
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The electronic structure of quasi-free-standing germanene on monolayer MX (M = Ga, In; X = S, Se, Te)†
Zeyuan
Ni
,
Emi
Minamitani
,
Yasunobu
Ando
and
Satoshi
Watanabe
*
Department of Materials Engineering, The University of Tokyo, Tokyo 113-8656, Japan. E-mail: zeyuan_ni@cello.t.u-tokyo.ac.jp; watanabe@cello.t.u-tokyo.ac.jp
First published on 23rd June 2015
Abstract
For the first time by using the ab initio density functional theory, the stability and electronic structures of germanene on monolayer GaS, GaSe, GaTe and InSe have been investigated. Germanene preserves its buckled-honeycomb structure on all the studied substrates similar to the free-standing case. Moreover, germanene stays neutral and preserves its Dirac-cone-like band structure on monolayer GaTe and InSe. In these two cases, a bandgap of 0.14–0.16 eV opens at the Dirac point of germanene, while the effective masses remain as small as 0.05–0.06 times the free-electron mass. The estimated carrier mobility is up to 2.2 × 105 cm2 V−1 s−1. These features show that monolayer GaTe and InSe are promising as substrates for germanene devices.
Introduction
Graphene, the first 2-dimentional (2D) material, is known for its ultrahigh carrier mobility up to 2 × 105 cm2 V−1 s−1 in suspended samples1 and 4 × 104 cm2 V−1 s−1 on a SiO2 substrate,2 which makes it attractive for application in electronic devices. Unfortunately, it remains challenging to open a band gap in graphene and preserve its extreme carrier mobility at the same time. Such a dilemma stimulates the search for other 2D materials, for example silicene and germanene, the Si and Ge cousins of graphene.3,4 Silicene and germanene have been successfully synthesized recently on several substrates (e.g. Ag(111),5–9 Ir(111)10 and ZrB211,12 for silicene; Pt(111),13 Au(111)14 and Al(111)15 for germanene) in experiments, and the first silicene field-effect transistor (FET) has been fabricated.16 Free-standing silicene and germanene are predicted to be new members of Dirac materials and share most of the amazing properties of graphene, e.g. the ultrahigh carrier mobility and the quantum spin Hall effect (QSHE).17 Additionally, due to their unique buckled structure, monolayer (ML) silicene and germanene show many behaviors different from those of the ML graphene, such as the tunable bandgap by electric-field18,19 or surface atom adsorption,20,21 and much stronger spin–orbit interaction,22,23 displaying great potential in electronic and spintronic applications. Among the three mentioned materials, germanene is predicted to have the highest intrinsic carrier mobility in theory, which is about twice as high as that of graphene.24 Germanene is also predicted to have the strongest spin–orbit (SO) interaction among the three with a SO gap over 23 meV,22,23 which makes the high-temperature QSHE possible,22 while the SO gap of silicene and graphene is only 1.55 meV22 and 8 μeV,25 respectively.However, germanene shares a problem with silicene: one cannot utilize germanene's ultimate properties unless suitable substrates are found for germanene. Currently, ML germanene can only be synthesized on metal surfaces, such as Au(111)14 and Pt(111),13 but electronic devices such as FETs require substrates with a large band gap. Moreover, many semiconducting substrates such as bare SiO2 and GaAs will strongly interact with germanene and destroy its Dirac cone.18,26 Although germanene is reported to have weak interaction with the fully hydrogenated GaAs (0001) surface,26 it is hard in practice to prepare an atomically flat and perfectly hydrogenated GaAs surface – the existence of defects may still ruin germanene. In addition, free-standing germanene changes from Dirac material to normal metal when the tensile or compressive strain reaches 5%,27,28 indicating the sensitivity of germanene to structural deformation. From the above, we can say that germanene still lacks an appropriate substrate to serve as a promising electronic material.
On the other hand, ML group III monochalcogenides MX (M = Ga, In; X = S, Se, Te), a new family of 2D materials, are predicted to have a large bandgap of 2–3 eV.29 Recently, ML and few-layer GaS,30,31 GaSe30,32 and InSe33 with an atomically flat surface have been successfully fabricated in experiment. Some of the MX like GaS and GaSe are predicted to have weak interaction with silicene in theory.34,35 Although MX seem promising as the suitable substrates for germanene, the interaction between them and germanene is yet to be investigated.
In this article, we study the structural and electronic behaviors of ML germanene on ML GaS, GaSe, GaTe and InSe for the first time by using the density functional theory (DFT). The Dirac-cone-like band structure of germanene on these substrates is preserved. Germanene is found to be semiconducting on GaTe and InSe with a band gap above 0.1 eV, while its effective masses at the Dirac point remain as small as 0.05–0.06 m0 (free electron mass), leading to an ultrahigh carrier mobility estimated to be around 1.5–2.2 × 105 cm2 V−1 s−1.
We use the DFT method implemented in Quantum ESPRESSO36 to perform geometry optimization and electronic structure calculation and VASP37 to perform hybrid functional calculation. In Quantum ESPRESSO calculation, ultrasoft pseudopotentials with nonlinear core correction are employed. The optB86b-vdw38–41 exchange–correlation functional is used in geometry optimization to take the van der Waals interaction into account. The generalized gradient approximation (GGA) exchange–correlation functional of the Perdew, Burke, and Ernzerhof (PBE) parametrization42 is used in electronic structure calculation with spin–orbital coupling (SOC). After the convergence test, the wave function cutoffs are chosen to be 952, 952, 1360, 816 eV for GaS, GaSe, GaTe and InSe, respectively, and the charge density cutoffs are chosen to be 10 times of them. A Monkhorst–Pack43 (MP) k-point grid of 21 × 21 × 1 is chosen. The dipole correction44 is engaged and found to have a negligible influence in our system. In VASP calculation, the projected augmented wave (PAW)45 pseudopotential is employed. The Heyd–Scuseria–Ernzerhof (HSE06) functional46–48 is used in hybrid functional calculation without SOC due to computational overburden. The corresponding MP k-point and q-point grids are both 27 × 27 × 1.
The initial structures of free standing ML MX (GaS, GaSe, GaTe and InSe) and germanene are adopted from previous literature17,29 (Fig. 1(a) and (b)). They have similar honeycomb structures and lattice constants of 3.58–4.06 Å, not far from that of germanene (4.02 Å), so only the 1 × 1 stacking (Fig. 1e) is considered in this work. There are three high symmetric points in one hexagonal cell and two different atoms in germanene after stacking, so there are 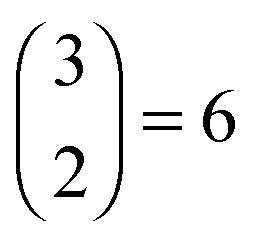 high symmetric stacking configurations in total (Fig. 1(c)). The six configurations are characterized by their stacking mode (AA or AB) and the type of the Ge atom (top “t” or bottom “b”) which overlaps the substrate atom (metal M or chalcogen X) in the top view: AA-t (AA-X-t or AA-M-b), AA-b (AA-X-b or AA-M-t), AB-M-t, AB-M-b, AB-X-t, and AB-X-b.
high symmetric stacking configurations in total (Fig. 1(c)). The six configurations are characterized by their stacking mode (AA or AB) and the type of the Ge atom (top “t” or bottom “b”) which overlaps the substrate atom (metal M or chalcogen X) in the top view: AA-t (AA-X-t or AA-M-b), AA-b (AA-X-b or AA-M-t), AB-M-t, AB-M-b, AB-X-t, and AB-X-b.
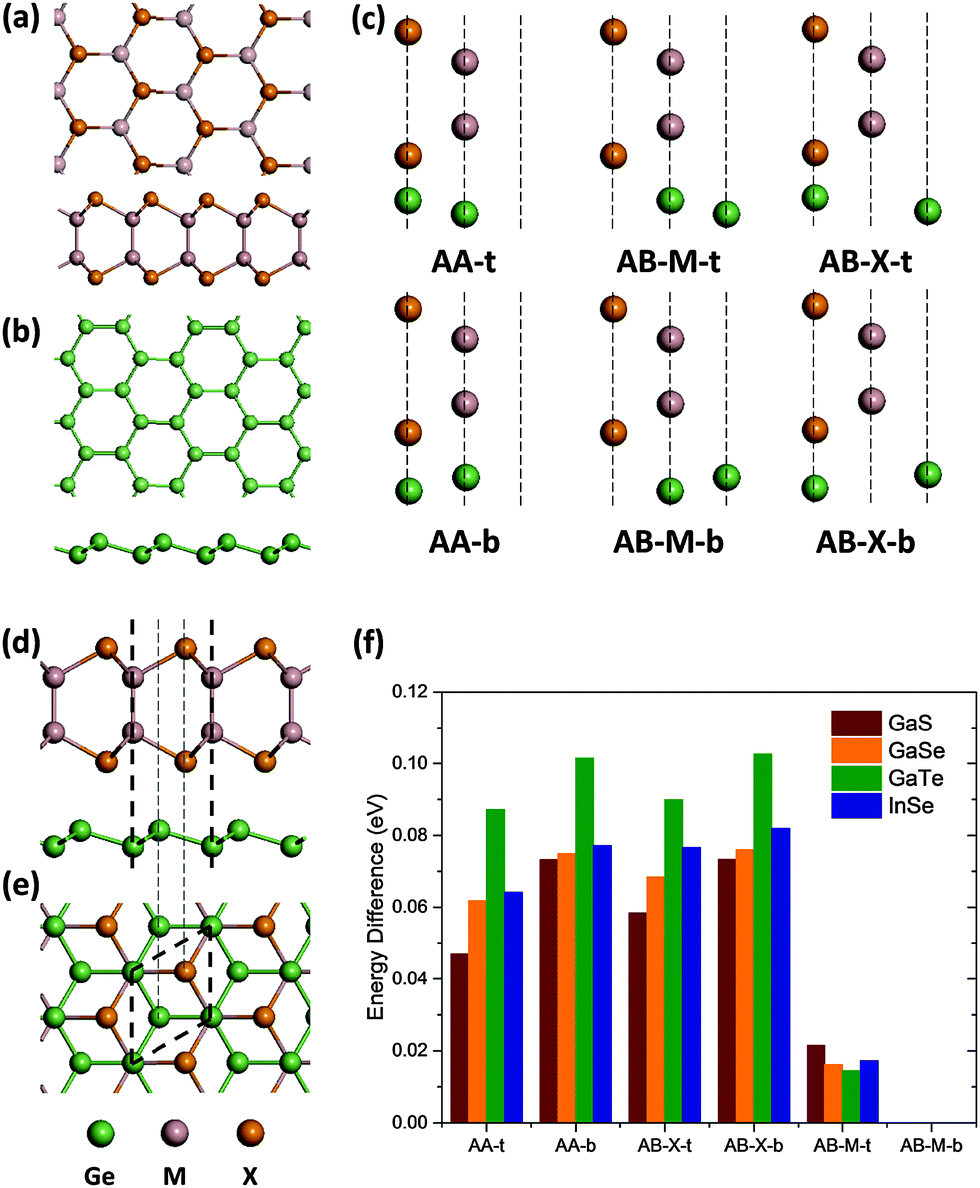 | ||
| Fig. 1 Top and side views of free standing ML (a) MX and (b) germanene. (c) All the six high symmetric stacking configurations of germanene on MX. (d) Side and (e) top views of 1 × 1 stacked ML germanene and MX with the most preferable stacking. Thick dashed lines denote the lattice, and thin dashed lines denote the high symmetric positions in hexagonal cells. M = Ga, In; X = S, Se, Te. (f) Energy difference between AB-M-b configuration and the other configurations. | ||
We begin from the investigation of the most energetically favorable configurations of germanene on ML MX. First, the lattice constant is determined. We choose one stacking configuration (AA-t) to obtain the most preferable lattice constant and apply it to the other configurations. The optimized lattice constant is shown in Table 1. GaS and GaSe have smaller lattice constant with germanene than in their free standing or bulk case, while GaTe and InSe are almost unchanged. This is probably due to the fact that the free standing germanene is predicted to have a lattice constant of 3.97–4.03 Å,17,24 larger than those of GaS and GaSe and close to those of GaTe and InSe. The lattice constant of germanene on GaTe with AB-M-b configuration is also examined and the result is similar to the above. Second, we fix the lattice constant and optimize the geometry using all the six configurations as the initial ones. The configuration with the lowest energy is found to be the same for all the four MX, which is shown in Fig. 1(d) and (e) and is named as the AB-M-b configuration (AB stacking, M atom above b-Ge). The total energy differences between the most favorable configuration and the others, which are presented in Fig. 1(f), are about 0.1 eV except for the AB-M-t configuration. Although the energy difference between AB-M-b and AB-M-t configurations is only ∼0.02 eV, they cannot be easily transformed into each other. This is because they are not related by any in-plane translation but a space inversion of the germanene part, and the transformation energy barrier between them is about 0.3 eV (Fig. S1, ESI†). Other non-high-symmetric configurations are also considered for germanene on GaTe and are found to be less favorable (Fig. S3, ESI†). Hence, we focus on the AB-M-b stacking of germanene on all kinds of ML MX in the following. The vertical distance between MX and germanene is 2.90–3.05 Å, and the nearest distance between the Ge atom and the X atom is about 3.63–3.85 Å. Such a far distance suggests the weak interaction between germanene and MX substrates. The buckling of germanene is enhanced from the free-standing value of 0.64–0.69 Å17,18,24 to 0.77–0.89 Å on GaS and GaSe, which can be understood easily from the smaller lattice constant than in the free-standing case. However, the buckling of germanene is still slightly enhanced to 0.71 Å on GaTe and InSe, where the lattice constants are larger than that of the free-standing germanene. This implies that the interaction is non-negligible.
| MX only | Germanene–MX | ||||
|---|---|---|---|---|---|
| Lattice (Å) | Lattice (Å) | z-Distance (Å) | Germanene buckling (Å) | ||
| Bulk | ML DFT | ML | |||
| Experiment | (HSE) | This work | |||
| GaS | 3.59 | 3.58 | 3.78 | 2.90 | 0.89 |
| GaSe | 3.74–3.76 | 3.75 | 3.89 | 2.99 | 0.77 |
| GaTe | 4.06 | 4.06 | 4.06 | 3.05 | 0.71 |
| InSe | 4.00–4.05 | 4.02 | 4.03 | 2.92 | 0.71 |
Next we investigate whether germanene also has a well-preserved electronic structure. The charge transfer from germanene to MX estimated using the Bader charge analysis49–51 with the PBE functional is as small as 0.02 (GaTe and GaSe), 0.03 (InSe) and 0.04 (GaS) electrons. The charge transfer estimated with the HSE functional give similar results (Table 2). Since the total dipole is mainly induced by the charge transfer, the strength of the dipole correction in the Hartree potential is in the same sequence as above, specifically 0.03 (GaTe) < 0.08 (GaSe) < 0.14 (InSe) < 0.23 (GaS) eV Å−1 (Table 2 and Fig. S4, ESI†). The differential charge densities ρdiff = ρgermanene–MX − ρgermanene − ρMX (shown in Fig. S5, ESI†) reveal that the charge density in the interspace between germanene and MX increases, while the density near both Ge and X atoms decreases.
| GaS | GaSe | GaTe | InSe | ||
|---|---|---|---|---|---|
| Charge transfer (e) | PBE | 0.04 | 0.02 | 0.02 | 0.03 |
| HSE | 0.03 | 0.03 | 0.02 | 0.04 | |
| Dipole correction (eV Å−1) | PBE | 0.23 | 0.08 | 0.03 | 0.14 |
| Dirac-gap (eV) | PBE | 0.14 | 0.13 | 0.12 | 0.11 |
| PBE + SOC | 0.11 | 0.10 | 0.10 | 0.08 | |
| HSE | 0.18 | 0.16 | 0.16 | 0.14 | |
| SOC-split (eV) | Conduction band | 0.030 | 0.024 | 0.015 | 0.021 |
| Valence band | 0.034 | 0.028 | 0.024 | 0.042 | |
| m*ΓK (m0) | m*e_h | 0.089 | 0.078 | 0.081 | 0.080 |
| m*e_l | 0.061 | 0.056 | 0.062 | 0.050 | |
| m*h_l | 0.060 | 0.055 | 0.059 | 0.049 | |
| m*h_h | 0.088 | 0.076 | 0.076 | 0.076 | |
| m KM * (m0) | m*e_h | 0.087 | 0.078 | 0.078 | 0.078 |
| m*e_l | 0.057 | 0.051 | 0.058 | 0.044 | |
| m*h_l | 0.056 | 0.051 | 0.054 | 0.043 | |
| m*h_h | 0.088 | 0.075 | 0.073 | 0.074 | |
The band structures of germanene on ML MX are presented in Fig. 2. All of the four compound systems have Dirac-cone-like band structures at the K-point with a “gap” opened (named as the Dirac-gap to prevent confusion with the band gap). Such a Dirac-gap can be attributed to germanene according to the projected band structure shown in the left panel of Fig. 2, which shows the Ge contribution is over 89% at the Dirac point of germanene and rules out the possibility that these states mainly come from the MX substrates. The Dirac-gap appears in germanene on all kinds of MX, but only germanene on GaTe and InSe is actually semiconducting. Germanene on GaS and GaSe is metallic due to the bands crossing the Fermi level near the Γ-point. This metallic behavior is caused by the cooperation of the deformation of germanene and the interaction with the MX substrate. Germanene on GaS and GaSe in a 1 × 1 cell has a compressive strain of 6% and 3%, respectively. As mentioned previously, germanene changes from Dirac material to metal at a compressive strain beyond 5% due to the band lifting at the Γ-point (“self-induced doping”), though no Dirac-gap opens.27 The band structures of the germanene-part of the germanene–GaS and –GaSe systems have similar band lifting at the Γ-point (Fig. S6, ESI†). It is worth mentioning that germanene on GaTe and InSe also has a little band lifting at the Γ-point in spite of the fact that the strain is no larger than 1% (Fig. S6, ESI†), which can only be explained by the interaction between germanene and MX.
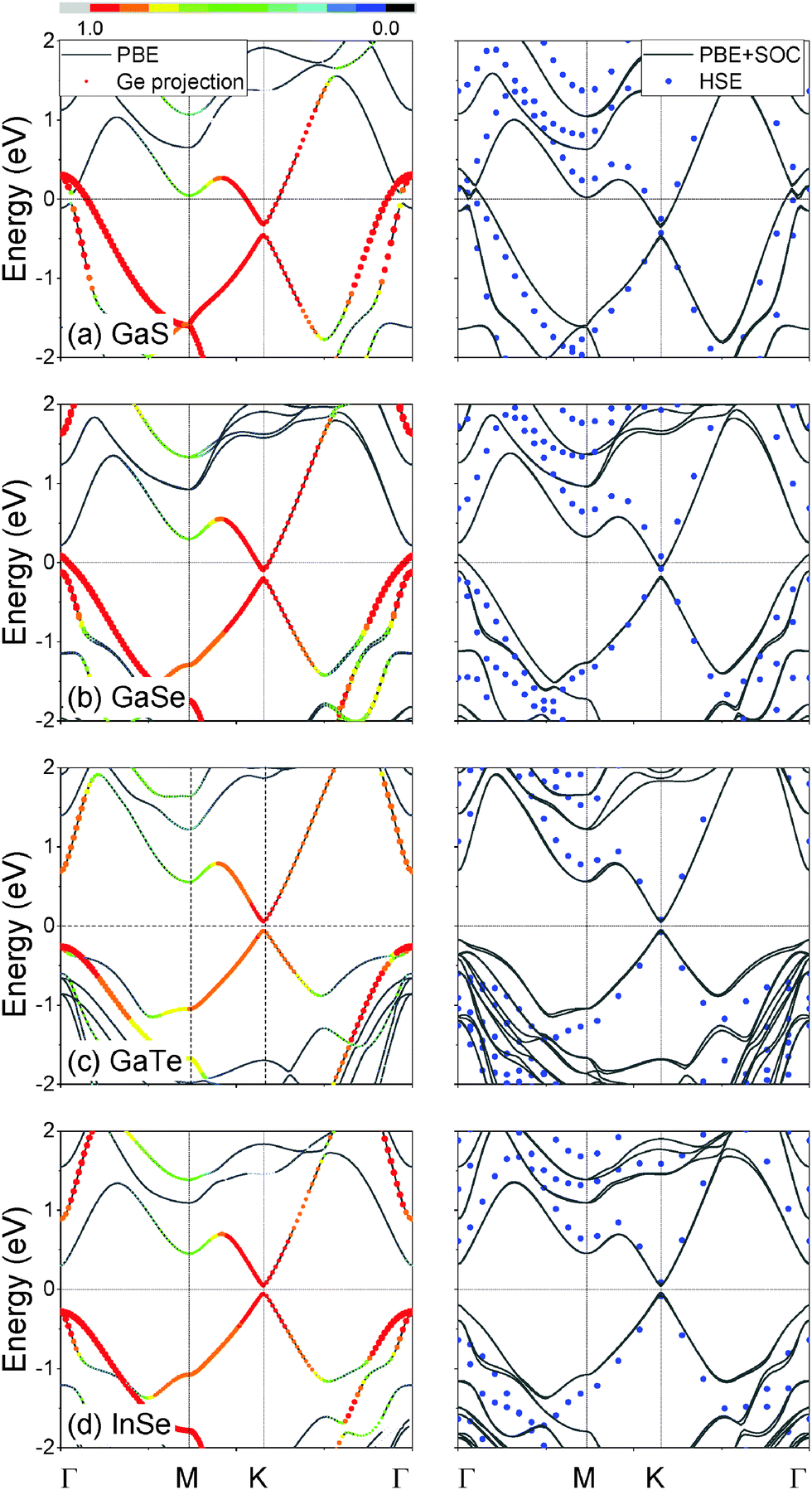 | ||
| Fig. 2 Band structures of germanene on (a) GaS, (b) GaSe, (c) GaTe and (d) InSe. The left panel is the band structures obtained using the PBE functional (black lines) and the Ge projections (size indicates the absolute projection value, and color indicates the percentage of Ge contribution in total with red standing for 90–100%), and the right panel is the band structures obtained using the PBE + SOC (black lines) and HSE without SOC (blue dots). | ||
The size of the Dirac-gaps in germanene on different MX are summarized in Table 2. In general, the Dirac-gap of germanene on MX decreases in the order of GaS > GaSe > GaTe > InSe from 0.14 to 0.11 eV when estimated using the PBE functional without SOC. The opening of the Dirac-gap can be attributed to the breaking of the inversion symmetry by introducing the MX substrate, similar to the band gap opening in germanene by the vertical electric field.18 If the SOC is considered in calculations with the PBE functional, the conduction and valence bands split into two bands by 0.02–0.04 eV (Table 2, Fig. 2 and 3), respectively, involving the Dirac-gap decrease by 0.02–0.03 eV. Moreover, it is well known that DFT can underestimate the actual band gap of semiconductors by up to 50%. The use of the hybrid functional, which includes a certain amount of the Hartree–Fock exchange, can yield much improved band gap values compared with the GGA functionals.46 As shown in Table 2, the Dirac-gap given by the HSE hybrid functional is about 0.16–0.18 eV, ∼25% larger than the PBE cases. The actual band gap of germanene on MX should be a little smaller than the HSE band gap due to the SOC splitting. Regretfully, the HSE calculation including SOC is not feasible within our available computational resources. Assuming that the SOC splitting is the same as the PBE calculation results, we estimate the actual Dirac gap to be around 0.1 eV. In the cases of germanene on GaTe and InSe, the 0.1 eV Dirac-gap also corresponds to the band gap. If combined with other band-gap-opening techniques, such as the application of vertical electric field18 and the surface atom adsorption,21 the band gap in these cases could possibly reach 0.4 eV, which is the minimum requirement as the channel material of field effect transistors.52 Note that contrary to the result given by the PBE functional, germanene on GaSe is predicted to be also semiconducting by the HSE calculation with a band gap of 0.16 eV. The reason is that the valence band at the Γ point is lowered to about 0.2 eV below the Fermi level in the HSE calculation, while the same band in the PBE calculation is above the Fermi level. Such a phenomenon of the lowered valence band at the Γ point in HSE calculations can also be found in other MX substrates (right panel of Fig. 2). Note that we have confirmed the reliability of our calculations by examining the HSE band structure of the free-standing germanene (Fig. S7, ESI†): it agrees well with the previous literature.53 We have also tried the GW calculation, but found it computationally too heavy within our available computational resources.
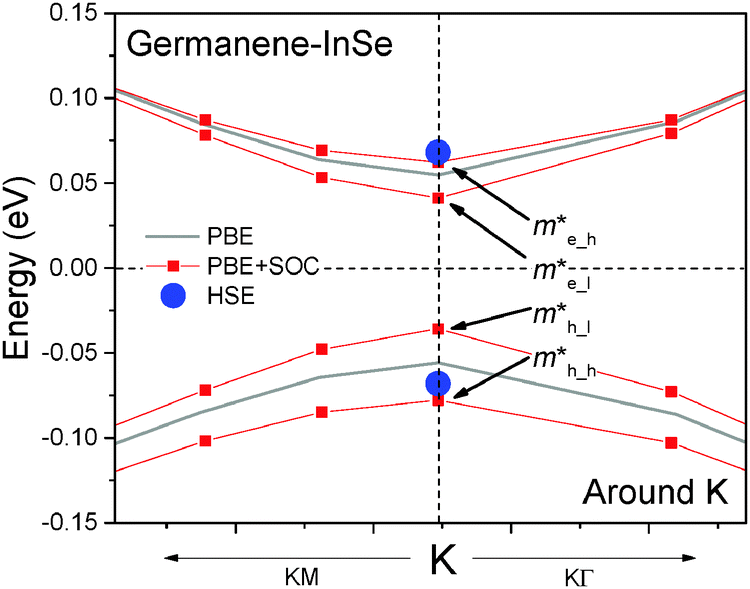 | ||
| Fig. 3 Details of the band structure of germanene on InSe near the Fermi level (dashed horizontal line) and K point. | ||
Although a band gap opens, germanene still has ultrahigh carrier mobility on GaTe and InSe as shown below. The SOC splitting introduces extra effective masses compared to non-SOC cases, namely the heavy hole/electron masses m*h_h/m*e_h and light hole/electron masses m*h_l/m*e_l along the KM and KΓ directions (Fig. 3). The effective masses calculated by quadratic polynomial fitting of the band structure are listed in Table 2. The light m* along KM remains as small as ∼0.05 m0, where m0 is the free electron mass, on GaTe and ∼0.04 m0 on InSe, and the heavy m* are just about 0.01 m0 higher. The m* along KΓ is only 0.004–0.006 m0 larger than the corresponding m* along KM, so they can be treated as almost the same. The relaxation time τ of free-standing germanene at room temperature, estimated using the deformation potential method including the electron-acoustic phonon scattering mechanism,24 is 5.3 ps. Considering the fact that graphene and graphite have similar phonon dispersion54,55 and relaxation time24,56 and that germanene and MX are connected through van der Waals interaction as in the case of graphite, it would be acceptable in the first rough estimation of the mobility to assume that germanene has similar τ on MX to that of the free-standing case. Given τ is the same as the free-standing case, the mobility in germanene calculated by μ = eτ/m* can be up to 1.5 and 2.2 × 105 cm2 V−1 s−1 for light carriers, and still above 1.2 × 105 cm2 V−1 s−1 for heavy carriers. The light carrier mobility is close to the best value of graphene obtained in suspended samples in experiment, around 2 × 105 cm2 V−1 s−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1, and the intrinsic carrier mobility of free-standing graphene (3.2–3.5 × 105 cm2 V−1 s−1) and silicene (2.2–2.6 × 105 cm2 V−1 s−1) in theory.24 If graphene is put on a substrate like SiO2, its carrier mobility will drop to 104 cm2 V−1 s−1 or lower.2 In addition, the high mobility in graphene and silicene will degrade significantly if a band gap of over 0.1 eV is opened.57,58
1, and the intrinsic carrier mobility of free-standing graphene (3.2–3.5 × 105 cm2 V−1 s−1) and silicene (2.2–2.6 × 105 cm2 V−1 s−1) in theory.24 If graphene is put on a substrate like SiO2, its carrier mobility will drop to 104 cm2 V−1 s−1 or lower.2 In addition, the high mobility in graphene and silicene will degrade significantly if a band gap of over 0.1 eV is opened.57,58
Admittedly, the relaxation time of germanene could be affected by the existence of a MX substrate, so the above estimation of carrier mobility might be just an estimation for the order of magnitude. More elaborated estimation of carrier mobility is left as a future problem. In doing so, methods like the deformation potential model24,59 can be used to examine the relaxation time of MX-supported germanene. Note that optical phonon may have to be taken into account in addition to the acoustic phonon. We say so because our preliminary molecular dynamics (MD) simulation shown in the ESI† indicates that the magnitude of ZO phonon is large at high temperature (Fig. S8, ESI†). On the other hand, we would like to note that our MD simulation has been performed just to see whether germanene can preserve its hexagonal lattice well on GaTe at 500 K, and strong phonon modes do not necessarily have strong electron–phonon coupling.
It should also be noted that in experiment, the ML MX itself will need a substrate. However, it does not affect our conclusions due to the following two reasons. First, the lattice constant of ML MX can be preserved on some common substrates like SiO2. For example, atomic layers of InSe on SiO2 have a lattice constant of 0.40 nm,33 similar to the free-standing case and our result. Hence, the advantage of small lattice mismatch between germanene and InSe will be preserved even if InSe is put on SiO2. Second, germanene can also retain its Dirac cone on thicker few-layer MX, and the thickness can screen out the effect of the substrate under MX. According to our calculation, germanenes on ML and trilayer InSe have similar band structures (Fig. S10, ESI†), probably because InSe layers are stacked with the weak van der Waals interaction. Using the bulk MX itself as the substrate is probably another good choice. If a clean and flat (001) surface of the bulk MX is made with little defects, it may also serve as a suitable substrate for germanene.
In conclusion, for the first time by using the density function theory, we predict that germanene can preserve its low-buckled honeycomb structure and the Dirac-cone-like band structure similar to the free-standing case. Furthermore, germanene is predicted to be semiconducting on GaTe and InSe with a band gap of over 0.1 eV, while an ultrahigh carrier mobility estimated up to 2.2 × 105 cm2 V−1 s−1 is preserved. The band splitting caused by the SOC can be up to 42 meV. Hence, we believe germanene on GaTe and InSe has potential in electronic and spintronic applications. Our research would stimulate the synthesis of high-performance germanene and its FET in the future.
Acknowledgements
This work has been supported by a Grant-in-Aid for Scientific Research on Innovative Areas No. 26107514 from MEXT, Japan.References
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS PubMed.
- J.-H. Chen, C. Jang, S. Xiao, M. Ishigami and M. S. Fuhrer, Nat. Nanotechnol., 2008, 3, 206–209 CrossRef CAS PubMed.
- M. Xu, T. Liang, M. Shi and H. Chen, Chem. Rev., 2013, 113, 3766–3798 CrossRef CAS PubMed.
- P. Miró, M. Audiffred and T. Heine, Chem. Soc. Rev., 2014, 43, 6537 RSC.
- B. Feng, Z. Ding, S. Meng, Y. Yao, X. He, P. Cheng, L. Chen and K. Wu, Nano Lett., 2012, 12, 3507–3511 CrossRef CAS PubMed.
- P. Vogt, P. De Padova, C. Quaresima, J. Avila, E. Frantzeskakis, M. C. Asensio, A. Resta, B. Ealet and G. Le Lay, Phys. Rev. Lett., 2012, 108, 155501 CrossRef.
- A. Resta, T. Leoni, C. Barth, A. Ranguis, C. Becker, T. Bruhn, P. Vogt and G. Le Lay, Sci. Rep., 2013, 3, 2399 Search PubMed.
- C. L. Lin, R. Arafune, K. Kawahara, M. Kanno, N. Tsukahara, E. Minamitani, Y. Kim, M. Kawai and N. Takagi, Phys. Rev. Lett., 2013, 110, 076801 CrossRef.
- C. L. Lin, R. Arafune, K. Kawahara, N. Tsukahara, E. Minamitani, Y. Kim, N. Takagi and M. Kawai, Appl. Phys. Express, 2012, 5, 045802 CrossRef.
- L. Meng, Y. Wang, L. Zhang, S. Du, R. Wu, L. Li, Y. Zhang, G. Li, H. Zhou, W. A. Hofer and H. J. Gao, Nano Lett., 2013, 13, 685–690 CrossRef CAS PubMed.
- A. Fleurence, R. Friedlein, T. Ozaki, H. Kawai, Y. Wang and Y. Yamada-Takamura, Phys. Rev. Lett., 2012, 108, 245501 CrossRef.
- A. Fleurence, Y. Yoshida, C. C. Lee, T. Ozaki, Y. Yamada-Takamura and Y. Hasegawa, Appl. Phys. Lett., 2014, 104, 021605 CrossRef PubMed.
- L. Li, S. Z. Lu, J. Pan, Z. Qin, Y. Wang, Y. Wang, G. Cao, S. Du and H. Gao, Adv. Mater., 2014, 26, 4820–4824 CrossRef CAS PubMed.
- M. E. Dávila, L. Xian, S. Cahangirov, A. Rubio and G. Le Lay, New J. Phys., 2014, 16, 095002 CrossRef.
- M. Derivaz, D. Dentel, R. Stephan, M.-C. Hanf, A. Mehdaoui, P. Sonnet and C. Pirri, Nano Lett., 2015, 15, 2510–2516 CrossRef CAS PubMed.
- L. Tao, E. Cinquanta, D. Chiappe, C. Grazianetti, M. Fanciulli, M. Dubey, A. Molle and D. Akinwande, Nat. Nanotechnol., 2015, 10, 227–231 CrossRef CAS PubMed.
- S. Cahangirov, M. Topsakal, E. Aktürk, H. Šahin and S. Ciraci, Phys. Rev. Lett., 2009, 102, 236804 CrossRef CAS.
- Z. Ni, Q. Liu, K. Tang, J. Zheng, J. Zhou, R. Qin, Z. Gao, D. Yu and J. Lu, Nano Lett., 2012, 12, 113–118 CrossRef CAS PubMed.
- N. D. Drummond, V. Zólyomi and V. I. Fal'Ko, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 075423 CrossRef.
- R. Quhe, R. Fei, Q. Liu, J. Zheng, H. Li, C. Xu, Z. Ni, Y. Wang, D. Yu, Z. Gao and J. Lu, Sci. Rep., 2012, 2, 853 Search PubMed.
- M. Ye, R. Quhe, J. Zheng, Z. Ni, Y. Wang, Y. Yuan, G. Tse, J. Shi, Z. Gao and J. Lu, Phys. E, 2014, 59, 60–65 CrossRef CAS PubMed.
- C. C. Liu, W. Feng and Y. Yao, Phys. Rev. Lett., 2011, 107, 076802 CrossRef.
- M. Ezawa, Phys. Rev. Lett., 2012, 109, 055502 CrossRef.
- X.-S. Ye, Z.-G. Shao, H. Zhao, L. Yang and C.-L. Wang, RSC Adv., 2014, 4, 21216 RSC.
- Y. Yao, F. Ye, X. L. Qi, S. C. Zhang and Z. Fang, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 041401 CrossRef.
- T. P. Kaloni and U. Schwingenschlögl, J. Appl. Phys., 2013, 114, 184307 CrossRef PubMed.
- Y. Wang and Y. Ding, Solid State Commun., 2013, 155, 6–11 CrossRef CAS PubMed.
- T. P. Kaloni and U. Schwingenschlögl, Chem. Phys. Lett., 2013, 583, 137–140 CrossRef CAS PubMed.
- H. L. Zhuang and R. G. Hennig, Chem. Mater., 2013, 25, 3232–3238 CrossRef CAS.
- D. J. Late, B. Liu, H. S. S. R. Matte, C. N. R. Rao and V. P. Dravid, Adv. Funct. Mater., 2012, 22, 1894–1905 CrossRef CAS PubMed.
- P. Hu, L. Wang, M. Yoon, J. Zhang, W. Feng, X. Wang, Z. Wen, J. C. Idrobo, Y. Miyamoto, D. B. Geohegan and K. Xiao, Nano Lett., 2013, 13, 1649–1654 CAS.
- S. Lei, L. Ge, Z. Liu, S. Najmaei, G. Shi, G. You, J. Lou, R. Vajtai and P. M. Ajayan, Nano Lett., 2013, 13, 2777–2781 CrossRef CAS PubMed.
- S. Lei, L. Ge, S. Najmaei, A. George, R. Kappera, J. Lou, M. Chhowalla, H. Yamaguchi, G. Gupta, R. Vajtai, A. D. Mohite and P. M. Ajayan, ACS Nano, 2014, 8, 1263–1272 CrossRef CAS PubMed.
- Y. Ding and Y. Wang, Appl. Phys. Lett., 2013, 103, 043114 CrossRef PubMed.
- E. Scalise, M. Houssa, E. Cinquanta, C. Grazianetti, B. van den Broek, G. Pourtois, A. Stesmans, M. Fanciulli and A. Molle, 2D Mater., 2014, 1, 011010 CrossRef.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- J. Klime, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- T. Thonhauser, V. R. Cooper, S. Li, A. Puzder, P. Hyldgaard and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 125112 CrossRef.
- G. Román-Pérez and J. M. Soler, Phys. Rev. Lett., 2009, 103, 096102 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- L. Bengtsson, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 12301–12304 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef PubMed.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 121, 1187–1192 CrossRef CAS PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef PubMed.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- F. Schwierz, Nat. Nanotechnol., 2010, 5, 487–496 CrossRef CAS PubMed.
- L. Matthes, O. Pulci and F. Bechstedt, J. Phys.: Condens. Matter, 2013, 25, 395305 CrossRef PubMed.
- G. Kresse, J. Furthmüller and J. Hafner, Europhys. Lett., 2007, 32, 729–734 CrossRef.
- O. Dubay and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 035401 CrossRef.
- J. Jiang, R. Saito, a. Grüneis, G. Dresselhaus and M. S. Dresselhaus, Chem. Phys. Lett., 2004, 392, 383–389 CrossRef CAS PubMed.
- M. W. Lin, C. Ling, L. A. Agapito, N. Kioussis, Y. Zhang, M. M.-C. Cheng, W. L. Wang, E. Kaxiras and Z. Zhou, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 125411 CrossRef.
- J. Wang, R. Zhao, M. Yang, Z. Liu and Z. Liu, J. Chem. Phys., 2013, 138, 084701 CrossRef PubMed.
- Y. He and G. Galli, Chem. Mater., 2014, 26, 5394–5400 CrossRef CAS.
- A. Belsky, M. Hellenbrandt, V. L. Karen and P. Luksch, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 364–369 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp02428e |
| This journal is © the Owner Societies 2015 |