
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Predicting near-UV electronic circular dichroism in nucleosomal DNA by means of DFT response theory†
Patrick
Norman
a,
Joseph
Parello
b,
Prasad L.
Polavarapu
b and
Mathieu
Linares
*a
aDepartment of Physics, Chemistry and Biology, Linköping University, SE-581 83 Linköping, Sweden. E-mail: mathieu@ifm.liu.se
bDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235, USA
First published on 23rd July 2015
Abstract
It is demonstrated that time-dependent density functional theory (DFT) calculations can accurately predict changes in near-UV electronic circular dichroism (ECD) spectra of DNA as the structure is altered from the linear (free) B-DNA form to the supercoiled N-DNA form found in nucleosome core particles. At the DFT/B3LYP level of theory, the ECD signal response is reduced by a factor of 6.7 in going from the B-DNA to the N-DNA form, and it is illustrated how more than 90% of the individual base-pair dimers contribute to this strong hypochromic effect. Of the several inter-base pair parameters, an increase in twist angles is identified as to strongly contribute to a reduced ellipticity. The present work provides first evidence that first-principles calculations can elucidate changes in DNA dichroism due to the supramolecular organization of the nucleoprotein particle and associates these changes with the local structural features of nucleosomal DNA.
I. Introduction
If a prize were to be awarded for the most significant molecular structure revelation of the 20th century, it should be bestowed on DNA. Watson and Crick's paper from 19531 changed the course of science by providing a fundamental understanding of the storage of the genetic code. It was later found that DNA can take on a number of structural forms: in addition to the right-handed A- and B-DNA forms, the left-handed Z-DNA form and the π-stacked double helical DNA structures have also entered as candidates in the technical science fields of nanotechnology and microelectronics.2,3In addition to the aforementioned fundamental variations, the molecular structure of DNA is also very sensitive to the environment in terms of temperature, concentration, counter ions, cationic polymers, and bio- and supramolecular assemblies.4–10 In this context, the interactions between B-DNA and histones are of paramount importance, leading to the formation of nucleosome core particles in chromatin that represents the fundamental packaging unit of the genome in eukaryotic cells. In this molecular assembly, a DNA sequence of 147 base-pairs coils 1.67 turns in a left-handed fashion around a histone core of four protein pairs.8 This supercoiled form of DNA is referred to as N-DNA.
The study of conformational changes in DNA is well suited for electronic circular dichroism (ECD) spectroscopy due to the fact that it is concentration-sensitive and able to elucidate the in situ supramolecular arrangement.11 In addition, it is a fortunate fact that the low-energy spectroscopic bands of the nucleobases do not significantly overlap with those attributed to the proteins at higher energy, which means that ECD spectra in the region of 250–300 nm act as fingerprints of conformational changes in DNA sequences.12 A most striking example is provided by the conformational change from B-DNA to N-DNA, an event which is known to be accompanied by a decrease in the ECD signal response of the low energy lying strong positive band by a factor of 3–4.13
As evidenced by a number of reviews,14–18 there exist numerous studies devoted to the calculation of UV absorption characteristics of the nucleic acid bases, nucleosides, nucleotides, and base-pair fragments. The characterization of the UV-induced photophysical excitation and relaxation channels is of course of highest concern as to understand the mechanisms behind photodamage in DNA. When it comes to theoretical work addressing ECD spectroscopy, the situation is quite different for reasons of computational complexity, both in terms of model system sizes and also requirements on the wave function (or density) parametrization. In a review by Kypr et al.12 from 2009, the state of affairs was summarized as: “The theoretical (i.e. quantum chemical) description of CD spectra of molecules as large as DNA is very complex, so the method is not able to provide structural information on the molecules at the atomic level”. It is clear that, in theoretical work on ECD, it is not relevant to study model systems in terms of bases or base-pairs due to their achiral nature. Instead the dichroism found in DNA originates from the π-stacking of base-pairs at defined twist angles, which means the smallest model system of real interest is a base-pair dimer. A successful theoretical elucidation of the ECD spectrum of the adenine–thymine homo-oligonucleotide was only recently presented by Di Meo et al.19 It was demonstrated that, with careful consideration of molecular structure parameters and with the use of extended basis sets, an accurate assignment of the experimental ECD spectrum could be obtained at the level of time-dependent density functional theory using hybrid functionals.19 The accuracy of the dimer model system was confirmed against large benchmark calculations for a trimer system, a result which will form the foundation for the present work.
The purpose of the present work is to take the simulations of ECD spectra to the next level and address hetero-nucleotide systems, i.e., real DNA, in the conformations of B-DNA and N-DNA.
II. Methodology
For an isotropic sample, the circular dichroism at a given wavelength is described by the ellipticity.20 Circular dichroism and linear absorption21 can be evaluated directly using standard electronic structure theory methods in the complex polarization framework22–25 or indirectly by determining oscillator and rotatory strengths from residues of linear response functions and subsequent employment of spectral broadening functions.26 The oscillator (fn) and rotatory strengths (Rn) for an electronic transition from the ground |0〉 to the excited state |n〉 are equal to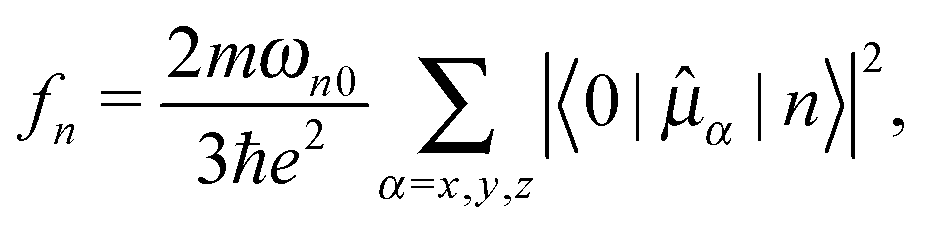 | (1) |
Rn = Im〈0|![[small mu, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b3.gif) α|n〉〈n| α|n〉〈n|![[m with combining circumflex]](https://www.rsc.org/images/entities/i_char_006d_0302.gif) β|0〉, β|0〉, | (2) |
α and β are the electric and magnetic dipole moment operators along the molecular axes α and β, respectively, and the excitation energy is given by ℏωn0 = En − E0.
III. Computational details
Unless stated otherwise, our calculations are based on molecular structures extracted from the X-ray crystal structure at 1.9 Å resolution of the nucleosome core particle containing 147 base-pairs (PDB file 1KX5).8 The experimental structure data are supplemented with hydrogen atoms that are added at C–H, N–H, and O–H bond distances of 1.090, 1.090, and 0.974 Å, respectively. Hydrogens belonging to aromatic rings or amino groups are added in a configuration as to preserve the planarity, see Fig. 1. The O–P bonds were cut and phosphorus atoms replaced by hydrogen. The sugar part was replaced by a methyl group. The added methyl group is oriented as to maintain the plane of symmetry in the system.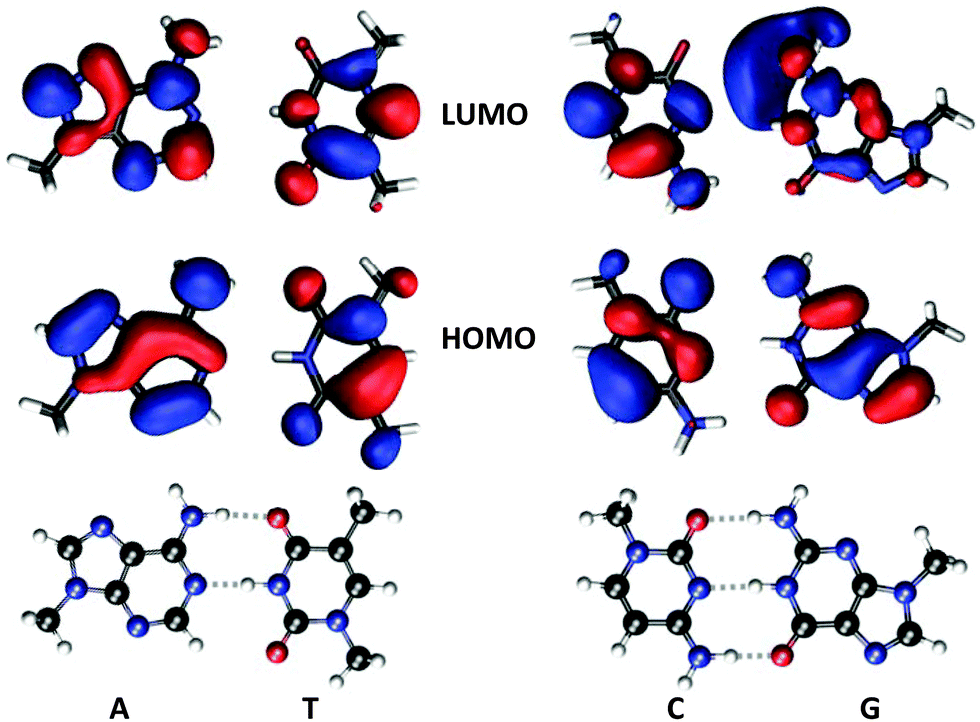 | ||
| Fig. 1 Molecular structures of methyl-capped nucleobases together with iso-density surfaces of HOMOs and LUMOs of the isolated systems (not base-pairs). The HOMO–LUMO transitions are of ππ*-character and provide the dominant contribution to the most intense electronic transitions addressed in Table 1. | ||
For comparison, calculations are also performed with use of molecular structures that are optimized at the level of density functional theory (DFT) in conjunction with the hybrid B3LYP27 exchange–correlation functional and Dunning's correlation consistent double-ζ (cc-pVDZ) basis set.28 The molecular structure optimizations are performed with use of the Gaussian program.29
All property calculations are carried out with use of the Dalton program30 through which gauge-origin independent circular dichroism data are obtained by means of London orbitals.31 In these calculations, we adopt the B3LYP27 and Dunning's augmented basis sets of double-ζ and triple-ζ quality (aug-cc-pVDZ and aug-cc-pVTZ).28 To evaluate the contributions from charge transfer states, test calculations were conducted for B-DNA with the CAM-B3LYP functional. In all spectrum calculations, we have converged 40 electronically excited states of singlet spin symmetry to a relative residual norm of 1.0 × 10−4. The combination of perturbation-dependent high-quality basis sets and tight numerical convergence leaves us certain that, within the approximation of the electronic structure method at hand, we present well converged spectra. Absorption and ECD spectra are obtained as sums of Gaussian profiles multiplied by oscillator and rotatory strengths, respectively. In both cases, the half-width at half-maximum was set equal to 0.083 eV.
IV. Results and discussion
A. Isolated and individual base-pairs of nucleobases and nucleosides
A comprehensive experimental study of the UV absorption spectra of nucleotides, polynucleotides, and nucleic acids is found in the work of Voet et al.32 from 1963 and in Fig. 1 of their paper one finds the spectra of the isolated DNA nucleobases in the wavelength region of 180–300 nm. The experimental UV absorption spectrum of cytosine (but not those of others) is strongly dependent on pH conditions. We are concerned with the basic chromophores, disregarding issues of protonation/deprotonation, so we will use the spectra recorded under neutral (or as close as possible) pH conditions as references for our calculations.| System | ΔE | λ | f | R | |
|---|---|---|---|---|---|
| a Nucleobase (methyl capped); B3LYP; aug-cc-pVDZ; non-optimized geometry from N-DNA. b As a but with B3LYP/cc-pVDZ optimized geometry. c Nucleoside (deoxyribose); parameters as a. d As c but at level B3LYP/cc-pVTZ. | |||||
| A | 4.89 | 254 | 0.013 | −14.4 | |
| 4.91 | 253 | 0.201 | 10.9 | ||
| 4.83 | 256 | 0.194 | 36.9 | ||
| 4.92 | 252 | 0.013 | −35.0 | ||
| 4.81 | 258 | 0.012 | −6.2 | ||
| 4.96 | 250 | 0.228 | −10.2 | ||
| 4.82 | 257 | 0.013 | −6.5 | ||
| 4.96 | 250 | 0.233 | −10.2 | ||
| T | 4.54 | 273 | 0.000 | 3.8 | |
| 4.64 | 267 | 0.163 | −1.7 | ||
| 4.67 | 265 | 0.000 | 0.1 | ||
| 4.71 | 263 | 0.174 | −0.1 | ||
| 4.52 | 274 | 0.000 | 4.8 | ||
| 4.70 | 264 | 0.175 | −18.5 | ||
| 4.53 | 274 | 0.000 | 4.8 | ||
| 4.71 | 263 | 0.177 | −18.5 | ||
| A–T | 4.60 | 269 | 0.137 | −14.6 | |
| 4.86 | 255 | 0.234 | 2.9 | ||
| 4.67 | 265 | 0.155 | −8.5 | ||
| 4.91 | 253 | 0.210 | 0.3 | ||
| G | 4.76 | 260 | 0.033 | −7.8 | |
| 4.76 | 260 | 0.096 | −4.8 | ||
| 4.86 | 255 | 0.009 | 7.5 | ||
| 4.96 | 250 | 0.197 | −7.5 | ||
| 4.44 | 279 | 0.020 | 12.5 | ||
| 4.77 | 260 | 0.130 | −16.3 | ||
| 4.99 | 249 | 0.012 | −1.4 | ||
| 5.01 | 247 | 0.173 | 12.3 | ||
| 4.68 | 265 | 0.001 | −0.9 | ||
| 4.73 | 262 | 0.122 | −8.8 | ||
| 4.87 | 255 | 0.007 | 7.6 | ||
| 5.00 | 248 | 0.189 | −13.9 | ||
| 4.68 | 265 | 0.001 | −1.2 | ||
| 4.71 | 263 | 0.123 | −8.0 | ||
| 4.87 | 255 | 0.006 | 6.8 | ||
| 4.99 | 248 | 0.184 | −14.4 | ||
| C | 4.34 | 286 | 0.046 | 0.9 | |
| 5.21 | 238 | 0.107 | −3.9 | ||
| 5.22 | 237 | 0.017 | −2.5 | ||
| 4.55 | 272 | 0.077 | −0.5 | ||
| 5.33 | 233 | 0.088 | 8.7 | ||
| 5.43 | 228 | 0.016 | −10.6 | ||
| 4.36 | 284 | 0.047 | −5.5 | ||
| 5.25 | 236 | 0.135 | −25.7 | ||
| 5.43 | 228 | 0.015 | 16.9 | ||
| 4.36 | 284 | 0.047 | −5.4 | ||
| 5.25 | 236 | 0.138 | −25.1 | ||
| 5.44 | 228 | 0.015 | 16.1 | ||
| G–C | 4.24 | 292 | 0.009 | −2.6 | |
| 4.63 | 268 | 0.106 | −61.7 | ||
| 4.65 | 267 | 0.019 | 10.2 | ||
| 4.67 | 265 | 0.064 | 29.6 | ||
| 4.78 | 259 | 0.027 | −2.3 | ||
| 4.88 | 254 | 0.101 | −24.3 | ||
| 4.89 | 253 | 0.088 | 6.2 | ||
| 4.08 | 304 | 0.009 | −1.7 | ||
| 4.60 | 269 | 0.018 | −5.9 | ||
| 4.66 | 266 | 0.082 | −43.8 | ||
| 4.73 | 262 | 0.017 | 7.0 | ||
| 4.76 | 260 | 0.102 | 6.5 | ||
| 4.84 | 256 | 0.057 | −13.1 | ||
| 4.88 | 254 | 0.012 | 1.4 | ||
It is deemed that the discrepancy between theory and experiment is well within the error bounds to be expected in the theoretical calculations as due to (i) neglect of the solvent, (ii) neglect of vibrational effects, and (iii) the limited accuracy of the adopted exchange–correlation functional, and we feel confident that we have a reasonably good description of the electronic structure of adenine and, as we shall discuss below, also of other bases. At the same time, we acknowledge the fact that, regardless of the choice made among the standard functionals, the time-dependent DFT approach cannot be expected to provide all aspects of the excitation spectra fully correct as has been demonstrated by Bartlett and co-workers in their benchmarking work using coupled cluster wave function theory with up to triple excitations in the cluster operator.38–40 The use of high-level wave function correlated methods comes at a price, however, not only in terms of an increased computational scaling with the size of the system but also in terms of stronger basis set requirements. For instance, the main absorption band of adenine is found at 5.66 and 5.47 eV (or 219 and 227 nm, respectively) in the CCSD and CCSD(T) calculations, respectively,40 which is some 0.7–0.9 eV higher than the reported band maximum in the experiment.33 These discrepancies are expected to be at least partly associated with basis set limitations. More extensive basis sets were applied by Ovchinnikov and Sundholm41 for the coupled cluster calculation of UV absorption spectra of a wide selection of the lowest tautomers of the nucleobases, but at the price of not making full inclusion of double excitations. It is argued, however, that the somewhat limited CC2 model is expected to perform well for these systems,41 and, for adenine, a CC2 excitation energy of 5.26 eV (or 236 nm) was reported using a high-quality quadruple-ζ basis set. The authors also reported a corresponding B3LYP result, which was found to be 0.29 eV lower in energy and thus in close agreement with the results in the present work. The overall assessment made by the authors concludes that in all but a few cases, the B3LYP results agree well with CC2, as well as with the experimental results.41
Our goals in the present work reach far beyond a few single point calculations of absorption spectra, so we are forced to accept the limitations of the more approximate DFT methods, and, arguably, the DFT/B3LYP level of theory represents a good compromise in between computational efficiency and accuracy in the present case.
The molecular structure used in our calculations (see footnote a of Table 1) is that of adenine in the crystal structure of the strand I of base-pair −1 in the palindromic sequence of 147 base-pairs in N-DNA (base-pair 0 is the central base-pair). We expect our results for optical transition data to be quite insensitive to the small intra-molecular structure differences in between different base-pairs. In fact, the X-ray crystallographic structure is based on fits of rigid molecular fragments to the observed electron density so intra-molecular distance parameters are not directly determined from the X-ray experiment of the N-DNA crystal but rather in combination with high-resolution data for the molecular fragments. A measure of the sensitivity of our theoretical calculations to the adopted molecular structures is provided by a comparison of the results obtained from experimental and theoretically optimized structures—results of the latter kind are labeled as footnote b in Table 1. It is clear from a comparison of a and b that the absorption characteristics are well preserved for this band, and any conclusion made about major changes in optical spectra will be insensitive to the issue of intra-molecular coordinates adopted in the calculations.
The reader may note that we choose to present not one but two electronic states for the discussion of the lowest absorption band, despite the fact that only one of them acquires a large oscillator strength. The reason for this choice becomes clear only after looking at the rotatory strengths. It is of course to be expected that the essentially planar nucleobases do not demonstrate ECD responses. The lowest absorption band in adenine (a and b in Table 1) is the result of two near-lying transitions and the oscillator strength is unevenly divided in between the two states (in a/b the second/first transition dominates the absorption). If we consider the optical activity, it is noticed that both states have rotatory strengths of similar magnitudes but of opposite signs. The ECD response from adenine will thereby be vanishingly small, as to be expected. The magnitudes of the rotatory strengths for the two individual states are larger in the optimized structure (36.9 and −35.0 × 10−40 esu2 cm2) as compared to when the structures are extracted from N-DNA (−14.4 and 10.9). The reason for this is that the amine hydrogen atoms that we are forced to add to the list of atoms in the crystallographic data are put in a planar configuration, whereas in the optimized structure the amine moiety becomes slightly pyramidal (and thereby also chiral). In the real system it is to be expected that a motion of inversion takes place on a relatively short time-scale and we therefore adopt the intermediate planar structure as to reflect a conformational average.
In DNA, the nucleobases are attached to deoxyribose, forming nucleosides. The sugar will induce chirality in the system since the plane of symmetry in the nucleobase is lost. However, we expect that the influence of the sugar on both the absorptive and chiral properties of the lowest UV bands is small. We address this issue in sections labeled c in Table 1, which present data for the nucleosides. When a comparison is made for the results for adenine, it becomes clear that the near UV absorption characteristics are very much unperturbed by the deoxyribose, as already established in the work of Improta and Barone,34 and the ECD response remains very small (although there is no longer a cancellation of the contributions from the two states).
Finally, with regard to isolated adenine, we turn to section d in Table 1 that presents results for the nucleoside as obtained with a large augmented triple-ζ basis set. We can simply confirm that the much smaller augmented double-ζ basis set is perfectly adequate and not associated with any limitations of concern in the present work.
An issue of concern to the present study is to what extent the UV bands of the nucleobases are affected by hydrogen bonding in the base-pairs. Adenine forms base-pairs with thymine for which computational results are presented further down in Table 1, at the position of the system label A–T. As discussed above, the dominant absorption band in isolated adenine is found at transition wavelengths of 253 and 256 nm with oscillator strengths of 0.201 and 0.194 for the nucleobase and nucleoside, respectively. In the base-pair environment, the corresponding absorption data become equal to 255 and 253 nm with values of f equal to 0.234 and 0.210, respectively. The conclusions from these results are that the absorption characteristics of adenine are not much affected by the formation of a base-pair with thymine and that the optical activity remains very small.
The rotatory strengths of thymine are very small and the main reason is of course the fact that the molecular structure is highly planar (more so than adenine). The two methyl groups are added in a way so that they adopt the plane of symmetry and this conformation is stable upon structure optimization, showing only positive vibrational frequencies. The plane of symmetry is broken by the addition of deoxyribose so that the rotatory strength of the dominant UV state reaches a value of −18.5 × 10−40 esu2 cm2, whereas the transition wavelength is not significantly affected by the sugar group (being equal to 267/264 nm without/with sugar).
In the data for the A–T base-pair in Table 1, it is clear that the band at 269 nm with f = 0.137 (section a without sugar) is due to thymine and that it is not much affected by hydrogen bonding. This base-pair is number −1, which of course means that the structure for thymine comes from the strand II and that it is not necessarily identical to the thymine structure of the strand I of base-pair +1. This is a further indication that the optical properties of the base-pairs in DNA are very much determined by the optical properties of the nucleobases themselves and that the optical activity remains small for the individual base-pairs.
The rotatory strengths in isolated guanine are all small or moderate. They are larger in the case of the optimized structure due to the pyramidalization of the amino group. We also note that the addition of deoxyribose does not inflict any significant changes in the near UV absorption or ECD spectrum.
The two absorption bands in guanine can be clearly identified in the G–C base-pair as well, referring to calculations performed on base-pair number −1. The lowest band (at 260 nm with f = 0.129 in isolated guanine) is found at 265–268 nm in the base-pair with f = 0.189 and R = −21.9 (the sum of contributions from three states) and the second band (at 250 nm with f = 0.197 in isolated guanine) is found at 253–254 nm in the base-pair with f = 0.189 and R = −18.1 (the sum of contributions from two states). So, in regard to base-pair formation, the results for guanine fall in line with those of adenine and thymine namely that nucleobase absorption characteristics are preserved and the ECD remains small.
B. Base pair dimers of nucleobases
Based on experimental43 and theoretical44 evidence, it appears that linear B-DNA free in solution is most stable with about 10.5 base pairs per turn rather than 10 as observed in the solid state, whereas superhelical DNA in chromatin is most stable with about 10 base pairs per turn. B-DNA is thus associated with an average twist angle of 360/10.5 = 34.3° while nucleosomal DNA corresponds to an average twist angle of 36°. We note here that the average twist in NCP147DNA in the 1KX5 crystal structure is 34.6° while 34.7° is the average twist of NCP146DNA in the 1EQZ crystal structure (values inferred from Cluster+ analysis): it is possible that the isolated NCP is not sufficiently representative of the DNA superhelical fold in native chromatin in which the core particles are part of nucleosomes connected through additional DNA including a linker (we will address this issue in more detail in Section D). The average rise distance is about 3.4 Å, which is a prototypical separation distance in systems showing effective π-stacking. These values for the twist angle and the rise distance will produce a helical system with a strong chirality that is expected to outperform the contributions discussed so far from the individual base-pairs. The stacking of the aromatic rings will give rise to alterations of the base-pair absorption bands along the lines of exciton coupling theory, as discussed in more detail in ref. 45.In the present section, we will study the exciton coupling effects of the lowest energy absorption band when stacking base-pairs A–T, T–A, G–C, and C–G. We restrict the study to base-pair dimers with geometries taken from N-DNA in the 1KX5 crystal structure. As seen in section a of Table 1 for base-pair A–T, there is a strong absorption band found around 269 nm and which is primarily associated with thymine. In the same wavelength region (265–268 nm), the G–C base-pair displays a strong absorption band that is primarily associated with guanine. These two bands and stacking interactions among them are likely to be responsible for the absorption and dichroism characteristics in the near UV region.
Since orientation will matter, there are 16 different ways to stack the two base-pairs A–T and G–C. For instance, it is obvious that the A–T/A–T and A–T/T–A dimers will be distinctly different, since, in the former case, π-systems of the same kind are stacked on top of one another and this is expected to give rise to a more pronounced exciton coupling. On a more subtle level, the defined direction in a DNA sequence is to read base-pairs from the 5′ to the 3′ carbons in deoxyribose so that dimers A–T/T–A and T–A/A–T are different, but this distinction is largely lost in our molecular models that exclude the sugar moieties.
In Table 2, we present absorption and ECD data for the dominant near UV bands of dimers of methyl-capped base-pairs of nucleobases. In the A–T/G–C dimer, thymine and guanine do not interact strongly and their respective near UV absorption bands are found in between 270 and 273 nm, yielding a combined positive rotatory strength of 86.0 × 10−40 esu2 cm2.
| System | ΔE | λ | f | R | |
|---|---|---|---|---|---|
| a Base pairs A–T (no. −57) and G–C (no. −56) of N-DNA. b Base pairs A–T (no. −1) and A–T (no. 0) of N-DNA. c Base pairs T–A (no. 18) and T–A (no. 19) of N-DNA. d Base pairs A–T (no. 12) and T–A (no. 13) of N-DNA. e Base pairs G–C (no. 24) and G–C (no. 25) of N-DNA. The numbering of the base pairs follows the PDB file 1KX5. | |||||
| A–T/G–C | 4.54 | 273 | 0.019 | 30.5 | |
| 4.56 | 272 | 0.074 | 19.5 | ||
| 4.60 | 270 | 0.057 | 36.0 | ||
| A–T/A–T | 4.55 | 272 | 0.038 | 78.4 | |
| 4.58 | 271 | 0.007 | 27.5 | ||
| 4.68 | 265 | 0.075 | 26.1 | ||
| 4.69 | 264 | 0.094 | −107.0 | ||
| 4.80 | 258 | 0.073 | 59.1 | ||
| 4.83 | 257 | 0.197 | −93.6 | ||
| T–A/T–A | 4.53 | 274 | 0.034 | 93.0 | |
| 4.67 | 265 | 0.171 | −94.6 | ||
| 4.75 | 261 | 0.055 | 49.2 | ||
| 4.83 | 257 | 0.225 | −97.8 | ||
| A–T/T–A | 4.52 | 274 | 0.161 | −251.6 | |
| 4.53 | 274 | 0.132 | 161.7 | ||
| 4.54 | 273 | 0.086 | 218.6 | ||
| 4.80 | 258 | 0.063 | −6.1 | ||
| 4.87 | 255 | 0.081 | 4.3 | ||
| G–C/G–C | 4.55 | 272 | 0.051 | 61.5 | |
| 4.56 | 272 | 0.005 | 18.5 | ||
| 4.57 | 271 | 0.012 | −44.2 | ||
| 4.59 | 270 | 0.002 | 26.5 | ||
This behavior stands in stark contrast to the case of the A–T/A–T dimer in which the stacking of the thymines leads to a strong exciton coupling for the lowest energy transition. As a consequence the band splits into two components. The low energy one of the two becomes less absorptive in comparison with the other, which is in agreement with standard exciton coupling theory, and they are separated by about 0.10–0.15 eV. The magnitudes of the rotatory strengths are enhanced by the exciton coupling and the signs alternate for the two components of the near UV band. The lower energy component of the exciton reaches a positive rotatory strength of 105.9 × 10−40 esu2 cm2 (the sum of two contributions) whereas the upper energy component shows a negative rotatory strength of −80.9 × 10−40 esu2 cm2 (also the sum of two contributions). The absorption band due to adenine is found at around 257–258 nm with an exciton splitting as small as 0.03 eV. Also in this case the lower energy component of the absorption band is the less absorptive one and it is found to give rise to a positive rotatory strength.
The properties of the T–A/T–A dimer are expected to be very similar to those of the A–T/A–T dimer. When comparing sets of data, the ECD of individual dimers will of course differ in its fine details due to differences in molecular configurations but the general characteristics must be the same for reasons discussed above. The lowest band in the T–A/T–A dimer is associated with thymine and shows an exciton splitting of 0.14 eV, with the two transition wavelengths being equal to 265 and 274 nm, respectively. The low-energy component of the pair of states is weakly absorbing with f = 0.034 but demonstrates a large positive dichroism with R = 93.0 × 10−40 esu2 cm2. The high-energy component, on the other hand, is strongly absorbing with f = 0.171 and contributes a large negative dichroism with R = −94.6 × 10−40 esu2 cm2. The exciton splitting of the adenine band in the T–A/T–A dimer is found to be equal to 0.08 eV, with the two transition wavelengths being equal to 261 and 257 nm, respectively. Also in this case the lower energy component of the absorption band is the less absorptive one and it is found to give rise to a positive rotatory strength. The results for the selected A–T/A–T and T–A/T–A dimers provides ample evidence that the two dimer types contribute to the dichroism of a DNA sequence in the same manner and that the stacking of thymines provides a key contribution to the lowest energy band in a DNA sequence.
In the A–T/T–A dimer, it is expected that the electronic interaction is weaker since different nucleobases are stacked on top of one another. As a consequence, the three reported transitions that contribute to the thymine absorption band are close in energy (transition wavelengths fall in the interval of 273–274 nm). The spread in transition wavelengths for the adenine band is also small, with the two dominant transition wavelengths being equal to 255 and 258 nm. As further evidence of the absence of exciton coupling in the case of the A–T/T–A dimer, we note that there is no pairing of transitions with low- and high-energy components that results in very different oscillator strengths. In the present case, the contributing transitions share more equally the total oscillator strength of the respective absorption bands. The total rotatory strength of the thymine band is large and positive, amounting to R = 191.8 × 10−40 esu2 cm2, whereas the ECD associated with the adenine band is negligible in comparison to what is found for the A–T/A–T and T–A/T–A dimers.
In the G–C/G–C dimer, the part of the system that is relevant for the formation of the lowest energy near the UV band in DNA sequences is the stack of the two guanines. From the results reported in Table 2, however, it is clear that the stacking of guanine does not induce as strong a chirality as the stacking of thymines. Exciton bands are not observed and the total rotatory strength of the guanine band in the G–C/G–C dimer amounts to R = 62.3 × 10−40 esu2 cm2. This is less significant a contribution to the overall ECD response as compared to contributions from dimers involving the stacking of thymine.
C. Circular dichroism in B-DNA and N-DNA
Experimental ECD spectra of DNA were reported by Gratzer et al. in 1970.46 The spectra from two different samples are provided in their work, with samples differing in the proportional amounts of A–T base pairs (28% and 67%). The first ECD band is positive with a peak maximum at around 270–280 nm and the second is negative with a peak minimum at around 245–250 nm. From our theoretical study of dimers presented in the previous section, it appears reasonable to conclude that these two bands are very much influenced by the exciton coupling of the A–T/A–T (and T–A/T–A) dimers but that, at the same time, important positive non-excitonic contributions to the first ECD band also stem from basically all the other studied dimers. In light of the theoretical findings, it appears likely that the larger negative ECD in the experimental spectrum of the 67%-sample as compared to the 28%-sample is an effect of the larger number of excitonic contributions to the signal in the former case.Our main concern in the present work is to obtain a microscopic understanding for the observed strong decrease in the dichroism of DNA due to a change in the 3D-fold going from the linear form (B-DNA) to the super-helical form seen in chromatin (N-DNA). The 147 base-pair long DNA sequence coils around the core particle in 1.67 left-handed turns, which obviously gives rise to changes in inter base-pair structure parameters, and it is the averaged effect of these structural changes that is read off in the comparison of ECD signal responses for B-DNA and N-DNA. Since no DNA sequence has been crystallized in both forms, we are forced to simulate the structure in the B-DNA form and we do so by (i) performing a molecular structure optimization of the respective base-pairs and (ii) creating a “perfect” base-pair dimer by taking two optimized base-pairs and separating them with a rise and twist. For each type of base-pair dimer we use the average rise and twist measured for N-DNA (1KX5), consequently our “perfect” dimer does not exhibit any slide, shift, roll or tilt. We will refer to these optimized molecular structures as being in the B-DNA conformation. If we focus on the ECD response signal in the vicinity of the wavelength for the experimental band maximum, we note a strong dependence of the dichroism on the twist angle. Fig. 2 shows this dependence for the A–T/A–T base-pair dimer at a wavelength of 272.5 nm with a y-axis scale chosen relative to the signal response for a twist angle of 33°. It should be noted that the overtwist will strongly reduce the ECD signal and, as an example, it is seen in the figure plot that an overtwist of six degrees causes a signal attenuation of more than 40%. Our prediction is in line with an experimental report that correlated an hypochromic effect on the positive near-UV band of the ECD spectrum of calf thymus DNA with duplex overwinding.47 The duplex winding angle per base-pair is equal to 360° divided by the pitch, which for a pitch of 10.5 amounts to 34.3° per base-pair, and it is known to decrease almost linearly with temperature in the interval from 0 to 83 °C.48 So the duplex winding angle referred to in the experiment is equal to the average twist angle in our work and it is also known to vary with the type and concentration of cations in the solution. It is observed that the magnitude of the positive band above 260 nm decreases in a linear manner—in the range of a few degrees about a winding angle of 36 degrees—as the duplex winding angle increases47 although with apparently a much steeper attenuation than in our theoretical prediction with the A–T/A–T base-pair dimer (Fig. 2). At a semi-quantitative level, this is a remarkable fit between the experimental data and our theoretical predictions. We note here that the experimental ECD spectra were obtained with linear calf thymus DNA under varying conditions of salt concentration and temperature and the dependence between these conditions and the duplex winding angle of circular PM-2 DNA was available from other studies. Normalization of the twist values in the linear DNA therefore rests on the correlation between the superstructure of circular DNA and its mean twist angle between two consecutive base pairs. It is anticipated that the strong dependence of the positive ellipticity above 260 nm on twist, as suggested by our theoretical data in Fig. 2, might allow future analyses to be carried out by ECD in the 280 nm region about the effects of non-histone protein binding to nucleosomes on the deformation of N-DNA. Strictly speaking our calculations involve a modest twist increase in average, from 34.3° to 34.6° for B- to N-DNA, respectively, thus suggesting (based on Fig. 2) that a general hypochromic effect is to be expected when going from B- to N-DNA, in line with our predictions (Fig. 6). We note that large overtwist values (>40 degrees in 1KX5 NCP147DNA, i.e. 24 bp dimers over 146 in total; Curves+ analysis49) are associated with varied bp dimers and their dependence on twist might markedly differ from the one reported in this work in the case of the A–T/A–T bp dimer (Fig. 2). Finally, the general hypochromism from B- to N-DNA originates from the variations of other DNA parameters besides twist. Roll and tilt variations (as found in kinked DNA topologies within N-DNA) might have a strong influence on the ECD response.
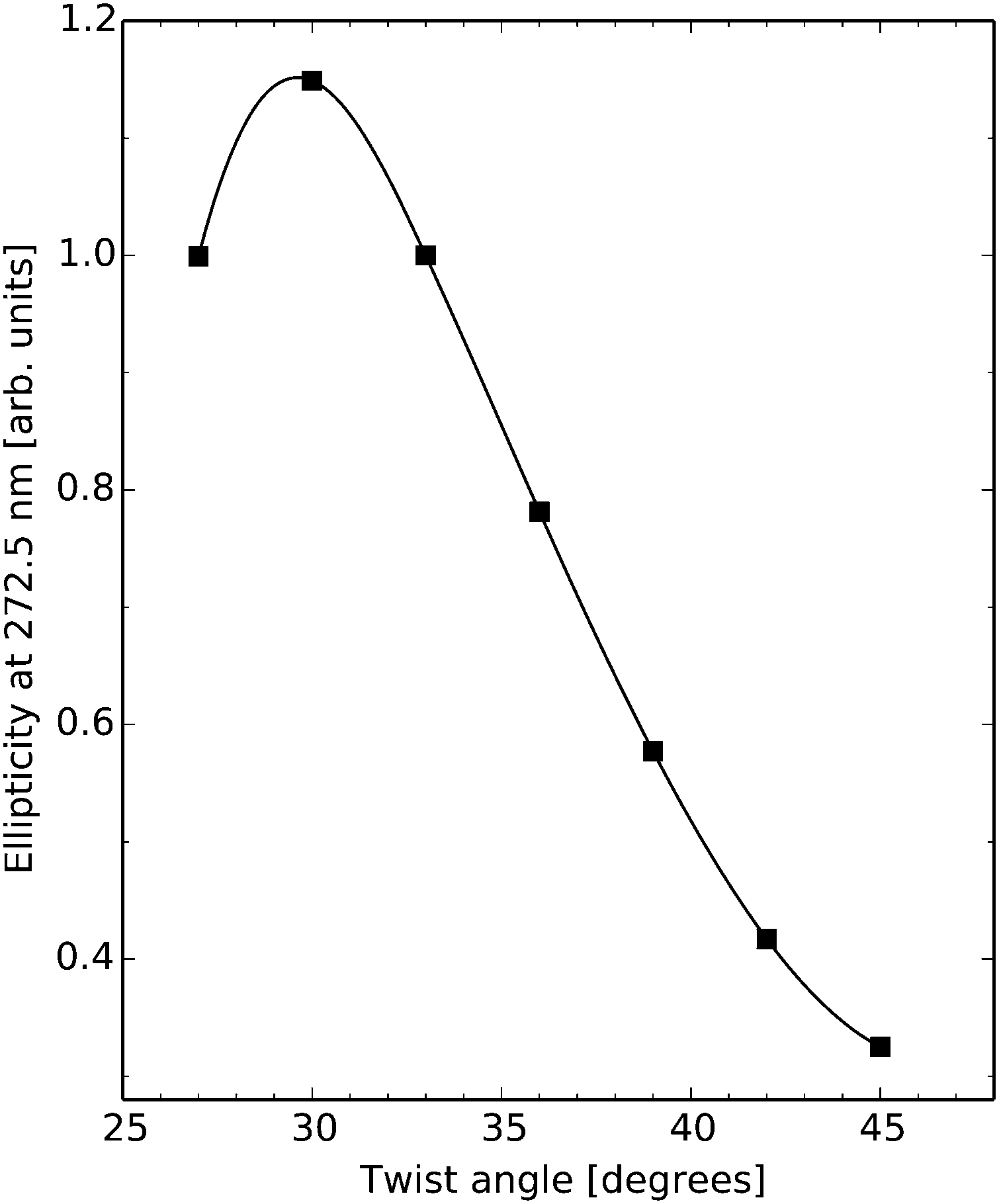 | ||
| Fig. 2 Electronic circular dichroism response at wavelength 272.5 nm for the A–T/A–T base-pair dimer as a function of twist angle in the B-DNA molecular configuration. | ||
There are ten unique base-pair dimers as listed in Table 3 and their frequency in the 1KX5 sequence is also detailed in the table. As can be seen, there are no base-pairs of type C–G/G–C appearing in the 1KX5 sequence and the two most frequent types are C–G/A–T and A–T/A–T with percentage representations of 19.2% and 17.2%, respectively. The circular dichroism spectra of the nine base-pair dimers of interest in their idealized B-form are shown in Fig. 3. Around 270 nm, the most important contributions to the positive band are provided by the base-pair dimers A–T/A–T and A–T/T–A, which together make up for 27.4% of the dimers in the 1KX5 sequence. The most abundant dimer C–G/A–T, being represented at 19.2%, contributes only weakly to the total spectrum since its signal response is seen to be small in the entire spectral region. It should also be noted that both A–T/A–T and A–T/T–A dimers have a weak negative ECD at long wavelengths, which will be apparent below in the total ECD spectra of N-DNA and B-DNA (Fig. 6).
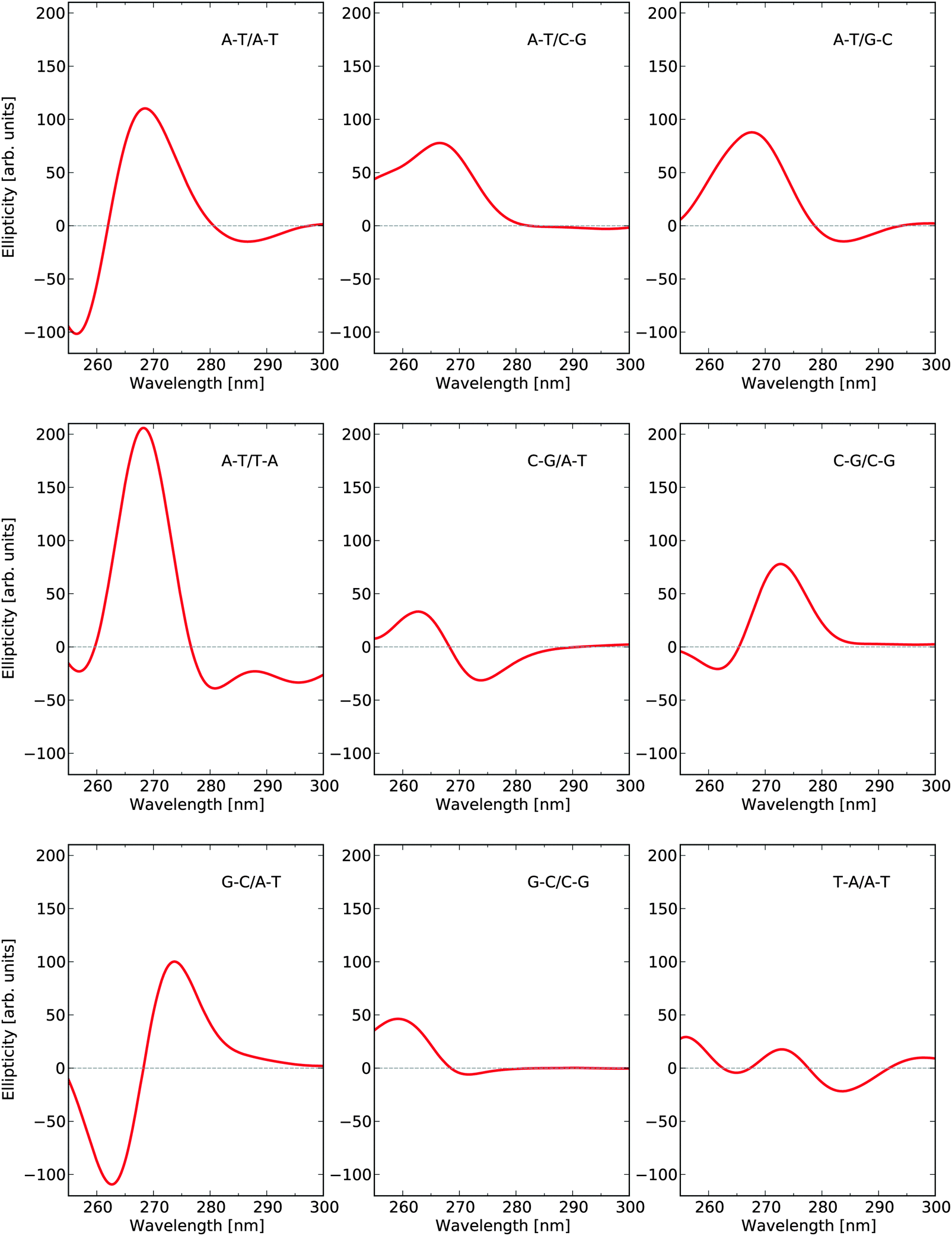 | ||
| Fig. 3 Electronic circular dichroism responses for base-pair dimers at their respective optimized molecular structures as to model B-DNA. | ||
| Base-pair dimer (stacked bb; ref. 54) | Base-pair dimer (stacked bp/bp; this work) | Frequency | Percentage |
|---|---|---|---|
| AA | A–T/A–T + T–A/T–A | 25 | 17.1 |
| AC | A–T/C–G + G–C/T–A | 14 | 9.6 |
| AG | A–T/G–C + C–G/T–A | 18 | 12.3 |
| AT | A–T/T–A | 15 | 10.3 |
| CA | C–G/A–T + T–A/G–C | 28 | 19.2 |
| CC | C–G/C–G + G–C/G–C | 14 | 9.6 |
| CG | C–G/G–C | 0 | 0.0 |
| GA | G–C/A–T + T–A/C–G | 16 | 11.0 |
| GC | G–C/C–G | 8 | 5.5 |
| TA | T–A/A–T | 8 | 5.5 |
| Total | 146 | 100 |
Fig. 4 shows the ECD spectra for eight selected base-pair dimers (along the 1KX5 NCP structure) in their N-DNA conformations, as well as in their idealized B-DNA conformations, as inferred from Fig. 3. This sample clearly demonstrates that base-pair dimers in the N-DNA conformation can (i) be closely related to B-DNA and display only small spectral differences, as illustrated by dimers 25–26 and 73–74; (ii) show signal depletion, as illustrated by dimers 1–2 and 146–147 (note that these two A–T/T–A base-pair dimers are related by the 2-fold axis of symmetry in the 1KX5 NCP 3D-structure with 147 base-pairs in total thus accounting for the nearly identical ECD spectra predicted theoretically); (iii) lose chiral responses in the entire spectral region, as illustrated by dimers 98–99 and 122–123; or (iv) even revert sign of the dichroism, as illustrated by dimer 49–50.
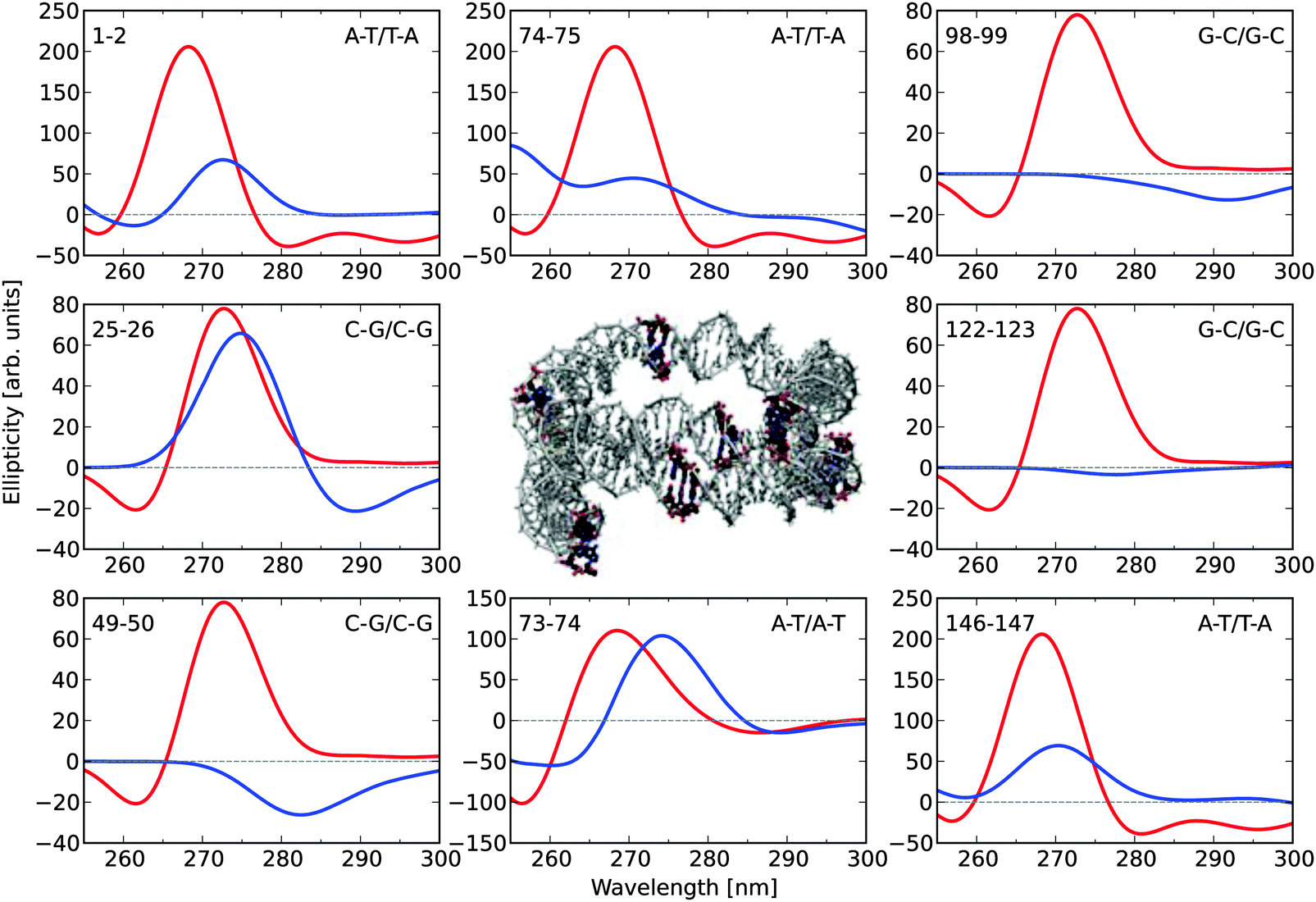 | ||
| Fig. 4 Electronic circular dichroism responses for eight selected base-pair dimers in the 1KX5 sequence of 147 base-pairs adopting (i) coordinates from the PDB file (blue) and (ii) optimized molecular structures (red). The base-pair dimers are numbered according to the output of the Curves+ program.49 The two extreme base-pair dimers, 1–2 and 146–147, therefore correspond to the base-pair steps, −73/−72 i.e. A–T/T–A and 146/147, i.e. A–T/T–A, respectively, in the 1KX5 PDB file, as read on strand I (for the first base on each of the two base pairs in the dimer). The two central base-pair dimers, 73–74 and 74–75, therefore correspond to the base-pair steps, −1/0 and 0/1, respectively. Positions of the selected base-pair dimers are highlighted in the central graphical illustration. | ||
In Fig. 5, we summarize the effects of all 146 base-pair dimers on the ECD ellipticity value at 272.5 nm when going from the conformation of B-DNA to that of N-DNA. We have here chosen to focus on the wavelength of 272.5 nm, which is close to the theoretical band maximum. The lower panel of the figure clearly shows that although in a restricted number of cases the signal responses are slightly larger in N-DNA as compared to B-DNA, the converse is true for the large majority of base-pair dimers. One way of interpreting the results presented in Fig. 5 is that we are sampling an “ECD surface” in a multi-dimensional configuration space given by the entire set of inter-base pair parameters, and our 146 samples indicate that the B-DNA conformations represent maxima so that moving away from these points in all (or at least most) directions leads to a reduction in the dichroism. This conceptual way of thinking was illustrated in Fig. 2, where we moved along a single dimension namely the twist angle, and it is tempting to try to parametrize the “ECD surface” with respect to the multi-dimensional degrees of freedom in the spirit of creating a force field parametrization of a regular potential energy surface. We have, however, been unsuccessful in such attempts and found it virtually impossible to disentangle the intricate dependencies of all the inter-base pair parameters on each other.
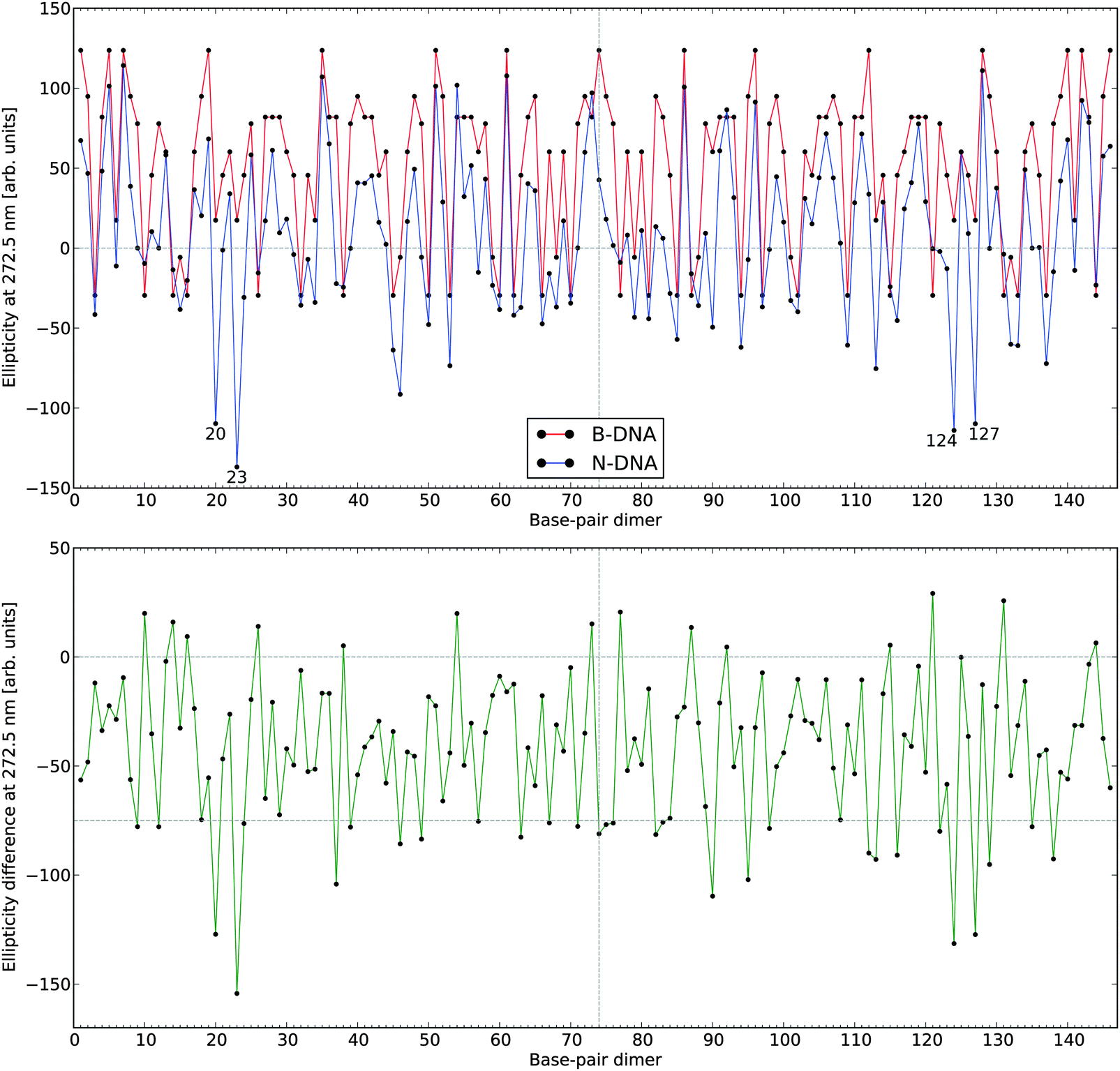 | ||
| Fig. 5 Electronic circular dichroism responses at 272.5 nm for the 146 base-pair dimers in N-DNA and B-DNA. The lower panel depicts CD signal differences with negative values referring to a hypochromic effect in going from B-DNA to N-DNA. The numbering of the base-pair dimers is made using the number of the first base pair (see definitions in legend to Fig. 4). | ||
However, we found that among the largest reductions in the ellipticity values observed in Fig. 5 (lower panel), the geometrical features of the corresponding base-pair dimers (i.e., base-pair dimers 20–21 and 23–24 as well as the symmetry related base-pair dimers 127–128 and 124–125, respectively) are highly distorted. Twist and roll are in regions of extrema for these parameters and, in this regard, we note that undertwisting combined with roll for two adjacent base-pairs closely corresponds to the definition of the DNA kinky helix as initially reported by Crick and Klug,50 with roll approximately defining the kinking angle. However, it should be noted that the kinking angles in NCP are of modest amplitude in comparison to the larger kinks observed in a variety of DNA–protein complexes. It is clear that the base-pair dimers with the largest hypochromic contributions correspond to highly distorted (kink-like) loci in the 1KX5 crystallographic structure. These loci are close to base-pair positions +5 and −5 along the supercoiled DNA.
Another example of the interplay between the different inter-base pair parameters and the ECD response is afforded by the base-pair dimer 25–26. As shown in Fig. 4, this C–G/C–G dimer displays rather similar ECD spectra in its N-DNA and B-DNA forms. In particular, the positive maxima at around 272–275 nm display closely related ellipticity values. Interestingly, this base-pair dimer displays a rather low twist angle of 29.8°—the mean value is 34.6° in 1KX5, with a minimum and maximum of 24.0 and 50.3°, respectively—so that a strong hypochromic effect in comparison to B-DNA is to be anticipated, in accordance with the curve plot in Fig. 2 (determined for A–T/A–T). However, this is not the case. We note that two other inter-base pair parameters, namely roll and tilt, are strongly shifted from their B-DNA values of zero (roll and tilt are 8.4 and 6.1°, respectively), while the remaining inter-base pair parameters (shift, slide and rise) are closely related to the B-DNA form. It is possible that the changes in the three inter-base pair parameters (twist, roll, and tilt) can result in compensatory effects at the level of the main positive band.
Base-pair dimer 122–123 is symmetrically positioned in comparison with the discussed C–G/C–G dimer 25–26 and it is therefore expected to display a very similar ECD response. But, as shown in Fig. 4, this is not at all the case. Instead the spectrum arising from dimer 122–123 in the N-DNA form is nearly vanishing in the entire 250–300 nm region. Both base-pair dimers, 25–26 and 122–123, display similar twist values of 29.8 and 28.9 degrees, respectively, and similar roll values of 8.4 and 10.0 degrees, respectively, but very different tilt values of 6.1 and −0.6 degrees, respectively. Also the remaining inter-base pair parameters are in close agreement comparing the two dimers, with shifts of 0.11 and 0.05 Å, respectively, slides of 0.23 and 0.00 Å, respectively, and rises of 3.39 and 3.33 Å, respectively. This comparison of the symmetrically related G–C/G–C base-pair dimers, 25–26 and 122–123, suggests that tilt variations are likely to play a major role in shaping the ECD response of a given base-pair dimer.
Adopting the building-block principle, the final B-DNA and N-DNA spectra for the full sequence of base-pairs are obtained by summing all the 146 individual base-pair dimer contributions in the B-DNA and N-DNA conformations, respectively. The resulting UV absorption and dichroism spectra are shown in Fig. 6. The UV absorption spectra for B-DNA and N-DNA display lambda-max values of 263.9 and 267.2 nm, respectively, and there is 25% decrease in the maximum absorption cross section in going from linear to nucleosomal DNA. The corresponding ECD spectra show small negative dichroism in the long wavelength region and strong positive bands with peaks at 269.3 and 272.5 nm for B-DNA and N-DNA, respectively (this weak negative band is likely to originate from the AT–TA and AT–TA dimers which represent as much as 27.4% of the total dimers, as mentioned above). The ratio between the maxima of the positive ECD signal intensities is determined to be equal to 6.7 in the theoretical spectra.
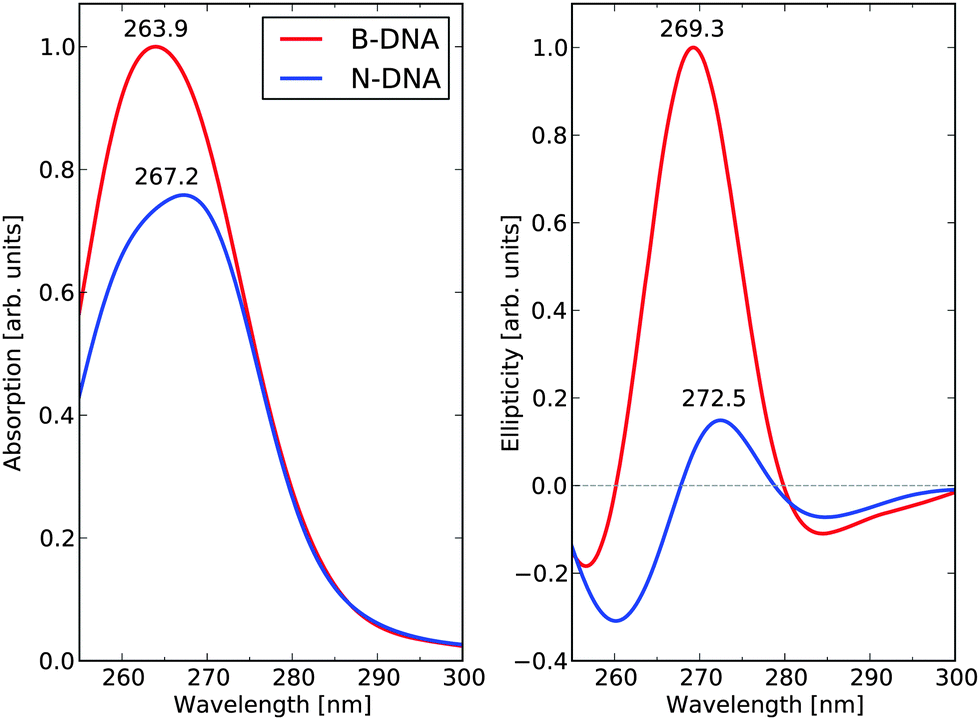 | ||
| Fig. 6 UV absorption and electronic circular dichroism spectra of the 1KX5 sequence in N-DNA and B-DNA conformations. | ||
The successful use of the B3LYP functional in this case relies on the assumption that the charge transfer (CT) states are not contributing strongly to the CD spectrum in the wavelength region of interest, but rather that it is the Frenkel excitonic states that are of predominant interest (as discussed in more detail in ref. 45). To obtain a more quantitative argument, we have also determined the B-DNA spectrum at the CAM-B3LYP level of theory and we include the comparison of the B3LYP and CAM-B3LYP spectra in Fig. S1 in the ESI.† As can be seen in this figure, the spectral shapes and intensities are in close agreement but one notices an overall spectral blueshift of some 10 nm in going from B3LYP to CAM-B3LYP. Such a shift is a direct consequence of the increased amount of Hartree–Fock exchange, and the similarities between both spectra show that there is no indication that the CT states should play an important role.
In a study of rat liver chromatin, Dumuis-Kervabon et al.13 used ECD spectroscopy to follow the structural organization of the nucleosomal core particle upon selective proteolysis of the core histones at the level of their N-terminal tails. In Fig. 4 of their work, the spectra for free B-DNA and the native core particle are presented as base references. In the near-UV region, there is a negligible influence of the proteins on the ECD spectrum and it is therefore reasonable to associate the signal response of the native core particle to N-DNA and make a reference to our theoretical work. Before making a direct comparison, however, it should be remembered that the N-DNA structure data in the 1KX5 PDB file that we employ are based on the synthesis and crystallization of a palindromic sequence of 147 base-pairs that obviously differs in base-pair composition from that of rat liver chromatin used in the experimental study.13 But a comparison is still warranted since the main characteristic ECD spectral changes in going from linear to nucleosomal DNA are well-known and have been documented by several groups under varying conditions.51,52 There is a striking agreement in the characteristics of our theoretical ECD spectra with the experimental spectra.13 The predicted red-shift of the maxima of the positive ECD band amounts to 3.2 nm in our theoretical study, which is on top of the corresponding result in the experiment. We want to draw the attention of the reader on the fact that such an accuracy is fortuitous and not within the scope of our model.
We need to consider that our calculations also predict a hypochromic effect in the near-UV spectrum, see the left panel of Fig. 6. The absorptivity value of DNA in its N-DNA form is not known accurately. In NCPs, the UV maximum at 258 nm includes the absorption of the DNA bases as well as the absorption of the histone tyrosine (Tyr) residues (30 in total in NCP). It is well known that the Tyr residues in NCP undergo a large hyperchromic effect in comparison to free Tyr—a hyperchromicity of up to 37% is recorded (Parello and Banères, unpublished data). However, the Tyr chromophores only represent a low contribution to the total absorptivity of NCP at 258 nm (not exceeding 5%). According to our theoretical predictions, it is anticipated that DNA itself in NCP undergoes an intrinsic hypochromic effect. To our knowledge, this effect has not been assessed experimentally. The theoretical spectra suffer from an overall spectral blue-shift of some 8–10 nm, which represents as good an agreement as one can hope for given the approximations that we are forced to adopt in the electronic structure method and systems models. The reported hypochromicity in the experimental ECD spectra is about 3 times comparing the results for the B-DNA and N-DNA conformations, with N-DNA in this case being dissolved under conditions of low counterion concentrations. Our value of 6.7 for this ratio is clearly overshooting the experimental result but, at the same time, there can be no doubt that our model calculations have captured the essential underlying microscopic reasons for the observed hypochromic effect. The exaggerated hypochromicity is likely due to a combination of reasons. First, we have adopted a fully optimized molecular structures to represent B-DNA, while a sampling of conformations based on a molecular dynamics simulation at room temperature would be more realistic. But due to the exceedingly high computational cost associated with such a procedure, we have been forced not to follow this route. Second, the 1KX5 structure is determined under conditions of full Mn2+ saturation and it appears that the ECD hypochromicity of N-DNA associated with spermidine3+ saturation amounts to about 2 times (in preparation). If we make the reasonable assumption that saturation of spermidine3+ and Mn2+ will affect ECD spectra of N-DNA in rather the same manner, then one would expect that an experimental result comparing B-DNA and N-DNA under full Mn2+ saturation would result in a hypochromicity factor of 3 × 2 = 6, which is very close to the theoretical prediction.
D. Structural and biological significance of the theoretical simulations
As stated above and summarized in Fig. 6, the main result of our theoretical work is that the predicted ratio B-DNA (269.3 nm)/N-DNA (272.5 nm) of 6.7 in the near-UV region of the ECD spectrum stands in very good agreement with available experimental results. Besides this, the wavelength shifts between the two ECD spectra are also well captured in the theoretical results. We need to emphasize, however, that the ECD response calculated in this work for NCP147DNA in the 1KX5 crystal structure is linked to a unique nucleotide sequence (i.e., a 147 base-pair palindromic sequence of DNA, derived from human alpha-satellite) associated with the recombinant histones from Xenopus laevis. In contrast, the experimental ECD spectra of NCP from natural origin correspond to a large variety of nucleotide sequences as found in the chromatin of different species studied so far (from avian, bovine, human, rodent origin, among others) with each individual NCP likely having a different nucleotide sequence as part of the genome of each species. Nevertheless, all ECD spectra of natural NCP show closely related profiles with a hypochromic attenuation of the positive band above 260 nm by a factor of 3–5, see ref. 53 and references therein.To take an example,51 the ECD of human NCP from HeLa cells was measured and showed a molecular ellipticity of 1.807 degrees cm2 dmol−1 at the band maximum of 283 nm. The corresponding ECD spectrum of B-DNA was obtained after addition of solid sodium dodecyl sulfate leading to the dissociation of the DNA–histone complex in situ and showed a molecular ellipticity of 7.734 (same units) at the band maximum of 277 nm. The ellipticity ratio, B-DNA (277 nm)/N-DNA (283 nm), thus amounts to 4.3 in this specific case. Based on our simulations, it can be concluded that even subtle differences in DNA sequences at selected loci in the N-DNA duplexes will be sensed spectroscopically and will contribute to differences in the B-DNA/N-DNA hypochromic ratio. In other words, the ECD spectrum of NCP from natural origin is to be viewed as a mean value over a large variety of ECD responses from the different core particles. This reinforces the validity of our calculated hypochromic effect (by a 6.7 factor) in the case of a specific DNA sequence as observed in the 1KX5 structure—to our knowledge, there is no experimental ECD information available in the literature about 1KX5 NCP itself.
An important question to ask at this point is whether ECD is sufficiently sensitive to the details of the supramolecular arrangement of DNA within the nucleosome core particle in order to monitor changes in the supramolecularity of the particle by recording the near-UV region of the spectrum where the contribution of the DNA chromophores largely exceeds those from the proteins—the latter have only very weak ECD contributions from aromatic residues and disulfide bridges. It has for example been reported that controlled trypsin proteolysis of NCP (during which all N-terminal regions of the octameric histones are cleaved) leads to a marked reduction of the hypochromic ratio.51 To what extent theoretical calculations can provide a microscopic insight to such more subtle changes in structure based on simulations of ECD spectra remains largely to be seen. But it stands clear that our present work represents an important step in this direction, leaving us with the perspective to study complex supramolecular assemblies that are involved in chromatin structure and function.
V. Conclusions
A building-block principle based on base-pair dimers has been adopted and applied for the calculation of circular dichroism spectra of linear, free, B-DNA and coiled, super-helical, N-DNA as found in nucleosome core particles. We demonstrate that the main ECD response is due to the inter-base pair coupling with the strongest contributions to the near-UV positive ECD band at around 270 nm originating from base-pair dimers A–T/A–T and A–T/T–A. There is a clear exciton coupling character of the dichroism in A–T/A–T giving rise to a strong negative band at shorter wavelengths.Our N-DNA calculations are based on the high-resolution crystal structure data found in the 1KX5 PDB file for a palindromic sequence of 147 base-pairs. Free B-DNA with an identical base-pair sequence is simulated by performing molecular structure optimizations of base-pairs with subsequent application of a base-pair separation and a twist angle. By comparing dichroism contributions from individual base-pairs for B-DNA and N-DNA, it is revealed that, in more than 90% of the cases, there is a hypochromic effect associated with the structural deformation of DNA that originates from the formation of the supramolecular assembly of the nucleosome core particle.
The theoretical ECD spectra for the full sequence of base-pairs in the B-DNA and N-DNA conformations are in good agreement with experiments performed on core particles from rat liver chromatin.13 In both cases there is an observed red-shift for the positive near-UV band of N-DNA as compared to B-DNA and a very strong decrease in the signal response.
By demonstrating that theoretical calculations are able to correctly predict the ECD spectral trends associated with changes in the supramolecular organization of DNA in chromatin, we open the field for exciting new scientific discoveries using ECD as the probing tool of the structure of DNA in varying external environments such as temperature, pH, ion-concentration, as well as chromatin interacting with functional non-histone proteins.
Acknowledgements
Financial support from the Swedish Research Council (Grant No. 621-2014-4646) and SeRC (Swedish e-Science Research Center) is acknowledged as well as a grant for computing time from National Supercomputer Centre (NSC), Sweden.References
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737 CrossRef CAS PubMed.
- N. Seeman, Mol. Biotechnol., 2007, 37, 246 CrossRef CAS PubMed.
- R. G. Endres, D. L. Cox and R. R. P. Singh, Rev. Mod. Phys., 2004, 76, 195 CrossRef CAS.
- A. Pérez, F. J. Luque and M. Orozco, Acc. Chem. Res., 2012, 45, 196 CrossRef PubMed.
- J. Garrec, C. Patel, U. Rothlisberger and E. Dumont, J. Am. Chem. Soc., 2012, 134, 2111 CrossRef CAS PubMed.
- S. Meyer, D. Jost, N. Theodorakopoulos, M. Peyrard, R. Lavery and R. Everaers, Biophys. J., 2013, 105, 1904 CrossRef CAS PubMed.
- N. Sugimoto, P. Wu, H. Hara and Y. Kawamoto, Biochemistry, 2001, 40, 9396 CrossRef CAS PubMed.
- C. A. Davey, D. F. Sargent, K. Luger, A. W. Maeder and T. J. Richmond, J. Mol. Biol., 2002, 319, 1097 CrossRef CAS.
- D. Gray, J.-D. Wen, C. Gray, R. Repges, C. Repges, G. Raabe and J. Fleischhauer, Chirality, 2008, 20, 431 CrossRef CAS PubMed.
- J. Rubio-Magnieto, A. Thomas, S. Richeter, A. Mehdi, P. Dubois, R. Lazzaroni, S. Clément and M. Surin, Chem. Commun., 2013, 49, 5483 RSC.
- Comprehensive Chiroptical Spectroscopy, ed. N. Berova, P. L. Polavarapu, K. Nakanishi and R. W. Woody, Wiley, 2012 Search PubMed.
- J. Kypr, I. Kejnovská, D. Renčiuk and M. Vorlčková, Nucleic Acids Res., 2009, 37, 1713 CrossRef CAS PubMed.
- A. Dumuis-Kervabon, I. Encontre, G. Etienne, J. Jauregui-Adell, J. Mery, D. Mesnier and J. Parello, EMBO J., 1986, 5, 1735 CAS.
- L. González, D. Escudero and L. Serrano-Andrés, ChemPhysChem, 2012, 13, 28 CrossRef PubMed.
- M. K. Shukla and J. Leszczynski, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 637 CrossRef CAS PubMed.
- K. Kleinermanns, D. Nachtigallová and M. S. de Vries, Int. Rev. Phys. Chem., 2013, 32, 308 CrossRef CAS PubMed.
- W. Domcke and D. R. Yarkony, Annu. Rev. Phys. Chem., 2012, 63, 325 CrossRef CAS PubMed.
- M. S. de Vries and P. Hobza, Annu. Rev. Phys. Chem., 2007, 58, 585 CrossRef CAS PubMed.
- F. Di Meo, M. N. Pedersen, J. Rubio-Magnieto, M. Surin, M. Linares and P. Norman, J. Phys. Chem. Lett., 2015, 6, 355 CrossRef CAS.
- L. D. Barron, Molecular Light Scattering and Optical Activity, Cambridge University Press, Cambridge, 2004 Search PubMed.
- R. W. Boyd, Nonlinear Optics, Academic Press, 2nd edn, 2003 Search PubMed.
- P. Norman, Phys. Chem. Chem. Phys., 2011, 13, 20519 RSC.
- A. Jiemchooroj and P. Norman, J. Chem. Phys., 2007, 126, 134102 CrossRef PubMed.
- P. Norman, D. M. Bishop, H. J. A. Jensen and J. Oddershede, J. Chem. Phys., 2005, 123, 194103 CrossRef PubMed.
- P. Norman, D. M. Bishop, H. J. A. Jensen and J. Oddershede, J. Chem. Phys., 2001, 115, 10323 CrossRef CAS PubMed.
- J. Olsen and P. Jørgensen, J. Chem. Phys., 1985, 82, 3235 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision A.1, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekström, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenæs, S. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansk, H. J. A. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjærgaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnæs, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Sałek, C. C. M. Samson, A. S. de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 269 CrossRef CAS PubMed.
- T. Helgaker, K. Ruud, K. L. Bak, P. Jørgensen and J. Olsen, Faraday Discuss., 1994, 99, 165 RSC.
- D. Voet, W. B. Gratzer, R. A. Cox and P. Doty, Biopolymers, 1963, 1, 193 CrossRef CAS PubMed.
- D. Onidas, D. Markovitsi, S. Marguet, A. Sharonov and T. Gustavsson, J. Phys. Chem. B, 2002, 106, 11367 CrossRef CAS.
- R. Improta and V. Barone, Theor. Chem. Acc., 2008, 120, 491 CrossRef CAS.
- L. Blancafort, J. Am. Chem. Soc., 2006, 128, 210 CrossRef CAS PubMed.
- L. Serrano-Andrés, M. Merchán and A. C. Borin, Chem. – Eur. J., 2006, 12, 6559 CrossRef PubMed.
- L. Serrano-Andrés, M. Merchán and A. C. Borin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8691 CrossRef PubMed.
- P. Szalay, T. Watson, A. Perera, V. Lotrich, G. Fogarasi and R. Bartlett, J. Phys. Chem. A, 2012, 116, 8851 CrossRef CAS PubMed.
- P. Szalay, T. Watson, A. Perera, V. Lotrich and R. Bartlett, J. Phys. Chem. A, 2012, 116, 6702 CrossRef CAS PubMed.
- P. Szalay, T. Watson, A. Perera, V. Lotrich and R. Bartlett, J. Phys. Chem. A, 2013, 117, 3149 CrossRef CAS PubMed.
- V. A. Ovchinnikov and D. Sundholm, Phys. Chem. Chem. Phys., 2014, 16, 6931 RSC.
- T. Gustavsson, A. Banyasz, E. Lazzarotto, D. Markovitsi, G. Scalmani, M. J. Frisch, V. Barone and R. Improta, J. Am. Chem. Soc., 2006, 128, 607 CrossRef CAS PubMed.
- A. Klug and L. C. Lutter, Nucleic Acids Res., 1981, 9, 4267 CrossRef CAS PubMed.
- M. Levitt, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 640 CrossRef CAS.
- P. Norman and M. Linares, Chirality, 2014, 26, 123 CrossRef PubMed.
- W. B. Gratzer, L. R. Hill and R. J. Owen, Eur. J. Biochem., 1970, 15, 209 CrossRef CAS PubMed.
- A. Chan, R. Kilkuskie and S. Hanlon, Biochemistry, 1979, 18, 84 CrossRef CAS.
- A. D. Bates and A. Maxwell, DNA topology, Oxford University Press, 2005 Search PubMed.
- R. Lavery, M. Moakher, J. H. Maddocks, D. Petkeviciute and K. Zakrzewska, J. Phys. Chem. Lett., 2015, 6, 355 CrossRef.
- F. Crick and A. Klug, Nature, 1975, 255, 530 CrossRef CAS PubMed.
- J. P. Whitlock Jr. and R. T. Simpson, J. Biol. Chem., 1977, 252, 6516 CAS.
- D. M. J. Lilley and K. Tatchell, Nucleic Acids Res., 1977, 4, 2039 CrossRef CAS PubMed.
- X. Wang, S. Moore, M. Laszckzak and J. Ausió, J. Biol. Chem., 2000, 275, 35013 CrossRef CAS PubMed.
- W. Kabsch, C. Sander and E. Trifonov, Nucleic Acids Res., 1982, 10, 1097 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp02481a |
| This journal is © the Owner Societies 2015 |