the Li+ diffusion barrier for layered structure with a progressive narrowing of the Li-slab distance during Li+ removal.41 For layered rocksalt LiVO2, cation migration from original octahedral sites to tetrahedral sites has been observed upon partial Li+ removal.42 However, so far no evidence has been found for such cation migration in Li2VO2F.18 Further work is on-going to clarify the local structural features of the materials upon Li+ removal in the DRS structure.
On the basis of the ab initio computations by Ceder et al.,19,26 it is anticipated that Li+ hopping from one octahedral site to its neighbouring octahedral site through a vacant interstitial tetrahedral site (i.e., o–t–o diffusion path, as denoted by the arrows in Fig. 7c) can proceed with large freedom for Li2VO2F and Li2VO3 with high overall Li+ concentration (2/3 of the cationic sites are occupied by Li+). The dilithium chemical composition with DRS structure ensures the percolation diffusion channel spanning the entire structure. The Li+ migration through the tetrahedral site, which shares face with the octahedral V cation, will be subjected to the electrostatic repulsion from the neighbouring V cation. Earlier ab initio computations on layered materials showed that the high-valent cation will have a negative impact on the activation energy for Li+ transport.43–45 It is a straightforward case that the electrostatic repulsion between the mobile Li+ and its neighbouring V4+ in Li2VO3 is higher than that between Li+ and V3+ in Li2VO2F (Fig. 7c). Li+ hopping through an o–t–o path in Li2VO2F is energetically more favourable than that in Li2VO3. Thus, in spite of the extremely high crystallographic similarities between Li2VO3 and Li2VO2F, the different anionic environment and the difference in the valence of vanadium lead not only to a different redox energy of vanadium, but also to a significant impact on the Li+ hopping behaviour in the lattice. In general, the size difference of anions can alter the dimension of the unit cell and affect the Li+ diffusivity.45 Additionally, anions with different electron charge densities and their local spatial distributions in the crystal structures can contribute to considerable differences in the activation barrier. These issues need to be addressed in future work. Divergences in the bonding character, atomic charges, electron density and electrostatic interaction across two isostructural samples with a DRS structure illustrate how material properties can vary largely depending on the anion environment.
4. Conclusions
In summary, we correlated the chemical/electronic nature to the electrochemical/kinetic behaviour of intercalation materials Li2VO3 and Li2VO2F with an isostructural DRS structure. Detailed crystallographic and spectroscopic characterizations were performed to identify the structure of the studied materials. Besides the higher theoretical and practical capacity of the oxyfluoride (Li2VO2F), improved electrochemical performance (higher voltage, lower polarization and better Li+ diffusivity) was also observed over its oxide analogue (Li2VO3). These comparative studies demonstrate how to enhance cathode performance by simply controlling the anion chemistry through incorporating a high electronegativity anion ligand into the crystal structure. In addition, our further work has also proven that even higher voltage and accordingly higher energy density can be achieved in such an oxyfluoride system.46 The combination of DRS structure and mixed anion chemistry would be of general interest for designing new high-performance energy storage materials.Acknowledgements
We acknowledge the allocated synchrotron beamtime at the ALBA Synchrotron Light Source, Spain. We thank I. Peral for fruitful PDF discussions and S. Lebedkin for assisting the Raman measurements. A.G. would like to thank support from Russian Ministry of Education and Science (MK-3206.2014.2). R.W. thanks the EU Fund, Estonian Research Council, TUT and KBFI (MTT68, SF0690034s09, B618 and PUT126).Notes and references
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J.-M. Tarascon, Nature, 2000, 407, 496 CrossRef CAS PubMed.
- K. Mizushima, P. C. Jones, P. J. Wiseman and J. B. Goodenough, Mater. Res. Bull., 1980, 15, 783 CrossRef CAS.
- T. Ohzuku and Y. Makimura, Chem. Lett., 2001, 642 CrossRef CAS.
- M. M. Thackeray, P. J. Johnson, L. A. de Picciotto, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1984, 19, 179 CrossRef CAS.
- R. Chen, M. Knapp, M. Yavuz, R. Heinzmann, D. Wang, S. Ren, V. Trouillet, S. Lebedkin, S. Doyle, H. Hahn, H. Ehrenberg and S. Indris, J. Phys. Chem. C, 2014, 118, 12608 CAS.
- B. C. Melot and J.-M. Tarascon, Acc. Chem. Res., 2013, 46, 1226 CrossRef CAS PubMed.
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. Van Schalkwijk, Nature Mater., 2005, 4, 366 CrossRef PubMed.
- N. Recham, J.-N. Chotard, L. Dupont, C. Delacourt, W. Walker, M. Armand and J.-M. Tarascon, Nature Mater., 2010, 9, 68 CrossRef CAS PubMed.
- N. Pereira, F. Badway, M. Wartelsky, S. Gunn and G. G. Amatucci, J. Electrochem. Soc., 2009, 156, A407 CrossRef CAS PubMed.
- M. Leblanc, V. Maisonneuve and A. Tressaud, Chem. Rev., 2015, 115, 1191 CrossRef CAS PubMed.
- D. Dambournet, K. W. Chapman, P. J. Chupas, R. E. Gerald II, N. Penin, C. Labrugere, A. Demourgues, A. Tressaud and K. Amine, J. Am. Chem. Soc., 2011, 133, 13240 CrossRef CAS PubMed.
- M. Duttine, D. Dambournet, N. Penin, D. Carlier, L. Bourgeois, A. Wattiaux, K. W. Chapman, P. J. Chupas, H. Groult, E. Durand and A. Demourgues, Chem. Mater., 2014, 26, 4190 CrossRef CAS.
- G. Hautier, A. Jain, H. Chen, C. Moore, S. P. Ong and G. Ceder, J. Mater. Chem., 2011, 21, 17147 RSC.
- K. M. Wiaderek, O. J. Borkiewicz, E. Castillo-Martínez, R. Robert, N. Pereira, G. G. Amatucci, C. P. Grey, P. J. Chupas and K. W. Chapman, J. Am. Chem. Soc., 2013, 135, 4070 CrossRef CAS PubMed.
- N. A. Chernova, M. Roppolo, A. C. Dillon and M. S. Whittingham, J. Mater. Chem., 2009, 19, 2526 RSC.
- R. Chen, S. Ren, M. Knapp, D. Wang, R. Witter, M. Fichtner and H. Hahn, Adv. Energy Mater., 2015, 5, 1401814 Search PubMed; R. Chen, S. Ren, S. Indris, M. Fichtner and H. Hahn, European Pat., EP 14160894.3, 2014 Search PubMed.
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519 CrossRef CAS PubMed.
- N. Yabuuchi, M. Takeuchi, D. Endo, T. Ozaki, T. Inamasu, J.-Y. Son, Y.-T. Cui, H. Oji and S. Komaba, 224th ECS Meeting (The Electrochemical Society, 2013).
- L. Zhang, H. Noguchi, D. Li, T. Muta, X. Wang, M. Yoshio and I. Taniguchi, J. Power Sources, 2008, 185, 534 CrossRef CAS PubMed.
- M. Küzma, R. Dominko, A. Meden, D. Makovec, M. Bele, J. Jamnik and M. Gaberšček, J. Power Sources, 2009, 189, 81 CrossRef PubMed.
- M. Yang, X. Zhao, Y. Bian, L. Ma, Y. Ding and X. Shen, J. Mater. Chem., 2012, 22, 6200 RSC.
- R. Dominko, C. Vidal-Abraca Garrido, M. Bele, M. Kuezma, I. Arcon and M. Gaberscek, J. Power Sources, 2011, 196, 6856 CrossRef CAS PubMed.
- M. N. Obrovac, O. Mao and J. R. Dahn, Solid State Ionics, 1998, 112, 9 CrossRef CAS.
- A. Urban, J. Lee and G. Ceder, Adv. Energy Mater., 2014, 4, 1400478 Search PubMed.
- J. Rodríguez-Carvajal, Physica B, 1993, 192, 55 CrossRef.
- A. P. Hammersley, S. O. Svenson, M. Hanfland and D. Hauserman, High Pressure Res., 1996, 14, 235 CrossRef PubMed.
- P. Juhás, T. Davis, C. L. Farrow and S. J. L. Billinge, J. Appl. Crystallogr., 2013, 46, 560 CrossRef.
- Th. Proffen and S. J. L. Billinge, J. Appl. Crystallogr., 1999, 32, 572 CrossRef CAS.
- V. Pralong, V. Gopal, V. Caignaert, V. Duffort and B. Raveau, Chem. Mater., 2012, 24, 12 CrossRef CAS.
- C. Chieh, B. L. Chamberland and A. F. Wells, Acta Crystallogr. B, 1981, 37, 1813 CrossRef.
- S. Hayakawa, T. Yoko and S. Sakka, J. Solid State Chem., 1994, 112, 329 CrossRef CAS.
- A. Grzechnik and P. F. McMillan, J. Phys. Chem. Solids, 1995, 56, 159 CrossRef CAS.
- R. Chen, M. Knapp, M. Yavuz, S. Ren, R. Witte, R. Heinzmann, H. Hahn, H. Ehrenberg and S. Indris, Phys. Chem. Chem. Phys., 2015, 17, 1482 RSC.
- C. P. Poole and H. A. Farach, The Theory of Magnetic Resonance, Wiley-Interscience, New York, 1972 Search PubMed.
- J. Wong, F. W. Lytle, R. P. Messmer and D. H. Maylotte, Phys. Rev. B, 1984, 30, 5596 CrossRef CAS.
- X. M. Jian, J. P. Tu, Y. Q. Qiao, Y. Lu, X. L. Wang and C. D. Gu, J. Power Sources, 2013, 236, 33 CrossRef CAS PubMed.
- M. D. Levi, K. Gamolsky, D. Aurbach, U. Heider and R. Oesten, Electrochim. Acta, 2000, 45, 1781 CrossRef CAS.
- C. Ho, I. D. Raistrick and R. A. Huggins, J. Electrochem. Soc., 1980, 127, 343 CrossRef CAS PubMed.
- A. Van der Ven, J. Bhattacharya and A. A. Belak, Acc. Chem. Res., 2013, 46, 1216 CrossRef CAS PubMed.
- M. M. Thackeray, L. A. de Picciotto, W. I. F. David, P. G. Bruce and J. B. Goodenough, J. Solid State Chem., 1987, 67, 285 CrossRef CAS.
- K. Kang, Y. S. Meng, J. Bréger, C. P. Grey and G. Ceder, Science, 2006, 311, 977 CrossRef CAS PubMed.
- A. Van der Ven and G. Ceder, J. Power Sources, 2001, 97–98, 529 CrossRef CAS.
- K. Kang and G. Ceder, Phys. Rev. B, 2006, 74, 094105 CrossRef.
- S. Ren, R. Chen, E. Maawad, O. Dolotko, A. A. Guda, V. Shapovalov, D. Wang, H. Hahn and M. Fichtner, Adv. Sci., 2015 DOI:10.1002/advs.201500128.
Footnote |
| † Electronic supplementary information (ESI) available: Rietveld refinement, PDF, Raman, NMR, EPR and EIS data. See DOI: 10.1039/c5cp02505b |
| This journal is © the Owner Societies 2015 |