
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComplexes of peracetylated cyclodextrin in a non-aqueous aprotic medium: the role of residual water†
Laszlo
Jicsinszky
*a,
Katia
Martina
a,
Marina
Caporaso
a,
Pedro
Cintas
b,
Andrea
Zanichelli
c and
Giancarlo
Cravotto
*a
aDipartimento di Scienza e Tecnologia del Farmaco, University of Turin, Via P. Giuria, 9, 10125, Turin, Italy. E-mail: ljicsinszky@gmail.com; giancarlo.cravotto@unito.it
bDepartamento de Química Orgánica e Inorgánica, Facultad de Ciencias-UEX, Avda. de Elvas s/n, E-06006 Badajoz, Spain
cResilia SRL, Via Milano 201 - 21017, Samarate, VA, Italy
First published on 16th June 2015
Abstract
This paper describes the interaction between aromatic esters and peracetylated cyclodextrins (CDs) studied by NMR spectroscopy in deuterochloroform (CDCl3). The observed chemical shift changes highlight the existence of interactions between an aromatic alkyl ester, water and peracetylated CDs. In some cases, substituent chemical shift determination was influenced by the low water content of CDCl3 and/or the host molecule. Higher CD concentrations resulted in water signal drifts in all studied cases. It was not possible to obtain a completely dry sample of peracetyl γCD: ∼1 mol of water remained and the water signal showed reversed movement, with respect to the other two CD analogues, upon increasing host concentration. The estimated 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stability constants for the water:peracetyl CD complexes are in the 50–150 M−1 range in CDCl3, but show a relatively large calculation error. The calculated 1:1 stability constants for the peracetyl CD:ester complexes are also in this range, but 1:2 and 2:1 complex compositions are also possible. Overall, our results highlight dynamic aspects of water nanoconfined in a highly hydrophobic environment, thus mimicking biological recognition where a few water molecules often play a pivotal role.
:1 stability constants for the water:peracetyl CD complexes are in the 50–150 M−1 range in CDCl3, but show a relatively large calculation error. The calculated 1:1 stability constants for the peracetyl CD:ester complexes are also in this range, but 1:2 and 2:1 complex compositions are also possible. Overall, our results highlight dynamic aspects of water nanoconfined in a highly hydrophobic environment, thus mimicking biological recognition where a few water molecules often play a pivotal role.
1. Introduction
Cyclic maltooligosaccharides, commonly known as cyclodextrins (CDs), are macrocycles of α(1 → 4) linked anhydroglucopyranoside units in a 4C1 conformation. In practice, the 6-, 7- and 8-membered versions are known as α-, β-, and γ-cyclodextrins (αCD, βCD, and γCD), respectively, and have all been well studied. Their low commercial price, particularly when bought in bulk, means that they are used worldwide in a variety of fields. The circular hydrogen bonds between the secondary hydroxyl groups on adjacent rings leads to a rigid truncated cone shape, where the hydroxyls form a highly hydrophilic rim and the glycosidic oxygens, together with a number of methine groups, create a considerably less hydrophilic environment inside the cavity. However, long-term inclusion normally occurs and rigid symmetric structures are frozen in the crystalline state only, in solutions a dynamic equilibrium exists between association and dissociation. Native CDs, except βCD, are easily soluble in water, while apolar guests easily displace the water molecules in the cavity via complexation resulting in a thermodynamically more favorable state. Inclusion complexes and the sandwich-like molecular associations are also often stabilized by hydrogen bonds.Once all hydroxyls are substituted, particularly as alkyl ethers or esters, the CD lipophilicity profile is completely reversed and the cavity becomes more hydrophilic than the outer surfaces, while substitution also enlarges CD cavity dimensions. However, substituted oxygens, particularly glycosidic oxygens, are still able to form hydrogen bonds with appropriate hydrogen bond donors and lipophilic interactions become dominant, particularly at the edges of the enhanced cavities. It is also generally accepted that some smaller substituents, such as methyl or acetyl group, can considerably deform the structures in the absence of a guest, as has been described for peracetylated β-1 and -γ-cyclodextrins2 (PAcβCD and PAcγCD, respectively). The crystal structures of peracetyl-α-cyclodextrin (PAcαCD),3 and probably -βCD,4 are somehow symmetrical and stabilized by crystal water, although it is also true that these substituted CDs rarely reproduce the usual truncated cone structure of unsubstituted CDs. The more flexible structures obtained by breaking hydrogen bonds or even completely losing the hydrogen-bond belt mean that the glucopyranose rings are able to rotate around the glycosidic linkages. In fact, some isolated units have been found to distort from the usual 4C1 chair conformation to skew or almost completely inverted 1C4 conformers.1 The larger macrocycle in γCD derivatives allows for greater flexibility and results in the most distorted crystal structures, as reported by Caira et al.2 Their solid-state structures have been described as elliptically distorted macrocycles where glucose units undergo significant rotation around the glycosidic connections and deform to create twisted conformers.4 Derivatization may not only create an enlarged cavity but can also lead to the self-inclusion of functional groups.
NMR techniques are currently the leading non-destructive methods for studying complexation processes in solution. Although various approaches have been formulated, two principal objectives are at the heart of this NMR titration based method. These two methodologies require two conflicting approximations. Determination of complex stoichiometry needs the sum of molar concentrations, a boundary condition for the most commonly used Job's plot method, to be constant. Calculation of complex stability constants, however, usually requires guest concentration being held constant in order to eliminate concentration dependent chemical shift drifts.5,6 In both cases, the equation for measured chemical shift (δobs), as a weighted average of the chemical shifts of the uncomplexed (δfree) and complexed (δbound) species, is combined with the equation for the association constant according to complex stoichiometry. A comprehensive review by Uccello-Barretta et al. traces the development and historical milestones in NMR technology's study of cyclodextrin complexes.7
Several exhaustive reviews have been dedicated to the use of NMR methods on cyclodextrins and their complexes,5,8–10 while Chankvetadze has published a fundamental review11 on enantiodiscrimination studies. He also pointed out that the multiple forces involved in analyte–CD interactions mean that combined molecular modeling-spectroscopy approaches are required if we are to shed more light on the molecular basis of chiral recognition processes.
The presence of self-association phenomena can also be evaluated by analyzing the NMR spectra of the pure component in progressively diluted solutions; the self-association constant can be determined when chemical shifts depend on concentration. Preliminary information on intermolecular interactions can be obtained directly from an analysis of the complexation shifts (Δδ = δobs − δfree), measured for all the species nuclei, in 1H NMR spectra. However, the most important conformational analysis method when defining binding geometry is the detection of dipolar interactions (NOEs).5 This method furnishes information on the spatial proximity between nuclei and, therefore, highlights proximity constraints between nuclei that are either located inside the same molecule (intramolecular dipolar interactions) or belong to a different one (intermolecular dipolar interactions). This technique is also an important tool in chiral discrimination research, as intermolecular dipolar interactions make it possible to define the relative stereochemistry of the chiral auxiliary and one, or other, of the enantiomers in diastereoisomeric complexes. Both native and modified CDs are effectively used as chiral selectors in the majority of enantioseparation techniques.7,11
Although proton NMR measurements are the most common ways to investigate molecular associations, carbon NMR can also be used, but it is less feasible due to the less natural abundancy of NMR active 13C and longer relaxation time of carbon. Other geometrical parameters, such as dihedral angles, can be determined using empirical relationships to analyze vicinal coupling constant dependence on dihedral angles.12,13 State-of-the-art high resolution NMR equipments also enable the scientists to study the effects of complexation on vicinal coupling constants. However, this effect is usually considerably less intense and more compound dependent than simple chemical shift analyses.
The enantioselective inclusion phenomena of lipophilic CD derivatives were reported in the pioneering work of König et al.,14 who described the interaction between (R,S)-methyl-2-chloropropionate and heptakis(3-O-acetyl-2,6-di-O-pentyl)-βCD (Lipodex D). NMR spectra recorded in cyclohexane-d1215–17 indicated that the guest enantiomeric methine proton displayed very high differentiation. Complexation shifts were in accordance with GC retention data and a molecular dynamics simulation based chiral recognition rational was devised. Numerous publications that used regiospecific acetylated CDs, mainly per(6-TBDMS-2,3-di-O-acetyl) CDs, then appeared.15–17
While H-3 and H-5 protons are the most heavily involved in the inclusion processes in unsubstituted CDs, hydroxyl group substitution increases the number of moieties to be investigated. In addition, the resulting spectra sometimes show very complex chemical shift patterns which are due to the normally distorted structures in the un-complexed states discussed above. However, the spectra of unsubstituted and persubstituted CDs are much simpler than those of the partially derivatized compounds.
Persubstituted versions are the simplest of the acetylated CD derivatives to prepare. They can be easily produced even on a ten-hundred-kg scale and are available as fine chemicals as well. These CDs are highly lipophilic compounds and are soluble in various organic solvents such as methylene chloride, chloroform, toluene, ethyl acetate and acetone, for example. In toluene, they form inclusion complexes, which precipitate after dissolution of the CD. They are chemically inert and the acetyl groups cause the cavity of the cyclic structure to widen. The steric stress inherent in these macrocycles makes it possible for anhydrous acids or Lewis acids to cleave the glycosidic bonds, while acetyl groups can usually be cleaved under basic conditions, like in the majority of organic esters.
Although peracetylated CDs were among the first CDs to be prepared,18 their complexation properties have only been sporadically studied. However, the discovery that even polar compounds, such as organic alkali metal salts, can be complexed by PAcβCD, despite the cavity's reversed polarity, is not novel.19 This same phenomenon was also observed in peracetylated 3,6-anhydro-CDs. However it remained but a curiosity despite the possible advantages it may offer to the routinely used analytical method, TLC.20,21 Despite some attempts made to use these complexation properties in capillary GC, the unfavorably high melting point of peracetyl CDs (∼235 °C, ∼198 °C and ∼248 °C for peracetyl-α-, -β- and -γCD, respectively) has been an obstacle to the wider penetration of these derivatives into the analytical repertoire.22
Enhanced lipophilicity and widened cavities would appear to make peracetylated CD use in the controlled release of hydrophilic drug obvious. Their potential has been known for 25 years,23 however after the initial advantageous attempts, only a few publications can be found. Some enantioseparations, both on analytical and preparative scales,24–28 have also been published and a couple of solid complexes have been prepared using various techniques.29–31
The investigation of soluble complexes in solution, particularly in apolar solvents, is rare and only a few examples are available in technical papers.4,19,24,32 Guest molecules in those cases also carry some polar components, apart from a species studied in a recent analytical impacted publication by Kopytin et al.22 The effect of αCD peracylation on its molecular structure has been investigated using single crystal X-ray analysis,3 and only revealed the presence of at least two associated water molecules in the solid state, despite the crystalline complex being prepared from aqueous 2-propanol. Crystals of a heptakis(2,3,6-tri-O-acetyl)-βCD methanol complex, as well as other peracylated CDs, were found to contain elliptically distorted molecules with non-planar, boat-shaped structures.4 Steric interactions between adjacent acyl chains in these molecules result in the severe tilting of the glucose units relative to the glycosidic O(4) planes. Additionally, significant acyl residue self-inclusion was found in those peracylated molecules. This self-inclusion means that the guest inclusion, occurring in native host molecules, is unfavorable both in the solid state and in solution. The structure of fully acetylated γCD also showed extensive macrocyclic distortion, in this case accompanied by the self-inclusion of three acetyl residues, while the complexation of alcohols occurred together with water molecules.2 A unique feature of the structure is the partitioning, by these residues, of the cavity region into two distinct sub-cavities that accommodate water and ethanol molecules. This unusual type of solvent dual inclusion was discovered thanks to powder X-ray diffraction experiments on PAcγCD and a series of solvents, such as acetone, n-propanol and ethanol.2
The complete assignment of peracetylated βCD signals has been carried out by Uccello-Barretta et al. who have also determined proximity constraints between PAcβCD protons using 2D ROESY analysis in CDCl3 and CD3OD.1 It was also found that H-3, H-2 and H-6a,6b protons correlate with the carbonyl groups on the same sites which, in turn, correlate with the respective methyl protons. The proton J-coupling patterns were found to be similar to those in the unsubstituted systems. The acetyl on the O2-site in the distorted glucopyranose ring(s) is in proximity to H-5 and hence pseudo axial, making H-2 pseudo equatorial, on the same site. The same extent of average distortion can also be found in alkylated, benzylated and benzoylated CDs.33 The stereochemical distortions found in solution are similar to the symmetry perturbations detected in CDs bearing benzoyl, at least on the secondary sites. These distortions were explained by the loss of the strong hydrogen-bond network on the secondary sites of the adjacent units and the need to minimize the repulsive steric interactions between the bulk substituents. Glucopyranoside units are found in an intermediate conformation, as they are between the two chairs. These ring deformations affect some units only and the undistorted 4C1 chairs were simultaneously detected. The truncated cone shape is heavily perturbed because of these distortions and the polarity differences between the outside and the inside of the cavity can be affected significantly, as can the solubility of derivatized CDs and their complexation properties. Similar conformations have been found in both CDCl3 and CD3OD.1
Complex formation between a structural analogue, 1,4-bis(2-ethylhexyl)benzene-1,2-dicarboxylate (our model compound is 1,4-dicarboxylate), and perbenzoylated βCD has been investigated by Yu et al.,34 who concluded that the host–guest complex was stabilized by π–π interactions between the aromatic ester and the perbenzoylated βCD.
In this paper, we report our studies on the interaction between a branched alkyl chain ester of terephthalic acid and peracetylated CDs. The presence of incompletely and possibly inhomogeneously dried products – due either to the drying procedure or the adsorption of small amounts of water from the air – is to be expected when bulk quantities are handled. It is worth pointing out that polar solvent molecules can be retained within the inner cavity of native CDs. Molecular dynamics evidence that the average residence time for residual DMSO lies in the nanosecond range, which is at least one order of magnitude longer than the one found for water.35 Molecular dynamics was also used to study the solvent polarity effect on the formation of CD dimers and emphasized the importance of hydrogen bonds in the formation. Although, it is also true that in the simulation the authors used solvents too, in which the native CDs are practically insoluble, and/or form complexes. Losing the strong hydrogen bonds by the substitution of the electrostatic and lipophilic interactions become dominant.36 Having noticed the difficulties of water removal from the lipophilic CDs, the possible effect of water on solution complexation in the non-polar solvent, CDCl3, was also investigated.
2. Experimental
2.1. Materials
Acetic anhydride, methanol, FeCl3·6H2O and methylene chloride were purchased from Sigma-Aldrich, while α-, β- and γCDs were generous gifts from Roquette, France. CDCl3 is a product of EurIsotop (D007H). Racemic 1,4-bis(2-ethylhexyl)benzene-1,4-dicarboxylate was obtained from Giganplast, Italy.2.2. Methods
NMR stock solutions were prepared in CDCl3 at fixed 16–25 ± 1 mM concentrations (except for water complexation experiments) and the guest was added via a microliter syringe. In order to determine apparent complex stability constants, CD concentrations were kept almost constant to avoid concentration dependent shift drifts. The dried peracetylated CDs were weighed and added gradually to the water saturated CDCl3 solution in the aqueous experiments.
NMR spectra were recorded on a Bruker Avance 300 MHz at 22.6 ± 0.1 °C. Residual chloroform of CDCl3 was used as an internal standard and chemical shifts were calibrated to the residual CHCl3 (δH = 7.260). Chemical shifts (δ) are given in ppm.
To reduce the possible errors, pure components were always recorded at the beginning of the experiments in each series. All experiments of the same run had been recorded on same days without interruption, using 64 transients, in a 4496.40 Hz spectral width. The 1H NMR data were processed using an ACD/NMR Processor,42 using a 4-fold zero filling and Lorentzian–Gaussian window function (LB = −0.3, GF = 0.3) prior to Fourier transformations.
2.3. Synthesis
3. Results and discussion
3.1. Peracetyl cyclodextrin–water systems
The X-ray structure of peracetylated αCD contains two moles of water as reported by Harata.3 One of these water molecules is found inside the CD cavity at the primary rim, which is not surprising considering that the acetyl groups introduced to the macrocycle reversed its lipophilicity and that carbonyl oxygens can stabilize the water in an apolar environment via hydrogen bonding. Harata, unlike Caira with PAcγCD,2 was able to prepare pure water–PAcαCD associates although he crystallized PAcαCD from 80% aqueous 2-propanol.NMR measurements show that even dried peracetylated CDs (Scheme 1a) contain water. The weight percentage content of water is 1–3% in the air-dried products, which is in the 1–2 molar equivalent range of peracetylated CDs due to the ca. 95–130-fold difference in molecular weight between peracetylated CDs and water. It was found that simple heat assisted drying did not usually result in water-free materials. It was possible to completely dry peracetylated α- and βCDs by azeotropic distillations. However, a relatively large amount of organic solvents used means that this method is not feasible for drying in bulk quantities. PAcγCD showed ∼1 mol residual water after treatment. Air-dried compounds were used to study the effects of CD concentration on water chemical shifts for the NMR titrations of 1,4-bis(2-ethylhexyl)benzene-1,4-dicarboxylate (Scheme 1b). It was also found that the water content did not considerably affect NMR analyses, except for PAcγCD which produced overlapping water and guest methine proton signals. The water signals smoothly drifted with increasing guest concentration and finally moved away from the methine signal.
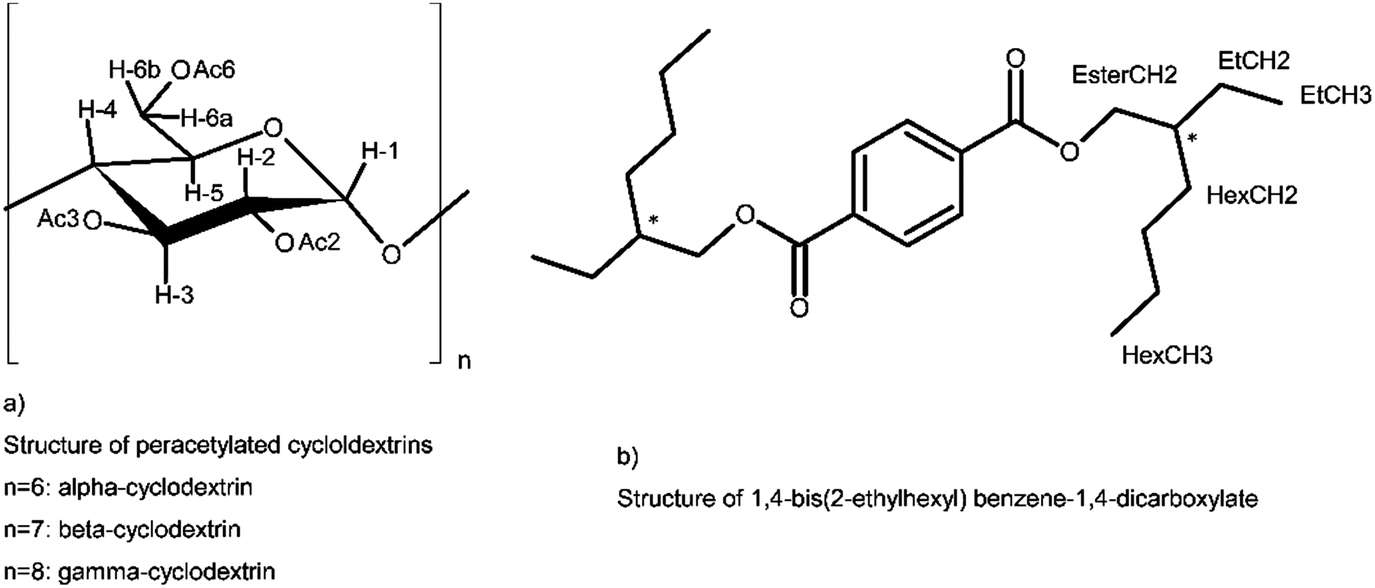 | ||
| Scheme 1 Structures of host and guest and labels for NMR identification. | ||
From the experimental data of 2-ethylhexyl benzoate,38 the methyl and some methylene protons from the guest molecule could be tentatively assigned, which enabled us to use their chemical shifts for our determination of complex composition and calculation of apparent complex stability constants. H-6 protons practically gave single signals in PAcαCD, while the methylene protons were split and well separated in both PAcβCD and PAcγCD (H-6a and H-6b). Overlapping signals were also observed in the H-5 protons of peracetyl-αCD, the H-6a protons of peracetyl β- and γ-CDs and the ester methylene protons of the guest. Despite these overlapping signals, the chemical shifts of H-5, H-6a and the ester methylene protons were determined, but with relatively large errors. In worst cases, the methyl and methylene protons of the 2-ethylhexyl moiety, the most separated signals were used instead of the multiplet chemical shifts. Although, this method is not absolutely correct because it assumes that the coupling constants remain constant upon the noncovalent interaction of the molecules. Changes in coupling constants may also be indicative of complexation,39 but now they are in the range of experimental errors at the NMR frequency used.
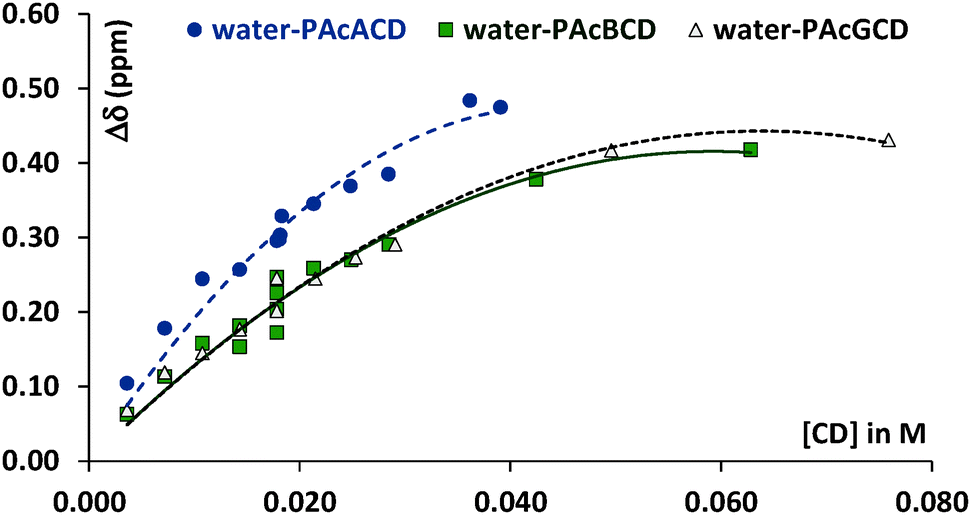 | ||
| Fig. 1 Movement of water proton signals upon increasing CD concentration. | ||
Water proton chemical shift differences (Δδ) show larger deviances at almost identical peracetylated CD molar concentrations, but at different molar fractions. This is probably due to experimental weaknesses, such as uncertainties in weighing and changing sample concentrations upon the transfer of solutions to the tubes. There are enough points to obtain a trend for PAcαCD, but the measurements still show large deviations, as seen in Fig. 2.
 | ||
| Fig. 2 Water proton chemical shift differences at almost identical molar concentrations but different peracetylated CD molar fractions. | ||
Water signals moved and finally overlapped with the acetyl signals of the CD derivative and this fact prevented the exact determination of the water content at high CD/water ratios. This also means that the absence or presence of water cannot be seen at high peracetyl CD–water molar ratios. When water was replaced with deuterium oxide, the HDO signals moved away from the acetyl proton signals, in PAcαCD and PAcβCD, but not in PAcγCD (see the ESI†). The water content of CDCl3 (≤0.01%) is in the same molar range as the CDs. Owing to ambiguous water content in the peracetylated CDs, the water content of the test solutions and the water content of “absolute” CDCl3, the boundary conditions of Job's method (constant sum of molar concentrations) could not be fulfilled.
However, Job's plot-like chart can be created despite these weaknesses8–10,40 (Fig. 3). When Job's plot is created at a non-constant sum of moles, the extremity is shifted to higher host molar fractions.
 | ||
| Fig. 3 Job's plot-like graph of water proton shift differences as a function of water molar fraction. | ||
Considering this effect we concluded the existence of an almost equimolar association between water and peracetylated CDs. This is also confirmed by the nonlinear curve fitting and stability constant calculations.
 | ||
| Fig. 4 Plots of selected chemical shift differences vs. molar concentration and vs. molar fraction of peracetylated CDs (a = alpha, b = beta, g = gamma CD, numbers refer to the appropriate proton as shown in Scheme 1a). | ||
In PAcαCD, methylene (H-6) protons are indistinguishable at low CD/high water concentrations. As the CD/water ratio decreased, at first shoulder appeared only then the signal was divided into two overlapping peaks, as seen in Fig. 5.
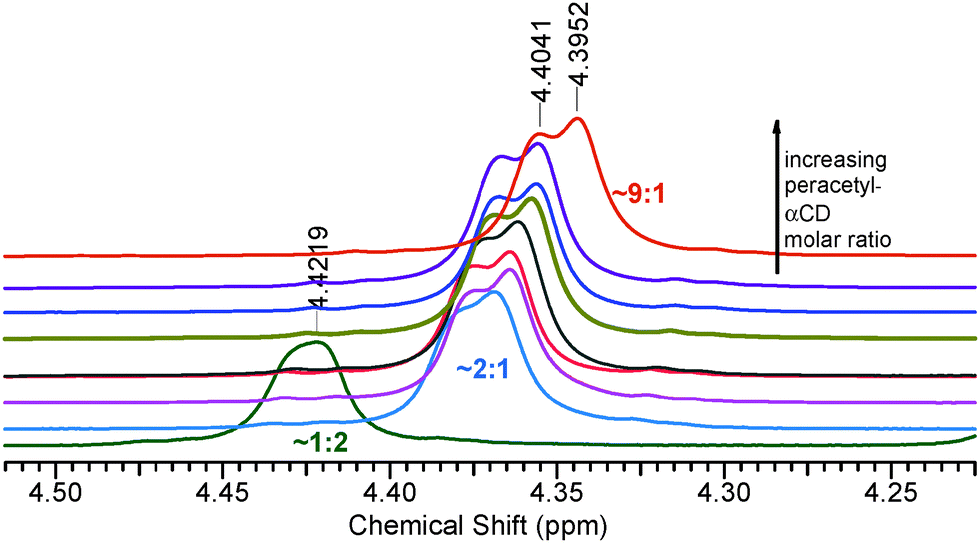 | ||
| Fig. 5 Split of methylene protons in PAcαCD when increasing the CD:water molar ratio. | ||
CD:water molar ratio values were found to be at around 0.4 in our samples. The high chemical shift drift in the mM concentration range indicated the presence of a strong interaction, according to the theoretical complex calculations by Fielding.5 The apparent 1:1 complex stability constants are in the range of 101–102 M−1.
The a priori determination of complex stoichiometry is not necessary if we use the nonlinear curve fitting procedure. WinEQNMR26,41 is a suitable program for calculating 1:1, 1:2 and 2:1 complex stability constants from NMR data. Though this program cannot calculate higher host:guest stoichiometries, however the peracetyl-α- and -γCD X-ray data suggest that these compositions are sufficient for the calculations. The goodness of parameters obtained from a nonlinear curve fitting is always challenging, particularly when the experiment number is limited.
Only the 1:1 peracetyl CD–water complexes in CDCl3 gave results that fulfill acceptance criteria. The calculated stability constants are tabulated in Table 1. Calculated concentrations and chemical shift are given in the ESI,† Fig. S3.1 and S3.2. Values for 1:2 and 2:1 complexes were in the range of thousands. However, both their relative errors and poor estimations of uncomplexed water shifts suggest a much lower probability that such associations form.
:1 complex stability constants for peracetyl CD–water systems in CDCl3
| K11 [M−1] | Estimated uncomplexed water shifta | Estimated complexed water shift | |
|---|---|---|---|
| a In ppm, measured value: 1.556 ± 0.005 ppm at ∼0.005 M water concentration. | |||
| Peracetyl-αCD | 114.3 ± 31.2 | 1.494 ± 0.029 | 2.125 ± 0.036 |
| Peracetyl-βCD | 94.2 ± 14.1 | 1.523 ± 0.015 | 2.061 ± 0.014 |
| Peracetyl-γCD | 58.2 ± 9.6 | 1.545 ± 0.032 | 2.234 ± 0.068 |
3.2. 1,4-Bis(2-ethylhexyl)benzene-1,4-dicarboxylate complex
The two-arm structure of the ester allows two CDs to interact with the guest molecule. Possible association modes are depicted in Scheme 2, where the additional rings represent the macrocycle being enlarged by the acetyl substituents. The free rotation of the ester moiety means that the longer alkyl chain can also approach closer to the macrocycle cavity, while the coordination of the second CD is sterically more hindered in this case. The molecular dimensions of the peracetyl-γCD allow for deeper intrusion into the cyclodextrin cavity and even the complete cross is geometrically allowed. | ||
| Scheme 2 Possible structures for various peracetylated CDs and 1,4-bis(2-ethylhexyl)benzene-1,4-dicarboxylate complexes. | ||
 | ||
| Fig. 6 Water proton signal movement as a function of the molar ratio. | ||
A section of proton spectra is shown in Fig. 7 in order to demonstrate water proton movements as a function of the host:guest ratio.
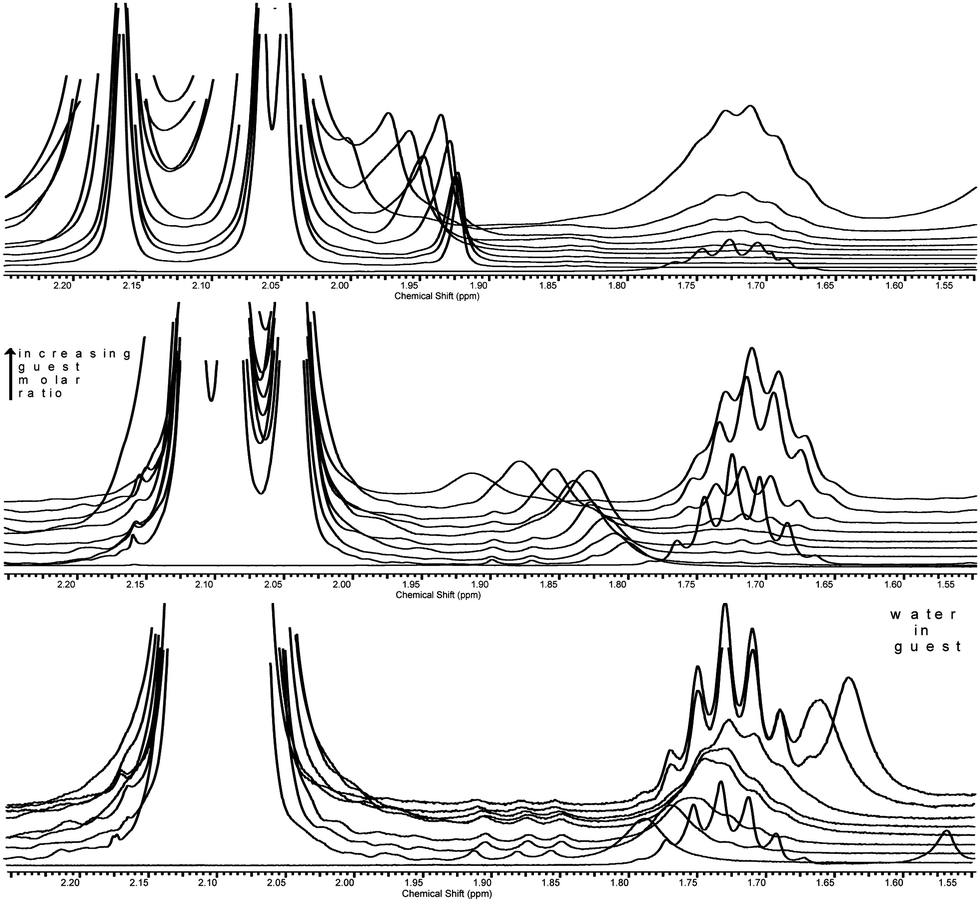 | ||
| Fig. 7 Water proton signal movement upon increasing the guest molecule molar ratio. Top: PerAcαCD, middle: PerAcβCD, bottom: PerAcγCD. Guest molar ratio increases from bottom to top in each spectrum; some curves have been removed to improve visibility. | ||
Calculation of complex stability constants for water in our three-component systems (neglecting the solvent) gave very high errors in all cases, as can be concluded from Table 2. The peracetylated cyclodextrins contain water and in the complexation experiments, the water concentrations varied in a very tight range. Additionally, in the case of PAcγCD due to the water signal overlaps with the methine signal of the organic guest and its chemical shifts are uncertain and integration is not satisfactory. For the PAcαCD case overlapping water and Ac3 methyl signals cause similar difficulties. Because the estimated stability constants are out of acceptance, the complexed/free water ratio cannot be well estimated. From Fig. 6 it can be concluded that the guest molecule cannot exclude water from PAcαCD and PAβCD, the lower limit of 1:1 apparent stability constant is at around 100 M−1, while for the PAγCD system the upper limit is around 200 M−1. These values are in good agreement with the values of Table 3 for PAcα-and -βCDs.
:1 complex stability constants for peracetyl CD–water-1,4-bis(2-ethylhexyl)benzene-1,4-dicarboxylate systems in CDCl3
| K11 [M−1] | Estimated K11 [M−1] (based on Table 3) | |
|---|---|---|
| Peracetyl-αCD | 4.1 ± 38.2 | >93 (lower limit) |
| Peracetyl-βCD | 6.2 ± 4.6 | >111 (lower limit) |
| Peracetyl-γCD | 34.2 ± 37.6 | <182 (upper limit) |
:guest molar ratio)
| K11a [M−1] | K11b [M−1] | K12b [M−2] | K11c [M−1] | K21c [M−2] | |
|---|---|---|---|---|---|
|
a The results are from the 1:1 (host:guest) curve fitting procedure.
b The results are from the 1:2 (host:guest) curve fitting procedure.
c The results are from the 2:1 (host:guest) curve fitting procedure.
|
|||||
| Peracetyl-αCD | 96.7 ± 3.4 | 8.6 ± 1.3 | 272 ± 75.6 | 28.2 ± 0.9 | 404.3 ± 0.0 |
| Peracetyl-βCD | 114.8 ± 3.0 | 28.3 ± 1.6 | 51.3 ± 2.0 | 37.7 ± 1.5 | 25.4 ± 10.5 |
| Peracetyl-γCD | 187.4 ± 3.7 | 68.1 ± 1.7 | 277.9 ± 129 | 49.8 ± 1.3 | 190.6 ± 102.7 |
Almost all CD protons moved, but usually without characteristic extremities in Job's plots, upon changing the concentration of the host molecule. It is assumed that these missing extremities are due to the opposite movement or the large distances between interacting and non-interacting moieties. The reproducibility of chemical shifts at identical concentrations was typically in the 0.001–0.004 range. Although PAcαCD H-5 protons overlap the ester methylene protons, it was possible to determine individual chemical shifts in most cases. This was not true for PAcβCD and PAcγCD where the H-6a protons completely overlapped the ester methylene protons at a higher CD molar ratio and it was impossible to determine the individual chemical shifts.
Although the acetyl protons are in huge excess in the CDs and their methyl groups can be very far from each other, the outcome of their shift changes are above experimental errors, as can be seen in their Job's plots, Fig. 8. The curves show large fluctuations, at ester molar fractions of around 0.25–0.3, 0.45–0.5 and 0.67, while definite minima – corresponding to 2:1 (or 3:1, due to inherent uncertainty in Job's plot), 1:1 and 1:2 host:guest ratios – can be found in the case of PAcαCD and PAcβCD. Only one minimum is found for PAcγCD at ∼0.3 guest molar fraction. The acetyl groups are far from the cavity and the observed chemical shift drifts can give information about the host and guest interaction centers. In the case of PAcαCD, the secondary acetyl substituents show an almost equal but smaller interaction than the primary rim substituent, where the 0.25–0.3 molar ratio minimum is missing. An interaction between the host and guest is therefore assumed to exist at the secondary rim, while the extremities point to a secondary rim interaction for 1:1, and a head-to-head association for the 2:1 host:guest ratio. PAcβCD shows almost equal participation from the O(3)- and O(6) acetyl groups in complexation, while the largest differences can be found in all PAcγCD acetyl groups which affect both sides of the host molecule. The dimensions of both molecules allow the guest to pass through the CD cavity in the latter case. This can also be confirmed by the migration of water protons in the opposite direction suggesting that the ester is able to move water from a relatively polar environment to a less polar one. NOESY and/or ROESY experiments may have given a more realistic picture, however these experiments failed due to the high number of almost chemically equivalent protons combined with the chirality of the ester.
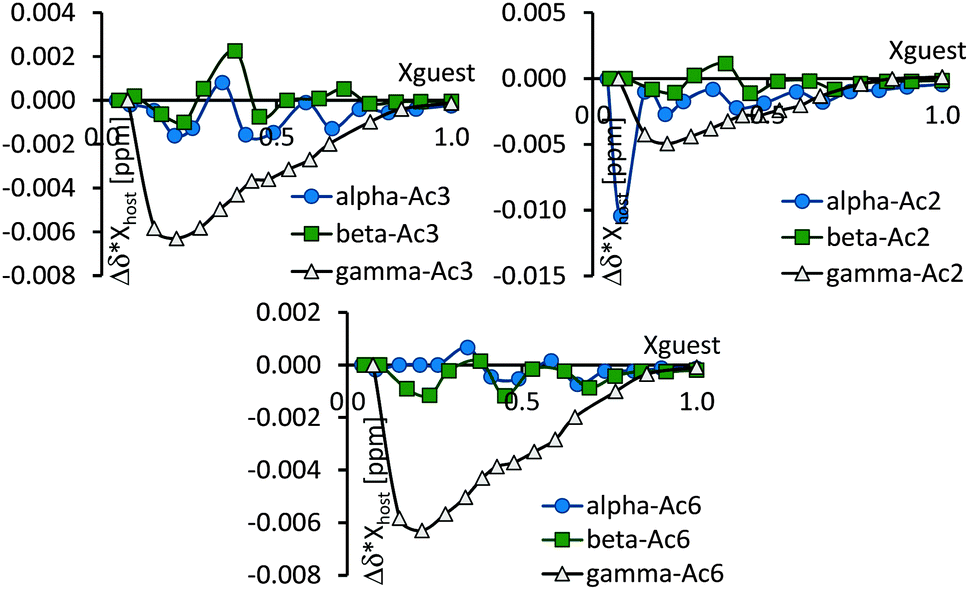 | ||
| Fig. 8 Job's plot of CD acetyl methyl protons. | ||
:1 host:guest associations. The 1:1 complex became dominant upon increasing guest concentration, as is illustrated in Scheme 2. The branched alkyl chain can penetrate deeper into the CD cavity allowing the more polar carbonyl group(s) to interact with the similar CD groups. Unfortunately, the guest molecule's most sensitive proton signals, the ester methylene and branch methine proton, overlap with some CD signals and also with water signals, in the case of PAcγCD. Additionally, the host methine group's chiral center brings new complexities as a potential enantiomer-selective complexation. Chemical shift determinations are ambiguous as a consequence of these bad constellations and, as such, conclusions should be handled carefully. Furthermore, CD Job's plots do not completely confirm the a priori assumed association stoichiometry. Job's plots of ester methylene and the methine protons can be seen in Fig. 9.
 | ||
| Fig. 9 Job's plot of guest methine and ester methylene protons. | ||
The aromatic region of the guest molecule is the only part of the molecule which has no signals that overlap with CD and water protons. Job's plot created from the data given in Fig. 10 is not absolutely clear, however the signal shape of the aromatic protons shows essential changes upon host:guest molar ratio changes, as is demonstrated in Fig. 11. The large extremities at the ∼0.3 molar ratio of the guest suggest 2:1 CD:ester associations for PAcαCD and PAcβCD. In the PAcγCD case, where water can be more readily excluded, a minimum can be found at around the 1:1 molar ratio. Water exclusion by the host is also confirmed by the higher complex stability constants seen in Table 3. The reasonable 1:2 host guest ratio also supports the idea that the terephthalate ester can pass through the PAcγCD cavity. The weak minima, which rather seem to be shoulders in the other two cases, suggest the existence of the 1:1 and 1:2 host:guest associations for the PAcγCD and 1:1 and 2:1 for the other two CDs. Only loose associations can be made from all figures. According to the literature,5 artificial complex simulations gave the apparent complex stability constants to be in the 10–100 M−1 range. To calculate the stability constants, the three-component systems were reduced to two components as water was neglected and the multicomponent curve fitting procedure from WinEQNMR26,41 was used.
 | ||
| Fig. 10 Job's plot of aromatic protons. | ||
 | ||
| Fig. 11 Aromatic protons at low, medium, and high guest ratios (black: no CD, red: low, green: middle, blue: high peracetyl CD:guest molar ratio, respectively). | ||
The stability constants calculated from the aromatic protons of the guest are summarized in Table 3. Simulated concentration curves and chemical shifts can be found in the ESI,† Fig. S3.3 and S3.4. Calculations for other than ester aromatic protons failed to give reasonable results, which is partially the consequence of the relatively large error in the chemical shifts of the overlapped signals in the complex concentration range. An additional source of the failed calculations can be found from the fact that the two identical arms of the guest the 1:1, 1:2 and 2:1 host:guest ratios are practically indistinguishable in the alkyl region.
The calculated stability constants show that 1:1 and 2:1 complexes are the most probable associations in the case of PAcαCD, while 1:1 and both 1:2 and 2:1 associations for PAcβCD and PAcγCD are reasonable but in these cases, the 1:1 association is the most probable. These results are also in accordance with the molecular dimensions of CDs. The large error suggests that the 1:2 complex, for PAcαCD, and 1:2 and 2:1 complexes, for PAcγCD, are less adequate models for the association by handling the peracetyl CD–water as one unit.
Similar calculations for the CD protons pointed to the glucose units participating differently in the complexation and showed considerable geometrical changes upon association. Data for the calculations can be found in the ESI.†
4. Conclusions
Water interacts strongly with the polar moieties of peracetyl CDs. The water content of peracetylated CDs can control the complexation of apolar organic molecules. Although the limited solubility of water in chloroform results in very low water content in the solvent, this residual amount of water is still in the equimolar range, as compared to the low dissolved CD concentration. Complete water removal from the peracetylated CDs is difficult and far from feasibility in bulk quantities. Approximately equimolar amounts of water remained in peracetyl-γCD even after azeotropic removal, which was the only successful method for α- and βCDs. The water signal at higher CD:water molar ratios overlapped with the acetyl methyl signals. Complexation means that the water content of chloroform can be considerably higher than when without peracetylated CDs.
Host and guest proton signals, and often water signals, overlap which made the determination of the chemical shifts of the interacting moieties difficult and prone to large relative errors. Structural changes in CDs upon interaction with the guest molecule affect all CD protons, regardless of their spatial interactions in the complexation process. The water proton NMR signal showed different shift directions upon increasing the aromatic ester molar ratio: a downfield shift in the cases of PAcαCD and PAcβCD and an upfield shift in the case of PAcγCD.
The interaction between apolar CD derivatives and polar (water) or apolar guests (1,4-bis(2-ethylhexyl)benzene-1,4-dicarboxylate) was confirmed via NMR. The peracetyl CD–water complexes are dominantly found in a 1:1 molar ratio with moderate complex stability constants. Both peracetyl-α- and -βCDs seem to form three component complexes with 1:1, 1:2, and 2:1 molecular ratios between CDs and esters, while the guest can exclude water from the peracetyl-γCD cavity by forming dominantly 1:1 molecular associations.
Acknowledgements
This research was kindly funded by the European Community's Seventh Framework Program through the EU project MAPSYN: Microwave, Acoustic and Plasma Syntheses, under grant agreement No. CP-IP 309376. P. Cintas thanks the Ministry of Economy and Competitiveness (Project CTQ2013-44787-P) and the Junta de Extremadura-FEDER (Grant GR15022) for financial support. Financial support from Resilia SRL (Samarate, Italy) is acknowledged. The authors acknowledge the assistance and suggestions of Orsolya Töke (MTA-TKK, Budapest, Hungary) in NMR experiments, and the invaluable help of Éva Fenyvesi (Cyclolab Ltd, Budapest, Hungary) in manuscript preparation.References
- G. Uccello-Barretta, G. Sicoli, F. Balzano and P. Salvadori, Carbohydr. Res., 2003, 338, 1103–1107 CrossRef CAS.
- M. R. Caira, G. Bettinetti, M. Sorrenti, L. Catenacci, D. Cruickshank and K. Davies, Chem. Commun., 2007, 1221–1223 RSC.
- K. Harata, Chem. Lett., 1998, 589–590 CrossRef CAS.
- M. Añibarro, K. Gessler, I. Uson, G. M. Sheldrick, K. Harata, K. Uekama, F. Hirayama, Y. Abe and W. Saenger, J. Am. Chem. Soc., 2001, 123, 11854–11862 CrossRef PubMed.
- L. Fielding, Tetrahedron, 2000, 56, 6151–6170 CrossRef CAS.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
- G. Uccello-Barretta, L. Vanni and F. Balzano, J. Chromatogr. A, 2010, 1217, 928–940 CrossRef CAS PubMed.
- H. Dodziuk, W. Kozminski and A. Ejchart, Chirality, 2004, 16, 90–105 CrossRef CAS PubMed.
- B. Chankvetadze, G. Endresz and G. Blaschke, Chem. Soc. Rev., 1996, 25, 141–153 RSC.
- H. J. Schneider, F. Hacket and V. Rüdiger, Chem. Rev., 1998, 98, 1755–1785 CrossRef CAS PubMed.
- B. Chankvetadze, Chem. Soc. Rev., 2004, 33, 337–347 RSC.
- M. Karplus, J. Am. Chem. Soc., 1963, 85, 2870–2871 CrossRef CAS.
- C. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1973, 95, 2333–2344 CrossRef CAS.
- J. E. H. Köhler, M. Hohla, M. Richters and W. König, Angew. Chem., Int. Ed. Engl., 1992, 31, 319–320 CrossRef PubMed.
- T. Köpke, H. G. Schmarr and A. Mosandl, Flavour Fragrance J., 1992, 7, 205–211 CrossRef PubMed.
- I. Abe, N. Fujimoto and T. Nakahara, J. Chromatogr. A, 1994, 676, 469–473 CrossRef CAS.
- C. Bicchi, V. Manzin, A. D'Amato and P. Rubiolo, Flavour Fragrance J., 1995, 10, 127–137 CrossRef CAS PubMed.
- K. Freudenberg and R. Jacobi, Justus Liebigs Ann. Chem., 1935, 518, 102–108 CrossRef CAS PubMed.
- M. Komiyama, H. Yamamoto and H. Hirai, Chem. Lett., 1984, 1081–1084 CrossRef CAS.
- C. Baudin, B. Perly and A. Gadelle, EP Pat., 787744 A1 970806, 1997 Search PubMed.
- C. Baudin, F. Tardy, J.-P. Dalbiez, C. Jankowski, C. Fajolles, G. Leclair, B. Amekraz, B. Perly and L. Mauclaire, Carbohydr. Res., 2005, 340, 131–138 CrossRef CAS PubMed.
- K. A. Kopytin, S. Y. Kudryashov and L. A. Onuchak, Russ. J. Phys. Chem. A, 2012, 86, 147–150 CrossRef CAS.
- K. Matsubara, T. Kuriki, H. Arima, K. Wakamatsu, T. Irie and K. Uekama, Minutes of the 5th International Symposium on Cyclodextrins, Paris, 1990, p. 491.
- M. Thunhorst and U. Holzgrabe, Magn. Reson. Chem., 1998, 36, 211–216 CrossRef CAS.
- J. Zmitek, D. Fercej-Temeljotov, K. Verhnjak, S. Kotnik and M. Kovacic, EP Pat., 578231 A1, 1994 Search PubMed.
- T. Kitagawa, A. Okamoto, T. Kanai, K. Shibayama, T. Aoki and S. Yamakawa, EP Pat., 970936, 2000 Search PubMed.
- L. Kim, A.-D. Stancu, E. Diacu, H.-J. Buschmann and L. Mutihac, Supramol. Chem., 2009, 21, 131–134 CrossRef CAS PubMed.
- E. Diacu, L. Mutihac, E. Ruse and M. M. Ceausescu, J. Inclusion Phenom. Macrocyclic Chem., 2011, 71, 339–342 CrossRef CAS PubMed.
- Z. P. Onuchak, L. A. Burmatnova, T. S. Spiryaeva, E. A. Kuraeva and Y. G. Belousova, Russ. J. Phys. Chem. A, 2012, 86, 1308–1317 CrossRef.
- C. M. Buchanan, D. W. Dixon, R. J. Offermann, J. Szejtli, L. Szente and M. Vikmon, Minutes of the 10th International Symposium on Cyclodextrins, Michigan, 2000, p. 586.
- M. Y. Lee, H. S. Ganapathy and K. T. Lim, J. Phys. Chem. Solids, 2010, 71, 630–633 CrossRef CAS PubMed.
- G. Gattuso, C. Gargiulli and M. F. Parisi, Int. J. Mol. Sci., 2007, 8, 1052–1063 CrossRef CAS PubMed.
- G. Uccello-Barretta, F. Balzano, A. Cuzzola, R. Menicagli and P. Salvadori, Eur. J. Org. Chem., 2000, 449–453 CrossRef CAS.
- B. Z. Yu, J. W. Chung and S.-Y. Kwak, Environ. Sci. Technol., 2008, 42, 7522–7527 CrossRef CAS.
- J. Rodriguez, D. Hernán Rico, L. Domenianni and D. Laria, J. Phys. Chem. B, 2008, 112, 7522–7529 CrossRef CAS PubMed.
- H. Zhang, T. Tan, W. Feng and D. van der Spoel, J. Phys. Chem. B, 2012, 116, 12684–12693 CrossRef CAS PubMed.
- L. Jicsinszky and J. Szejtli, Minutes of the 6th International Symposium on Cyclodextrins, Chicago, 1992, p. 96.
- 2-Ethylhexyl benzoate: SDBS No. 17041HSP-43-349, http://sdbs.db.aist.go.jp. (Accessed on September, 2014).
- S. Bekiroglu, L. Kenne and C. Sandström, J. Org. Chem., 2003, 68, 1671–1678 CrossRef CAS PubMed.
- S. Lee, D.-H. Yi and S. Jung, Bull. Korean Chem. Soc., 2004, 25, 216–220 CrossRef CAS.
- Current version of WinEQNMR (v2.0) can be obtained free of charge from Michel J. Hynes, National University of Ireland, Department of Chemistry, Galway, Ireland; http://michael.j.hynes@nuigalway.ie.
- ACD/NMR Processor Academic Edition Release 12.00 product version 12.01 (Build 39104), Advanced Chemistry Development Inc. (http://www.acdlabs.com).
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of pure compounds and complexes at low, middle, and high peracetyl CD molar ratios, tables of proton chemical shifts, and figures of calculated concentrations and chemical shifts. See DOI: 10.1039/c5cp02379c |
| This journal is © the Owner Societies 2015 |