
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational simulation and interpretation of the low-lying excited electronic states and electronic spectrum of thioanisole†
Shaohong L.
Li
 ,
Xuefei
Xu
and
Donald G.
Truhlar
*
,
Xuefei
Xu
and
Donald G.
Truhlar
*
Department of Chemistry, Chemical Theory Center, and Supercomputing Institute, University of Minnesota, Minneapolis, Minnesota 55455, USA. E-mail: truhlar@umn.edu
First published on 5th June 2015
Abstract
Three singlet states, namely a closed-shell ground state and two excited states with 1ππ* and 1nσ* character, have been suggested to be responsible for the radiationless decay or photochemical reaction of photoexcited thioanisole. The correct interpretation of the electronic spectrum is critical for understanding the character of these low-lying excited states, but the experimental spectrum is yet to be fully interpreted. In the work reported here, we investigated the nature of those three states and a fourth singlet state of thioanisole using electronic structure calculations by multireference perturbation theory, by completely-renormalized equation-of-motion coupled cluster theory with single and double excitations and noniterative inclusion of connected triples (CR-EOM-CCSD(T)), and by linear-response time-dependent density functional theory (TDDFT). We clarified the assignment of the electronic spectrum by simulating it using a normal-mode sampling approach combined with TDDFT in the Tamm–Dancoff approximation (TDA). The understanding of the electronic states and of the accuracy of the electronic structure methods lays the foundation of our future work of constructing potential energy surfaces.
1 Introduction
As discussed by Sobolewski, Domcke, and coworkers,1–4 many organic molecules with aromatic rings and heteroatoms have photoinduced hydrogen detachment channels in the gas phase and proton transfer channels in protic solvents that are mediated by a repulsive 1πσ* or 1nσ* state. Among these, phenol and thiophenol, as well as their derivatives anisole and thioanisole, exhibit qualitatively similar potential energy profiles that may be considered prototypes of this behavior.5–13 For thioanisole, the existence and nature of the 1nσ* state has been identified and discussed in previous work.13,14 It was conjectured, based on complete active space self-consistent field (CASSCF) calculations and spectroscopic studies, that the dynamics of the photodissociation of thioanisole into thiophenoxyl and methyl radicals is facilitated by the coupling of the optically accessible 1ππ* state to the dark 1nσ* state. The detailed mechanism of the reaction is, however, still open to question.Our ultimate goal is to use theory and computation to gain more insight into the mechanism, by constructing the potential energy surfaces of the relevant ground and excited states and performing dynamical simulation on the coupled surfaces. As the first step, in this work we study the ultraviolet absorption spectrum of thioanisole to obtain more information about the character of the electronic excited states in the Franck–Condon region and to validate electronic structure methods for calculating these states. A vapor-phase experimental spectrum was reported in a recent article14 but has yet to be fully interpreted and understood. Instead of assigning states to the spectral bands simply according to their vertical excitation energies and transition probabilities, we attempt to simulate the spectrum using a normal-mode sampling strategy as detailed below so as to directly compare the simulated profile to the experimental spectrum.
The rest of this paper will proceed as follows. After describing the computational details in Section 2, we discuss in Section 3 the optimized geometries of S0 and S1, the character of the excited states, and the interpretation of the electronic spectrum based on our simulation. Finally we conclude our discussion in Section 4.
2 Computational details
2.1 Basis sets, electronic structure methods, and software
We used five basis sets. In order of decreasing completeness, they are:• augmented correlation-consistent polarized valence triple zeta with an additional tight d function for sulfur: aug-cc-pV(T+d)Z;15–17
• minimally augmented, multiply polarized valence triple zeta: MG3S;18
• minimally augmented, polarized valence triple zeta (multiply polarized for sulfur), denoted as MB hereafter: 6-311+G(d)19,20 for carbon and hydrogen and MG3S for sulfur;
• partially augmented, correlation-consistent polarized valence double zeta with an additional spherical harmonic set of tight d functions for sulfur: jun-cc-pV(D+d)Z;15–17,21
• minimally augmented, polarized valence double zeta: 6-31+G(d);20,22–24
We used both Kohn–Sham density functional theory (KS-DFT) and wave function theory (WFT) for electronic structure calculations. The levels of theory and their abbreviations are listed in Table 1 along with ref. 25–42 that explain each level of theory.
| Type | Abbreviation | Name of method | Ref. |
|---|---|---|---|
a All KS-DFT calculations in this paper used an ultrafine (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 590) grid with 99 radial shells and 590 grid points per shell for numerical integration except the TDA-TDDFT calculations for simulating the electronic spectrum, which used a fine (75302) grid.
b SA(N)-CASSCF denotes that the average is over N states. In this article, state averages are always carried out with equal weights for all states averaged.
c All MC-QDPT and XMC-QDPT calculations were carried out with intruder state avoidance56 with an energy denominator shift of 0.02Eh.
d In these calculations, the 1s orbitals on H and C and the 1s, 2s, and 2p orbitals on S were not correlated.
e All CR-EOM-CCSD(T) calculations were carried out with variant IA of the method for the so-called δ-corrected excitation energies which incorporates a noniterative connected triples correction to EOM-CCSD excitation energies rather than to state energies. 590) grid with 99 radial shells and 590 grid points per shell for numerical integration except the TDA-TDDFT calculations for simulating the electronic spectrum, which used a fine (75302) grid.
b SA(N)-CASSCF denotes that the average is over N states. In this article, state averages are always carried out with equal weights for all states averaged.
c All MC-QDPT and XMC-QDPT calculations were carried out with intruder state avoidance56 with an energy denominator shift of 0.02Eh.
d In these calculations, the 1s orbitals on H and C and the 1s, 2s, and 2p orbitals on S were not correlated.
e All CR-EOM-CCSD(T) calculations were carried out with variant IA of the method for the so-called δ-corrected excitation energies which incorporates a noniterative connected triples correction to EOM-CCSD excitation energies rather than to state energies.
|
|||
| KS-DFTa | M06-2X | M06-2X exchange–correlation functional | 25 |
| TD-M06-2X | Linear-response time-dependent density functional theory (TDDFT) with the M06-2X exchange–correlation functional | 26 | |
| 25 | |||
| TD-B3LYP | TDDFT with the B3LYP exchange–correlation functional | 27 | |
| TDA-τ-HCTHhyb | TDA-TDDFT (TDDFT with the Tamm–Dancoff approximation) with the τ-HCTHhyb exchange–correlation functional | 28–31 | |
| WFT | CASSCF(n,m) | Single-state complete active space self-consistent-field theory with n active electrons in m active orbitals | 32 |
| SA-CASSCF(n,m)b | State-averaged complete-active-space self-consistent-field theory with n active electrons in m active orbitals | 33 | |
| MC-QDPTc | Multi-configurational quasi-degenerate perturbation theory | 34, 35 | |
| XMC-QDPTc | Extended multi-configurational quasi-degenerate perturbation theory | 36 | |
| EOM-CCSDd | Equation-of-motion coupled cluster theory with singles and doubles | 37, 38 | |
| CR-EOM-CCSD(T)d,e | Completely-renormalized equation-of-motion coupled cluster theory with singles, doubles, and noniterative connected triples | 39, 40 | |
| PM3 | Parametrized model 3 | 41, 42 | |
We used the following software to perform various types of calculations: Gaussian 09,43GAMESS,44,45NWChem,46ANT (incorporating the MOPAC 5.021 mn code),47,48 and Multiwfn.49 The calculations done with each software package are listed in Table 2.
| Software | Ref. for software | Calculations done with the software |
|---|---|---|
| Gaussian 09 | 43 | KS-DFT, TDDFT, TDA-TDDFT |
| GAMESS | 44, 45 | CASSCF, SA-CASSCF, MC-QDPT, XMC-QDPT |
| NWChem | 46 | EOM-CCSD, CR-EOM-CCSD(T) |
| ANT | 47, 48 | Geometry sampling for the simulation of electronic spectrum |
| Multiwfn | 49 | Gaussian broadening for the simulation of electronic spectrum |
2.2 Coordinates and geometry optimization
The equilibrium geometry of both S0 and S1 has Cs symmetry with the symmetry plane in the phenyl ring. The equilibrium geometry of S0 is shown in Fig. 1, which also illustrates our convention for numbering the carbon atoms. The molecular orientation is defined by putting the C2, C1, and S atoms in the xy plane with the C1–S bond pointing to the positive x direction. | ||
| Fig. 1 Equilibrium geometry of thioanisole with Cs symmetry as optimized by M06-2X/MG3S. Orientation of the x and y axes and numbering of the carbon atoms for convenience of description are also shown. | ||
The ground-state equilibrium geometry was optimized by M06-2X/MG3S and by CASSCF(12,11)/MB.50 The equilibrium geometry of S1 was also optimized by CASSCF(12,11)/MB. The 11 active orbitals used in the CASSCF(12,11) calculations nominally correspond to three π and three π* on the phenyl ring, two σC–S and two σC–S* on the C1–S and S–C7 bonds, and n(pz) on the sulfur (see Fig. 2). All the active orbitals delocalize to some extent; for example, the two σC–S have components on both the C1–S and S–C7 bonds, and the orbital we label as n(pz) has both a component on S and a π component on the ring.
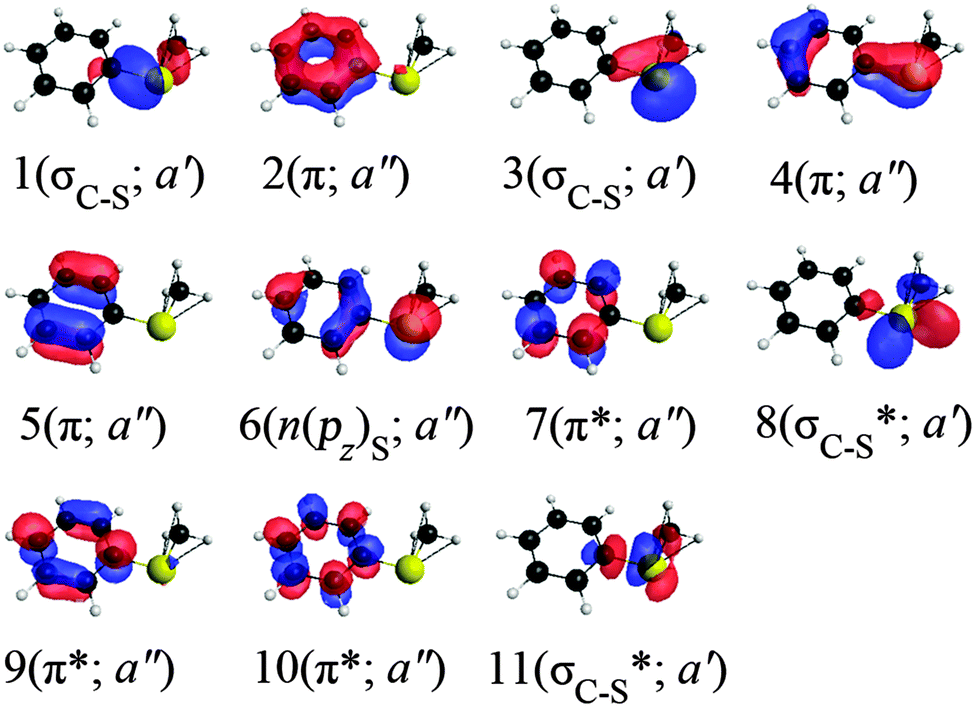 | ||
| Fig. 2 Schematic of three-state-averaged active orbitals at the equilibrium geometry with Cs symmetry from SA-CASSCF calculations. The character and the irrep of the orbitals are given in parentheses. | ||
2.3 Excitation energies and simulation of the electronic absorption spectrum in the vapor phase
The vertical excitation energies (VEEs) to the two or three lowest singlet excited states in the Franck–Condon region were calculated by MC-QDPT, XMC-QDPT, EOM-CCSD, CR-EOM-CCSD(T), TDDFT, and TDA-TDDFT. (X)MC-QDPT calculations were carried out with a three-dimensional model space based on an SA(3)-CASSCF(12,11) reference. The 11-orbital active space has the same character as described in Section 2.2.Our strategy to simulate the spectrum of the vapor-phase thioanisole is to sample a certain number of representative geometries according to the ground-state vibrational distribution, calculate their contributions to the spectrum, and add the contributions. In particular, we first optimized the ground-state geometry of thioanisole and performed normal mode analysis by the semiempirical PM3 method. We then sampled 200 geometries with a ground-state harmonic oscillator distribution along each Cartesian normal coordinate. For each sampled geometry, we calculated the VEEs and oscillator strengths of the four lowest excited states using TDA-τ-HCTHhyb/6-31+G(d). These VEEs and oscillator strengths from the sampled geometries were collected to generate a stick spectrum. This stick spectrum accounts for the broadening and peak shift of the spectrum due to the statistical distribution of geometries generated by ground-state molecular vibrations, taking into account also the dependence of transition probabilities on geometry. Finally, we broadened the sticks with Gaussian functions with half width at half maximum (HWHM) equal to 0.15 eV to produce a smooth profile of electronic spectrum. The Gaussian broadening mimics the missing effects such as broadening due to lifetime broadening, thermally excited vibrations, rotational broadening, anharmonic broadening, and instrumental resolution. At the longest wavelengths, the resolution is presumably limited by spectral congestion (the jet-cooled excitation spectrum in ref. 51 shows resolved rovibrational structure). At shorter wavelengths, lifetime broadening probably dominates.
3 Results and discussion
3.1 Optimized geometries of S0 and S1
The optimized geometry of S0 has Cs symmetry with phenyl, S, C7, and one H of the methyl group in a plane and the other two hydrogens of the methyl group above and below the plane. (See the ESI† for the coordinates and vibrational frequencies of the optimized geometries.) The length of the C1–S and S–C7 bonds given by M06-2X (by CASSCF) are 1.76 Å (1.80 Å) and 1.80 Å (1.83 Å) respectively. The C1–S–C7 bond angle given by M06-2X (by CASSCF) is 102.88° (103.84°). The length of the C–C bonds in the phenyl ring is in the range of 1.38–1.40 Å by either method. Originally there was debate on whether the C2–C1–S–C7 torsion at the ground-state equilibrium of thioanisole is zero or not, but recent experiments and electronic structure calculations support the former,51,52 with which our results are consistent. Since M06-2X is more reliable for geometries than CASSCF (because the latter lacks dynamic correlation), we use the M06-2X geometry as the equilibrium geometry for S0 in the following calculations, except for the calculation of adiabatic excitation energy and the simulation of spectrum as will be discussed below.The optimized geometry of S1 by CASSCF(12,11)/MB is similar to the S0 equilibrium geometry optimized at the same level, with the C–C bonds in the phenyl ring lengthened by ∼0.03 Å, the C1–S bond shortened by 0.03 Å, the S–C7 bond lengthened by 0.02 Å, and the C1–S–C7 bond angle increased by 1°, which is also consistent with previous work.51
3.2 Nature of the four lowest singlet states and electronic spectroscopy
For the photochemistry of thioanisole with the low excitation energies (∼4.2–4.5 eV) used in the experiments,13,14 only the two lowest singlet electronically excited states (S1 and S2) are deemed to be important. However, the third excited singlet state (S3) makes an important contribution to the spectrum, so in this section we consider the four lowest singlet states. At the equilibrium geometry of the ground state with Cs symmetry, S0 is a closed-shell state belonging to the A′ irreducible representation (irrep), S1 and S3 are 1ππ* states (simply to be called ππ*) belonging to the A′ irrep, and S2 is a 1nσ* state (simply to be called nσ*) belonging to the A′′ irrep. The dominant configurations of the four states given by SA-CASSCF are listed in Table 3. Table 3 actually shows that one of the dominant configurations of both S1 and S3 has nπ* character, but considering the facts that the other dominant configuration is ππ* and that the n orbital also has π character, we simply follow other authors13,14 and label S1 and S3 as ππ*.| State | Dominant configuration(s)b | CI coefficient |
|---|---|---|
| a Geometry optimized by M06-2X/MG3S. b The occupation numbers of the dominant configurations correspond to orbitals in Fig. 2 in the same order. | ||
| S0 | 222222 00000 | 0.93 |
| S1 | 222221 10000 | 0.62 |
| 222212 00100 | 0.58 | |
| S2 | 222221 01000 | 0.84 |
| 222122 01000 | 0.33 | |
| S3 | 222221 00100 | 0.86 |
| 222122 00100 | 0.26 | |
The vertical excitation energies (VEEs) calculated by a variety of electronic structure methods are listed in Table 4. There are several messages we can take from this table. Firstly, MC-QDPT and XMC-QDPT based on the same SA-CASSCF reference give essentially the same VEEs at the S0 equilibrium geometry. Secondly, SA(3)-MC-QDPT(12,11) and CR-EOM-CCSD(T), which are very different kinds of high-level methods, give very similar VEEs when used with the extensive aug-cc-pV(T+d)Z basis set, which suggests that both should be quite accurate. Thirdly, TDA-τ-HCTHhyb with a small 6-31+G* basis set gives VEEs close to our best estimate values from CR-EOM-CCSD(T)/aug-cc-pV(T+d)Z. Because of the accuracy and low computational cost of TDA-τ-HCTHhyb/6-31+G*, we use it for the simulation of electronic spectrum discussed later.
| Method | Basis set | S1 (2 A′) | S2 (1 A′′) | S3 (3 A′) |
|---|---|---|---|---|
| a All data are from this work, calculated at the equilibrium geometry optimized by M06-2X/MG3S, unless specified otherwise. b Ref. 14. | ||||
| EOM-CCSD | aug-cc-pV(T+d)Z | 4.84 | 5.21 | 5.52 |
| CR-EOM-CCSD(T) | aug-cc-pV(T+d)Z | 4.53 | 5.03 | 5.25 |
| TD-M06-2X | MG3S | 4.94 | 5.05 | 5.30 |
| TD-B3LYP | MB | 4.60 | 4.82 | 5.00 |
| TDA-τ-HCTHhyb | 6-31+G* | 4.65 | 4.97 | 5.18 |
| TD-CAM-B3LYPb | aug-cc-pVTZ | 4.95 | 5.14 | 5.31 |
| SA(3)-MC-QDPT(12,11) | MB | 4.64 | 5.13 | — |
| SA(3)-XMC-QDPT(12,11) | MB | 4.64 | 5.13 | — |
| SA(3)-MC-QDPT(12,11) | aug-cc-pV(T+d)Z | 4.52 | 5.02 | — |
To further validate the methods we use, we calculate the S1 ← S0 adiabatic excitation energy by using the following formula:
| ΔE(0–0)(S1 ← S0) = Ve(S1) + ZPE(S1) − Ve(S0) − ZPE(S0) | (1) |
The S1 state is often called a “bright” state;14 even though this description makes sense as compared to the dark nσ* state, some readers may think it corresponds to the bright band of the experimental vapor-phase UV absorption spectrum which peaks at 4.96 eV [see Fig. 3(a)]. This view may be strengthened by our EOM-CCSD/aug-cc-pV(T+d)Z calculation, which gives the S1 ← S0 VEE equal to 4.84 eV (see Table 4). However, if we compare our best estimate of the S1 ← S0 VEE by CR-EOM-CCSD(T) (4.53 eV) to the maximum of the experimental spectrum (4.96 eV), the discrepancy is as large as 0.4 eV. On the other hand, the experimental adiabatic excitation energy of 4.28 eV13,14 also differs significantly from the maximum of the spectral band. These facts imply that the experimentally bright band at 4.96 eV may not correspond to the S1 ← S0 excitation.
 | ||
| Fig. 3 (a) Experimental vapor-phase UV spectrum of thioanisole. (Adapted from Fig. S6 in the Electronic Supplementary Information of ref. 14 with the permission of the authors.) (b) Simulated electronic spectrum of thioanisole (red curve) and the position of vertical excitation energies (blue sticks) as calculated by TDA-τ-HCTHhyb/6-31+G*. | ||
There have been disagreements in the literature about the nature of states and the interpretation of the electronic spectrum. For instance, Lim et al.13 and Roberts et al.14 reported S1 as a bright ππ* state and S2 a dark nσ* state, and Roberts et al. assigned the strong absorption band peaked at 4.96 eV to S3 ← S0 transition based on their single-point TD-CAM-B3LYP, LR-CCSD, and CCSDR(3) calculations, although their calculated S3 ← S0 VEE is higher than 5.3 eV. Liu et al.,53 on the other hand, reported S1 as a dark ππ* state, S2 a bright ππ* state, and S3 a dark nσ* state based on their TD-B3LYP/6-31G(d) calculation, assigning the experimental band to the S2 ← S0 transition. Our calculations of VEEs (Table 4) and oscillator strengths (Table 5) suggest the same assignment as ref. 13 and 14. Interestingly, our TD-B3LYP/MB calculation disagrees with the TD-B3LYP/6-31G(d) calculation in ref. 53 on the nature of the states while our calculation agrees with ref. 14. The reason why the TD-B3LYP/6-31G(d) calculation in ref. 53 gave incorrect results is that the lack of diffuse functions in the 6-31G(d) basis set fails to properly describe the nσ* state because it has a significant amount of Rydberg character, which makes their calculated excitation energy for the first nσ* state (which should be S2) higher than the second ππ* state (which should be S3). We confirmed this explanation of the source of error in their calculation by performing a TD-B3LYP/6-31+G(d) calculation, which gives the first nσ* state lower than the second ππ* state.
| Method | Basis set | S1 (2 A′) | S2 (1 A′′) | S3 (3 A′) |
|---|---|---|---|---|
| a All data are from this work, calculated at the equilibrium geometry optimized by M06-2X/MG3S, unless specified otherwise. b Ref. 14. | ||||
| EOM-CCSD | jun-cc-pV(D+d)Z | 0.0086 | 0.0000 | 0.2745 |
| TD-M06-2X | MG3S | 0.0177 | 0.0001 | 0.2628 |
| TD-B3LYP | MB | 0.0158 | 0.0001 | 0.2433 |
| TDA-τ-HCTHhyb | 6-31+G* | 0.0177 | 0.0001 | 0.2938 |
| TD-CAM-B3LYPb | aug-cc-pVTZ | 0.0154 | 0.0005 | 0.2311 |
| SA(4)-CASSCF(12,11) | MB | 0.0005 | 0.0014 | 0.3832 |
To further clarify the nature of states and the interpretation of the electronic spectrum, we simulated the spectrum in the vapor phase using a normal-mode sampling approach as described in Section 2.3. The experimental spectrum shown in Fig. 3(a), as a reproduction of Fig. S6 in the Electronic Supplementary Information of ref. 14, has two closely located peaks at 239 nm (5.19 eV) and 250 nm (4.96 eV) respectively. We aim to simulate the lower-energy peak at 250 nm only, in order to verify our understanding of the lowest excited states (but we will discuss the other peak later in this section). We are interested in the three lowest excitations because we expect that, according to our calculated vertical excitations, the second ππ* state (S3 at the equilibrium geometry) may be responsible for the strong band at 250 nm. In practice we calculated the four lowest excitations because at some sampled geometries the second ππ* state is S4.
The simulated spectrum shown in Fig. 3(b) has a strong band peaked at 243 nm (5.10 eV) and a tail extending to longer wavelengths. Compared to Fig. 3(a), Fig. 3(b) has an overall shift to higher energy, but the profile reasonably resembles the experiment. Although the peak of the experimental spectrum is at 250 nm (4.96 eV), 0.14 eV is an acceptable error for this type of simulation. The other peak of Fig. 3(a) at higher energy does not appear in the simulation, which will be discussed further later. Despite the small quantitative differences, this simulation further supports our characterization of the excited states and our interpretation of the electronic spectrum.
Another important datum here is that the peak of the simulated spectrum (5.10 eV) shifts to longer wavelength compared to the VEE calculated at the same TDA-τ-HCTChyb/6-31+G* level (5.18 eV) by a non-negligible amount (0.08 eV). This result is computed with harmonic vibrations, and if anharmonicity were taken into account the shift could be even larger.54 We also note that calculated transition moments can be hugely influenced by vibronic interaction, as illustrated in a recent theoretical study of the electronic absorption spectrum pyrrole.55 It is therefore worthwhile to state, although it is not a new observation, that one should not take the wavelength of the peak of an unresolved electronic spectrum as a “benchmark” of VEE, a conclusion that has long been recognized but is still often ignored.
A noteworthy point is that our TDDFT (with B3LYP and τ-HCTHhyb) and SA-CASSCF calculations do not reveal another electronic state energetically near the bright ππ* state that has comparable transition probability to that state, at either the equilibrium geometry or nearby geometries in the Franck–Condon region. Accordingly the two peaks of the experimental spectrum at 239 and 250 nm may correspond to the same electronic state but be split vibronically, although the splitting is rather large (0.23 eV). More study will be needed to clarify why there are two peaks. We emphasize that the aim of the present simulation is to clarify the nature of the excited states and the spectrum—not to quantitatively reproduce the experimental band shape. Achieving a quantitative simulation of the band shape, including reproducing the fine features at ∼280–290 nm, may require more realistic sampling of the ground-state potential energy surface, explicitly taking into account the anharmonicity of the surfaces and hot bands, and accounting for the quantization of vibrations.
The transitions between the states can be further understood by examining their oscillator strengths and transition dipole moments. All of these transitions are allowed since in the Cs symmetry the x and y components of the dipole operator transform as the A′ irrep and the z component as the A′′ irrep, and accordingly the x and y components of the dipole operator couple S1 and S3 (A′) to S0 (A′) and the z component couples S2 (A′′) to S0. As displayed in Table 5, SA-CASSCF, EOM-CCSD, and TDDFT qualitatively agree on the large oscillator strength of the S3 ← S0 transition. EOM-CCSD and TDDFT agree as well on the oscillator strengths of S1 ← S0 being one order of magnitude smaller than that of S3 ← S0 and on the oscillator strength of S2 ← S0 being negligible. The small oscillator strength of the S2 ← S0 transition may be attributed to the relatively small spatial overlap of the n(pZ) and σ* orbitals involved in the transition. SA-CASSCF predicts the oscillator strength of S1 ← S0 to be smaller than of S2 ← S0, both two orders of magnitudes smaller than that of S3 ← S0, which is inaccurate due to lack of dynamical correlation.
4 Concluding remarks
In this paper, we clarified the nature of the four lowest singlet states of thioanisole at the equilibrium geometry using various wave function and density functional methods. We simulated the electronic absorption spectrum using a normal mode sampling approach, and its good agreement with experiment confirms our understanding of the spectrum, which is as follows. The transitions from S0 (closed-shell) to S1 (ππ*) and to S2 (nσ*) both have relatively small probability and contribute to the low-energy tail of the spectrum, whereas S3 ← S0 is mainly responsible for the bright band at 250 nm. We also confirmed that TDDFT, CR-EOM-CCSD(T), MC-QDPT, and XMC-QDPT are all reasonably accurate for computing the excitation energies. For potential energy surfaces, however, MC-QDPT and XMC-QDPT are more desirable at the current stage of development. Since XMC-QDPT generates smoother potential energy surfaces near state intersections,36 it appears suitable for future work of constructing the potential energy surfaces of thioanisole.Acknowledgements
The authors are grateful to Dr Ke R. Yang, Dr Jingjing Zheng, and Professor Piotr Piecuch for many helpful discussions. We also thank Professor Vasilios G. Stavros and Mr Jamie D. Young for providing us with the experimental data of the UV/Vis spectrum of thioanisole. This work was supported in part by the U. S. Department of Energy, Office of Basic Energy Sciences, under grant no. DE-SC0008666. S.L.L. was also supported by the Dr V. Pothapragada Excellence Fellowship in Chemistry at the University of Minnesota.References
- A. L. Sobolewski and W. Domcke, Chem. Phys. Lett., 1999, 315, 293–298 CrossRef CAS.
- A. L. Sobolewski and W. Domcke, Chem. Phys., 2000, 259, 181–191 CrossRef CAS.
- A. L. Sobolewski and W. Domcke, J. Phys. Chem. A, 2001, 105, 9275–9283 CrossRef CAS.
- A. L. Sobolewski, W. Domcke, C. Dedonder-Lardeux and C. Jouvet, Phys. Chem. Chem. Phys., 2002, 4, 1093–1100 RSC.
- Z. Lan, W. Domcke, V. Vallet, A. L. Sobolewski and S. Mahapatra, J. Chem. Phys., 2005, 122, 224315 CrossRef PubMed.
- M. G. D. Nix, A. L. Devine, B. Cronin, R. N. Dixon and M. N. R. Ashfold, J. Chem. Phys., 2006, 125, 133318 CrossRef PubMed.
- O. P. J. Vieuxmaire, Z. Lan, A. L. Sobolewski and W. Domcke, J. Chem. Phys., 2008, 129, 224307 CrossRef PubMed.
- X. Xu, K. R. Yang and D. G. Truhlar, J. Chem. Theory Comput., 2013, 9, 3612–3625 CrossRef CAS.
- X. Xu, J. Zheng, K. R. Yang and D. G. Truhlar, J. Am. Chem. Soc., 2014, 136, 16378–16386 CrossRef CAS PubMed.
- K. R. Yang, X. Xu, J. Zheng and D. G. Truhlar, Chem. Sci., 2014, 5, 4661–4680 RSC.
- I. S. Lim, J. S. Lim, Y. S. Lee and S. K. Kim, J. Chem. Phys., 2007, 126, 034306 CrossRef PubMed.
- R. Omidyan and H. Rezaei, Phys. Chem. Chem. Phys., 2014, 16, 11679–11689 RSC.
- J. S. Lim and S. K. Kim, Nat. Chem., 2010, 2, 627–632 CrossRef CAS PubMed.
- G. M. Roberts, D. J. Hadden, L. T. Bergendahl, A. M. Wenge, S. J. Harris, T. N. V. Karsili, M. N. R. Ashfold, M. J. Paterson and V. G. Stavros, Chem. Sci., 2013, 4, 993–1001 RSC.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS PubMed.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS PubMed.
- T. H. Dunning, K. A. Peterson and A. K. Wilson, J. Chem. Phys., 2001, 114, 9244–9253 CrossRef CAS PubMed.
- B. J. Lynch, Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2003, 107, 1384–1388 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS PubMed.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS PubMed.
- E. Papajak, J. Zheng, X. Xu, H. R. Leverentz and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 3027–3034 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS PubMed.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- M. E. Casida, in Recent Advances in Density Functional Methods, Part I, ed. D. P. Chong, World Scientific, Singapore, 1995, pp. 155–192 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS; (b) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (c) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS; (d) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- I. Tamm, J. Phys., 1945, 9, 449–460 Search PubMed.
- S. M. Dancoff, Phys. Rev., 1950, 78, 382–385 CrossRef.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- A. D. Boese and N. C. Handy, J. Chem. Phys., 2002, 116, 9559–9569 CrossRef CAS PubMed.
- M. W. Schmidt and M. S. Gordon, Annu. Rev. Phys. Chem., 1998, 49, 233–266 CrossRef CAS PubMed.
- H.-J. Werner and W. Meyer, J. Chem. Phys., 1981, 74, 5794–5801 CrossRef CAS PubMed.
- H. Nakano, J. Chem. Phys., 1993, 99, 7983 CrossRef CAS PubMed.
- H. Nakano, Chem. Phys. Lett., 1993, 207, 372–378 CrossRef CAS.
- A. A. Granovsky, J. Chem. Phys., 2011, 134, 214113 CrossRef PubMed.
- H. Koch and P. Jørgensen, J. Chem. Phys., 1990, 93, 3333–3344 CrossRef CAS PubMed.
- J. F. Stanton and R. J. Bartlett, J. Chem. Phys., 1993, 98, 7029–7039 CrossRef CAS PubMed.
- K. Kowalski and P. Piecuch, J. Chem. Phys., 2004, 120, 1715–1738 CrossRef CAS PubMed.
- K. Kowalski, J. Chem. Phys., 2009, 130, 194110 CrossRef PubMed.
- J. J. P. Stewart, J. Comput. Chem., 1989, 10, 209–220 CrossRef CAS PubMed.
- J. J. P. Stewart, J. Comput. Chem., 1989, 10, 221–264 CrossRef CAS PubMed.
- M. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS PubMed.
- M. S. Gordon and M. W. Schmidt, in Theory and Applications of Computational Chemistry: The First Forty Years, ed. C. E. Dykstra, G. Frenking, K. S. Kim and G. E. Scuseria, 2005, pp. 1167–1189 Search PubMed.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. de Jong, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS PubMed.
- J. Zheng, Z.-H. Li, A. W. Jasper, D. A. Bonhommeau, R. Valero, R. Meana-Pañeda and D. G. Truhlar, ANT, version 2014-2, University of Minnesota, Minneapolis, 2015, http://comp.chem.umn.edu/ant Search PubMed.
- J. J. P. Stewart, L. Fiedler, P. Zhang, J. Zheng, I. Rossi, W.-P. Hu, G. C. Lynch, Y.-P. Liu, Y.-Y. Chuang, J. Pu, J. Li, C. J. Cramer, P. L. Fast, J. Gao and D. G. Truhlar, MOPAC, version 5.021mn, University of Minnesota, Minneapolis, MN, 55455, 2013 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- MG3S is a well-established basis set for ground-state geometry optimization with DFT, but to check for basis set effects we have also performed the optimization with another well-established basis set, namely the Turbomole basis set def2-TZVP, and the optimized coordinates differ from the ones with MG3S by only ∼0.001 Å.
- M. Hoshino-Nagasaka, T. Suzuki, T. Ichimura, S. Kasahara, M. Baba and S. Kawauchi, Phys. Chem. Chem. Phys., 2010, 12, 13243–13247 RSC.
- M. Nagasaka-Hoshino, T. Isozaki, T. Suzuki, T. Ichimura and S. Kawauchi, Chem. Phys. Lett., 2008, 457, 58–61 CrossRef CAS PubMed.
- M.-X. Liu, B.-B. Xie, M.-J. Li, Y.-Y. Zhao, K.-M. Pei, H.-G. Wang and X. Zheng, J. Raman Spectrosc., 2013, 44, 440–446 CrossRef CAS PubMed.
- B. Lasorne, J. Jornet-Somoza, H. D. Meyer, D. Lauvergnat, M. A. Robb and F. Gatti, Spectrochim. Acta, Part A, 2014, 119, 52–58 CrossRef CAS PubMed.
- G. Wu, S. P. Neville, O. Schalk, T. Sekikawa, M. N. R. Ashfold, G. A. Worth and A. Stolow, J. Chem. Phys., 2015, 142, 074302 CrossRef PubMed.
- H. A. Witek, Y.-K. Choe, J. P. Finley and K. Hirao, J. Comput. Chem., 2002, 23, 957–965 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp02461g |
| This journal is © the Owner Societies 2015 |