
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Concepts in bio-molecular spectroscopy: vibrational case studies on metalloenzymes
M.
Horch
*,
P.
Hildebrandt
and
I.
Zebger
*
Technische Universität Berlin, Institut für Chemie, Sekr. PC14, Straße des 17. Juni 135, D-10623 Berlin, Germany. E-mail: marius.horch@gmx.de; ingo.zebger@tu-berlin.de
First published on 17th June 2015
Abstract
Spectroscopic techniques play a major role in the elucidation of structure–function relationships of biological macromolecules. Here we describe an integrated approach for bio-molecular spectroscopy that takes into account the special characteristics of such compounds. The underlying fundamental concepts will be exemplarily illustrated by means of selected case studies on biocatalysts, namely hydrogenase and superoxide reductase. The treatise will be concluded with an overview of challenges and future prospects, laying emphasis on functional dynamics, in vivo studies, and computational spectroscopy.
 M. Horch | Marius Horch received his degree in Biotechnology from the Technische Universität Berlin (Germany) in 2010. Since then he has worked as a researcher in the group of Prof. Peter Hildebrandt at the Max-Volmer Laboratory for Biophysical Chemistry where he prepared his PhD thesis on structure–function relationships of metalloenzymes. His current research interests focus on protein functional dynamics and vibrational spectroscopy as well as experimental and theoretical aspects of biological transition metal catalysis. |
 P. Hildebrandt | Peter Hildebrandt received his PhD in Chemistry from the Universität Göttingen in 1985. After postdoctoral and research positions at the Max-Planck-Institut für Biophysikalische Chemie Göttingen, Princeton University, the Max-Planck-Institut für Strahlenchemie Mülheim, and the Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa, he became full professor for physical chemistry and biophysical chemistry at the Technische Universität Berlin. He is mainly interested in the analysis of structure–dynamics–function relationships of redox proteins and photoreceptors in solution and at interfaces, employing static, time-resolved, and spatially resolved spectroscopic methods, specifically Raman and IR spectroscopic techniques. |
 I. Zebger | Ingo Zebger obtained his PhD in Physical Chemistry for his work on the IR spectroscopic characterization of photo-addressable polymers at the Universität Essen, Germany in 2000. Afterwards he moved to Aarhus Universitet, Denmark, as a postdoc to monitor singlet oxygen in polymeric and biological systems. Since 2003 he works as a senior scientist at the department of Biophysical Chemistry at the Technische Universität Berlin, Germany. His research interests center on the vibrational spectroscopic characterization of molecular processes in redox proteins and metalloenzymes, such as hydrogenases, under the influence of external perturbations, thereby elucidating (dynamic) structure–function relationships. |
Motivation and scope of the article
A molecular understanding of biochemical reactions is essential for both fundamental and applied life sciences. Like every living being, mankind has an ongoing (and increasing) demand for efficient processes that accomplish energy conversion and the synthesis of tailored functional compounds. In principle, nature provides a rich and dedicated biocatalytic machinery for these tasks that can be utilised, optimised, and emulated for human demands, given that the underlying processes are understood on a conceptual level. With this in mind, relationships between structure and function are highly important for understanding elementary molecular principles.In the current perspective article special requirements and fundamental concepts for a spectroscopic elucidation of structure–function relationships in biological macromolecules will be outlined. Besides a general treatise, this approach will be illustrated by using the example of vibrational spectroscopic studies on metalloenzymes involved in the transformation of small molecules. Finally, we will provide a perspective for future studies, covering both explicit challenges and conceptual extensions.
A molecular approach to biological reactions
Characteristics of biological macromolecules
Since the second half of the twentieth century, the molecular aspects of biological systems and processes have strongly come into focus, and intellectual and experimental concepts of classical chemistry and physics have been adopted for their study. While this is a natural and justified approach, certain care has to be taken since functional biomolecules exhibit a number of specialities that can be largely traced back to two fundamental properties (see Fig. 1). (1) The biological systems involved in energy conversion and biosynthesis are modular macromolecular entities, usually enzymes, whose number of atoms N may exceed the magnitude of classical chemical compounds by orders of magnitude. (2) Biological reaction networks usually involve a large number of molecules and ions that may interact on different levels, both concomitantly and successively. While these two aspects appear to be quantitative in nature, they have a considerable impact on the conceptual and experimental treatment of these systems, e.g. in spectroscopic studies.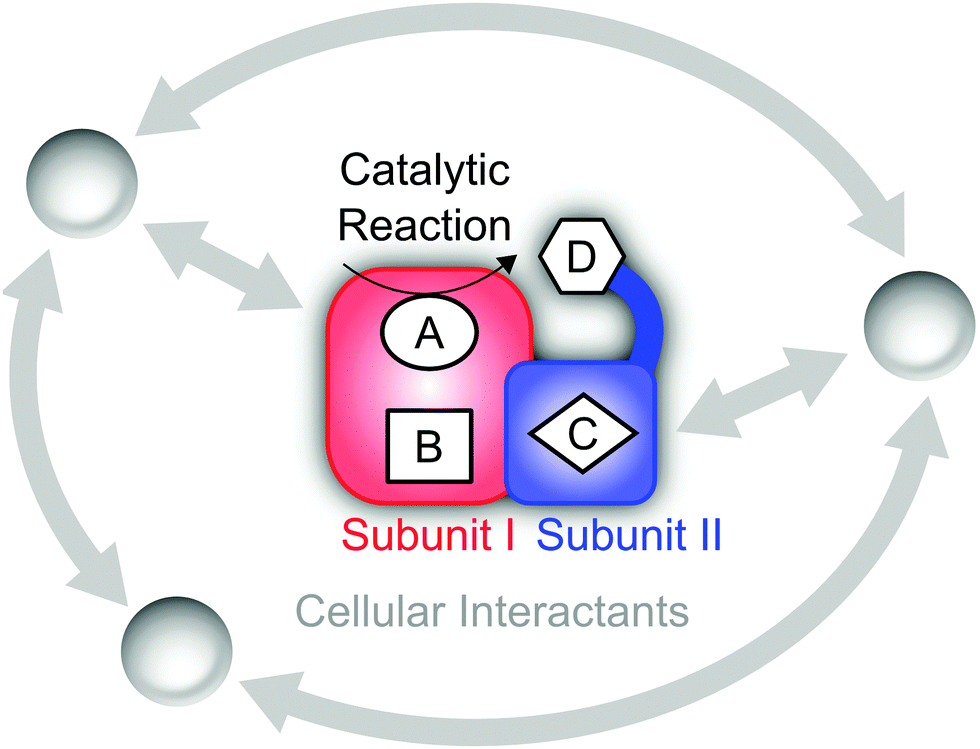 | ||
| Fig. 1 Schematic representation of a hypothetical enzyme consisting of two subunits that may represent distinct functional modules. Explicit functional sites embedded in the protein matrix (red and blue) are labelled A–D. Here, the active site A catalyses a chemical reaction, while D is located within a flexible domain, which allows functionally relevant movements, e.g. contacts with A. Interactions of cellular factors with each other and functional sites of the central biomolecule are shown in grey. | ||
Referring to (1), the size of typical biocatalysts implicates the necessity to identify and selectively address the functional sites of interest, may it be catalytic centres or molecular moieties that are involved, for instance, in proton or electron transfer (see Fig. 1). These functional sites can be often associated with few amino acids or certain cofactors. However, due to the assembly of several polypeptide chains and the presence of multiple domains and cofactors an a priori assignment might be ambiguous. Even more, the differentiation between a functional site and the surrounding protein matrix may be challenging. Often a clear cut between both regimes is ad hoc not possible, and parts of the protein matrix provide a dynamic and anisotropic multilayer environment for the explicit functional sites. Taking this thought to an extreme leads to the question about a possible function of the protein matrix as a whole that goes beyond stabilization and protection. In this respect, it should be noted that molecules are dynamic entities, which often exhibit a certain amount of intramolecular motions (normal modes) that are excited at ambient temperature (298 K). This statement is highly relevant for proteins, which exhibit numerous degrees of freedom (3N − 6) and an accumulation of normal modes in the thermal energy regime below 207 cm−1 = kT (see Fig. 2A).1,8–10 Given the alternation of flexible and rigid segments, these properties predestine proteins for the involvement of functional movements. In line with this statement, thermal protein motions have been reported to integrate distant functional sites or facilitate mechanistically relevant atom displacements (see Fig. 1), and this possibility has been extensively discussed.11–35
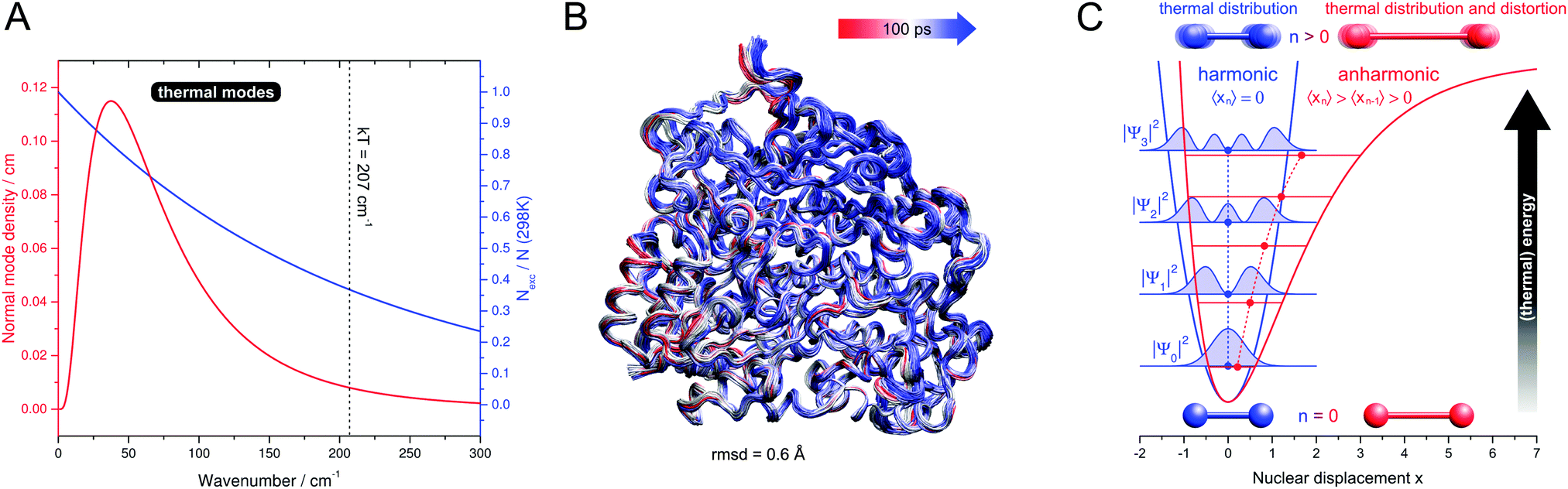 | ||
| Fig. 2 (A) Functionally relevant vibrational properties as a function of the normal mode energy. Red: low-frequency normal mode density (normalized number of modes per energy interval) of typical globular proteins, plotted as a log-normal distribution according to ref. 1. Blue: Boltzmann ambient temperature population of vibrationally excited states, calculated for a harmonic oscillator with an infinite number of non-degenerate, equally spaced levels.2Nexc – number of vibrationally excited molecules. N – total number of molecules. k – Boltzmann constant. T – temperature. (B) Cα-tube representation of protein thermal fluctuations (300 K) as observed in a molecular dynamics trajectory of membrane-bound hydrogenase from Ralstonia eutropha. The simulation was performed as described in ref. 3, and the picture was created by using VMD.4 rmsd – root mean square deviation. (C) Potential curves, energy levels n, and spatial expectation values 〈xn〉 of one-dimensional harmonic (blue) and anharmonic (red) oscillators.5–7 Spatial probability densities |ψn|2 are shown for the harmonic case as filled blue curves.5 For a vibrating diatomic molecule with a displacement x from equilibrium bond length, 〈xn〉 represents the average displacement, and |ψn|2 can be interpreted as the probability to observe a given displacement. Effects on these properties are sketched for vibrational ground and excited states, and for thermal modes these two scenarios give a simplified illustration of the situations at 0 K and ambient temperature. All values are in arbitrary units and shown for visualization purposes only. | ||
Considering aspect (2), the large number of possible interaction partners of a biological macromolecule actually extends the above challenge to identify functional determinants. In fact, a holistic view on such a biomolecule may involve all physiological factors that affect its structural and functional state. These factors may be perceived as a functional environment or complementary partners in a superordinate functional network of high elasticity (see Fig. 1).
Structure, function, and spectroscopy
To outline the importance and challenges of spectroscopic studies on biological macromolecules we will first specify the implicitly introduced termini structure and function. Notably, the advancement of X-ray diffraction techniques has considerably improved the three-dimensional perception of macromolecules in the life sciences.36 At the same time, however, the representation of crystal structure data as sets of atomic coordinates or schematic drawings has partly led to a misconception of chemical structure that neither includes dynamics nor electronic aspects in an explicit manner. In contrast to this perception, we use the term structure in the most general sense, i.e. with regard to the distribution of nuclei and electrons in both space and time. According to this definition, the entirety of structural information is only accessible by a combination of crystallography and spectroscopy, the latter providing insights into structural details that exceed the spatial and temporal resolution of typical X-ray diffraction data. In this context, it should be stressed that spectroscopic methods are conceptually different from other experimental techniques insofar as they provide direct insights into the quantised energy levels of a molecule, which are intimately linked to chemical structure.With this in mind, the structure of a molecule can be perceived as a multidimensional physical property that is explicitly encoded in (and partly extractable from) the spectroscopic data. In contrast, function is a more ambiguous terminus, which has to be defined with respect to a certain reference, e.g. the fulfilment of a cellular requirement or transformation of a substrate. In this respect, functional aspects are strictly speaking not inherent to spectroscopic observables. However, their relation to structure can be explored by spectroscopy, in particular if a system of interest is adequately manipulated during or between measurements. In conclusion, spectroscopic techniques provide access to structural factors and structure–function relationships, given that the functional aspects in question have been properly defined on a molecular level.
Concepts in bio-molecular spectroscopy
Considering the outlined specialties of biological macromolecules, a number of technical and conceptual requirements have to be fulfilled in order to extract valuable information from the corresponding experimental data. In the following, fundamental principles for the spectroscopic elucidation of structure–function relationships in such systems will be presented.Control
With increasing size and complexity of a molecule the interplay with external factors that affect the structure will become more complex as well. As a consequence, the detailed characterization of biological macromolecules requires the control of preparative and experimental parameters in a strict and adequate manner. This includes the adjustment of beneficial and functionally relevant determinants, if possible as a function of time, as well as the exclusion of deleterious or interfering factors.Representativeness
A second notable aspect, closely related to the first, is the choice of experimental conditions that are biologically representative. In this respect, the characterization of biological macromolecules in their native plasma or membrane environment is strongly recommended, especially if whole living cells can be probed.37,38 This applies especially to systems that are sensitivity towards oxygen or tightly interacting with other cellular factors.Selectivity
As stated above, biological macromolecules are large atomic ensembles that often involve several functional sites, a manifold of relevant structural properties, and a number of interaction partners. Consequently, the selectivity of the spectroscopic methods used is a prerequisite for the elucidation of individual aspects in these molecules. The requirement of specific spectroscopic markers is particularly important, if an individual molecular system has to be investigated in an ensemble of reaction partners or, even more, within whole cells.38 In the absence of specific markers, reaction-induced difference spectroscopy can be applied to elucidate structural aspects related to a chemical reaction that can be selectively triggered (vide infra). Moreover, structural information on selected molecular moieties may be obtained by site-specific isotope or amino acid exchange.Complementarity
With increasing specificity of a spectroscopic technique the amount of accessible structural information will necessarily decrease. As a consequence, a set of complementary methods has to be applied in order to obtain a comprehensive spectroscopic signature of the molecular moiety in question. Special care in the design of experiments has to be taken, if individual methods require specific sample preparation or if measurement conditions may interfere with the probed molecular system.39,40Interpretability
The evaluation of experimental data can be considered a key determinant in bio-molecular spectroscopy. Even if all previous requirements are fulfilled, an understanding of spectroscopic data from complex and unexplored molecules ultimately relies on adequate experimental or theoretical models. Experimental models may include established synthetic and biochemical compounds, whose spectroscopic properties are understood and reported. Alternatively, tailored small molecule analogues may serve as potent spectroscopic and functional models as well. In most cases, the computation of spectroscopic parameters (computational spectroscopy) requires the application of sophisticated quantum mechanical (QM) models of bio-molecular functional sites, usually involving density functional theory (DFT).35,40–42 In addition, hybrid models including the QM modelling of functional sites and a molecular mechanic (MM) treatment for the rest of a macromolecule represent an alternative approach, given that three dimensional data from X-ray diffraction, nuclear magnetic resonance (NMR) spectroscopy, or homology modelling are available.3,39,43–47 In either case, the adequate design of the QM model is essential for both reliability and computational efficiency. In this respect, reliability may be considerably increased by evaluating relative changes rather than absolute values of spectroscopic observables.40,41,48 This is especially important for small computational models that are designed in view of computational efficiency or for lack of representative three-dimensional data.The integrated approach
Depending on the specific target, the type of question, and the methods involved, the feasibility and benefit of each concept has to be judged from case to case. In this respect, a weighted combination of the individual principles represents an integrated approach that may serve as a benchmark for the spectroscopic investigation of biological macromolecules. In the following sections, a set of spectroscopic case studies on selected metalloenzymes will be presented to illustrate this integrated approach and the underlying peculiarities of biomolecular structure–function relationships.Selected case studies on metalloenzymes
Resonance Raman spectroscopy in hydrogenase research
In this first case study we will demonstrate the manifold of structural information accessible from a spectroscopic technique and the challenges associated with its first application to a certain delicate biomolecule. Using the resonance Raman (RR) characterization of hydrogenase as an example, the benefits of computationally aided spectroscopy are presented. Moreover, the importance of selectivity and the necessity of complementary methods in the establishment of a novel technique will be highlighted.[NiFe] hydrogenases represent a class of enzymes that catalyse the reversible cleavage of hydrogen into protons and electrons.49 Given the demand for clean and sustainable energy conversion processes, an in-depth understanding of the underlying mechanism is essential to design bioengineered or synthetic catalysts for hydrogen conversion. Since the beginning, spectroscopic methods have played a major role in hydrogenase research, both in the de novo exploration of cofactors and the detailed characterization of individual reaction intermediates.49
Most [NiFe] hydrogenases consist of a small subunit that contains three FeS clusters for electron transfer purposes and a large subunit harbouring the deeply buried active site (Fig. 3A).50 The latter cofactor contains two metal ions, Ni and Fe, which are bridged by two cysteinyl thiolates (Fig. 4). Two further cysteinates bind as terminal ligands to the Ni, while the Fe ion is also coordinated by three unusual inorganic ligands, one CO and two CN−.51–53 A third bridging position between the two metals serves as the substrate binding site. Individual redox states of the [NiFe] catalytic centre are mostly defined by the Ni oxidation state and the chemical nature of the ligand in this third bridging position.49 While structural models have been proposed for all detectable intermediates, the exact geometries and electronic properties are often not known.
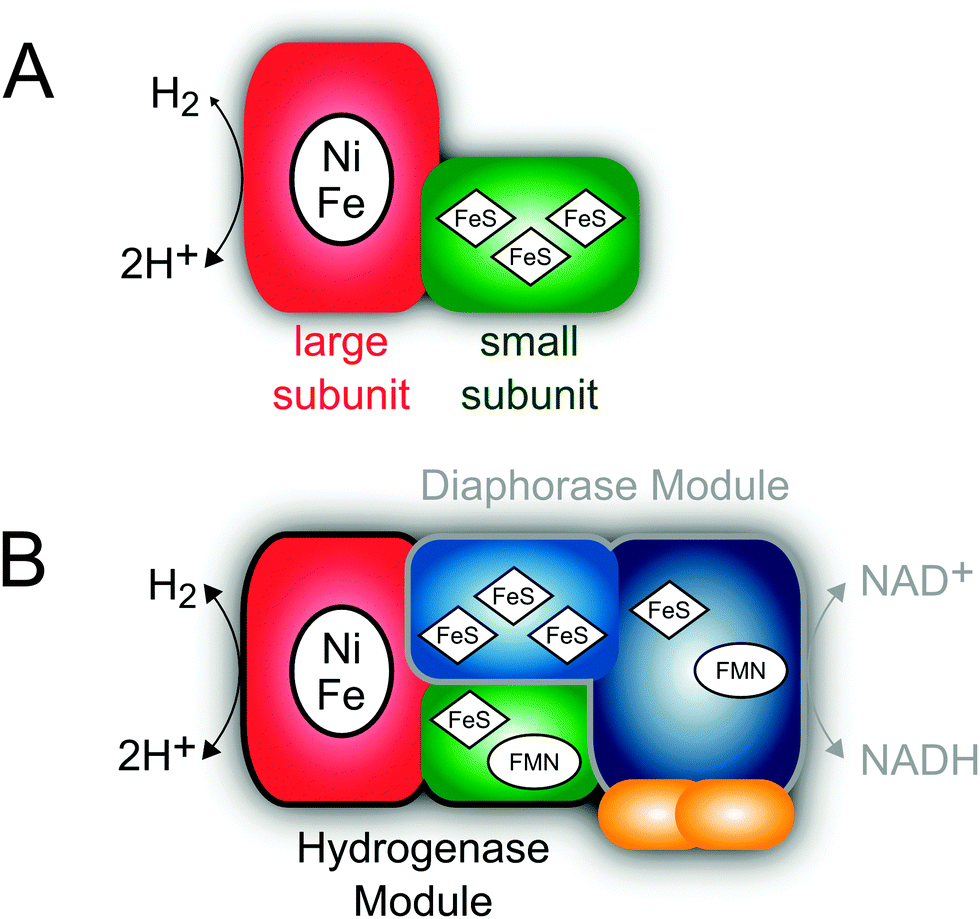 | ||
| Fig. 3 Subunit and cofactor composition of oxygen tolerant [NiFe] hydrogenases from Ralstonia eutropha. (A) Consensus depiction of the membrane-bound and regulatory hydrogenase. For the sake of simplicity, structural differences between both enzymes that are beyond the scope of this article have been ignored. (B) The NAD+-reducing soluble hydrogenase. | ||
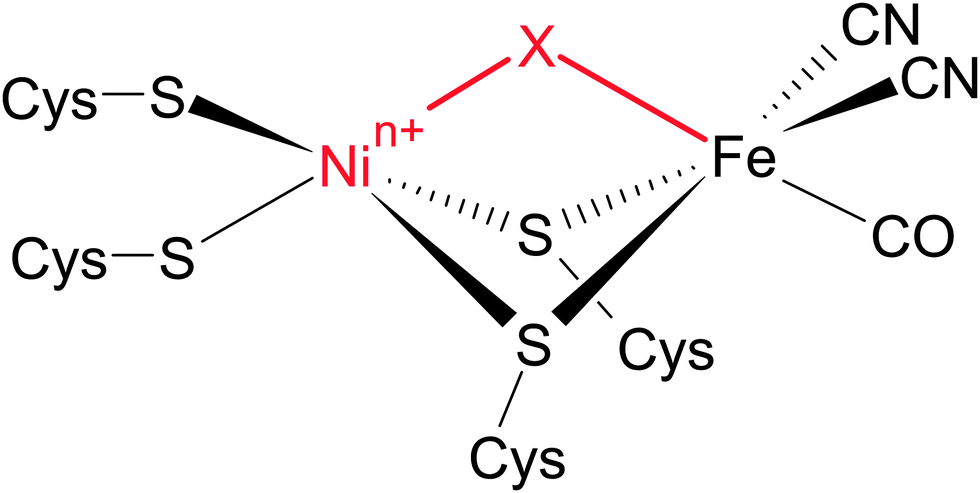 | ||
| Fig. 4 Simplified presentation of the active site structure of [NiFe] hydrogenases. Different redox states are mainly defined by the chemical nature of the third bridging ligand X and the oxidation state of the Ni ion n+. | ||
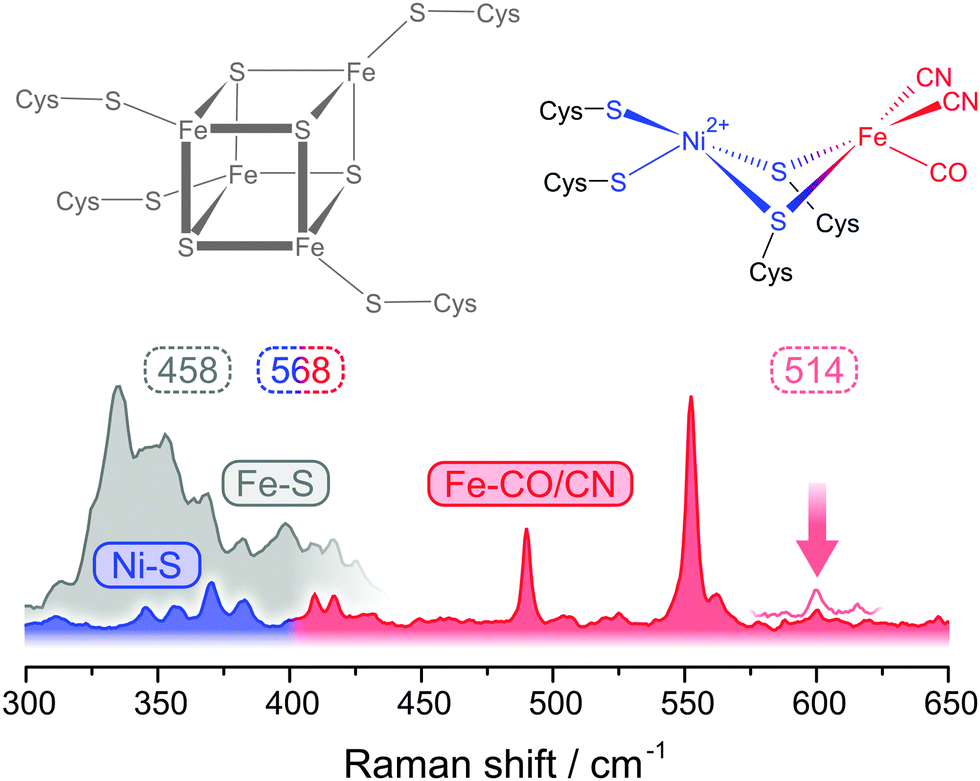
RR spectroscopy is a powerful vibrational spectroscopic technique for the selective probing of light-absorbing cofactors in large biomolecules, e.g. metalloenzymes.54 This technique provides direct insights into molecular coordinates, which can reflect structural properties as well as functional atom movements. In particular, metal–ligand coordinates that encode properties and interactions of the metal ion and its ligands can be probed. While the FeS clusters of hydrogenase have been investigated by RR spectroscopy for about three decades,55–58 the complex active site remained inaccessible by this technique.
Only recently, we were able to probe metal–ligand vibrations of both the FeS clusters and, for the first time, the [NiFe] active site by using low-temperature RR spectroscopy.39,40 While interfering signals from the protein matrix are absent in these spectra, the presence of several metal sites still necessitates a rigorous approach for the detection and assignment of individual metal–ligand vibrations. Besides potential markers for substrate and inhibitor binding,59–61 three major sets of structurally sensitive modes are expected from theoretical models and experimental studies on compounds resembling the cofactors of hydrogenase (Fig. 5).39,40,59,62–67 The [NiFe] active site may give rise to normal modes primarily reflecting Fe–CO/CN (400–650 cm−1) and Ni–S coordinates (<400 cm−1), while the FeS clusters will exhibit typical Fe–S modes (<400 cm−1). Clearly, both types of metal–sulphur modes will strongly overlap, and even the assignment of Fe–CO/CN modes is not straightforward, because signals are partly weak and might be misinterpreted as Fe–S overtones that exhibit about twice the fundamental frequencies. To clearly assign Fe–CO/CN modes we labelled an entire hydrogenase with 13C.39 According to a QM/MM model, pronounced isotopic shifts in the order of 10–15 cm−1 are solely expected for these modes. In the experimental spectra, all notable signals in the 400–650 cm−1 range showed isotopic shifts consistent with the calculated values, validating the assignment of Fe–CO/CN modes (Fig. 6, right).39
 | ||
| Fig. 5 Spectral regions for resonance Raman detected metal–ligand vibrations of [NiFe] hydrogenases accessible at different excitation wavelengths (in nm). Spectra and cofactor structures are shown for the oxidized state of the regulatory hydrogenase from Ralstonia eutropha (Nia-S).40 The vertical arrow indicates a sensitive reporter mode that was used as a structural marker (see Fig. 7). | ||
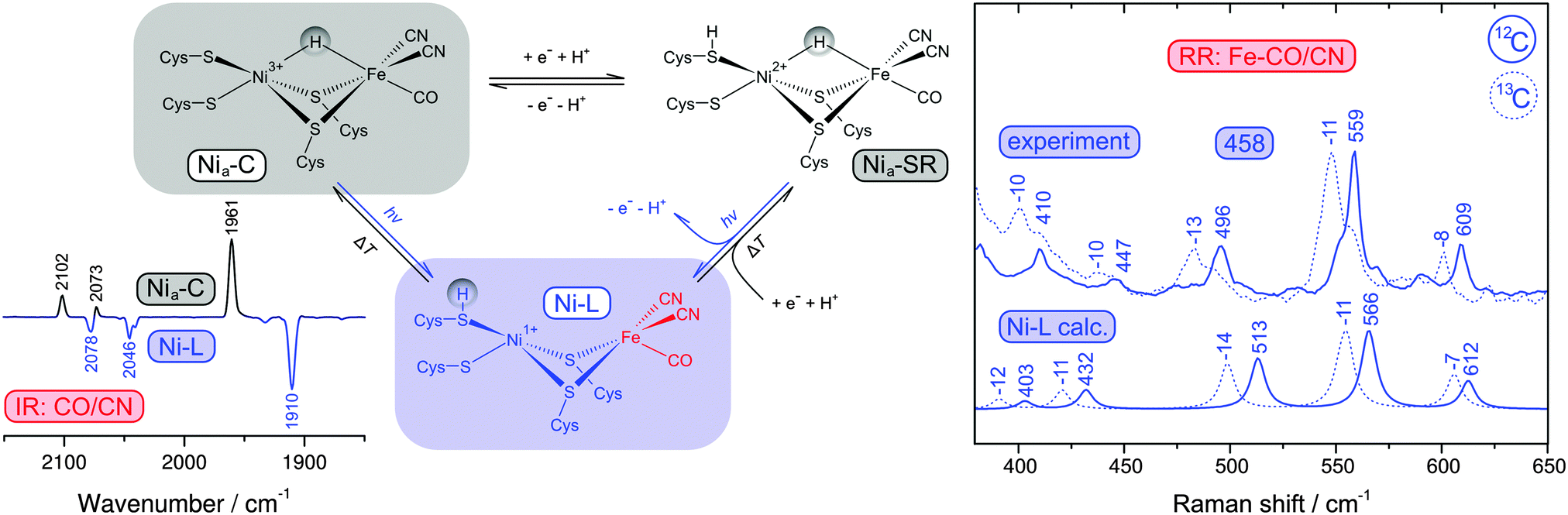 | ||
| Fig. 6 (left) Simplified scheme showing active site redox states of reduced [NiFe] hydrogenases and the corresponding thermal and photochemical equilibria. The inset shows an IR difference spectrum that illustrates the Nia-C → Ni-L photoconversion by following the CO and CN stretching vibrations of the active site. (right) Comparison of calculated Raman spectra of the Ni-L state with experimental resonance Raman spectra of reduced [NiFe] hydrogenase containing either 12C or 13C (using 458 nm excitation). Experimental and calculated spectra were obtained from the membrane-bound hydrogenase from Ralstonia eutropha and a corresponding QM/MM model.39 | ||
Catalytically active hydrogenase, obtained by reduction with hydrogen, usually exhibits a mixture of several [NiFe] redox states with different normal mode compositions and frequencies (Fig. 6, left).39,49 Under these conditions, a structural interpretation of the various Fe–CO/CN modes also requires an assignment to an individual redox state. Most reduced species of hydrogenase represent bridging hydride adducts of the [NiFe] site,49 which could be converted in the Raman probe beam to another potential catalytic intermediate (Ni-L), as shown in Fig. 6.39,40,68 Infrared (IR) and electron paramagnetic resonance (EPR) spectroscopy are established techniques in the field of hydrogenase research (vide infra), and so we used these methods to investigate possible photoreactions.39 Mimicking the conditions of the RR experiment, it could be shown that the central Nia-C intermediate is converted to Ni-L at low temperatures (79 K) and wavelengths close to the laser probe beam (458 nm), see inset of Fig. 6.39 Other low-potential hydride adducts, combined under the term Nia-SR, are generally assumed to be photo-stable.49 However, the photon densities of the RR probe beam exceed those of common light sources by orders of magnitude, which may open novel photochemical reaction pathways (Fig. 6, left). Indeed, the RR spectrum was found to be unaffected by moderate variations of the reduction potential as a function of the hydrogen partial pressure, suggesting that the Nia-SR subspecies may be converted to Ni-L as well.39 To test this assumption, we calculated RR spectra for several redox states of the [NiFe] centre using a QM/MM approach. Comparison with overall signatures, normal mode frequencies, and 13C isotopic shifts of these calculated data showed that the experimental spectrum is clearly dominated by the Ni-L state, confirming the redox state assignment and the photoconversion of Nia-SR subspecies (Fig. 6, right).39
In the low frequency regime, RR spectra of hydrogenase are usually dominated by FeS cluster signals that are strongly enhanced by S → Fe charge transfer transitions (Fig. 5).39,40,55–57 Therefore, Ni–S modes of the active site are normally not accessible, and special requirements have to be met for their detection. The absorbance of FeS clusters strongly decreases with increasing wavelength, which represents an opportunity to detect RR signals that are otherwise masked by spectral contributions from these cofactors.40 Excitation with 568 nm does not provide any resonance enhancement for the FeS cluster signals, but instead a novel set of weak bands could be observed (Fig. 5). Using 64Ni labelling in conjunction with DFT calculations, these bands could be clearly assigned to active site normal modes with major contributions of Ni–S coordinates.40
With the above assignments, one has a set of potent structural markers at hand. In particular, normal modes with predominant Fe–CO/CN character sense even subtle changes in electron density and, consequently, these modes represent valuable probes for structural variations at the entire [NiFe] active site.39,40 Using a computationally aided approach, we have used these Fe–CO/CN modes to gain insights into two catalytic intermediates of [NiFe] hydrogenases, Ni-L and Nia-S. Ni-L is kinetically stabilized as a photoproduct at low temperatures (vide supra) and presumably formed by removal of a bridging hydride, which is transferred as a proton to a nearby base (Fig. 6, left). However, neither the process of proton transfer nor the final acceptor has been experimentally verified. Using DFT, we could show that Fe–CO/CN modes would have contributions from Fe–H coordinates as well, if a bridging hydride was present.40 This would give rise to notable H/D isotopic shifts, in line with experimental data from small molecule analogues.59 In contrast, RR spectra of the Ni-L state of H2- and D2-reduced hydrogenase were identical, showing that the Fe–H bond is completely dissociated in N-L.40 Ni–H bond dissociation has been previously inferred from advanced EPR data,68 so that a complete removal of the bridging hydride is demonstrated by these two complementary techniques. Moreover, using a QM/MM model it could be shown that the experimental RR spectra are only compatible with a Ni-L structure that has a proton bound to one of the two terminal cysteinates (Fig. 6).39 In summary, RR spectroscopic data have proven both the structure and the photo-induced formation of Ni-L.
Nia-S is another catalytic intermediate, which is assumed to provide a free bridging site for initial hydrogen binding (Fig. 5).69 While this intermediate is a minor species in most hydrogenases, it clearly dominates the oxidized state of the regulatory hydrogenase (RH) from the ‘Knallgas’ bacterium Ralstonia eutropha,70,71 highlighting the importance of an adequate model system for spectroscopic studies. In line with predictions from DFT, RR spectra of the Nia-S state of the RH were shown to be similar to Ni-L,40 confirming a vacant third bridging position. Notably, the presence of a potential binding site between the two metals alone is insufficient to allow catalytic hydrogen turnover. Recent computational studies proposed that efficient hydrogen binding to Nia-S is only possible, if the NiII site exhibits a low spin (S = 0) electronic ground state and a seesaw geometry (Fig. 7).69 However, the real spin state and geometry is so far not known. To elucidate these properties, we used RR spectroscopy in conjunction with DFT.40 Due to the lack of a crystal structure for the RH, a compact [NiFe] computational model was constructed for the calculation of vibrational frequencies.40,41,47,48,72 Despite this simplification, valuable information can be extracted, if frequency changes rather than absolute values are evaluated.40,41,48 Therefore we compared calculated frequencies of several structural variants of Nia-S and Ni-L, the two major redox states of the RH at 79 K (Fig. 7).40 Focusing on a structurally sensitive Fe–CO stretching mode, the similarity of the two experimental spectra was only reproduced by the calculations, if Nia-S exhibits a seesaw-shaped NiIIS4 site with an S = 0 ground state,40 in line with the proposed functional significance of this configuration (vide supra).69 In conclusion, the combination of spectroscopy and theory has revealed all fundamental structural factors that determine the function of the central hydrogen-binding intermediate of hydrogenase.
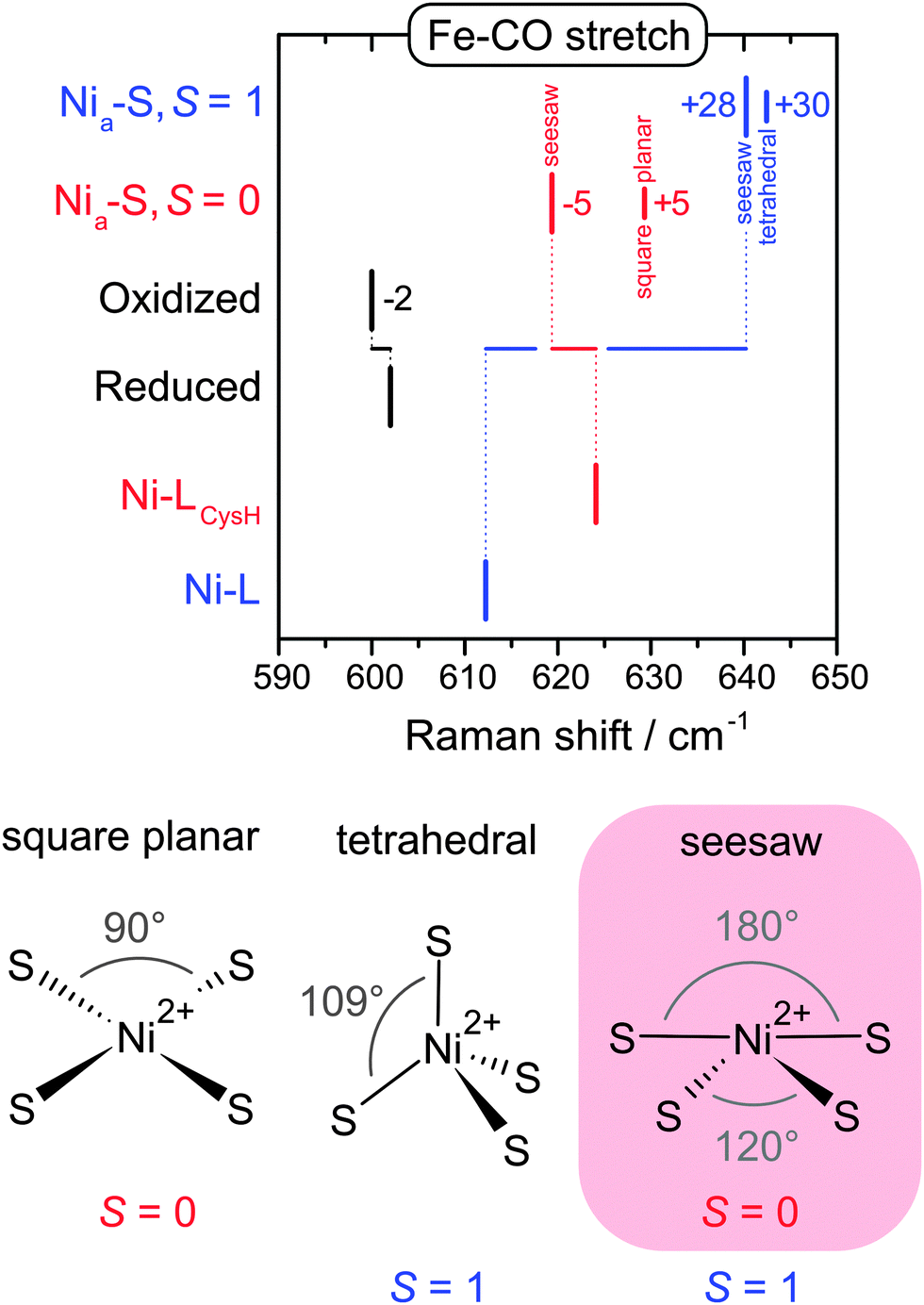 | ||
| Fig. 7 (top) Experimental band positions of oxidized and reduced regulatory hydrogenase from Ralstonia eutropha (black) compared with calculated Fe–CO stretching frequencies obtained for structural variants of the Nia-S and Ni-L redox states (red, blue).40 (bottom) Simplified presentations of the possible Ni geometries and spin states of Nia-S. | ||
Laying emphasis on the [NiFe] active site of hydrogenase, the capabilities of RR spectroscopy for the site-selective characterization of complex multi-cofactor enzymes have been demonstrated in the present section. We have shown how structural markers can be defined, validated, and used in order to extract valuable information by a concerted multi-spectroscopic and computational approach, even in the absence of crystallographic data. Photo-induced phenomena have been presented as challenging aspects in the interpretation of RR data. Such aspects should also be considered in other spectroscopic techniques that involve high photon energies or densities. While these processes are undesired in most cases, they can also provide access to otherwise sparse intermediates and ‘forbidden’ photochemical reactions.
Oxygen tolerance of a bifunctional hydrogenase
In the following, challenges and concepts in spectroscopy-based model building will be demonstrated. Here, structural determinants for oxygen tolerance and catalysis of a bifunctional hydrogenase will be used as mechanistic examples to highlight the importance of adequate experimental conditions and the pitfalls of data interpretation.The complexity of hydrogenases may considerably exceed the structure and composition described above. The soluble hydrogenase (SH) from Ralstonia eutropha is a heterohexameric enzyme, which couples the reversible cleavage of hydrogen to the reduction of NAD+.72–74 These two catalytic activities are accomplished by distinct functional modules, a truncated hydrogenase and a diaphorase moiety (Fig. 3B). Besides the [NiFe] active site the SH contains five FeS clusters and two FMN cofactors.72,74–87 Due to its bifunctional character this enzyme represents a valuable biocatalyst for photolytic hydrogen production in vivo and the regeneration of nucleotide cofactors.88–96
In contrast to most other hydrogenases, the SH is insensitive towards oxygen.72–74,87,97 Oxygen tolerance represents a prerequisite for technological applications and, thus, the underlying mechanisms are of major interest. The SH has been investigated for more than three decades,72–74 yet no crystal structure is available so far. Consequently, spectroscopic methods have played an outstanding role in the investigation of structural aspects, including those responsible for oxygen tolerance. IR spectroscopy represents a major technique in hydrogenase research, because it can monitor CO and CN stretching vibrations of the three inorganic ligands, which represent sensitive probes for the electron density at the active site.51,52,98,99 In this respect, it is complementary to RR spectroscopy, and together both techniques provide the complete vibrational signature of the Fe2+(CO)(CN−)2 moiety (Fig. 6). Usually, one CO and two CN stretching bands are observed in the IR spectrum of [NiFe] hydrogenases. In contrast, the IR spectrum of isolated SH exhibits four instead of two CN stretching bands (Fig. 8A, inset).48,82,84,100,101 In the past, this was interpreted in terms of additional CN− ligands, one of which was claimed to sterically shield the active site from oxygen attack.82,84,100,101 EPR spectroscopy represents another central technique for the characterization of hydrogenase, which inter alia probes paramagnetic Ni intermediates of the active site.102 In most in vitro studies, no Ni signals were observed for the SH.82,84,87,101 Based on this finding and the proposed presence of supernumerary CN− ligands, hydrogen cycling in this enzyme was presumed to take place at a terminal binding site of the apparently redox-inactive Ni ion.84,103 From a chemical point of view, the proposed structures are unlikely to be stable and, consistently, DFT calculations were unable to reproduce them.41 Therefore, we have reinvestigated the structural models for oxygen tolerance and catalysis, using two different approaches.
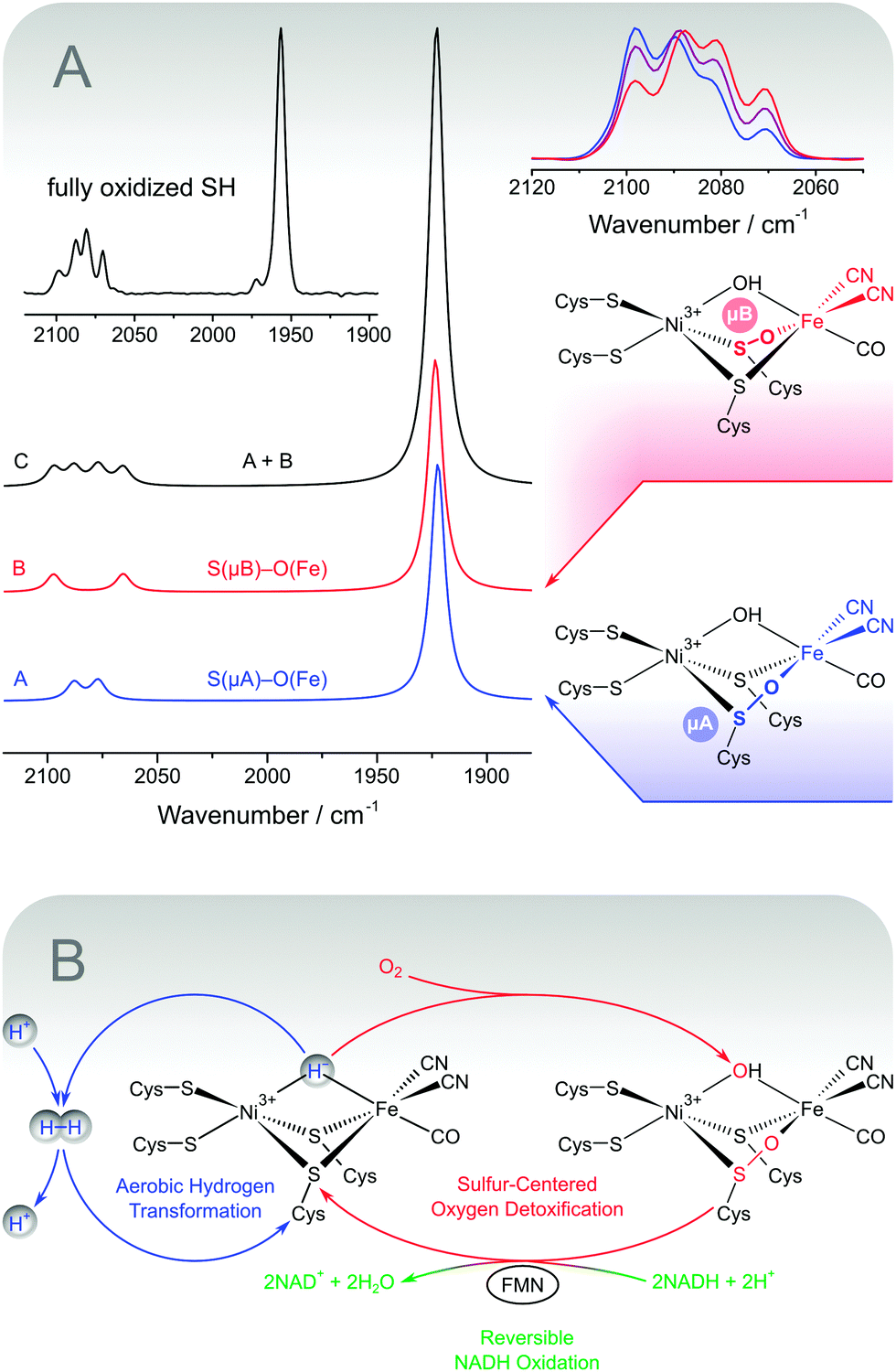 | ||
| Fig. 8 (A) Structures and DFT-calculated IR spectra for sulphoxygenated [NiFe] model compounds that mimic the experimental spectrum of fully oxidized soluble hydrogenase from Ralstonia eutropha (shown as an inset). Variations of the experimental CN stretching pattern are shown on the upper right.48 (B) Simplified scheme illustrating the proposed oxygen tolerance mechanism of the soluble hydrogenase, which involves three catalytic activities. Aerobic hydrogen transformation (blue) is enabled by the sulphur-centred detoxification of oxygen (red), which in turn depends on the reversible oxidation of NADH (green). | ||
First, the above structural models are based on in vitro spectroscopic data, which may not necessarily reflect the native state of the enzyme. Therefore, we characterized the SH within whole living cells using IR and EPR spectroscopy (Fig. 9).38 In this way, the reducing cytoplasmic environment of the enzyme is preserved, and possible artefacts from protein purification and sample treatment can be omitted. Notably, the feasibility of in vivo spectroscopy depends on a set of highly specific markers, which are distinguishable from those of other cellular components. In the case of hydrogenase, these are the typical EPR signatures of paramagnetic Ni species and the IR-detectable stretching vibrations of the biologically uncommon CO and CN− ligands, which are clearly separated from the frequencies of most other normal modes. Using this whole-cell approach, EPR spectroscopy was able to show that the SH active site resides mainly in the paramagnetic Nia-C state in vivo (Fig. 9, blue).38 Upon incubation with hydrogen, this redox species is reversibly converted to the typical, EPR-silent Nia-SR intermediates, probed by IR spectroscopy (Fig. 9, red).38
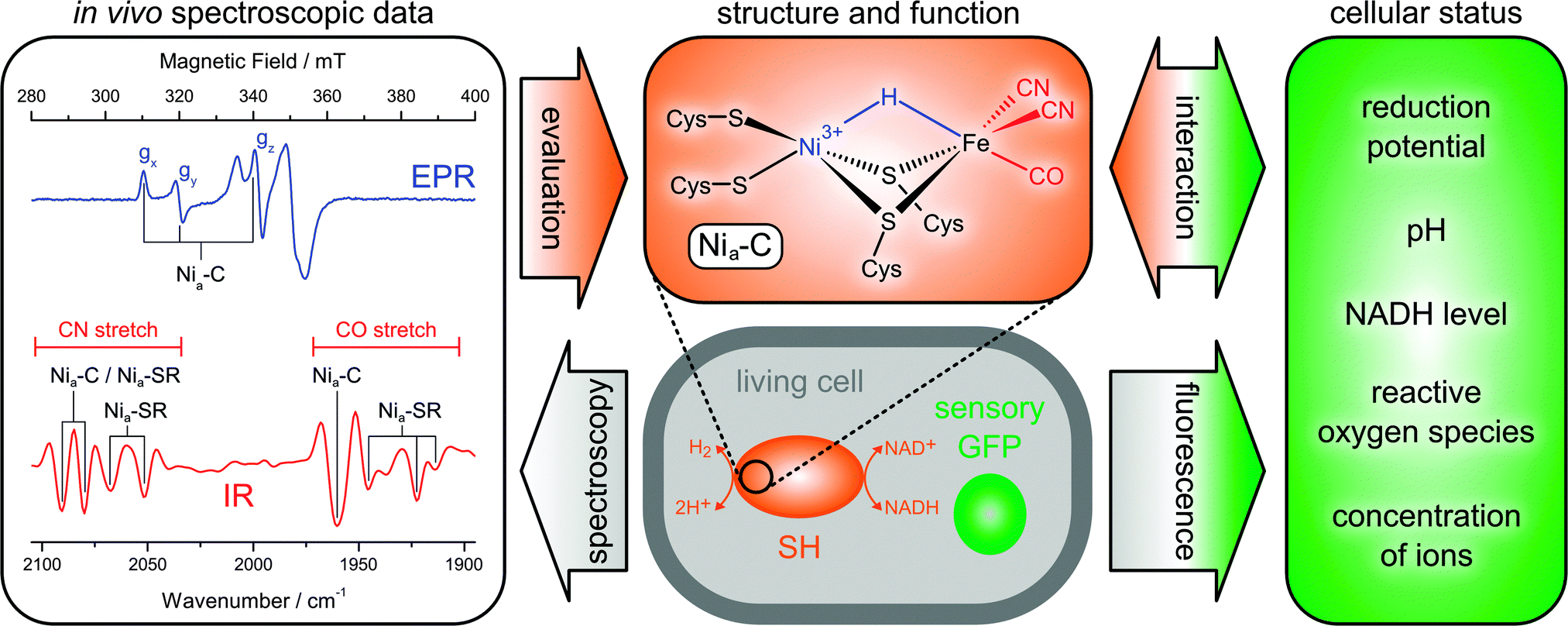 | ||
| Fig. 9 Principles of extended in vivo spectroscopy and selected results obtained for the soluble hydrogenase (SH) from Ralstonia eutropha.38 All fully reduced species of the SH are subsumed under the heading Nia-SR. A derivative of the green fluorescent protein (GFP) is shown as an example for genetically encoded fluorescent biosensors. | ||
These findings demonstrate that the catalytic process of the SH involves a redox-active Ni site and substrate binding at the usual third bridging site. Moreover, the IR detection of typical reaction intermediates shows that the [NiFe] site comprises a standard set of one CO and two CN− ligands.38 Notably, the spectroscopic signatures obtained in vivo can be reproduced in vitro by incubating the enzyme with an excess of NADH.48,83 This treatment mimics the cytoplasmic conditions experienced by the SH, highlighting the significance of adequate and representative sample treatment.
Besides the optimization of experimental conditions, a proper interpretation of spectroscopic data can also be ensured by theoretical considerations (vide supra). This second approach, complementary to the first, represents a more fundamental strategy, which can also be applied to test, if a certain interpretation could be legitimate at all. Using this methodology, we have revisited the previous structural proposals for the SH, showing that the old model for oxygen tolerance and catalysis is incompatible with the underlying spectroscopic data.41 These findings rule out the previous interpretations on a fundamental level, demonstrating that the additional CN stretching bands cannot reflect extra CN− ligands.
The two spectroscopic studies have shown that hydrogen cycling by the SH involves a usual [NiFe] site and typical intermediates. Ruling out the old model, the structural basis for the oxygen tolerance and the unusual IR spectrum of as-isolated SH remains to be elucidated. Notably, this unusual split-spectrum, exhibiting four instead of two CN stretching bands (Fig. 8A, inset), is only observed in the fully oxidized state of the enzyme, which may indicate oxidative modifications.48 Indeed, the split-spectrum disappears upon incubation with the cytoplasmic electron donor NADH and reappears upon re-oxidation.48 The re-formation of the fully oxidized state, reflected by the split-spectrum, is dependent on the presence of oxygen, demonstrating oxygenation of the [NiFe] site.48 In this respect, the observation of four CN stretching bands indicates a mixture of states, as also confirmed by preparation-dependent variations of the CN stretching pattern (Fig. 8A, top right).48,84,101 The appearance of a single high-intensity CO stretching band shows that the structural differences between the subspecies in this mixture affect the equatorial plane of the Fe site in a selective manner (Fig. 4). This rules out variations of an oxygen-derived ligand in the third bridging position and strongly indicates sulphoxygenation of active site thiolates, likely involving the bridging cysteines (Fig. 8A, right).48 To test this hypothesis, we calculated IR spectra of a large set of sulphoxygenated [NiFe] model compounds by DFT. The resulting data clearly show that cysteine oxygenation has a strong influence on the CN stretching vibrations, while the CO stretching vibration remains unaffected in most cases.48 In particular, an equimolar mixture of states that exhibit Fe-bound sulphenates at the bridging cysteines resembles the typical split-spectrum of fully oxidized SH (Fig. 8A). Thus, a structural model has been obtained, which is able to consistently explain all present and previous IR spectroscopic properties of the SH, thereby disentangling the seeming discrepancy between in vitro and in vivo data.
Besides an explanation of spectroscopic data, active site sulphenates also represent a potential key to the oxygen tolerance mechanism of the SH (Fig. 8B).48 In general, oxygen tolerant hydrogenases are assumed to prevent oxidative damage by the catalytic reduction of molecular oxygen. Sulphoxygenated active site species of the SH are formed under aerobic conditions (vide supra) and characterized by two oxygen atoms in the lowest oxidation state O−II. This means that molecular oxygen has been completely reduced, most likely including a peroxidase sub-reaction.48 To recover catalytic activity, active site sulphenates must be re-reduced in a dehydration reaction at potentials clearly below −100 mV.104 Most hydrogenases, however, are linked to the high-potential quinone pool in the cytoplasmic membrane (approximately +100 mV),105,106 so that sulphenates would represent irreversible oxidative modifications. In contrast, the SH is linked to the low-potential NADH pool of the cytoplasm (approximately −320 mV),72–74,79,107 and sulphenate formation is completely reversible in this enzyme. This statement is verified by the spectroscopic data (vide supra) and the NADH-driven release of oxygen-derived water.48,97 The latter reaction appears to depend on the presence of the FMN cofactor close to the active site, which can be explained by the fact that sulphenate reduction requires a two-electron carrier. Moreover, this functional model for oxygen tolerance of the SH can explain why aerobically isolated SH cannot react with hydrogen prior to re-activation by NADH.48,84,101,103
Using the example of a bifunctional [NiFe] hydrogenase, we have shown how spectroscopy and theory can be used to propose, validate, or reject models for biomolecular structure and function. In particular, the importance of the biochemical reaction network for the design of meaningful (in vivo) experiments and the functional interpretation of spectroscopic data has been emphasized. In this respect, the availability of highly selective spectroscopic markers is of central importance for the structural characterization of a biomolecule in its cellular environment. Another crucial aspect is an increased awareness of potentially misleading (in vitro) spectroscopic data, whose evaluation requires special care. In this respect, the present case study demonstrates the importance of an integrated approach that brings together the expertise from biological, chemical, and physical sciences.
The dynamic structure of superoxide reductase
Using the example of superoxide reductase, the last case study will demonstrate how reaction-induced difference spectroscopy can be used to gain both site-specific and global information on structural elasticity. A combined approach of X-ray diffraction, spectroscopy, and theoretical methods that can be used to provide detailed insights into the dynamic structure of biomolecules will be presented (Fig. 10).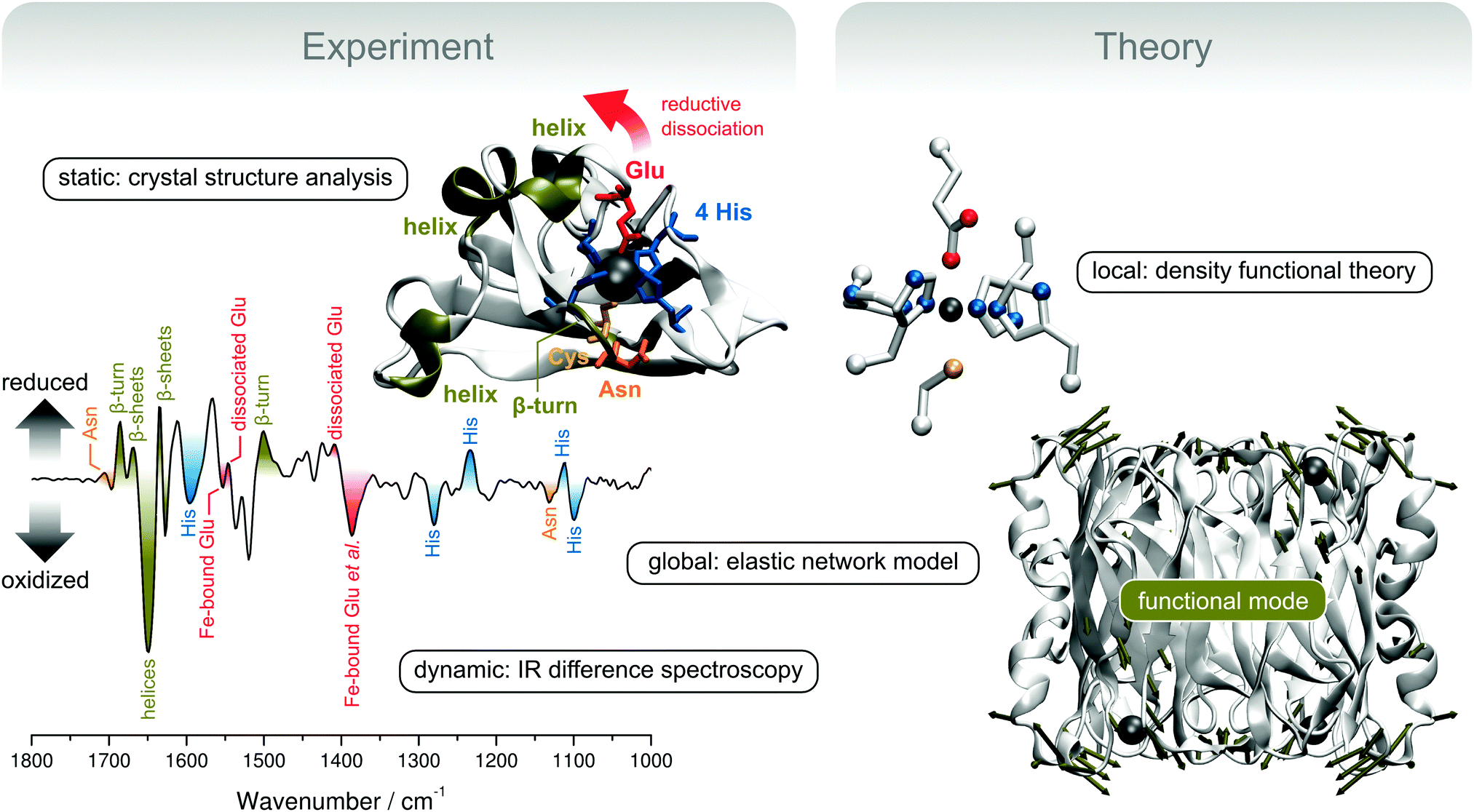 | ||
| Fig. 10 Experimental and theoretical concepts for the exploration of structural elasticity, exemplarily shown for the redox-dependent rearrangement of superoxide reductase (SOR).35 (top left) Crystal structure presentation of a single monomer of SOR from Ignicoccus hospitalis (Ih). Important structural motifs are coloured, except for the β-barrel in the background (represented by silver arrows). (bottom left) Redox-dependent IR difference spectrum of Ih SOR. Encoded structural changes are colour-coded according to the above crystal structure presentation. (top right) Computational SOR active site model used for the calculation and assignment of IR spectra. (bottom right) Crystal structure of the whole homotetrameric Ih SOR. Deformation by a delocalized normal mode that is proposed to guide the transition between the ferric and ferrous states is represented by dark green displacement vectors, which are shown for Cα atoms whose movements exceed the root mean square deviation (arbitrarily chosen as 2 Å). Molecular and normal mode presentations were created using VMD and the NMWiz plug-in.4,114 | ||
Superoxide O2˙− is a reactive oxygen species that may severely harm cellular compounds, e.g. FeS proteins.108 To cope with these hazards most living species harbour molecular systems for detoxification, and the underlying principles are important both from a biochemical and medical point of view.109 Superoxide reductase (SOR) is a non-heme iron enzyme that catalyses the reduction of superoxide to hydrogen peroxide by transferring one electron from its ferrous active site.108
In this study, we will deal with the canonical 1Fe-SOR from Ignicoccus hospitalis (Ih).35 Enzymes of this type are composed of four equal subunits (Fig. 10, bottom right), each of which harbours a non-heme iron active site in a β-barrel fold. In the ferrous state, the active site Fe is coordinated by four equatorial histidines and an axial cysteine in a square pyramidal fashion. This geometry provides a vacant coordination site for substrate binding, which is occupied by a glutamate ligand in the ferric state of several SOR enzymes.108,110 This coordination pattern is also reflected by a crystal structure of ferric Ih SOR (Fig. 10, top left). However, dissociation and displacement of the glutamate ligand has not been observed unambiguously upon reduction to the ferrous state.35 This finding demonstrates that X-ray diffraction is unable to properly monitor structural changes associated with the central step of reductive activation, most likely due to crystal packing effects. Therefore, we have used electrochemically triggered IR difference spectroscopy, which selectively monitors structural changes associated with the redox transition between the ferric and ferrous state (Fig. 10, bottom left).35 In contrast to X-ray diffraction studies on protein crystals, these experiments probe SOR in aqueous solution more closely related to the natural conditions, which is crucial to obtain representative information on the dynamic structure.
Using this technique and a crystal structure-based DFT model of the active site (Fig. 10, top right), it could be shown that the glutamate ligand is reversibly removed from the substrate binding site upon reduction of Ih SOR, which is mandatory for the enzymatic function (Fig. 10, bottom left).35 This information can be inferred from changes of the vibrational signature of the glutamate ligand (red) as well as side chain normal modes of the coordinating histidines (blue). Comparison with a mutant protein lacking the coordinating glutamate also showed that the dissociation of this ligand induces extended structural changes in the wild type enzyme, indicating high structural elasticity in SOR. Inter alia, these structural changes include perturbations of the protein backbone (dark green) and amino acid side chain residues in loop and helical regions close to the active site (orange and red). Even more, the spectroscopic signature of the wild type enzyme also revealed a redox-dependent structural reorganization of the central β-sheet core of the enzyme (dark green), remote from the active site. This is a remarkable finding, which shows that the addition of a single electron per subunit can trigger a global conformational transition in a protein consisting of thousands of atoms.
To proceed in a controlled manner, such a process needs to follow a well defined coordinate, which is determined by the molecular degrees of freedom and the associated forces. Remarkably, global structural transitions were shown to often depend on one or few delocalized, thermally excited normal modes (vide supra).13,14,111–113 Global modes of macromolecules can be calculated by using classical molecular mechanics. In this respect, normal mode analysis based on elastic network models (ENMs) is a robust technique, which provides reliable results at minimum computational cost.13,112,115–119 In contrast to other classical approaches, elaborate force field parameterization is not necessary, and the Hookean pair potentials of ENMs allow skipping the error-prone and time-consuming process of geometry optimization so that crystal structure data can be directly used as an input.115,117 Using this methodology120 and the crystal structure of ferric Ih SOR, we could show that normal modes with a considerable displacement of the glutamate ligand resemble the structural changes inferred from the IR difference spectra.35 In particular, a large glutamate displacement was observed for a normal mode that exhibits in-phase movements of helical and loop regions of all four subunits as well as the β-sheet core of the enzyme (Fig. 10, bottom right). This normal mode represents a reasonable candidate for thermal movements that guide the structural transition between ferric and ferrous SOR and, thus, a coarse-grained 3D depiction of the structural changes encoded in the IR difference spectrum. The underlying atom displacements proceed synchronously in all four subunits and, thus, the associated structural rearrangements can integrate the structural states of the four active site copies. Such a process might be functionally relevant by allowing reductive activation or catalysis to proceed in a cooperative manner.35
The example of superoxide reductase shows that structure should be perceived as a dynamic rather than a static property, especially in those cases where the molecular function is linked to considerable atom movements. In this respect, normal modes may serve both as structural markers in vibrational spectroscopy and determinants of functional dynamics. Moreover, the present case study demonstrates the feasibility of a complementary approach including both crystal structure analysis and computationally aided spectroscopy. The former provides an overall model of the static molecular structure and a basis for detailed spectroscopic and computational studies. In return, these latter studies extend the available structural information to the dynamic regime and conditions that are more closely related to the physiological environment. In this respect, the combination of IR difference spectroscopy and ENM analysis has been introduced as a powerful vibrational approach for the elucidation of reaction-specific structural rearrangements and the underlying molecular degrees of freedom.
Future prospects
Challenges and chances
We have outlined an integral approach for the spectroscopic characterization of biological macromolecules, which explicitly considers requirements arising from (1) size and complexity as well as (2) the native reaction network. While the underlying principles have been exemplarily demonstrated by selected case studies, we will now present some conceptual extensions to cope with the challenges in bio-molecular spectroscopy.The second case study on a complex hydrogenase has highlighted the importance of adequate experimental conditions to obtain representative information on bio-molecular structure and function.38 In this respect, the concept of in vivo spectroscopy has been introduced as a means to directly probe a system of interest in its native cellular environment. An extension of this approach is highly desirable in order to increase the amount of extractable information.
Structure and function of a molecule in vivo depend on the cellular status, which in turn responds to environmental triggers, e.g. hydrogen and oxygen partial pressures in the case of hydrogenase. In principle, these relations offer the possibility to manipulate a molecule of interest in a physiological manner. However, the underlying cellular reaction networks are complex, and an adjustment of external triggers alone is insufficient to precisely control and explore the biochemical factors that determine the status of a target molecule. To reach this goal, complementary information on the cellular status is required as well. In recent years, a number of sensory derivatives of fluorescent proteins have been designed.121–139 These genetically encoded biosensors can complement spectroscopic data of the target molecule by probing essential cytoplasmic properties in space and time, e.g. the concentration of ions and small molecules as well as pH and reduction potential (Fig. 9). Using these tools, an extended type of in vivo spectroscopy can provide structural information on a biomolecule of interest under various native conditions, and the underlying cellular determinants can be identified. Therefore, such studies could also help to elucidate adequate conditions for elaborate in vitro experiments and targets for optimizing biotechnological processes. With a strict control of extracellular triggers, this approach may additionally yield insights into the biochemical reaction network of the target molecule as a whole, which is important for both fundamental and applied sciences. For an extended in vivo analysis of the soluble hydrogenase (vide supra), we are currently exploring the possibilities of IR and fluorescence spectroscopy in an experimental approach that allows a tight control of atmospheric gas mixtures and a precise monitoring of the cellular NADH level.
Another special aspect of biological macromolecules that has been stressed in the case study on superoxide reductase is the potential importance of functional dynamics.35 Thermal motions could also play a role in the second model system hydrogenase, e.g. by facilitating hydrogen tunnelling. Such effects, discussed for other enzymes,15–33 have not been considered in the available catalytic models for hydrogenase, all of which are based on classical over-the-barrier reactions involving equilibrium structures.69,140–142
Remarkably, even global movements in proteins can be often traced back to few thermally excited normal modes.13,14,111–113 In principle, this may allow to enhance the molecular function by tuning the dynamic structure, given that the underlying intramolecular motions are properly understood. On a theoretical level, these phenomena can often be studied by using classical normal mode or essential dynamics analysis.13,14,112,113,115–119,143 Their experimental exploration by vibrational spectroscopy, however, represents a highly delicate issue. With increasing size of the molecule, the number of thermal modes will increase as well, and very slow motions become possible. While this might be beneficial from a functional point of view, the necessity of detecting one or few relevant modes amongst many others poses considerable challenges for the spectroscopic techniques used, especially in the low-frequency regime. While some information may be accessible by far-IR and terahertz spectroscopy,144,145 site-selective vibrational techniques are more suitable, given that the relevant atom movements can be associated with explicit functional sites. Besides RR spectroscopy, presented in the first case study, nuclear resonance vibrational spectroscopy (NRVS)146,147 and vibrational coherence spectroscopy (VCS)148 represent two further vibrational techniques that are inter alia suited for the site-selective probing of low-frequency modes.† All three methods can be used for the characterization of biological macromolecules, (see e.g.ref. 39, 40, 54, 64, 86, 146 and 149–161) and both RR and NRVS spectroscopy have been recently established in hydrogenase research.39,40,64,86,155 Despite technical advancements, the explicit application of vibrational spectroscopy to study functional dynamics is still sparse, the low-frequency modes of heme proteins being one of the few examples.34,158,161–163 At this point, it should be noted that molecular vibrations exhibit periods on the timescale of fast chemical processes and, consequently, valuable information may be obtained. In this respect, NRVS can be used for the selective and quantitative probing of normal modes involving Fe movements.146,147 This may provide a unique access to vibrational dynamics of Fe proteins,34,146,149–154e.g. the functional movements predicted for superoxide reductase (vide supra). Complementary, VCS and femtosecond stimulated Raman spectroscopy (FSRS) offer a time resolution that exceeds the frequencies of molecular vibrations.148,164 In this way, valuable insights into functional dynamics, non-equilibrium structural properties, and biochemical reaction coordinates may come into reach.
Besides their possible functional importance, vibrational dynamics also represent a challenge for the theoretical description of macromolecular properties, including spectroscopic observables. While technical aspects of computational chemistry are beyond the scope of this article, we like to stress that typical electronic structure calculations refer to the molecular equilibrium geometry, which approximates the situation at 0 K. Naturally, biochemical reactions proceed at ambient temperature or above, and under these conditions the probability to observe the equilibrium geometry is decreased to some extent, even in the absence of large scale domain movements (Fig. 2B). First, the thermal excitation of numerous low-frequency modes (vide supra) increases the probability of observing molecular geometries approaching the classical turning points of these oscillations (Fig. 2C, blue). Second, anharmonic motions may shift the average of the thermal distribution away from the equilibrium geometry, which implies a net distortion of the molecule (Fig. 2C, red). Indeed, anharmonicity of thermal protein motions has been reported, especially for very low-frequency modes with major contributions to the overall thermal motion.10,14,112,113,117,119,143,165 In summary, the properties of thermally excited macromolecules may considerably differ from those predicted for 0 K, as suggested by the temperature dependence of many spectroscopic observables. This notion may be particularly relevant for metalloproteins, whose functional sites comprise a number of relatively soft metal–ligand coordinates (vide supra). As a consequence, a future quantum chemistry approach to such systems may benefit from a more explicit consideration of thermal effects on the molecular geometry, especially with respect to anharmonic low-energy coordinates. Besides a more representative prediction of spectroscopic and other properties, this may also provide valuable insights into non-equilibrium structures and their possible impact on biochemical reactivity.
Conclusion and outlook
In the present article we have illustrated the challenges and capabilities of bio-molecular spectroscopy. Selected case studies have shown how structure–function relationships can be elucidated by computationally aided spectroscopy, preferably under physiological conditions. In a more general sense, these examples have highlighted the natural entanglement of spectroscopic observables and essential molecular properties. As demonstrated by these studies, this relation is normally utilized to explore the molecular structure from spectroscopic experiments. Using the example of functional vibrations, we have furthermore shown that certain subjects of spectroscopic research may also represent a direct link to molecular function that goes beyond their evaluation as structural markers. Expanding this thought, spectroscopic observables could also serve as sophisticated targets for the design and optimization of functional molecules. This paradigm shift from an analytical to a predictive expertise may represent a valuable perspective for the emerging field of computational spectroscopy. Indeed, similar conceptual expansions have proven valuable, including the extension of classical biology in terms of biotechnology and bio-inspired chemistry. In conclusion, we propose a dual future approach, which uses spectroscopic observables as classical markers for structure analysis as well as experimentally traceable targets for the computational optimization of tailored molecules.Acknowledgements
We thank Yvonne Rippers for providing the molecular dynamics trajectory that was used to prepare Fig. 2B. The work was supported by UniCat, funded by the Deutsche Forschungsgemeinschaft.Notes and references
- P. Etchegoin, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1998, 58, 845–848 CrossRef CAS.
- P. W. Atkins and J. de Paula, Physical Chemistry for the Life Sciences, Oxford University Press, Oxford, 1st edn, 2006 Search PubMed.
- Y. Rippers, T. Utesch, P. Hildebrandt, I. Zebger and M. A. Mroginski, Phys. Chem. Chem. Phys., 2011, 13, 16146–16149 RSC.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- D. A. McQuarrie, Quantum Chemistry, University Science Books, Sausalito, California, 2nd edn, 2008 Search PubMed.
- P. M. Morse, Phys. Rev., 1929, 34, 57–64 CrossRef CAS.
- V. S. Vasan and R. J. Cross, J. Chem. Phys., 1983, 78, 3869–3871 CrossRef CAS.
- D. Ben-Avraham, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 14559–14560 CrossRef CAS.
- D. Ben-Avraham and M. M. Tirion, Physica A, 1998, 249, 415–423 CrossRef.
- N. Go, T. Noguti and T. Nishikawa, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 3696–3700 CrossRef CAS.
- C. Xu, D. Tobi and I. Bahar, J. Mol. Biol., 2003, 333, 153–168 CrossRef CAS PubMed.
- M. Tekpinar and W. Zheng, Proteins, 2013, 81, 240–252 CrossRef CAS PubMed.
- I. Bahar, T. R. Lezon, L. W. Yang and E. Eyal, Annu. Rev. Biophys., 2010, 39, 23–42 CrossRef CAS PubMed.
- S. Hayward and N. Go, Annu. Rev. Phys. Chem., 1995, 46, 223–250 CrossRef CAS PubMed.
- S. Hammes-Schiffer, Biochemistry, 2002, 41, 13335–13343 CrossRef CAS PubMed.
- S. Hammes-Schiffer, Acc. Chem. Res., 2006, 39, 93–100 CrossRef CAS PubMed.
- S. Hammes-Schiffer and S. J. Benkovic, Annu. Rev. Biochem., 2006, 75, 519–541 CrossRef CAS PubMed.
- J. P. Layfield and S. Hammes-Schiffer, Chem. Rev., 2014, 114, 3466–3494 CrossRef CAS PubMed.
- B. J. Bahnson and J. P. Klinman, Methods Enzymol., 1995, 249, 373–397 CAS.
- A. Kohen and J. P. Klinman, Chem. Biol., 1999, 6, R191–R198 CrossRef CAS PubMed.
- Z. X. Liang and J. P. Klinman, Curr. Opin. Struct. Biol., 2004, 14, 648–655 CrossRef CAS PubMed.
- Z. D. Nagel and J. P. Klinman, Chem. Rev., 2006, 106, 3095–3118 CrossRef CAS PubMed.
- N. S. Scrutton, J. Basran and M. J. Sutcliffe, Eur. J. Biochem., 1999, 264, 666–671 CrossRef CAS PubMed.
- C. R. Pudney, S. Hay, C. Levy, J. Y. Pang, M. J. Sutcliffe, D. Leys and N. S. Scrutton, J. Am. Chem. Soc., 2009, 131, 17072–17073 CrossRef CAS PubMed.
- S. Hay, L. O. Johannissen, M. J. Sutcliffe and N. S. Scrutton, Biophys. J., 2010, 98, 121–128 CrossRef CAS PubMed.
- S. Hay and N. S. Scrutton, Nat. Chem., 2012, 4, 161–168 CrossRef CAS PubMed.
- C. R. Pudney, A. Guerriero, N. J. Baxter, L. O. Johannissen, J. P. Waltho, S. Hay and N. S. Scrutton, J. Am. Chem. Soc., 2013, 135, 2512–2517 CrossRef CAS PubMed.
- W. Bialek and J. N. Onuchic, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 5908–5912 CrossRef CAS.
- W. J. Bruno and W. Bialek, Biophys. J., 1992, 63, 689–699 CrossRef CAS PubMed.
- D. Antoniou, S. Caratzoulas, C. Kalyanaraman, J. S. Mincer and S. D. Schwartz, Eur. J. Biochem., 2002, 269, 3103–3112 CrossRef CAS PubMed.
- J. S. Mincer and S. D. Schwartz, J. Proteome Res., 2003, 2, 437–439 CrossRef PubMed.
- D. Antoniou and S. D. Schwartz, J. Phys. Chem. B, 2011, 115, 15147–15158 CrossRef CAS PubMed.
- D. Antoniou, J. Basner, S. Nunez and S. D. Schwartz, Chem. Rev., 2006, 106, 3170–3187 CrossRef CAS PubMed.
- J. T. Sage, S. M. Durbin, W. Sturhahn, D. C. Wharton, P. M. Champion, P. Hession, J. Sutter and E. E. Alp, Phys. Rev. Lett., 2001, 86, 4966–4969 CrossRef CAS PubMed.
- M. Horch, A. F. Pinto, T. Utesch, M. A. Mroginski, C. V. Romao, M. Teixeira, P. Hildebrandt and I. Zebger, Phys. Chem. Chem. Phys., 2014, 16, 14220–14230 RSC.
- M. Jaskolski, Z. Dauter and A. Wlodawer, FEBS J., 2014, 281, 3985–4009 CrossRef CAS PubMed.
- M. Saggu, I. Zebger, M. Ludwig, O. Lenz, B. Friedrich, P. Hildebrandt and F. Lendzian, J. Biol. Chem., 2009, 284, 16264–16276 CrossRef CAS PubMed.
- M. Horch, L. Lauterbach, M. Saggu, P. Hildebrandt, F. Lendzian, R. Bittl, O. Lenz and I. Zebger, Angew. Chem., Int. Ed., 2010, 49, 8026–8029 CrossRef CAS PubMed.
- E. Siebert, M. Horch, Y. Rippers, J. Fritsch, S. Frielingsdorf, O. Lenz, F. Velazquez Escobar, F. Siebert, L. Paasche, U. Kuhlmann, F. Lendzian, M. A. Mroginski, I. Zebger and P. Hildebrandt, Angew. Chem., Int. Ed., 2013, 52, 5162–5165 CrossRef CAS PubMed.
- M. Horch, J. Schoknecht, M. A. Mroginski, O. Lenz, P. Hildebrandt and I. Zebger, J. Am. Chem. Soc., 2014, 136, 9870–9873 CrossRef CAS PubMed.
- M. Horch, Y. Rippers, M. A. Mroginski, P. Hildebrandt and I. Zebger, ChemPhysChem, 2013, 14, 185–191 CrossRef CAS PubMed.
- F. Neese, Coord. Chem. Rev., 2009, 253, 526–563 CrossRef CAS.
- V. Barone, M. Biczysko and G. Brancato, Adv. Quantum Chem., 2010, 59, 17–57 CrossRef CAS.
- H. M. Senn and W. Thiel, Angew. Chem., Int. Ed., 2009, 48, 1198–1229 CrossRef CAS PubMed.
- H. M. Senn and W. Thiel, Curr. Opin. Chem. Biol., 2007, 11, 182–187 CrossRef CAS PubMed.
- H. M. Senn and W. Thiel, Top. Curr. Chem., 2007, 268, 173–290 CrossRef CAS.
- Y. Rippers, M. Horch, P. Hildebrandt, I. Zebger and M. A. Mroginski, ChemPhysChem, 2012, 13, 3852–3856 CrossRef CAS PubMed.
- M. Horch, L. Lauterbach, M. A. Mroginski, P. Hildebrandt, O. Lenz and I. Zebger, J. Am. Chem. Soc., 2015, 137, 2555–2564 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- A. Volbeda, M. H. Charon, C. Piras, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, Nature, 1995, 373, 580–587 CrossRef CAS PubMed.
- A. Volbeda, E. Garcin, C. Piras, A. L. De Lacey, V. M. Fernandez, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 1996, 118, 12989–12996 CrossRef CAS.
- R. P. Happe, W. Roseboom, A. J. Pierik, S. P. J. Albracht and K. A. Bagley, Nature, 1997, 385, 126 CrossRef CAS PubMed.
- A. J. Pierik, W. Roseboom, R. P. Happe, K. A. Bagley and S. P. J. Albracht, J. Biol. Chem., 1999, 274, 3331–3337 CrossRef CAS PubMed.
- T. G. Spiro and R. S. Czernuszewicz, Methods Enzymol., 1995, 246, 416–460 CAS.
- M. K. Johnson, R. S. Czernuszewicz, T. G. Spiro, R. R. Ramsay and T. P. Singer, J. Biol. Chem., 1983, 258, 12771–12774 CAS.
- K. A. Macor, R. S. Czernuszewicz, M. W. Adams and T. G. Spiro, J. Biol. Chem., 1987, 262, 9945–9947 CAS.
- W. Fu, P. M. Drozdzewski, T. V. Morgan, L. E. Mortenson, A. Juszczak, M. W. Adams, S. H. He, H. D. Peck, Jr., D. V. DerVartanian, J. Legall and M. K. Johnson, Biochemistry, 1993, 32, 4813–4819 CrossRef CAS PubMed.
- H. Furuichi, Y. Ozaki, K. Niki and H. Akutsu, J. Biochem., 1990, 108, 707–710 CAS.
- H. S. Shafaat, K. Weber, T. Petrenko, F. Neese and W. Lubitz, Inorg. Chem., 2012, 51, 11787–11797 CrossRef CAS PubMed.
- H. Ogata, Y. Mizoguchi, N. Mizuno, K. Miki, S. Adachi, N. Yasuoka, T. Yagi, O. Yamauchi, S. Hirota and Y. Higuchi, J. Am. Chem. Soc., 2002, 124, 11628–11635 CrossRef CAS PubMed.
- U. Bergmann, W. Sturhahn, D. E. Linn, Jr., F. E. Jenney, Jr., M. W. Adams, K. Rupnik, B. J. Hales, E. E. Alp, A. Mayse and S. P. Cramer, J. Am. Chem. Soc., 2003, 125, 4016–4017 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds - Applications in Coordination, Organometallic and Bioinorganic Chemistry, John Wiley and Sons, Inc, Hoboken, New Jersey, 6th edn, 2009 Search PubMed.
- Y. H. Huang, I. Moura, J. J. G. Moura, J. LeGall, J. B. Park, M. W. W. Adams and M. K. Johnson, Inorg. Chem., 1993, 32, 406–412 CrossRef CAS.
- S. Kamali, H. Wang, D. Mitra, H. Ogata, W. Lubitz, B. C. Manor, T. B. Rauchfuss, D. Byrne, V. Bonnefoy, F. E. Jenney, Jr., M. W. Adams, Y. Yoda, E. Alp, J. Zhao and S. P. Cramer, Angew. Chem., Int. Ed., 2013, 52, 724–728 CrossRef CAS PubMed.
- T. G. Spiro, R. S. Czernuszewicz and S. Han, in Biological Applications of Raman Spectroscopy, ed. T. G. Spiro, John Wiley & Sons, Inc., New York, 1988, ch. 12, vol. 3, pp. 523–553 Search PubMed.
- M. K. Johnson, R. E. Duderstadt and E. C. Duin, in Advances in Inorganic Chemistry, ed. A. G. Sykes, Academic Press, 1999, vol. 47, pp. 1–82 Search PubMed.
- R. S. Czernuszewicz, K. A. Macor, M. K. Johnson, A. Gewirth and T. G. Spiro, J. Am. Chem. Soc., 1987, 109, 7178–7187 CrossRef CAS.
- M. Brecht, G. M. van, T. Buhrke, B. Friedrich and W. Lubitz, J. Am. Chem. Soc., 2003, 125, 13075–13083 CrossRef CAS PubMed.
- M. Bruschi, M. Tiberti, A. Guerra and L. De Gioia, J. Am. Chem. Soc., 2014, 136, 1803–1814 CrossRef CAS PubMed.
- A. J. Pierik, M. Schmelz, O. Lenz, B. Friedrich and S. P. J. Albracht, FEBS Lett., 1998, 438, 231–235 CrossRef CAS PubMed.
- M. Bernhard, T. Buhrke, B. Bleijlevens, A. L. De Lacey, V. M. Fernandez, S. P. J. Albracht and B. Friedrich, J. Biol. Chem., 2001, 276, 15592–15597 CrossRef CAS PubMed.
- M. Horch, L. Lauterbach, O. Lenz, P. Hildebrandt and I. Zebger, FEBS Lett., 2012, 586, 545–556 CrossRef CAS PubMed.
- K. Schneider and H. G. Schlegel, Biochim. Biophys. Acta, 1976, 452, 66–80 CrossRef CAS.
- T. Burgdorf, O. Lenz, T. Buhrke, E. van der Linden, A. K. Jones, S. P. J. Albracht and B. Friedrich, J. Mol. Microbiol. Biotechnol., 2005, 10, 181–196 CrossRef CAS PubMed.
- S. P. J. Albracht, E. van der Linden and B. W. Faber, Biochim. Biophys. Acta, 2003, 1557, 41–49 CrossRef CAS.
- K. Schneider and H. G. Schlegel, Biochem. Biophys. Res. Commun., 1978, 84, 564–571 CrossRef CAS PubMed.
- E. van der Linden, B. W. Faber, B. Bleijlevens, T. Burgdorf, M. Bernhard, B. Friedrich and S. P. J. Albracht, Eur. J. Biochem., 2004, 271, 801–808 CrossRef CAS PubMed.
- S. P. J. Albracht, Biochim. Biophys. Acta, 1993, 1144, 221–224 CrossRef CAS.
- L. Lauterbach, J. Liu, M. Horch, P. Hummel, A. Schwarze, M. Haumann, K. A. Vincent, O. Lenz and I. Zebger, Eur. J. Inorg. Chem., 2011, 1067–1079 CrossRef CAS.
- S. J. Pilkington, J. M. Skehel, R. B. Gennis and J. E. Walker, Biochemistry, 1991, 30, 2166–2175 CrossRef CAS PubMed.
- M. Long, J. Liu, Z. Chen, B. Bleijlevens, W. Roseboom and S. P. J. Albracht, J. Biol. Inorg. Chem., 2007, 12, 62–78 CrossRef CAS PubMed.
- R. P. Happe, W. Roseboom, G. Egert, C. G. Friedrich, C. Massanz, B. Friedrich and S. P. J. Albracht, FEBS Lett., 2000, 466, 259–263 CrossRef CAS PubMed.
- A. Erkens, K. Schneider and A. Müller, J. Biol. Inorg. Chem., 1996, 1, 99–110 CrossRef CAS.
- E. van der Linden, T. Burgdorf, A. L. De Lacey, T. Buhrke, M. Scholte, V. M. Fernandez, B. Friedrich and S. P. J. Albracht, J. Biol. Inorg. Chem., 2006, 11, 247–260 CrossRef CAS PubMed.
- S. D. Patel, R. Aebersold and G. Attardi, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 4225–4229 CrossRef CAS.
- L. Lauterbach, H. Wang, M. Horch, L. B. Gee, Y. Yoda, Y. Tanaka, I. Zebger, O. Lenz and S. P. Cramer, Chem. Sci., 2015, 6, 1055–1060 RSC.
- K. Schneider, R. Cammack, H. G. Schlegel and D. O. Hall, Biochim. Biophys. Acta, 1979, 578, 445–461 CrossRef CAS.
- R. C. Prince and H. S. Kheshgi, Crit. Rev. Microbiol., 2005, 31, 19–31 CrossRef CAS PubMed.
- I. Okura, K. Otsuka, N. Nakada and F. Hasumi, Appl. Biochem. Biotechnol., 1990, 24–25, 425–430 CrossRef CAS PubMed.
- B. Payen, M. Segui, P. Monsan, K. Schneider, C. G. Friedrich and H. G. Schlegel, Biotechnol. Lett., 1983, 5, 463–468 CrossRef CAS.
- J. Cantet, A. Bergel, M. Comtat and J. L. Séris, J. Mol. Catal., 1992, 73, 371–380 CrossRef CAS.
- J. Cantet, A. Bergel and M. Comtat, J. Electroanal. Chem., 1992, 342, 475–486 CrossRef.
- J. Ratzka, L. Lauterbach, O. Lenz and M. B. Ansorge-Schumacher, Biocatal. Biotransform., 2011, 9, 246–252 Search PubMed.
- L. Lauterbach, O. Lenz and K. A. Vincent, FEBS J., 2013, 280, 3058–3068 CrossRef CAS PubMed.
- H. A. Reeve, L. Lauterbach, P. A. Ash, O. Lenz and K. A. Vincent, Chem. Commun., 2012, 48, 1589–1591 RSC.
- F. Hasumi, K. Fukuoka, S. Adachi, Y. Miyamoto and I. Okura, Appl. Biochem. Biotechnol., 1996, 56, 341–344 CrossRef CAS PubMed.
- L. Lauterbach and O. Lenz, J. Am. Chem. Soc., 2013, 135, 17897–17905 CrossRef CAS PubMed.
- K. A. Bagley, E. C. Duin, W. Roseboom, S. P. J. Albracht and W. H. Woodruff, Biochemistry, 1995, 34, 5527–5535 CrossRef CAS PubMed.
- M. Y. Darensbourg, E. J. Lyon and J. J. Smee, Coord. Chem. Rev., 2000, 206–207, 533–561 CrossRef CAS.
- B. Bleijlevens, T. Buhrke, E. van der Linden, B. Friedrich and S. P. J. Albracht, J. Biol. Chem., 2004, 279, 46686–46691 CrossRef CAS PubMed.
- E. van der Linden, T. Burgdorf, M. Bernhard, B. Bleijlevens, B. Friedrich and S. P. J. Albracht, J. Biol. Inorg. Chem., 2004, 9, 616–626 CrossRef CAS PubMed.
- W. Lubitz, E. Reijerse and M. van Gastel, Chem. Rev., 2007, 107, 4331–4365 CrossRef CAS PubMed.
- T. Burgdorf, S. Löscher, P. Liebisch, E. van der Linden, M. Galander, F. Lendzian, W. Meyer-Klaucke, S. P. J. Albracht, B. Friedrich, H. Dau and M. Haumann, J. Am. Chem. Soc., 2005, 127, 576–592 CrossRef CAS PubMed.
- V. Gupta and K. S. Carroll, Biochim. Biophys. Acta, 2014, 1840, 847–875 CrossRef CAS PubMed.
- P. M. Vignais, B. Billoud and J. Meyer, FEMS Microbiol. Rev., 2001, 25, 455–501 CrossRef CAS PubMed.
- P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS PubMed.
- T. Burgdorf, E. van der Linden, M. Bernhard, Q. Y. Yin, J. W. Back, A. F. Hartog, A. O. Muijsers, C. G. de Koster, S. P. J. Albracht and B. Friedrich, J. Bacteriol., 2005, 187, 3122–3132 CrossRef CAS PubMed.
- Y. Sheng, I. A. Abreu, D. E. Cabelli, M. J. Maroney, A. F. Miller, M. Teixeira and J. S. Valentine, Chem. Rev., 2014, 114, 3854–3918 CrossRef CAS PubMed.
- M. Valko, D. Leibfritz, J. Moncol, M. T. D. Cronin, M. Mazur and J. Telser, Int. J. Biochem. Cell Biol., 2007, 39, 44–84 CrossRef CAS PubMed.
- A. F. Pinto, J. V. Rodrigues and M. Teixeira, Biochim. Biophys. Acta, 2010, 1804, 285–297 CrossRef CAS PubMed.
- S. E. Dobbins, V. I. Lesk and M. J. E. Sternberg, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10390–10395 CrossRef CAS PubMed.
- E. C. Dykeman and O. F. Sankey, J. Phys.: Condens. Matter, 2010, 22, 423202 CrossRef PubMed.
- A. Amadei, A. B. Linssen and H. J. Berendsen, Proteins, 1993, 17, 412–425 CrossRef CAS PubMed.
- A. Bakan, L. M. Meireles and I. Bahar, Bioinformatics, 2011, 27, 1575–1577 CrossRef CAS PubMed.
- M. M. Tirion, Phys. Rev. Lett., 1996, 77, 1905–1908 CrossRef CAS PubMed.
- I. Bahar and A. J. Rader, Curr. Opin. Struct. Biol., 2005, 15, 586–592 CrossRef CAS PubMed.
- K. Hinsen, Proteins, 1998, 33, 417–429 CrossRef CAS.
- C. Chennubhotla, A. J. Rader, L. W. Yang and I. Bahar, Phys. Biol., 2005, 2, S173–S180 CrossRef CAS PubMed.
- S. Hayward and B. L. de Groot, Methods Mol. Biol., 2008, 443, 89–106 CAS.
- E. Lindahl, C. Azuara, P. Koehl and M. Delarue, Nucleic Acids Res., 2006, 34, W52–W56 CrossRef CAS PubMed.
- D. M. Chudakov, M. V. Matz, S. Lukyanov and K. A. Lukyanov, Physiol. Rev., 2010, 90, 1103–1163 CrossRef CAS PubMed.
- R. De Michele, F. Carimi and W. B. Frommer, Int. J. Biochem. Cell Biol., 2014, 48, 39–44 CrossRef CAS PubMed.
- K. Deuschle, M. Fehr, M. Hilpert, I. Lager, S. Lalonde, L. L. Looger, S. Okumoto, J. Persson, A. Schmidt and W. B. Frommer, Cytometry, Part A, 2005, 64, 3–9 CrossRef PubMed.
- M. Fehr, S. Okumoto, K. Deuschle, I. Lager, L. L. Looger, J. Persson, L. Kozhukh, S. Lalonde and W. B. Frommer, Biochem. Soc. Trans., 2005, 33, 287–290 CrossRef CAS PubMed.
- S. Okumoto, K. Deuschle, M. Fehr, M. Hilpert, I. Lager, S. Lalonde, L. L. Looger, J. Persson, A. Schmidt and W. B. Frommer, Soil Sci. Plant Nutr., 2004, 50, 947–953 CrossRef CAS.
- M. Fehr, D. W. Ehrhardt, S. Lalonde and W. B. Frommer, Curr. Opin. Plant Biol., 2004, 7, 345–351 CrossRef CAS PubMed.
- W. B. Frommer, M. W. Davidson and R. E. Campbell, Chem. Soc. Rev., 2009, 38, 2833–2841 RSC.
- A. M. Jones, G. Grossmann, J. A. H. Danielson, D. Sosso, L. Q. Chen, C. H. Ho and W. B. Frommer, Curr. Opin. Plant Biol., 2013, 16, 389–395 CrossRef CAS PubMed.
- S. Lalonde, D. W. Ehrhardt and W. B. Frommer, Curr. Opin. Plant Biol., 2005, 8, 574–581 CrossRef CAS PubMed.
- S. Okumoto, A. Jones and W. B. Frommer, Annu. Rev. Plant Biol., 2012, 63, 663–706 CrossRef CAS PubMed.
- S. K. Gjetting, A. Schulz and A. T. Fuglsang, Front. Plant Sci., 2013, 4, 234 Search PubMed.
- B. H. Hou, H. Takanaga, G. Grossmann, L. Q. Chen, X. Q. Qu, A. M. Jones, S. Lalonde, O. Schweissgut, W. Wiechert and W. B. Frommer, Nat. Protoc., 2011, 6, 1818–1833 CrossRef CAS PubMed.
- A. M. Jones, D. W. Ehrhardt and W. B. Frommer, BMC Biol., 2012, 10, 39 CrossRef CAS PubMed.
- S. Mehta and J. Zhang, Annu. Rev. Biochem., 2011, 80, 375–401 CrossRef CAS PubMed.
- A. Miyawaki, Nat. Rev. Mol. Cell Biol., 2011, 12, 656–668 CrossRef CAS PubMed.
- R. H. Newman, M. D. Fosbrink and J. Zhang, Chem. Rev., 2011, 111, 3614–3666 CrossRef CAS PubMed.
- A. E. Palmer, Y. Qin, J. G. Park and J. E. McCombs, Trends Biotechnol., 2011, 29, 144–152 CrossRef CAS PubMed.
- S. B. VanEngelenburg and A. E. Palmer, Curr. Opin. Chem. Biol., 2008, 12, 60–65 CrossRef CAS PubMed.
- W. Ren and H. W. Ai, Sensors, 2013, 13, 15422–15433 CrossRef PubMed.
- S. O. Lill and P. E. Siegbahn, Biochemistry, 2009, 48, 1056–1066 CrossRef PubMed.
- P. E. Siegbahn, J. W. Tye and M. B. Hall, Chem. Rev., 2007, 107, 4414–4435 CrossRef CAS PubMed.
- M. Bruschi, G. Zampella, P. Fantucci and L. De Gioia, Coord. Chem. Rev., 2005, 249, 1620–1640 CrossRef CAS.
- S. Hayward, A. Kitao and N. Go, Proteins, 1995, 23, 177–186 CrossRef CAS PubMed.
- C. Berthomieu, L. Marboutin, F. Dupeyrat and P. Bouyer, Biopolymers, 2006, 82, 363–367 CrossRef CAS PubMed.
- D. F. Plusquellic, K. Siegrist, E. J. Heilweil and O. Esenturk, ChemPhysChem, 2007, 8, 2412–2431 CrossRef CAS PubMed.
- W. R. Scheidt, S. M. Durbin and J. T. Sage, J. Inorg. Biochem., 2005, 99, 60–71 CrossRef CAS PubMed.
- W. Sturhahn, J. Phys.: Condens. Matter, 2004, 16, S497–S530 CrossRef CAS.
- L. Dhar, J. A. Rogers and K. A. Nelson, Chem. Rev., 1994, 94, 157–193 CrossRef CAS.
- Y. Xiao, M. L. Tan, T. Ichiye, H. Wang, Y. Guo, M. C. Smith, J. Meyer, W. Sturhahn, E. E. Alp, J. Zhao, Y. Yoda and S. P. Cramer, Biochemistry, 2008, 47, 6612–6627 CrossRef CAS PubMed.
- K. L. Adams, S. Tsoi, J. S. Yan, S. M. Durbin, A. K. Ramdas, W. A. Cramer, W. Sturhahn, E. E. Alp and C. Schulz, J. Phys. Chem. B, 2006, 110, 530–536 CrossRef CAS PubMed.
- S. P. Cramer, Y. M. Xiao, H. X. Wang, Y. S. Guo and M. C. Smith, Hyperfine Interact., 2006, 170, 47–54 CrossRef.
- D. Mitra, V. Pelmenschikov, Y. S. Guo, D. A. Case, H. X. Wang, W. B. Dong, M. L. Tan, T. Ichiye, F. E. Jenney, M. W. W. Adams, Y. Yoda, J. Y. Zhao and S. P. Cramer, Biochemistry, 2011, 50, 5220–5235 CrossRef CAS PubMed.
- Y. Xiao, H. Wang, S. J. George, M. C. Smith, M. W. Adams, F. E. Jenney, Jr., W. Sturhahn, E. E. Alp, J. Zhao, Y. Yoda, A. Dey, E. I. Solomon and S. P. Cramer, J. Am. Chem. Soc., 2005, 127, 14596–14606 CrossRef CAS PubMed.
- Y. M. Xiao, K. Fisher, M. C. Smith, W. E. Newton, D. A. Case, S. J. George, H. X. Wang, W. Sturhahn, E. E. Alp, J. Y. Zhao, Y. Yoda and S. P. Cramer, J. Am. Chem. Soc., 2006, 128, 7608–7612 CrossRef CAS PubMed.
- J. M. Kuchenreuther, Y. Guo, H. Wang, W. K. Myers, S. J. George, C. A. Boyke, Y. Yoda, E. E. Alp, J. Zhao, R. D. Britt, J. R. Swartz and S. P. Cramer, Biochemistry, 2013, 52, 818–826 CrossRef CAS PubMed.
- M. L. Tan, A. R. Bizzarri, Y. Xiao, S. Cannistraro, T. Ichiye, C. Manzoni, G. Cerullo, M. W. Adams, F. E. Jenney, Jr. and S. P. Cramer, J. Inorg. Biochem., 2007, 101, 375–384 CrossRef CAS PubMed.
- I. Delfino, G. Cerullo, S. Cannistraro, C. Manzoni, D. Polli, C. Dapper, W. E. Newton, Y. S. Guo and S. P. Cramer, Angew. Chem., Int. Ed., 2010, 49, 3912–3915 CrossRef CAS PubMed.
- Y. H. Sun, A. Benabbas, W. Q. Zeng, J. G. Kleingardner, K. L. Bren and P. M. Champion, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6570–6575 CrossRef CAS PubMed.
- F. Rosca, A. T. N. Kumar, D. Ionascu, X. Ye, A. A. Demidov, T. Sjodin, D. Wharton, D. Barrick, S. G. Sligar, T. Yonetani and P. M. Champion, J. Phys. Chem. A, 2002, 106, 3540–3552 CrossRef CAS.
- F. Gruia, M. Kubo, X. Ye and P. M. Champion, Biophys. J., 2008, 94, 2252–2268 CrossRef CAS PubMed.
- F. Gruia, M. Kubo, X. Ye, D. Ionascu, C. Lu, R. K. Poole, S. R. Yeh and P. M. Champion, J. Am. Chem. Soc., 2008, 130, 5231–5244 CrossRef CAS PubMed.
- M. Kubo, F. Gruia, A. Benabbas, A. Barabanschikov, W. R. Montfort, E. M. Maes and P. M. Champion, J. Am. Chem. Soc., 2008, 130, 9800–9811 CrossRef CAS PubMed.
- U. Liebl, G. Lipowski, M. Negrerie, J. C. Lambry, J. L. Martin and M. H. Vos, Nature, 1999, 401, 181–184 CrossRef CAS PubMed.
- P. Kukura, S. Yoon and R. A. Mathies, Anal. Chem., 2006, 78, 5952–5959 CrossRef CAS.
- W. Zheng, Biophys. J., 2010, 98, 3025–3034 CrossRef CAS PubMed.
Footnote |
| † NRVS has also been called nuclear resonant inelastic X-ray scattering (NRIXS). Alternative termini for VCS variants include femtosecond coherence spectroscopy (FCS), impulsive coherent vibrational spectroscopy (ICVS), and impulsive stimulated Raman scattering (ISRS). |
| This journal is © the Owner Societies 2015 |