
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface structured platinum electrodes for the electrochemical reduction of carbon dioxide in imidazolium based ionic liquids
Florin A.
Hanc-Scherer
a,
Miguel A.
Montiel
b,
Vicente
Montiel
b,
Enrique
Herrero
b and
Carlos M.
Sánchez-Sánchez
*cd
aBabes-Bolyai University, Faculty of Chemistry and Chemical Engineering, 11 Arany Janos Street, RO-400028 Cluj-Napoca, Romania
bInstituto Universitario de Electroquímica, Universidad de Alicante, Ap. 99, 03080, Alicante, Spain
cSorbonne Universités, UPMC Univ Paris 06, UMR 8235, Laboratoire Interfaces et Systèmes Electrochimiques, F-75005, Paris, France. E-mail: carlos.sanchez@upmc.fr; Fax: +33 144274074; Tel: +33 144274158
dCNRS, UMR 8235, LISE, F-75005, Paris, France
First published on 5th August 2015
Abstract
The direct CO2 electrochemical reduction on model platinum single crystal electrodes Pt(hkl) is studied in [C2mim+][NTf2−], a suitable room temperature ionic liquid (RTIL) medium due to its moderate viscosity, high CO2 solubility and conductivity. Single crystal electrodes represent the most convenient type of surface structured electrodes for studying the impact of RTIL ion adsorption on relevant electrocatalytic reactions, such as surface sensitive electrochemical CO2 reduction. We propose here based on cyclic voltammetry and in situ electrolysis measurements, for the first time, the formation of a stable adduct [C2mimH–CO2−] by a radical–radical coupling after the simultaneous reduction of CO2 and [C2mim+]. It means between the CO2 radical anion and the radical formed from the reduction of the cation [C2mim+] before forming the corresponding electrogenerated carbene. This is confirmed by the voltammetric study of a model imidazolium-2-carboxylate compound formed following the carbene pathway. The formation of that stable adduct [C2mimH–CO2−] blocks CO2 reduction after a single electron transfer and inhibits CO2 and imidazolium dimerization reactions. However, the electrochemical reduction of CO2 under those conditions provokes the electrochemical cathodic degradation of the imidazolium based RTIL. This important limitation in CO2 recycling by direct electrochemical reduction is overcome by adding a strong acid, [H+][NTf2−], into solution. Then, protons become preferentially adsorbed on the electrode surface by displacing the imidazolium cations and inhibiting their electrochemical reduction. This fact allows the surface sensitive electro-synthesis of HCOOH from CO2 reduction in [C2mim+][NTf2−], with Pt(110) being the most active electrode studied.
Introduction
Carbon dioxide (CO2) emission arising from human activities is considered as a major contributor to the global greenhouse effect. Thus, the CO2 conversion into useful chemical products and/or energy is a critical concern nowadays.1 CO2 reduction can yield a large variety of useful products, such as C1 species like carbon monoxide (CO), formic acid (HCOOH), methane (CH4) and methanol (CH3OH), or C2 products like oxalic acid (H2C2O4), ethane (C2H6), ethanol (C2H5OH), and ethylene (C2H4). Indeed, different alternatives have been studied to transform CO2,2,3 which include electro-synthesis of pharmacological or industrial valuable compounds,4,5 chemical reduction by heterogeneous catalysis,6,7 photocatalytic conversion,8,9 and electrochemical reduction, which comprises molecular catalysed electrochemical reduction of CO210–12 and direct electrocatalytic reduction of CO2.12–16In particular, direct electrochemical conversion and recycling of CO2 represents a promising pathway, where product selectivity strongly depends on two main factors; (i) the electrode material employed13,15 and (ii) the nature of the solvent (aqueous/non-aqueous) supporting electrolyte system.14,17 The type of solvent is an important factor for the electrochemical CO2 reduction, since most studies in aqueous media present some difficulties in controlling the concomitant side reaction of hydrogen evolution, which takes place at similar thermodynamic potentials.18 On the other hand, conventional aprotic non-aqueous media, such as dimethyl sulfoxide (DMSO), N,N-dimethyl formamide (DMF) or acetonitrile (AN), present several advantages, since they avoid hydrogen evolution and present high CO2 solubility (see Table 1). However, their main drawbacks are their high toxicity and volatility, the costly electrolytes employed and the difficulty in recycling the solvent and the electrolyte. A special category of non-aqueous solvents is comprised of room-temperature ionic liquids (RTILs), which have been pointed out as the most suitable solvents for carrying on the future developments in electrochemistry.19 RTILs are commonly defined as materials composed entirely of organic cations and organic or inorganic anions, which melt at or below 100 °C.20 This low melting point is their main difference versus molten salts, which require high working temperatures. Moreover, RTILs present a unique set of properties, which includes suitable ionic conductivity, high thermal stability, negligible vapor pressure and non-flammability.21 All these properties allow RTILs to perform simultaneously the role of a solvent and an electrolyte in electrochemical applications and rule out any safety concern. RTILs comprise a large number of different solvents, but depending on whether the cation employed presents an available proton or not, two main subdivisions are possible; protic ionic liquids (PILs)22 and aprotic ionic liquids (AILs).23 Imidazolium based cations represent an example of that, since depending on the degree of substitution within the imidazolium ring they belong either to the PILs (1,2-dialkyl-imidazoliums) or AILs (1,3-dialkyl-imidazoliums and 1,2,3-trialkylimidazoliums). One of the major drawbacks of RTILs in electrochemical applications is their high viscosity, since this produces low diffusion of the electroactive species and limits the maximum current density in the process. The viscosity of RTILs varies with the type of cation and anion. In particular, 1,3-dialkyl-imidazolium [Cnmim+] cations are one of the most common RTIL cations for electrochemical purposes, since they exhibit both high ionic conductivity and low viscosity values in comparison with all other RTIL cations. Conductivity and viscosity are inversely related, the alkyl chain length (n) of the imidazolium cation being responsible for limiting both. For this reason, the short alkyl chain (n = 2) cation, 1-ethyl-3-methylimidazolium [C2mim+], presents both low viscosity and high conductivity values and is selected as a suitable solvent in the present work. This cation is combined with an asymmetric anion like bis(trifluoromethylsulfonyl)imide [NTf2−], which also presents one of the lowest viscosity values among the most common anions in RTILs.21 Thus, [C2mim+][NTf2−] exhibits a low viscosity (29 cP at 298 K)21 combined with a high solubility of CO2, comparable to the one exhibited in non-aqueous solvents (see Table 1). In principle, the gas solubility in RTILs is mainly controlled by the nature of the RTIL anion. Thus, a strong interaction of CO2 with the RTIL anion is the best indicator of CO2 solubility, with [NTf2−] being the anion presenting one of the greatest affinities for CO2.24 Moreover, this hydrophobic anion allows [C2mim+][NTf2−] to be water immiscible, which is an advantage, since the water solubility in [C2mim+][NTf2−] limits its available electrochemical window (3400 ppm H2O under atmospheric conditions at 298 K).25 For all those reasons, its moderate viscosity, high CO2 solubility and conductivity, we propose herein [C2mim+][NTf2−] as the suitable RTIL for studying the direct CO2 electrochemical reduction on model surfaces such as platinum single crystal electrodes.
After solvent selection, the other key point controlling the product selectivity in the electrochemical reduction of CO2 is the electrocatalytic activity of the electrode material. Thus, CO2 interacts with the metal surface in the presence of [C2mim+][NTf2−] ionic pairs. In spite of that, only a few reports have approached that interaction pointing out structures such as the checkerboard-type sandwich arrangement with imidazolium cations and [NTf2−] anions absorbed alternatively next to each other on the electrode substrate.29,30 For this reason, single crystal electrodes represent the most convenient type of surface structured electrodes for studying the impact of RTIL ion adsorption on electrocatalytic reactions, since they provide site distribution control at the electrode surface. In particular, we propose studying the electrocatalytic activity for CO2 reduction as a function of surface sites using Pt single crystal (Pt(hkl)) electrodes, since it has been already proved that imidazolium cation adsorption is a surface-sensitive process31 and this may drastically affect the CO2 reduction activity at the electrode.
Electrochemical reduction of CO2 on Pt(hkl) electrodes in acid aqueous solution takes place at potentials where hydrogen adsorption also occurs. Moreover, this reaction presents high sensitivity towards the surface site available at the electrode surface by displaying low activity at highly ordered Pt(111) and Pt(100), but high activity at Pt(110) kinked and stepped surfaces.32–34 More recently, several Pt(hkl) electrodes selectively modified with adsorbed adatoms (Bi, Te and Sb) have also exhibited high catalytic activity for CO2 reduction in buffered neutral aqueous media.16 In contrast, electrocatalytic studies for CO2 reduction in different RTILs have been only reported using Pt polycrystalline surfaces.35,36 But, as far as the author's knowledge, the electrochemical reduction of CO2 on Pt(hkl) electrodes in a RTIL medium has not been reported yet. Only a few articles devoted to studying conducting polymers37 or electrochemical hydrogen38,39 and carbon monoxide40 oxidation reactions on Pt(hkl) electrodes in different RTILs can be found in the literature.
It is well established in the electrocatalytic CO2 reduction reaction that the first reaction step consists of the CO2 adsorption at the electrode surface and the subsequent electron transfer to form the corresponding radical-anion (CO2˙−), as described in eqn (1), where M stands for the metal electrocatalyst.15 Moreover, in liquid electrolytes it has been also described that CO2 may form adducts by coupling with different anions present in solution, such as BF3Cl− or 1-isoquinolinecarboxylic acid.35,41 The formation of those adducts weakens the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and makes the reduction of CO2 more favorable. For this reason, it has been recently suggested that a convenient way to lower the potential necessary for achieving the first electron transfer for CO2 reduction is decreasing the CO2˙− free energy of formation. This is achieved by forming a complex between the CO2˙− and an imidazolium based cation such as [C2mim+], since this will help to decrease the overall energy barrier.42 The formation of such a complex has been proposed to be mainly responsible for the highly active ionic-liquid mediated electrochemical conversion of CO2 into CO at low overpotentials in a mixture of water and an imidazolium based ionic liquid. After the first electron transfer forms CO2˙−, several possible reaction pathways may be followed depending on the solvent conditions. They can be mainly divided into those that need the abundant presence of available protons and a second electron transfer, which may form either HCOOH (eqn (2)), if the proton is attached to the CO2˙−, or CO (eqn (3)), if the proton removes one of the oxygen atoms within the CO2˙−. In contrast, if protons are not available, as usually happens in nonaqueous systems, the dimerization reaction mainly produces C2O42− (eqn (4)).
O bond and makes the reduction of CO2 more favorable. For this reason, it has been recently suggested that a convenient way to lower the potential necessary for achieving the first electron transfer for CO2 reduction is decreasing the CO2˙− free energy of formation. This is achieved by forming a complex between the CO2˙− and an imidazolium based cation such as [C2mim+], since this will help to decrease the overall energy barrier.42 The formation of such a complex has been proposed to be mainly responsible for the highly active ionic-liquid mediated electrochemical conversion of CO2 into CO at low overpotentials in a mixture of water and an imidazolium based ionic liquid. After the first electron transfer forms CO2˙−, several possible reaction pathways may be followed depending on the solvent conditions. They can be mainly divided into those that need the abundant presence of available protons and a second electron transfer, which may form either HCOOH (eqn (2)), if the proton is attached to the CO2˙−, or CO (eqn (3)), if the proton removes one of the oxygen atoms within the CO2˙−. In contrast, if protons are not available, as usually happens in nonaqueous systems, the dimerization reaction mainly produces C2O42− (eqn (4)).
| M + CO2 + e− → M⋯CO2˙− | (1) |
| CO2 + 2H+ + 2e− → HCOOH | (2) |
| CO2 + 2H+ + 2e− → CO | (3) |
| 2CO2 + 2e− → C2O42− | (4) |
Experimental
Chemicals and electrodes
The ionic liquid 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide [C2mim+][NTf2−] 98% was supplied by Aldrich. This was used straight from the supplier, and its water content was minimized by maintaining it under an argon atmosphere within a silica gel container. 1,3-Dimethylimidazolium-2-carboxylate (C6H8N2O2) > 80% was supplied by Aldrich. Bis(trifluoromethanesulfonyl)amine [H+][NTf2−] 95% supplied by Aldrich is a strong acid, which is fully dissociated when dissolved in [C2mim+][NTf2−]. Ar and CO2 ≥ 99.998 purity supplied by Air Liquide were used in all experiments.Pt polycrystalline electrodes and Pt(100), Pt(110) and Pt(111) single crystal electrodes43 were made from single crystal platinum beads (2.5 mm in diameter), obtained by fusion and subsequent slow crystallization of a 99.999% platinum wire (0.5 mm in diameter). For the Pt(hkl) electrodes, after careful cooling, the resulting single crystal beads were oriented, cut and polished following the procedure described by Clavilier et al. in ref. 44. Prior to any experiment, the Pt(hkl) electrodes were air flame-annealed45 for 20–30 s, cooled in a reductive atmosphere (H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ar, ratio 1:3 v/v), quenched in [C2mim+][NTf2−] in equilibrium with that atmosphere and finally immersed in the electrochemical cell under potential controlled conditions using a meniscus configuration.
:Ar, ratio 1:3 v/v), quenched in [C2mim+][NTf2−] in equilibrium with that atmosphere and finally immersed in the electrochemical cell under potential controlled conditions using a meniscus configuration.
Electrochemical characterization
A one compartment electrochemical glass cell with a three-electrode configuration at room temperature and atmospheric pressure was employed for performing cyclic voltammetry experiments. All measurements were made using a computer controlled Autolab PGSTAT30 potentiostat driven by NOVA software. The electrocatalytic activity for the CO2 reduction reaction was studied in the presence and the absence of a proton source using [C2mim+][NTf2−] as a solvent supporting electrolyte system. Prior to any experiment, the electrochemical cell was dried in hot air and Ar gas was flowed into the empty cell in order to remove any trace of water. A small volume of [C2mim+][NTf2−] (∼3.5 mL) was used in each experiment and this was saturated with either Ar or CO2 gas for at least 20 min before each experiment. A platinum wire, 0.5 mm diameter, was used as a counter electrode and the reference electrode was also another Pt wire (quasi-reference electrode). This reference was placed inside of a glass cylinder with a glass frit separator at the bottom and immersed in a solution identical to the one initially used within the electrochemical cell. This is necessary to isolate the quasi-reference electrode as much as possible from potential chemical interference derived from the faradic processes occurring in solution during cyclic voltammetry.46 This is a crucial issue here, since the highly negative potentials reached in voltammetries may provoke H2 evolution to some extent, which in contact with the Pt wire quasi-reference electrode may modify its open circuit potential and subsequently, the reference potential in the electrochemical cell.Results and discussion
RTILs generally exhibit a wide electrochemical potential window. But, in the absence of any other electroactive species, they may undergo electrochemical degradation. The electrochemical stability of the imidazolium cation limits the cathodic polarization window, since the cation [C2mim+] may be reduced to form the corresponding radical [C2mim˙]. The carbon atom between the two nitrogen atoms in the imidazolium ring is the most electron deficient atom and is hence the most active site for reduction as shown in eqn (5).47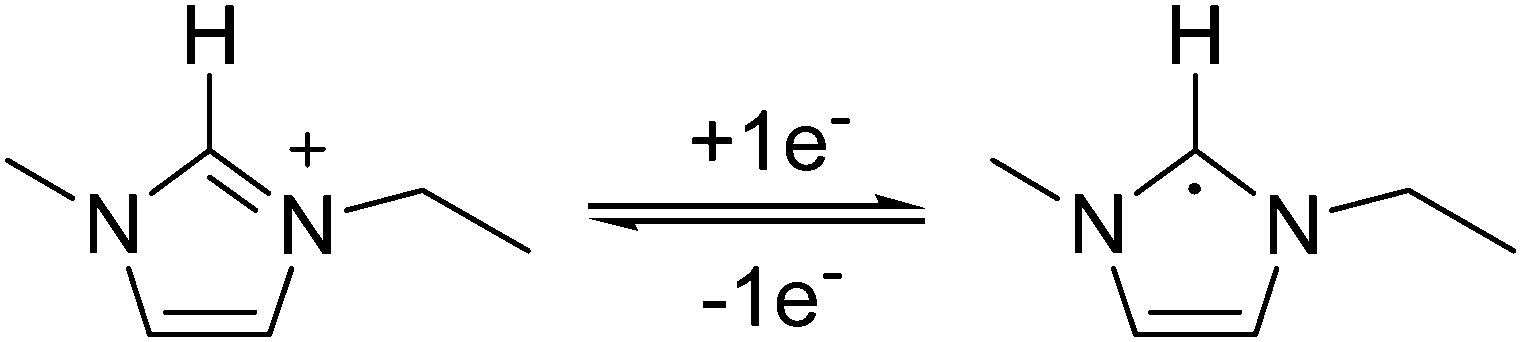 | (5) |
Fig. 1 shows the cyclic voltammogram of [C2mim+][NTf2−] between −1.3 and 1.0 V, where a well-defined quasi-reversible reduction–oxidation response is observed on the Pt(111) electrode, which proves high stability of the radical formed [C2mim˙], since this may be oxidized back to the initial cation. This behavior is observed in both argon and CO2 saturated [C2mim+][NTf2−]. Similar quasi-reversible voltammograms on the Pt(111) electrode were already reported in the literature using several imidazolium-based RTILs.38 However, if the cathodic limit of potential is extended up to −3.1 V, (Fig. 2), this quasi-reversible behaviour disappears. This has been already assigned in the literature to the formation of the corresponding imidazolium carbene by electrochemical reduction and simultaneous hydrogen abstraction following eqn (6)38 or (7).47–49
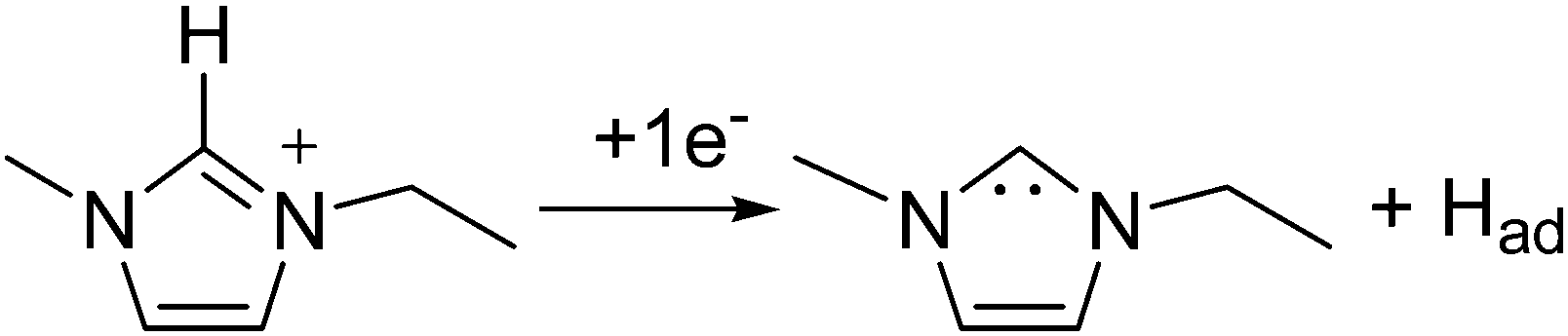 | (6) |
 | (7) |
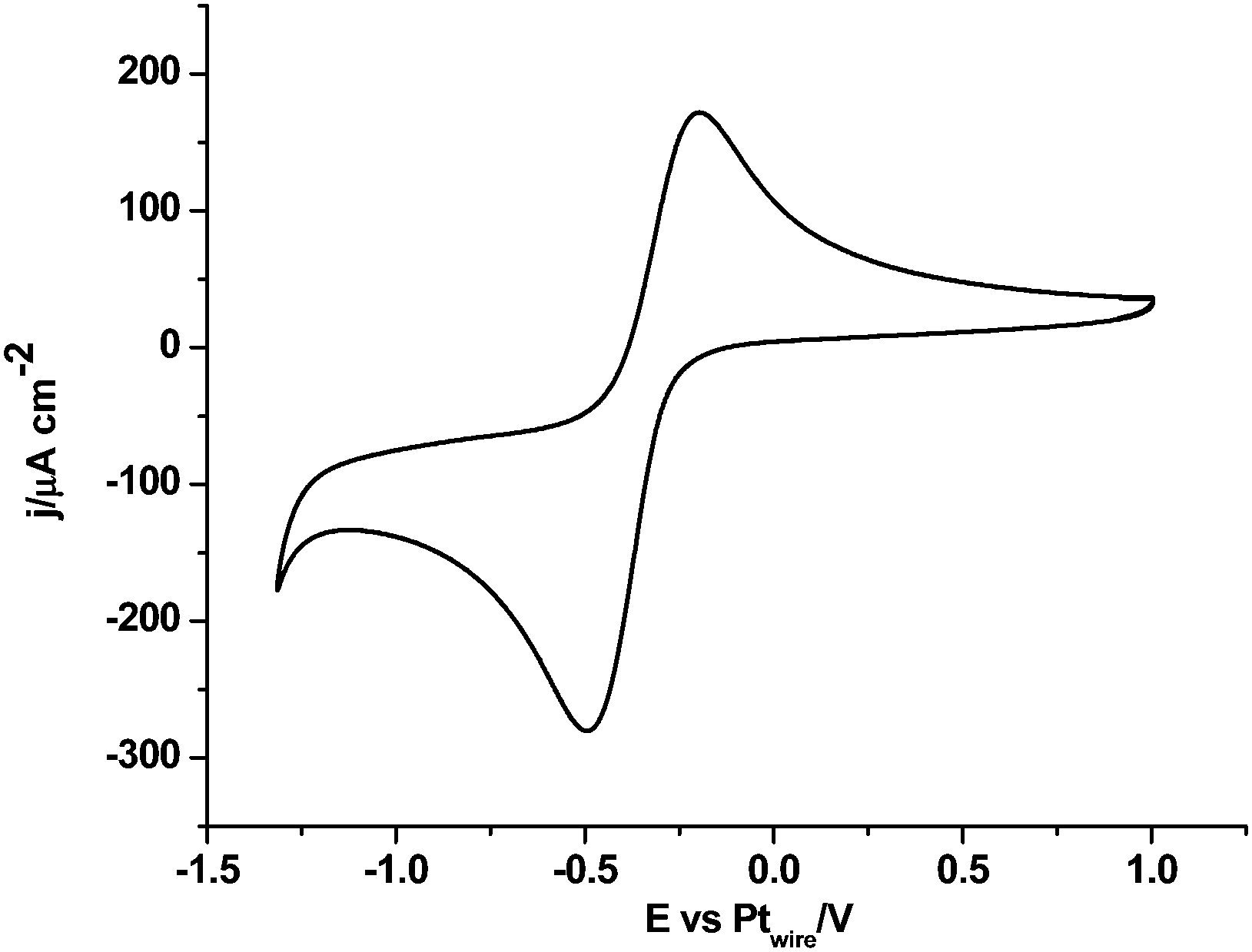 | ||
| Fig. 1 Cyclic voltammogram of Pt(111) in argon saturated [C2mim+][NTf2−]. Scan rate: 0.05 V s−1. | ||
 | ||
| Fig. 2 Cyclic voltammograms of Pt(111), black plot, Pt(110), red plot, and Pt(100), blue plot, in argon saturated [C2mim+][NTf2−]. Scan rate: 0.05 V s−1. | ||
Furthermore, it is possible to observe in Fig. 2 the H2 evolution reaction in all three low index planes of platinum. Oxidation of the newly formed H2 can be more clearly observed on Pt(100) and Pt(110) electrodes in the reverse potential scan (inset of Fig. 2, oxidation peak between −1.7 and −1.0 V). In contrast, the oxidation peak shown at −0.5 V in Fig. 2 has been assigned in the literature49 to the oxidation of the electrogenerated imidazolium carbene back to the initial cation. In particular, Feroci and co-workers have proposed the use of those electrogenerated imidazolium carbenes as a synthetic tool for capturing CO2 and forming an imidazolium-2-carboxylate product by a coupling reaction between the electrogenerated carbene and CO2 as described in eqn (8).49,50 This reported reaction takes place outside the electrochemical cell, by CO2 saturation of [C4mim+][BF4−] after performing a galvanostatic reductive electrolysis on glassy carbon, Pt, Pd or Au for electrogenerating the corresponding imidazolium carbene. This CO2 coupling reaction is reported to produce a diminution in the oxidation current at −0.5 V assigned to the carbene oxidation back to the initial cation when the voltammogram in the CO2 presence is compared with the one in the CO2 absence, since most of the carbene is consumed by reaction with CO2. For this reason, the imidazolium-2-carboxylate product formed in eqn (8) is reported as an electroinactive adduct,49 which may only give back CO2 and imidazolium cations by heating induced decomposition.
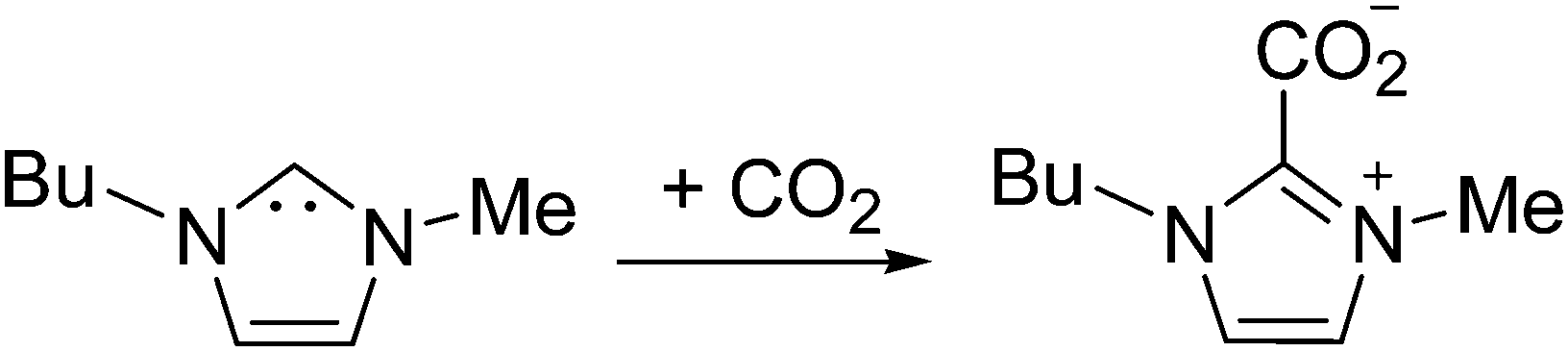 | (8) |
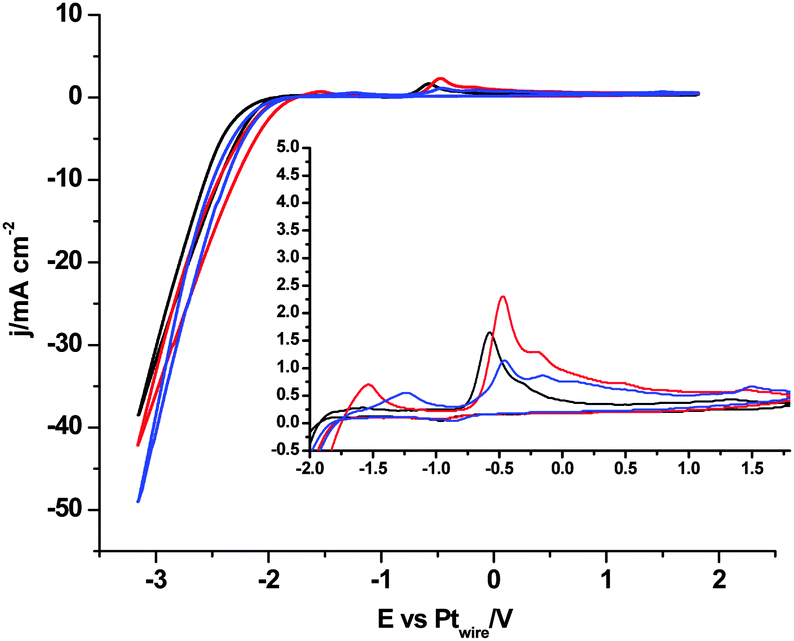
In contrast, when we compare the cyclic voltammograms reaching −3.1 V, in the absence of CO2 in [C2mim+][NTf2−] (Fig. 2) and in CO2 saturated [C2mim+][NTf2−] (Fig. 3), the oxidation peak current at −0.5 V increases for Pt(110) and Pt(100) in the presence of CO2, and decreases only for Pt(111), which points out that different processes are taking place. Firstly, H2 evolution is inhibited in all three Pt(hkl) electrodes when CO2 is present in the electrochemical cell, since no subsequent oxidation current peaks are displayed in the reverse potential scan between −1.7 and −1.0 V (see the inset of Fig. 3). Secondly, the comparison between the insets in Fig. 2 and 3 shows an important increase in the oxidation peak current displayed at −0.5 V for Pt(110) and Pt(100) electrodes when CO2 is present, but none for Pt(111). These facts indicate that the process taking place at potentials more negative than −2.0 V is not only the [C2mim+] reduction following eqn (6) or (7). In fact, this corresponds to the simultaneous reduction of [C2mim+] and CO2 species on the Pt(100) and Pt(110) electrodes, which produces the formation of a new stable species [C2mimH–CO2−] by a radical–radical coupling of both reaction products formed following eqn (1) (CO2˙−) and (5) [C2mim˙], as globally described in eqn (9) for the first time. In contrast, at the Pt(111) electrode, which has been already reported to be less active in acid aqueous solutions for the CO2 electrochemical reduction,34 this radical–radical coupling reaction may not take place in [C2mim+][NTf2−], since −3.1 V is not a cathodic enough potential for significantly reducing CO2 on Pt(111) following eqn (1). Thus, the reported coupling reaction by Feroci and co-workers between the electrogenerated carbene and CO2 described in eqn (8) takes place at Pt(111). However, the imidazolium-2-carboxylate formed is electroinactive and for this reason the peak current at −0.5 V decreases in the presence of CO2 (1.1 mA cm−2) with respect to the absence of CO2 (1.6 mA cm−2), as described by Feroci et al.49,50 In contrast, the new [C2mimH–CO2−] adduct described in eqn (9) and formed on Pt(110) and Pt(100) is electroactive and may be easily oxidized back to the initial imidazolium cation at −0.5 V. For this reason, the oxidation peak current for Pt(110) and Pt(100) displayed in the inset of Fig. 3 is 2–3 times larger than the one displayed in the inset of Fig. 2.
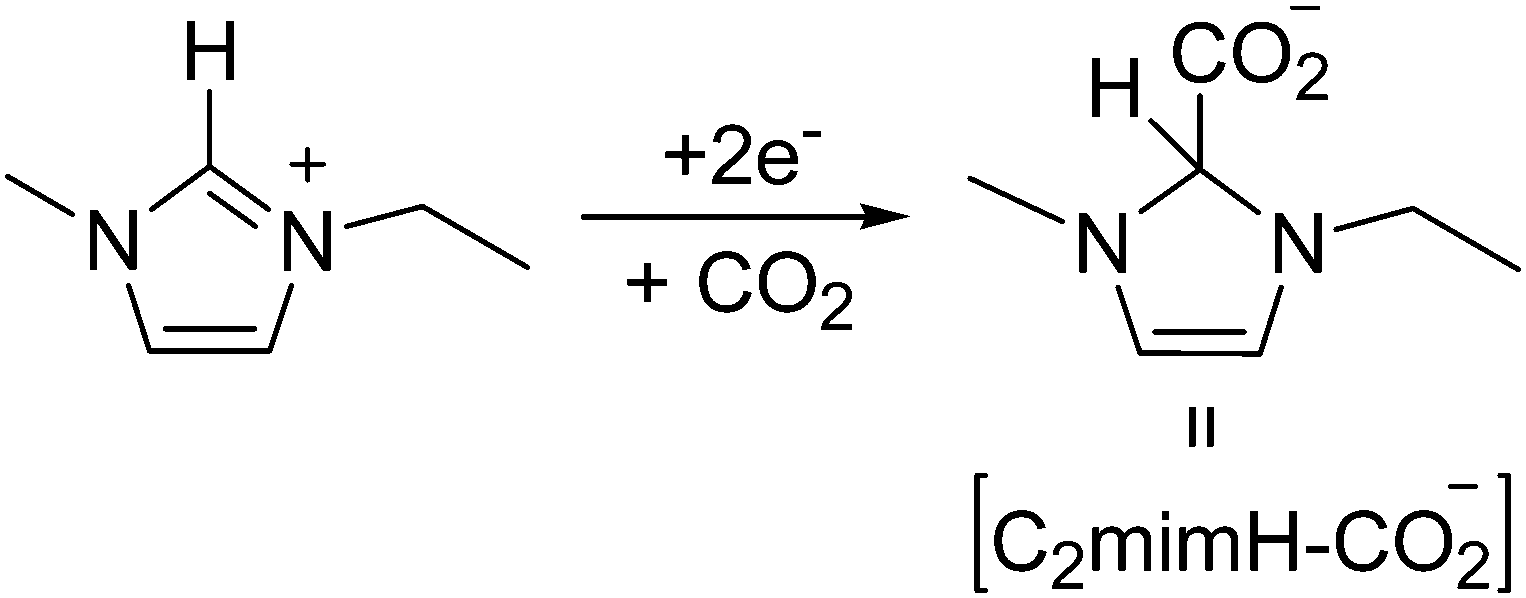 | (9) |
 | ||
| Fig. 3 Cyclic voltammograms of Pt(111), black plot, Pt(110), red plot, and Pt(100), blue plot, in CO2 saturated [C2mim+][NTf2−]. Scan rate: 0.05 V s−1. | ||
Fig. 4 shows the electrochemical behaviour of 1,3-dimethylimidazolium-2-carboxylate on the Pt polycrystalline electrode as a model carboxylic product of the CO2 coupling reaction reported in eqn (8). Fig. 4 shows the total electroinactivity of that compound within the potential range of interest, from −0.75 V to 1.0 V, in agreement with the literature, since the current density displayed in Fig. 4 is negligible in comparison with all other voltammograms and no oxidation or reduction peaks appear. This fact demonstrates that the product generated at −3.1 V by simultaneous reduction of [C2mim+] and CO2 at Pt(110) and Pt(100) electrodes does not follow eqn (8) and thus, it is not the imidazolium-2-carboxylate derivate already described in the literature.
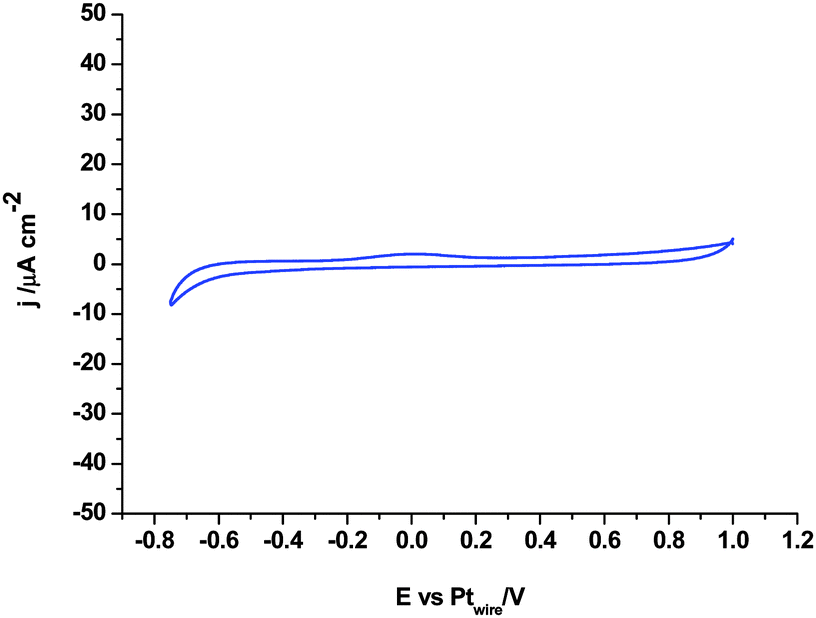 | ||
| Fig. 4 Cyclic voltammogram of the Pt polycrystalline electrode in argon saturated [C2mim+][NTf2−] containing 50 mM 1,3-dimethylimidazolium-2-carboxylate. Scan rate: 0.05 V s−1. | ||
In order to demonstrate that the formation of that new [C2mimH–CO2−] adduct requires the initial electrochemical reduction of both [C2mim+] and CO2, as described in eqn (9) and was never proposed before, Fig. 5 shows a short in situ electrolysis at −3.4 V in the presence and the absence of CO2 followed by a linear voltammogram from −3.4 V to 1.0 V. The peak current at −0.5 V in the argon saturated [C2mim+][NTf2−] does not increase with the electrolysis time (dashed plot in Fig. 5), but in the CO2 saturated [C2mim+][NTf2−] the peak current at −0.5 V exhibits a constant increase, pointing out a great reversibility of the [C2mimH–CO2−] adduct for releasing back [C2mim+] and CO2 under oxidative conditions. Thus, the formation of [C2mimH–CO2−] inhibits radical–radical dimerization reactions to form C2O42− as described in eqn (4) as well as the imidazolium radical dimerization reaction.47,48 However, the electrochemical reduction of CO2 under those conditions provokes the electrochemical degradation of the imidazolium based solvent and inhibits further electrochemical reduction of CO2.
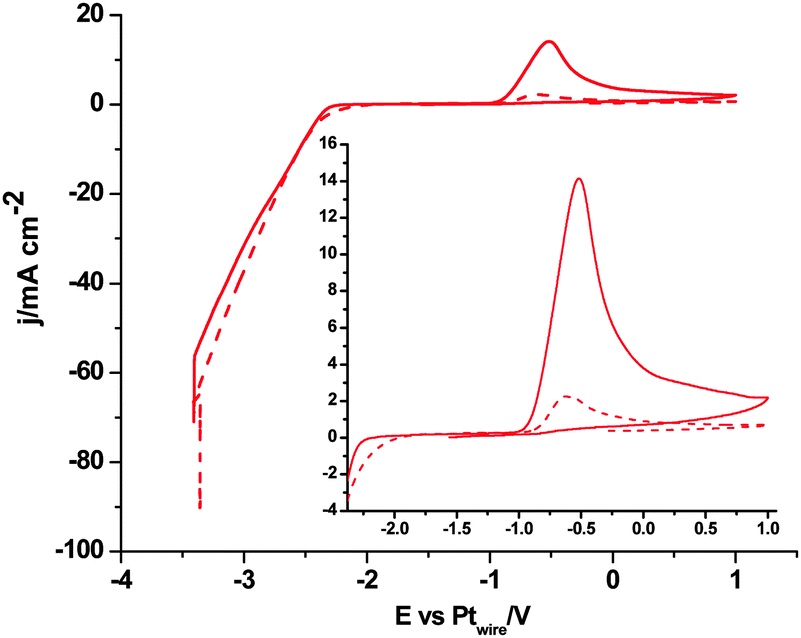 | ||
| Fig. 5 Linear voltammograms of Pt(110) in [C2mim+][NTf2−] after holding constant the potential at −3.4 V for 4 min. Dashed red plot, argon saturated and solid red plot, CO2 saturated solution. Scan rate: 0.05 V s−1. | ||
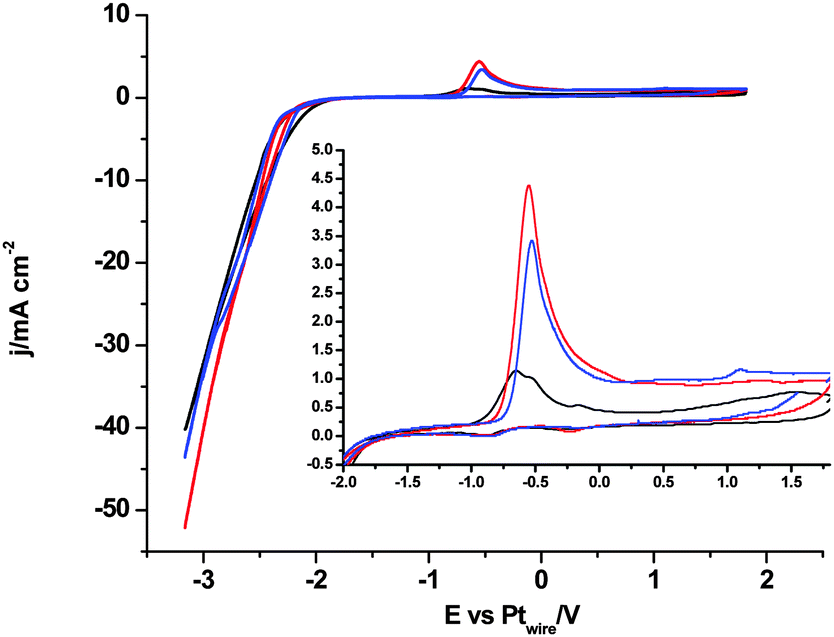
Thus, we think that all voltammograms and the in situ electrolysis reported here demonstrate that the CO2 reduction in [C2mim+][NTf2−] is blocked after a single electron transfer and the simultaneous degradation of the imidazolium based solvent is produced by the formation of a stable [C2mimH–CO2−] adduct. Then, a strong acid as a source of free protons, [H+][NTf2−], is added into solution to facilitate the electrochemical reduction of CO2 following a 2 electron pathway to form either HCOOH (eqn (2))36 or CO (eqn (3)).42 In the absence of CO2, Fig. 6 displays a clear reduction feature at −1.0 V, corresponding to the reduction of protons to form H2 at the Pt(hkl) electrode surface and its corresponding reversible oxidation on the reverse potential scan.38 This process seems to inhibit the [C2mim+] reduction, because no reduction peak is shown at −0.5 V. Thus, the electrode–RTIL interface has been drastically modified by the presence of a high concentration of available protons, which are preferentially adsorbed and have displaced the imidazolium cations from the surface of the electrode, catalyzing the proton reduction and inhibiting the [C2mim+] reduction.
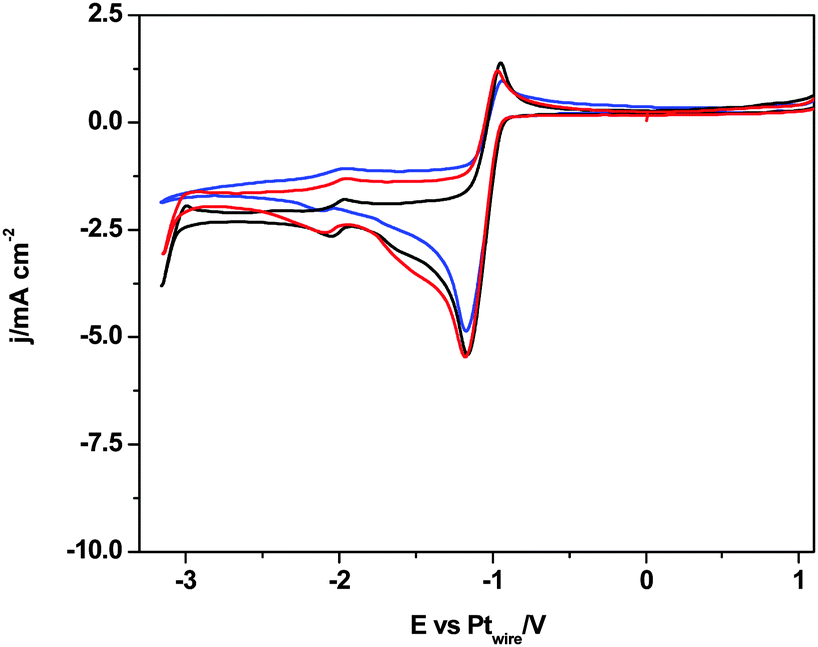 | ||
| Fig. 6 Cyclic voltammograms of Pt(111), black plot, Pt(110), red plot, and Pt(100), blue plot. Solution composition 270 mM [H+][NTf2−] in argon saturated [C2mim+][NTf2−]. Scan rate: 0.05 V s−1. | ||
In contrast, Fig. 7 shows the voltammetric profiles in the presence of dissolved CO2 and protons with different lower potential limits. When the lower potential limit is set at −2.5 V (Fig. 7A), no significant changes are observed in the presence of CO2 in solution, which indicates that the CO2 reduction reaction is not taking place. Only when the lower potential limit is set below −3.0 V, changes in the voltammogram are observed, which indicates that CO2 reduction reaction is taking place. Fig. 7B and C show a new oxidative peak appearing at 0.3 V (see the inset of Fig. 7B and C), which is univocally linked to the reduction of the CO2 species present in solution, since this peak is absent in an argon saturated solution (Fig. 6). This new oxidation peak does not fit our previous findings on the study of the CO electrooxidation reaction in [C2mim+][NTf2−], since the CO oxidation peak appears at more positive potentials.40 Furthermore, a very similar peak has been assigned in a recent paper by Compton's group36 to the oxidation of the HCO2H/HCO2− species formed at a polycrystalline Pt electrode surface after CO2 reduction in [C2mim+][NTf2−]. Thus, herein we prove that the electro-synthesis of HCOOH from CO2 reduction in [C2mim+][NTf2−] is a surface sensitive process, since the three different basal planes of Pt produce different amounts of HCOOH, which is denoted by the different peak area displayed in Fig. 7B and C and quantified in Table 2. Thus, the results in Table 2 show that the Pt(110) electrode is the most active basal plane for this reaction and Pt(111) and Pt(100) electrodes exhibit a similar low activity (3 times lower than that of the Pt(110) electrode at −3.5 V).
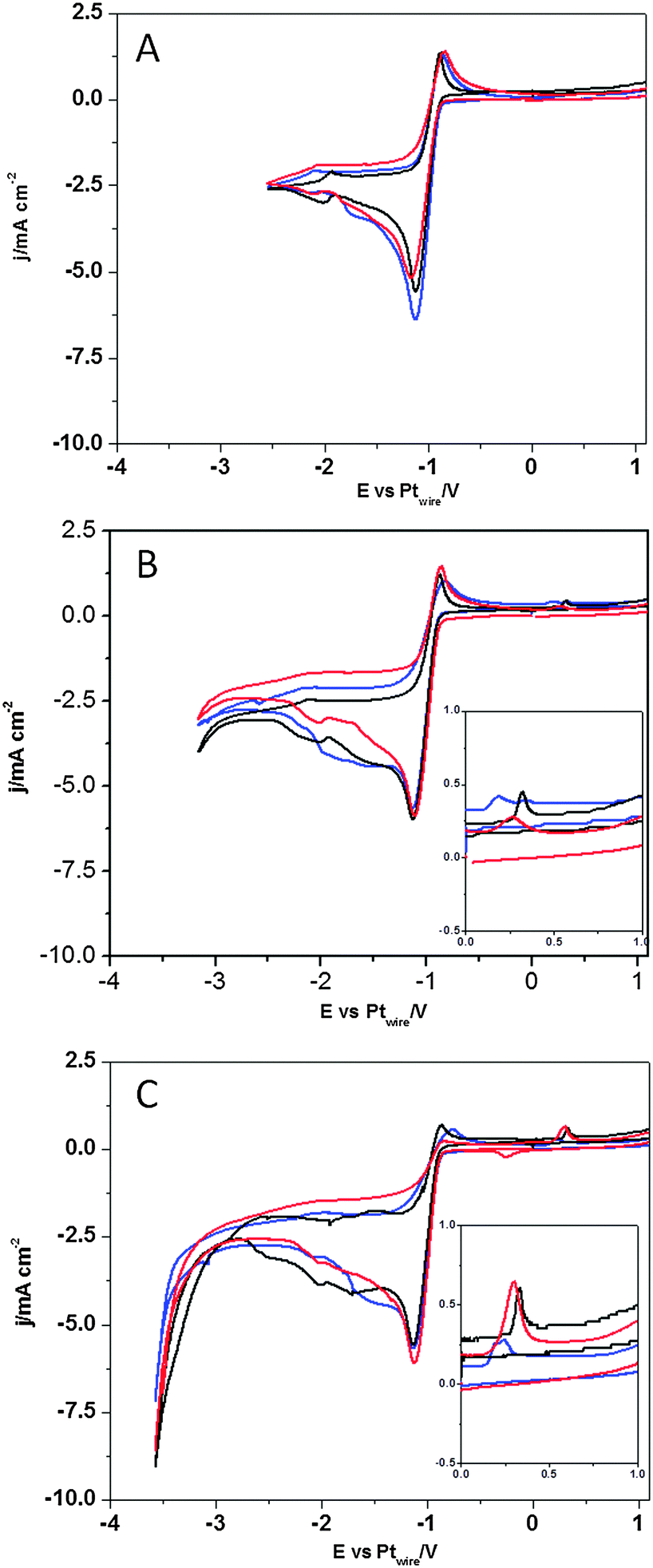 | ||
| Fig. 7 Cyclic voltammograms of Pt(111), black plot, Pt(110), red plot, and Pt(100), blue plot. Solution composition 270 mM [H+][NTf2−] in CO2 saturated [C2mim+][NTf2−]. Scan rate: 0.05 V s−1. Plots A, B and C represent the same experiment, but reaching a different limit in negative potential: −2.5 V for plot A, −3.1 V for plot B and −3.5 V for plot C. | ||
| Cathodic potential limit (V) | Q (μC cm−2) Pt(110) | Q (μC cm−2) Pt(111) | Q (μC cm−2) Pt(100) |
|---|---|---|---|
| −3.1 | 15.4 | 10.8 | 6.22 |
| −3.5 | 46.2 | 14.1 | 12.9 |
Conclusions
We study here the direct electrochemical conversion and recycling of CO2 in one of the most used AILs, [C2mim+][NTf2−], which is mainly hydrophobic and presents a suitable set of physical properties for electrochemical purposes such as moderate viscosity, high conductivity and high CO2 solubility. Moreover, we use Pt single crystal electrodes, which represent the most convenient type of surface structured electrodes for achieving mechanistic information on electrocatalytic reactions.We propose, for the first time, the formation of a stable adduct [C2mimH–CO2−] by a radical–radical coupling after the simultaneous reduction of CO2 and [C2mim+]. It means between the CO2 radical anion and the radical formed from the reduction of the cation [C2mim+]. This is especially relevant from a stability point of view, since this reaction provokes the electrochemical degradation of the RTIL employed. So far, only adducts formed by either the reduced form of 1,3-dialkyl-imidazolium cations (carbene) and CO2,49,50 bare RTIL anions and CO2,35 or the CO2 radical anion and bare 1,3-dialkyl-imidazolium cation42 were previously reported in the literature. However, radical–radical coupling reactions between two 1,3-dialkyl-imidazolium radicals to form a dimer were already reported.47,48
We proved by cyclic voltammetry and in situ electrolysis that the reduction of both CO2 and [C2mim+] is necessary in order to form the stable adduct [C2mimH–CO2−]. Thus, the formation of such a compound represents an important limitation in the future use of RTILs at the industrial scale, since it represents a new cathodic degradation pathway that limits their successful recycling and the extension of the electrochemical CO2 reduction reaction. Nevertheless, the recyclability of [C2mim+][NTf2−] and successful direct CO2 electrochemical reduction are achieved here by adding a proton source [H+][NTf2−] into solution. The presence of protons in high concentration modifies the electrode–RTIL interface by inhibiting the [C2mim+] reduction and favoring the HCOOH electro-synthesis. Proton abundance avoids [C2mim+] from being adsorbed at the Pt electrode surface and blocks its cathodic degradation.
Finally, we continue increasing the use of RTILs as a safe and non-flammable solvent for studying relevant electrocatalytic reactions from an energetic and environmental point of view. Moreover, we demonstrate that the electrochemical reduction of CO2 to form HCOOH at the Pt(hkl)-RTIL interface is considered as a surface sensitive reaction and exhibits the same activity trend as in aqueous acid media, since Pt(110) is the most active electrode studied.
Acknowledgements
This work has been partially financed by Generalitat Valenciana through Ayudas para la realización de proyectos de I+D para grupos de investigación emergentes (GV/2014/096) and by the MICINN (project CTQ2013-48280-C3-3-R). Dr Hanc-Scherer thanks his former supervisor Prof. Petru Ilea for his advice.References
- T. Faunce, S. Styring, M. R. Wasielewski, G. W. Brudvig, A. W. Rutherford, J. Messinger, A. F. Lee, C. L. Hill, H. deGroot, M. Fontecave, D. R. MacFarlane, B. Hankamer, D. G. Nocera, D. M. Tiede, H. Dau, W. Hillier, L. Wang and R. Amal, Energy Environ. Sci., 2013, 6, 1074–1076 Search PubMed.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 CAS.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- B. Batanero, F. Barba, C. M. Sánchez-Sánchez and A. Aldaz, J. Org. Chem., 2004, 69, 2423–2426 CrossRef CAS PubMed.
- A. Gennaro, C. M. Sánchez-Sánchez, A. A. Isse and V. Montiel, Electrochem. Commun., 2004, 6, 627–631 CrossRef CAS PubMed.
- Z. Zhang, Y. Xie, W. Li, S. Hu, J. Song, T. Jiang and B. Han, Angew. Chem., Int. Ed., 2008, 47, 1127–1129 CrossRef CAS PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, J. Org. Chem., 2009, 74, 487–498 CrossRef CAS PubMed.
- F. Sastre, A. V. Puga, L. Liu, A. Corma and H. Garcia, J. Am. Chem. Soc., 2014, 136, 6798–6801 CrossRef CAS PubMed.
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- C. Finn, S. Schnittger, L. J. Yellowlees and J. B. Love, Chem. Commun., 2012, 48, 1392–1399 RSC.
- R. J. Lim, M. Xie, M. A. Sk, J.-M. Lee, A. Fisher, X. Wang and K. H. Lim, Catal. Today, 2014, 233, 169–180 CrossRef CAS PubMed.
- M. Jitaru, D. A. Lowy, M. Toma, B. C. Toma and L. Oniciu, J. Appl. Electrochem., 1997, 27, 875–889 CrossRef CAS.
- C. M. Sánchez-Sánchez, V. Montiel, D. A. Tryk, A. Aldaz and A. Fujishima, Pure Appl. Chem., 2001, 73, 1917–1927 CrossRef.
- R. P. S. Chaplin and A. A. Wragg, J. Appl. Electrochem., 2003, 33, 1107–1123 CrossRef CAS.
- C. M. Sánchez-Sánchez, J. Souza-Garcia, E. Herrero and A. Aldaz, J. Electroanal. Chem., 2012, 668, 51–59 CrossRef PubMed.
- F. Zhou, S. Liu, B. Yang, P. Wang, A. S. Alshammari and Y. Deng, Electrochem. Commun., 2014, 46, 103–106 CrossRef CAS PubMed.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- Topics in Current Chemistry, Ionic Liquids, ed. B. Kirchner, Springer, Heidelberg, 2010, vol. 290 Search PubMed.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 CrossRef CAS PubMed.
- P. Hapiot and C. Lagrost, Chem. Rev., 2008, 108, 2238–2264 CrossRef CAS PubMed.
- C. Cadena, J. L. Anthony, J. K. Shah, T. I. Morrow, J. F. Brennecke and E. J. Maginn, J. Am. Chem. Soc., 2004, 126, 5300–5308 CrossRef CAS PubMed.
- A. M. O'Mahony, D. S. Silvester, L. Aldous, C. Hardacre and R. G. Compton, J. Chem. Eng. Data, 2008, 53, 2884–2891 CrossRef.
- K. Ohta, M. Kawamoto, T. Mizuno and D. A. Lowy, J. Appl. Electrochem., 1998, 28, 717–724 CrossRef CAS.
- A. Gennaro, A. A. Isse and E. Vianello, J. Electroanal. Chem., 1990, 289, 203–215 CrossRef CAS.
- S. S. Moganty and R. E. Baltus, Ind. Eng. Chem. Res., 2010, 49, 5846–5853 CrossRef CAS.
- H.-P. Steinrück and P. Wasserscheid, Catal. Lett., 2015, 145, 380–397 CrossRef.
- Y.-Z. Su, Y.-C. Fu, Y.-M. Wei, J.-W. Yan and B.-W. Mao, ChemPhysChem, 2010, 11, 2764–2778 CrossRef CAS PubMed.
- Y.-Z. Su, Y.-C. Fu, J.-W. Yan, Z.-B. Chen and B.-W. Mao, Angew. Chem., Int. Ed., 2009, 48, 5148–5151 CrossRef CAS PubMed.
- S. Taguchi and A. Aramata, Electrochim. Acta, 1994, 39, 2533–2537 CrossRef CAS.
- N. Hoshi, S. Kawatani, M. Kudo and Y. Hori, J. Electroanal. Chem., 1999, 467, 67–73 CrossRef.
- N. Hoshi and Y. Hori, Electrochim. Acta, 2000, 45, 4263–4270 CrossRef CAS.
- L. L. Snuffin, L. W. Whaley and L. Yu, J. Electrochem. Soc., 2011, 158, F155–F158 CrossRef CAS PubMed.
- B. C. M. Martindale and R. G. Compton, Chem. Commun., 2012, 48, 6487–6489 RSC.
- A. P. Sandoval, J. M. Feliu, R. M. Torresi and M. F. Suárez-Herrera, RSC Adv., 2014, 4, 3383–3391 RSC.
- A. M. Navarro-Suárez, J. C. Hidalgo-Acosta, L. Fadini, J. M. Feliu and M. F. Suárez-Herrera, J. Phys. Chem. C, 2011, 115, 11147–11155 Search PubMed.
- A. P. Sandoval, M. F. Suárez-Herrera and J. M. Feliu, Electrochem. Commun., 2014, 46, 84–86 CrossRef CAS PubMed.
- F. A. Hanc-Scherer, C. M. Sánchez-Sánchez, P. Ilea and E. Herrero, ACS Catal., 2013, 3, 2935–2938 CrossRef CAS.
- C. M. Sánchez-Sánchez, E. Expósito, B. Batanero, V. Montiel, F. Barba and A. Aldaz, Electrochem. Commun., 2004, 6, 595–599 CrossRef PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- V. Climent and J. M. Feliu, J. Solid State Electrochem., 2011, 15, 1297–1315 CrossRef CAS.
- J. Clavilier, D. Armand, S. G. Sun and M. Petit, J. Electroanal. Chem., 1986, 205, 267–277 CrossRef CAS.
- J. Clavilier, R. Faure, G. Guinet and R. Durand, J. Electroanal. Chem., 1980, 107, 205–209 CrossRef CAS.
- Electrochemical Aspects of Ionic Liquids, ed. H. Ohno, John Wiley and Sons, Hoboken, New Jersey, 2005, p. 30 Search PubMed.
- G. H. Lane, Electrochim. Acta, 2012, 83, 513–528 CrossRef CAS PubMed.
- N. De Vos, C. Maton and C. V. Stevens, ChemElectroChem, 2014, 1, 1258–1270 CrossRef CAS PubMed.
- M. Feroci, I. Chiarotto, S. V. Ciprioti and A. Inesi, Electrochim. Acta, 2013, 109, 95–101 CrossRef CAS PubMed.
- M. Feroci, I. Chiarotto, G. Forte and A. Inesi, J. CO2 Util., 2013, 2, 29–34 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2015 |