
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Adsorption of bovine serum albumin on silver surfaces enhances the release of silver at pH neutral conditions
X.
Wang
*a,
G.
Herting
a,
I.
Odnevall Wallinder
a and
E.
Blomberg
*ab
aKTH Royal Institute of Technology, School of Chemical Science and Engineering, Department of Chemistry, Surface and Corrosion Science, Drottning Kristinas väg 51, SE-100 44 Stockholm, Sweden. E-mail: blev@kth.se
bSP Technical Research Institute of Sweden, Chemistry, Material and Surface, P.O. Box 5607, SE-114 86 Stockholm, Sweden
First published on 19th June 2015
Abstract
Metallic biomaterials are widely used to replace and/or restore the function of damaged bodily parts. The use of silver as antibacterial coatings onto implants has recently gained large interest in medical applications. The extent of silver that can be released into different biological fluids from such coatings is, except for the surface characteristics of the coating, governed by parameters such as protein characteristics, adsorbed layer properties, formation of silver–protein complexes as well as concentrations of proteins in the solution. This study aims to relate the structure of adsorbed net negatively charged bovine serum albumin (BSA), which is the most abundant protein in serum, to the release of silver from metallic silver surfaces in order to elucidate if the net charge of the protein has any effect of the silver release. Simultaneous adsorption measurements were performed in real time on the very same surface using combined ellipsometry and quartz crystal microbalance with dissipation monitoring (QCM-D) measurements to provide a more comprehensive understanding on adsorption kinetics and layer structures. The amount of released silver into solution was measured by means of graphite furnace atomic absorption spectroscopy (GF-AAS). The structure of the adsorbed BSA layer largely influenced the amount of released silver, an enhancement that increased with BSA concentration. These observations are in complete contrast to the effect of net positively charged lysozyme (LSZ) adsorbed on silver, previously studied by the authors, for which a complete surface coverage suppressed the possibility for silver release. The underlying mechanisms behind the enhanced release of silver in the presence of BSA were mainly attributed to surface complexation between BSA and silver followed by an enhanced exchange rate of these surface complexes with BSA molecules in the solution, which in turn increase the amount of released silver in solution.
1. Introduction
Due to its broad-spectrum antibacterial property, silver is widely used in daily life and medical systems.1–4 Silver has been recognized for its abilities to prevent diseases for thousands of years. In the early 20th century, Halstead promoted metallic silver foil as an antibacterial wound dressing.5 Looping silver wire implants through the dorsal skin of rats efficiently reduced colonization of Staphylococus aureus bacteria near the interface.6 Recently, the use of silver as an effective antimicrobial agent has increased for a variety of medical devices. Silver is for instance applied as coatings of implants (e.g. prostheses) to inhibit adhesion of microorganisms and thereby reduce any biofilm-forming infection.1,2,7 Significantly reduced infection rates have been observed when using silver-coated prostheses compared with non-coated prostheses in patients with bone sarcoma.8Some studies claim that silver is generally toxic to bacteria as it destructs cell membranes, but relatively less toxic to humans.3,9,10 Other studies report that silver is toxic to human cells (e.g. leukocytes) and may at certain conditions be more toxic than mercury.11,12 From a long term use of silver-coated implants follows that silver not only locally accumulates in the surrounding fluids and tissues but also can spread systemically, effects that are associated with certain diseases.13,14 Potential risks for human adverse effects caused by the use of silver may hence increase due to its rapid use in novel applications on the market.
The underlying mechanisms of the antibacterial properties of silver, especially for silver nanoparticles are at present not completely understood. Studies show that zero-valent silver has no antibacterial properties, whereas the silver ion is remarkably reactive.15,16 This demonstrates the importance to quantitatively assess the release of silver from metallic silver and to study interactions with biological systems. Proteins will, as an example, spontaneously and rapidly adsorb on the surface of a silver coated implant material,17,18 and act as the real interface towards the biological environment. Previous studies by the authors, and literature findings show that proteins significantly influence the metal release process of metallic materials.19–22 Depending on material and surface properties, type of proteins and prevailing chemical conditions, the release of metals can either be enhanced or depressed. Few studies exist up to date on silver that assess these aspects.
In a previous study by the authors, the influence of adsorption of net positively charged LSZ on the release of silver was investigated using the same experimental procedure as in this study. From this study it was evident that a full surface coverage of LSZ with a bilayer structure efficiently suppresses the released amount of silver.22 This was attributed to the net positively charged protein/solution interface that efficiently hinders the transport of positively charged silver ions trough the protein layer due to electrostatic repulsion.
The objectives of this study are to investigate the adsorption of net negatively charged bovine serum albumin (BSA) on massive silver surfaces (mimicking e.g. a silver-coated implant surface) and its influence on the release of silver ions at pH neutral conditions in order to elucidate if the properties of the protein have any influence of the mechanism of silver release in the presence of proteins. Parallel investigations were made using the polypeptide, poly-L-glutamic acid (PGA), to gain more insight on whether the silver release process is solely influenced by surface adsorbed BSA and/or BSA in solution. The research goal was to deduce a plausible mechanism on protein-induced silver release based on the structure of the adsorbed BSA layer.
Adsorption of BSA on silver was monitored in real time by means of QCM-D and ellipsometry, and their combination. Ex situ time-dependent measurements of the extent of silver release into solution were performed by means of GF-AAS, both in the absence and presence of proteins/polypeptides of different concentration in solution.
2. Experimental
2.1 Chemicals, solutions and substrates preparation
Purified albumin, from bovine serum (BSA, A7906), poly-L-glutamic acid sodium salt (PGA, Mw 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–50000, P7906), ultrapure sodium nitrate (≥99.999%) for the adsorption studies, and sodium nitrate (≥99.5%) for the silver release studies were purchased from Sigma-Aldrich. Serum albumin is the most abundant blood protein in mammals. BSA is the analog of human serum albumin (HSA), which is abundant in plasma.23,24 BSA has a isoelectric point (iep) of 4.7–5.223,25 and is hence negatively charged in a pH neutral fluid. The polypeptide, PGA, was chosen for comparative reasons due to its similarity with BSA, i.e. PGA (pKa, 4.07–4.2526) is net negatively charged to BSA at pH 7 and glutamic acid is one of the most abundant amino acid in the amino acid sequence of BSA.
000–50000, P7906), ultrapure sodium nitrate (≥99.999%) for the adsorption studies, and sodium nitrate (≥99.5%) for the silver release studies were purchased from Sigma-Aldrich. Serum albumin is the most abundant blood protein in mammals. BSA is the analog of human serum albumin (HSA), which is abundant in plasma.23,24 BSA has a isoelectric point (iep) of 4.7–5.223,25 and is hence negatively charged in a pH neutral fluid. The polypeptide, PGA, was chosen for comparative reasons due to its similarity with BSA, i.e. PGA (pKa, 4.07–4.2526) is net negatively charged to BSA at pH 7 and glutamic acid is one of the most abundant amino acid in the amino acid sequence of BSA.
Ultra-pure water (18.2 MΩ cm, Millipore Sweden) was used for the preparation of all aqueous solutions. 0.15 M sodium nitrate solution (NaNO3) was used as background solution, considering the fact that it has a comparable ionic strength in solution with chloride in PBS buffer, and that it will not interfere in the analysis of released silver in solution. A series of protein solutions with different concentrations of BSA (0.001, 0.01, 0.1, and 1 g L−1) and a PGA solution (1 g L−1) were prepared by the direct dissolution of BSA or PGA into the NaNO3 solution. 1% sodium hydroxide (NaOH), diluted from 50% NaOH (BHD Prolabo, VWR), was used to adjust the solution pH to 7.
Silver-coated (100 nm thick silver layer) AT-cut quartz crystals (QSX 322, Q-sense AB, Sweden) with a surface area of approximately 1.5 cm2 were used for adsorption measurements. The substrates were cleaned by immersion in isopropanol and ethanol for 15 min, respectively. The crystals were subsequently rinsed in MilliQ water and dried under gentle flow of cold nitrogen gas.
Double sided massive silver coupons, approximately sized 2.5 × 1.0 × 0.05 cm, were used for the silver release measurements. Prior to exposure, all surfaces (6 sides) were abraded by 1200 grit SiC paper, followed by ultrasonic cleaning in isopropanol and ethanol, dried with nitrogen gas and pre-aged in a desiccator for 24 h. This was done to ensure comparable and reproducible initial surface characteristics.
2.2 Quartz crystal microbalance with dissipation monitoring (QCM-D)
A quartz crystal microbalance with dissipation monitoring (Q-Sense E-4 or E-1, Q-Sense AB, Sweden) was used to monitor the adsorption of BSA or the PGA on silver-coated QCM crystals. QCM-D allows simultaneous changes in resonance frequency (Δf) and energy dissipation (ΔD) at multiple odd overtones with a fundamental frequency of 5 MHz to be recorded. The solutions were injected into the measuring chamber by means of a peristaltic pump using a flow rate of 200 μL min−1 at a fixed temperature of 25 °C. The sensed mass of the adsorbed layer was estimated with the Sauerbrey relation27 (eqn (1)), since the energy dissipation was very small (<1 × 10−6), indicative of a rigid layer.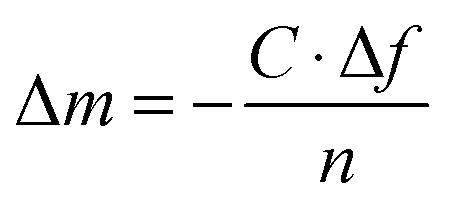 | (1) |
2.3 Ellipsometry combined with QCM-D
An ellipsometer was combined with QCM-D (Q-Sense E1 system) to simultaneously monitor the adsorption of BSA or PGA on the very same silver-coated substrate by using a Q-Sense ellipsometry module (from Q-Sense AB, Sweden). A Multiskop instrument (Optrel GdBR, Berlin, Germany) equipped with a 532 nm laser (Nd-YAG Laser), was used to perform Nullellipsometry measurements at an incidence angle of 65° and a flow rate of 200 μL min−1. A model of nullellipsometry kinetics was selected to study adsorption kinetics by recording the time evolution of ellipsometric angles in situ, delta (Δ) and psi (Ψ), every 10 s from the repeated cycles of two zone 1/3 measures. The de Feijter formula (eqn (2)) was used to estimate the mass of the adsorbed layer, expressed as:28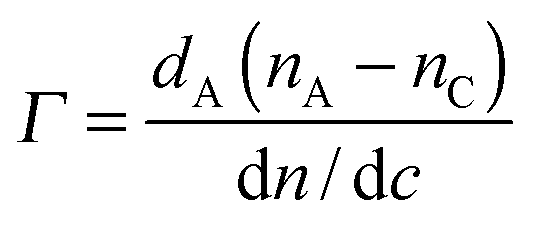 | (2) |
By utilizing the combined QCM-D and ellipsometry set-up, adsorption measurements were conducted on the very same surface with the same error signals deriving from the experimental cell and/or surface, and the same mass transport effect.32 The sensed mass (“wet” mass) of adsorbed proteins was measured by means of QCM-D, including trapped water (background solution) in the layer, and the temporal variation in molecular mass (“dry” mass) was simultaneously evaluated using ellipsometry.28,33 Variations in layer hydration, and hence information on layer structures, are yielded by comparing differences between the “wet” and “dry” mass according to eqn (3):34
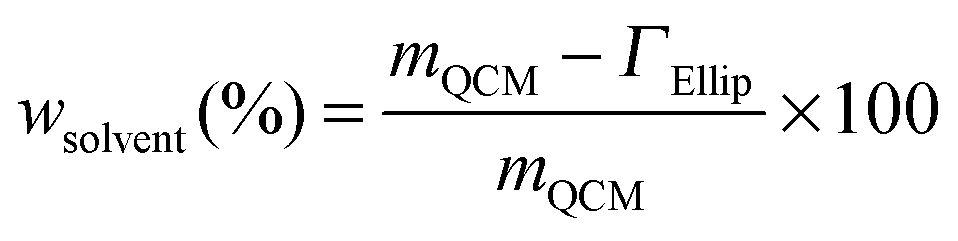 | (3) |
2.4 Silver release measurements
Cleaned and aged massive silver coupons were exposed to 0.15 M NaNO3 solutions (pH = 7 ± 0.2) containing different concentrations of BSA (0.01, 0.1 and 1 g L−1) or PGA (1 g L−1). The surface area/solution volume ratio was set to 1 cm2 mL−1. Exposures were conducted for 2 and 24 h in an incubator (Platform-Rocker incubator SI80, Stuart) at 25 ± 0.1 °C. After exposure, the coupons were immediately withdrawn from the test vessels and the exposed solutions acidified by addition of ultrapure 65 vol% HNO3 to a pH < 2, before stored for subsequent digestion and released silver concentration analysis. Triplicate samples and one blank (not exposed to silver) were exposed in parallel and analyzed to ensure data reliability.Silver coupons (triplicate) were alternately exposed to 0.15 M NaNO3 solutions without BSA (NA) and with 1 g L−1 BSA (BSA). Each coupon was immersed in each solution for 30 min, followed by its consecutive immersion into the next “fresh” solution (NA or BSA).22 Four different exposure sequences were investigated: (i) NaNO3 (NA–NA–NA–NA–NA–NA), (ii) 1 g L−1 BSA in NaNO3 (BSA–BSA–BSA–BSA–BSA–BSA), (iii) consecutive immersion in BSA and NaNO3 (BSA–NA–BSA–NA–BSA–NA), and (iv) consecutive immersion in BSA, NaNO3 and NaNO3 (BSA–NA–NA–BSA–NA–NA). After the final immersion step, the coupons were removed from solution, and each solution in every step and sequence was collected and acidified to pH < 2 for further determination of released silver. Triplicate solutions and one blank solution were evaluated for each step.
All vessels and tools were acid-cleaned prior to use to avoid contamination (immersion in 10% HNO3 for at least 24 h, rinsing four times with ultrapure water and drying using cold nitrogen gas).
2.5 Digestion procedure for analyses of silver in solution
All collected and acidified solutions were UV digested (705 UV Digester, Metrohm) to ensure measurements of the total concentration of silver in solution by means of GF-AAS (graphite furnace atomic absorption spectroscopy). Digestion was performed according to the following procedure: (i) 2 mL solution was mixed with 250 μL 30 vol% H2O2 (Fluka), 6.6 mL ultrapure water and 150 μL 30 vol% HCl (Fluka) and digested for 30 min, (ii) 1 mL H2O2 was added and the solution digested for another 30 min, (iii) another 1 mL H2O2 was added and the solution digested for 60 min. The digested solutions were allowed to cool down to the ambient laboratory temperature before stored in acid-cleaned containers for subsequent GF-AAS analyses. Since the collected solutions were diluted (approximately 5 times), the digested volumes were recorded and recalculated to assess the total silver concentration in each solution.2.6 Graphite furnace atomic absorption spectroscopy (GF-AAS)
Graphite furnace atomic absorption spectroscopy (GF-AAS, Perkin Elmer AAnalyst 800) was utilized to analyze the total concentration of silver in digested solutions. The calibration curve was generated from five standards in ultrapure water (0, 7.5, 15, 30, and 45 μg L−1). Measured concentrations were derived from triplicate measurements of each solution. The limit of detection (LOD) was evaluated with the Vogelsang and Hädrich method, considering grade linearity, recovery of the samples and solution matrix.35 The LOD was yielded from the linear range in concentrations of each solution, i.e. 0–45 μg L−1 for the NaNO3 and BSA solutions and 0–30 μg L−1 for the PGA solution based on calculations from the calibration curve and measurements in matrix solutions with standard concentrations of silver (0, 7.5, 15, 30, and 45 μg L−1). LODs were determined to 2.1, 2.8 and 1.0 μg L−1 in NaNO3, BSA and PGA solutions, respectively. All reported data are above LOD. The corresponding limits of quantification (LOQ) were 7.4, 9.7 and 3.3 μg L−1, respectively. Total released silver concentrations were recalculated considering the dilution factor multiplied with the measured concentration of each digested solution, subtracting the blank concentration (solution exposed in parallel without silver coupons).3. Results and discussion
3.1 BSA irreversibly adsorbs as a monolayer on silver in a side-on confirmation, and its layer structure is influenced by the protein concentration in solution
The adsorption of BSA (1 g L−1) in NaNO3 (0.15 M) with time on silver, monitored by QCM-D, is presented in Fig. 1a. The arrows marked t0 and t2 display the times for protein injection and subsequent rinsing with NaNO3 solution, respectively. The frequency changes (Δf) sharply shift down at t0, while the energy dissipation changes (ΔD) rapidly shifts up. This illustrates an immediate (within a few seconds, t0 → t1) and rapid adsorption of BSA onto the silver surface. Between t1 to t2 follows a linear increase in Δf, which is associated with a constant value of ΔD, clearly elucidated in Fig. 1b. This behavior is different from findings on BSA adsorption on stainless steel and chromium for which plateaus of both Δf and ΔD are simultaneously reached within a few minutes, i.e. the achievement of adsorption equilibrium.19 A change in energy dissipation lower than 1.0 × 10−6 implies the formation of a rigid protein layer on a surface.36 This is illustrated further by the ΔD/Δf curve in Fig. 1b that shows Δf increase without any increased dissipation, characteristic of a fairly rigid layer. The increase in magnitude of Δf is related to a reduction in sensed mass. The reasons could be manifold, e.g. overshoot adsorption followed by re-orientation of adsorbed proteins, desorption of trapped water (solvent) within the adsorbed layer, and/or release of silver from the substrate. An overshoot adsorption followed by a layer reorientation is usually seen as a rapid increase followed by a slower exponential-like decrease, indicative of a mass loss. Δf increases slightly upon injection and rinsing with protein-free NaNO3 (t2 in Fig. 1a). An initial rapid increase in Δf follows a more linear increase, however, at a lower rate compared with conditions with BSA in solution. Meanwhile, ΔD decreases and reaches a stable value after a rapid decrease. This illustrates desorption of a few protein molecules from the surface during rinsing, clearly shown by the ΔD/Δf plot (Fig. 1b) with a reduction in both mass (Δf) and ΔD during rinsing.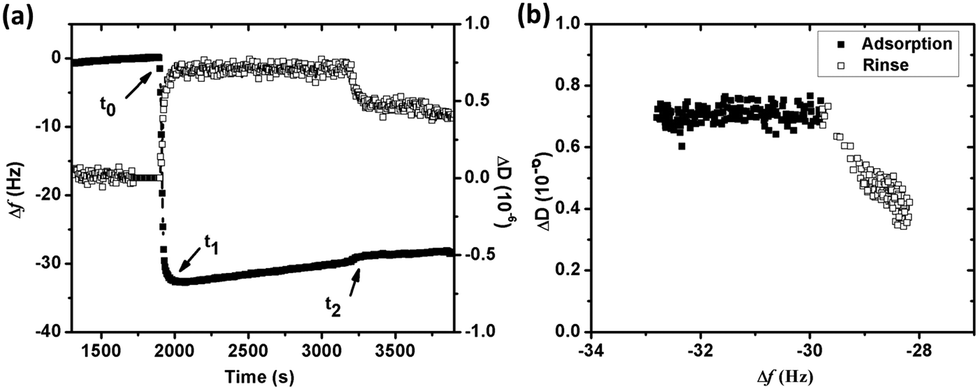 | ||
| Fig. 1 (a) Changes in frequency (Δf) (filled squares) and energy dissipation (ΔD) (open squares) versus time during adsorption of BSA (1 g L−1) onto silver in 0.15 M NaNO3 (pH 7.0 and 25 °C) monitored by QCM-D. BSA solution was injected at t0 and non-protein containing NaNO3 solution was used to rinse the surface at t2 (the time axis was cut to improve the visibility), (b) dissipation shift (ΔD) plotted versus the frequency shift (Δf) from t1 in (a) for overtone 5, randomly selected to illustrate observed changes. | ||
The linear increase of Δf with time observed in Fig. 1a is assumed to be attributed to the release of silver from the underlying surface. The same effect has been observed in a previous study by the authors on the adsorption of lysozyme on silver-coated surfaces.22 Changes in frequency as a function of time were therefore analyzed in more detail to obtain a deeper understanding of the adsorption behavior of BSA onto silver. Prior to the injection of BSA, the baseline, i.e. initial exposure of the substrate to non-protein containing NaNO3, exhibits a linear increase in Δf with time (well fitted with linear regression with a slope of 0.0010), Fig. 2. Similar drifts are observed during adsorption and upon rinsing. The measured Δf between t1 and t2 (i.e. during adsorption) is also linear (slope of 0.0026), a slope which more than two times higher compared with the slope of the baseline. The mass loss rate is reduced during rinsing, as shown by a lower slope (0.0007) that is similar to the slope of the baseline (0.0010). This indicates that the release of silver from the underlying surface most probably is the reason for the observed linear increase in Δf. This is further supported by the of silver in the collected solution before, during and after the adsorption process. However, the amount of silver release was not quantitatively assessed for these solutions due to the lack of a blank solution (without silver). A higher mass loss rate observed in the presence of BSA in solution indicates that more silver is released from the surface during adsorption, an effect further discussed in Section 3.2.
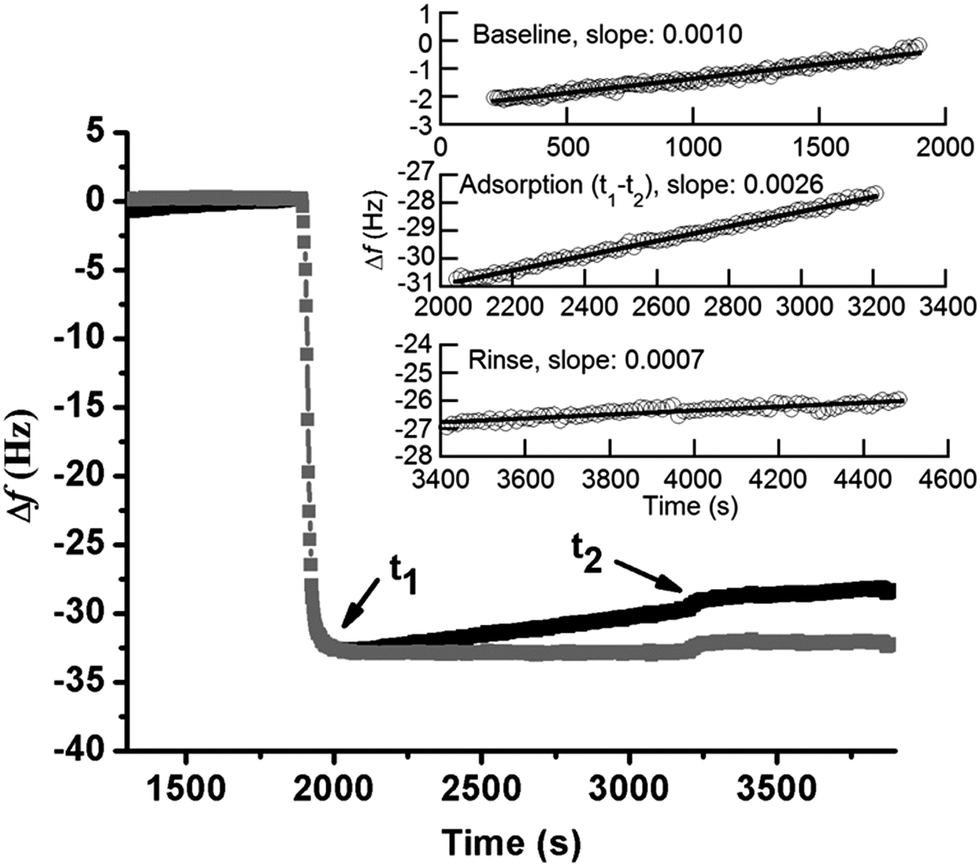 | ||
| Fig. 2 Monitored (black) and corrected (grey) frequency changes (Δf) with time by means of QCM-D for adsorbed BSA (1 g L−1, pH 7.0 and 25 °C) on silver (the time axis was cut to improve the visibility). Corrected data is calculated by offsetting the linear reduction, shown in the inserted figure, where the baseline and adsorption process between t1 and t2 and the rinsing step were linearly fitted (this correction does not change the measured Δf significantly). | ||
When subtracting the linear Δf shift of the baseline from the measured Δf, a frequency plateau is evident both during adsorption and rinsing, Fig. 2 (this correction does not change the measured Δf significantly). This observation shows that adsorption equilibrium is obtained within a few minutes after injection of BSA. Observed results are consistent with characteristic frequency changes during adsorption of BSA (0.5 g L−1) on gold, where complete and irreversible adsorption of BSA is reached within 2 min once it reaches the surface.37 Irreversible adsorption is also observed in this study where only a slight increase in Δf is observed upon rinsing (t2), see corrected curve in Fig. 2. Previous studies have shown that BSA readily adsorb onto almost any surface.19,38,39 If only considering electrostatic interactions, BSA would not adsorb onto the silver surface as BSA and the silver coated crystal both are negatively charged at pH 7.19,22 However, the high ionic strength of NaNO3 (0.15 M) screens the charges of the silver surface and BSA efficiently, and the electrostatic repulsion will hence be less long-ranged (Debye length, κ−1 = 0.304/C01/2 = 0.78 nm, for 1:1 NaNO3).40 This screening of surface charges, which reduces the repulsion both between BSA molecules as between BSA and the surface, is favorable for the adsorption of BSA. The strong binding of BSA to silver might also be related to the formation of sulfur–silver complexes as BSA contains 17 disulfide bridges and one free sulfhydryl (Cys-34).41 It is hence plausible that these groups will form strong bonds with the silver surface as is the case for other S–S containing proteins.23,42
Adsorption of BSA on silver is concentration dependent (from 0.001 to 1 g L−1), Fig. 3a. Since the adsorbed protein layer is assumed rigid due to small changes in energy dissipation (ΔD < 1 × 10−6) and small differences in Δf and ΔD between different overtones, Fig. 3b, the Sauerbrey model was used to evaluate the sensed mass (“wet” mass). All measurements were corrected for mass losses induced by released silver, Fig. 3a. The addition of BSA to the pH adjusted NaNO3 solution changed the pH from 7 to 5.5–6.6, depending on BSA concentration. This pH effect increased with reduced BSA concentration, and may be attributed both to the dissolution of CO2 from the air and to ionic dissociation of the negatively charged BSA molecule. The sensed mass of adsorbed BSA increases for these non pH-adjusted solutions, and gradually approaches a plateau with increasing protein concentration. The sensed masses correspond to approximately 3.1 mg m−2 in 0.001 g L−1 BSA, and to 5.7 mg m−2 in 1 g L−1 BSA, Fig. 3a. pH adjusted protein solutions (pH 7) reveal plateaus at 3.1 mg m−2 (0.001 g L−1 BSA) and 5.7 mg m−2 (1 g L−1 BSA), i.e. a marginal effect due to differences in pH. This observation is in line with the screening of the electrostatic interaction in high ionic strengths solutions, as discussed earlier.
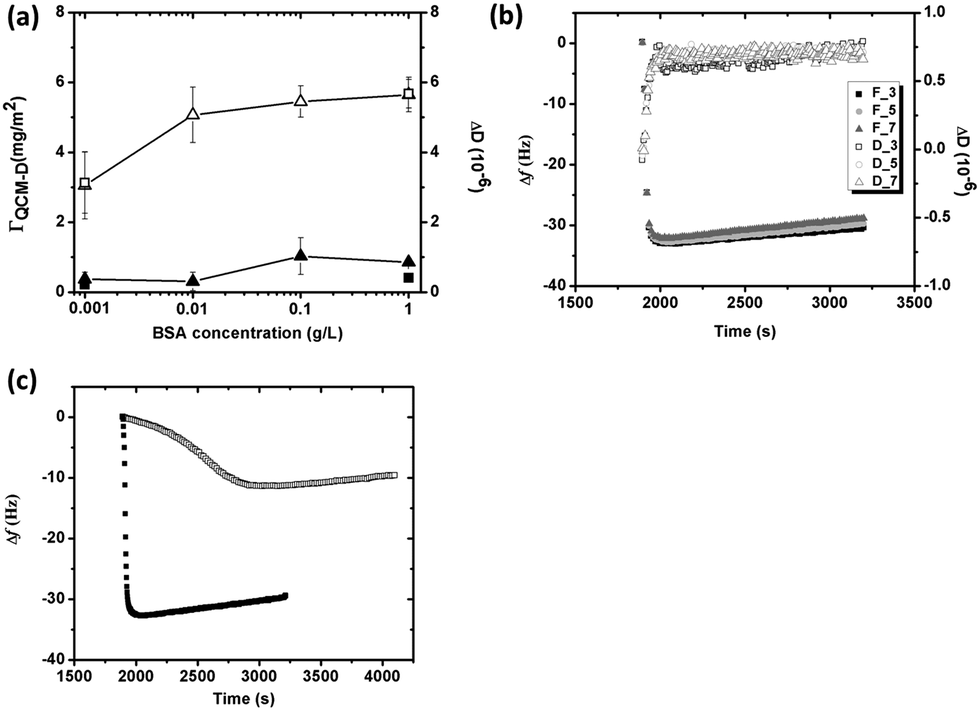 | ||
| Fig. 3 (a) Changes in sensed mass (unfilled symbols) and energy dissipation (filled symbols) versus BSA concentration (from 0.001 g L−1 to 1 g L−1) of non pH-adjusted (pH 5.5–6.6) (triangles) and pH adjusted (pH 7) NaNO3 solutions (squares) (error bars correspond to the deviation between at least two discrete experiments), (b) changes in frequency (Δf) and energy dissipation (ΔD) versus time in BSA (1 g L−1) at pH 7 illustrated by the response from overtones 3 (squares), 5 (circles) and 7 (triangles), (c) changes in frequency (Δf) as a function of time for the adsorption of BSA of two concentrations (0.001 g L−1, unfilled squares and 1 g L−1, filled squares) in 0.15 M NaNO3 at pH 7, 25 °C. The time axis was cut to improve the visibility. | ||
According to previous zeta potential measurements on silver crystals, the isoelectric point is between 5 and 6,22 and the surface hence negatively charged at pH 7. However, the surface maintains electric neutrality in solution due to the adsorption of counter ions that screen the effect of surface charges. Previous adsorption studies of HSA onto negatively charged mica show that the apparent interfacial potential is not influenced by the density of adsorbed protein, which indicates that a charge regulation mechanism that involves small ions is operating during adsorption.43 As proteins arrive to the surface, exclusion of the counterions (both at the protein and the silver surface) results in entropy gain, which is favorable for adsorption. BSA is further a globular protein of low internal conformation stability and surface-induced conformational changes occur during adsorption, properties that also result in entropy gain, primarily at lower BSA concentrations.18,43Fig. 3c clearly displays that the initial adsorption rate (dΔf/dt) of BSA at low concentrations (0.001 g L−1) is significantly lower compared with its rapid adsorption at high concentrations (1 g L−1), and hence more time available for spreading of the protein at lower concentrations, i.e. conformational changes during initial adsorption. Any loss of the secondary and/or the tertiary structure within the protein results in an entropy gain,18,44 which is favorable for protein adsorption. On the other hand, spreading of the protein molecules will occupy a larger surface area that hinders the adsorption of new molecules that reach the surface. The sensed mass and energy dissipation is as a consequence low, cf.Fig. 3a. At high BSA concentrations, the molecules rapidly occupy the surface and the time for their spreading is short. More protein hence adsorbs on the surface, observed as a higher sensed mass, Fig. 3a. The energy dissipation increases only slightly, which indicates that the protein layer remains rigid, even at the highest BSA concentration.
Changes in sensed mass and adsorbed amount of BSA (1 g L−1 in 0.15 M NaNO3) on silver were also monitored simultaneously in real time on the same substrate by using a combined ellipsometry/QCM-D setup, Fig. 4. The sensed mass is consistently higher than the adsorbed amount, both during adsorption and rinsing. The sensed mass and the adsorbed amount rapidly increase upon injection of BSA, followed by a gradual increase in the adsorbed amount (Fig. 4a). A plateau is reached after a few minutes, indicative of adsorption equilibrium. The lack of significant changes in sensed mass or adsorbed amount, observed during rinsing with protein-free NaNO3 solution, imply nearly irreversible adsorption of BSA. The adsorbed amount of BSA corresponds to approximately 2.2 ± 0.17 mg m−2. The maximum theoretically adsorbed amount is 2.3 mg m−2 (assuming prolated ellipsoids), or 2.6 mg m−2 (assuming heart-shaped triangular molecules), for a hexagonally close packed (HCP) monolayer of BSA molecules in a “side-on” orientation. Observed results show that the adsorbed layer of BSA corresponds to a HCP monolayer with a “side-on” orientation.
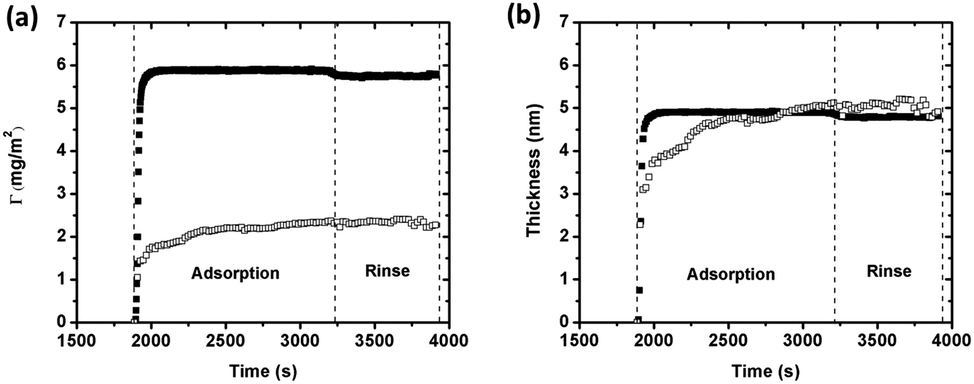 | ||
| Fig. 4 (a) Mass of adsorbed BSA (1 g L−1) on silver determined by means of QCM-D (filled squares) and combined ellipsometry (open squares) as a function of time (25 °C, pH 7). (b) Corresponding changes in calculated layer thicknesses. The time axis was cut to improve the visibility. | ||
If the density of the layer is known, the layer thickness can be estimated from the Sauerbrey equation (eqn (1)).45 Calculated thicknesses, based on the sensed mass (assuming a density of 1200 kg m−3),46 are presented in Fig. 4b together with the thickness determined by means of ellipsometry (assuming a fixed refractive index of 1.42).30 Estimated layer thicknesses are very similar, 4.7 ± 0.34 nm (QCM-D) and 4.8 ± 0.38 nm (ellipsometry). When calculating the effective (average) thickness of the protein layer from the mass uptake measured by means of QCM-D combined with ellipsometry,47 the layer thickness is 5.3 nm. This corresponds to a “hydrated” layer that includes trapped water and agrees with findings in Fig. 4b. Assuming the ellipsoidal dimension of BSA (14 nm × 4 nm × 4 nm), the observed thickness is slightly higher than the protein dimension in width and/or height (4 nm). Compared with the heart-shaped dimension of BSA, like an equilateral triangle with the dimension 9 nm × 5.5 nm,48 the observed layer thickness is slightly smaller. Since BSA consists of three rather compact units that are held together by short flexible regions, it is possible that BSA adsorbs onto the surface in its more elongated conformation (as prolated ellipsoids).23 The adsorbed BSA monolayer is hence most likely in a “side-on” elongated orientation with some protein molecules possibly orientated with a certain angle to the surface, as they anchor on the surface. Such a conformation is well consistent with observed mass adsorption results, Fig. 4a.
Fig. 5a displays the water content within the BSA layer as function of surface coverage of adsorbed BSA on silver (based on eqn (3)) during adsorption and rinsing. The surface coverage of BSA increases upon initial adsorption of BSA, and is followed by a concomitant sharp increase of the water content of the layer. At the break point, the water content reaches 74% and, the “dry” mass corresponds to 1.5 mg m−2, which is close to the theoretical amount of a random sequentially adsorbed (RSA) monolayer with a “side-on” orientation (1.3 mg m−2). This means that the proteins are randomly adsorbed and does not fully cover the surface, which enables enough space (voids) for the entrapment of water. This free space is reduced as adsorption increases, which hinders and excludes water from the layer, Fig. 5b. The water content is 60% within the layer before the surface is rinsed with the protein-free solution, Fig. 5b. Since the water content only slightly changes during rinsing (<5%) and stabilizes at a relatively constant level (±1%) as observed before rinsing, it can be concluded that the structure of adsorbed BSA layer remains unaffected.
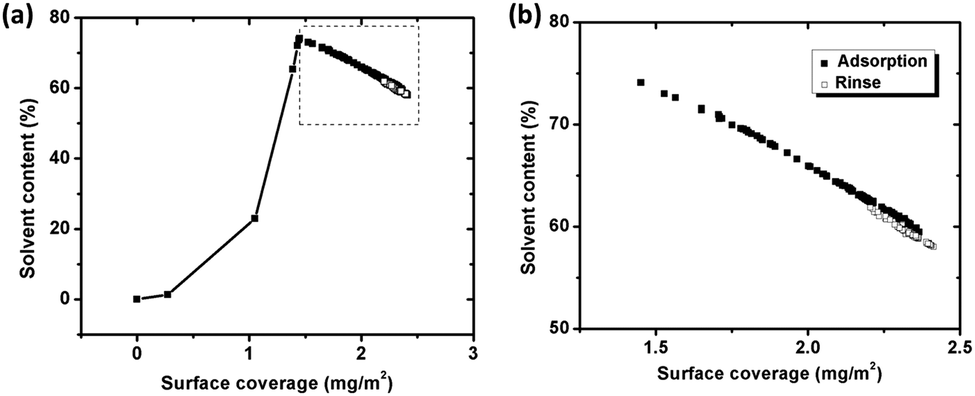 | ||
| Fig. 5 (a) Water (solvent) content trapped in the protein layer, calculated from simultaneous measurements of the sensed mass (QCM-D) and the adsorbed amount (ellipsometry), as a function of surface coverage of BSA on silver coated QCM crystals during adsorption (filled squares) and rinsing (unfilled squares). (b) Close up of segment in Fig. 5a. | ||
3.2 Adsorption of BSA of the highest concentration (1 g L−1) enhances the release of silver in pH neutral solutions (0.15 M NaNO3) due to surface complexation with silver
The QCM-D measurements show that silver ions are released from the underlying silver-coated crystals upon exposure in NaNO3 solutions, both with (more pronounced) and without BSA (1 g L−1). This observation is supported by quantitative measurements of the released amount of silver (up to 24 h) in each solution, Fig. 6. The amount of released silver generally increases with exposure time in both solutions, but can only be statistically ensured in the BSA solution. This means that silver is predominantly released during the initial 2 h of exposure in the non-protein containing NaNO3 solution, whereas the process continues up to 24 h in the BSA-containing solution. The highest released rates of silver are hence observed initially with subsequent rates that are reduced with time, and that measure 0.016 μg cm−2 h−1 in non-protein containing NaNO3 and 0.11 μg cm−2 h−1 in NaNO3 with BSA after 24 h. Similar findings are observed for stainless steels with an initially higher extent of released metals that is reduced with time due to surface passivation.20 Silver, in contrast to stainless steels, does not form any surface oxide at ambient conditions but atomic oxygen may physically adsorb on the surface.49,50 Ag2S and AgCl are the predominating corrosion products forming on silver in aqueous solutions of different pH and composition. This means that the interaction of BSA with the silver surface via disulfide linkages will most likely have an effect on the corrosion behavior and on the release of silver. Fig. 6 shows further that the presence of BSA enhances the release of silver compared with non-protein solutions for all time periods, 3-fold after 2 h and 7-fold after 24 h. The adsorption of BSA hence promotes rather than inhibits the release of silver from the substrate in contrast to short-term findings for carbon steel surfaces.20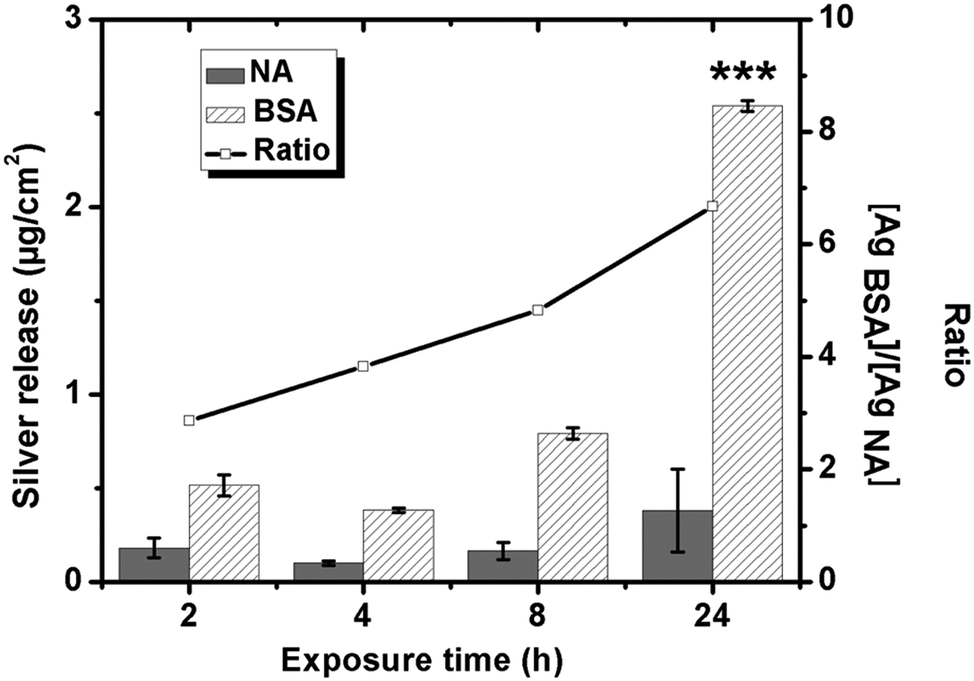 | ||
| Fig. 6 Released amounts of silver from massive silver exposed in 0.15 M NaNO3 (NA) and 0.15 M NaNO3 with 1 g L−1 BSA up to 24 h (pH 7, 25 °C) measured by means of GF-AAS. The inserted curve equals the ratio between released silver in the presence of BSA compared with non-protein containing NaNO3. Asterisks indicate a significant increase of released silver after 24 h compared with 2 h, as calculated by a student t-test, p < 0.001 (***). | ||
3.3 The adsorption of the negatively charged PGA polypeptide on silver is minor and reversible and has no effect on the extent of released silver into solution at pH neutral conditions
Due to molecular similarities, the adsorption of the PGA polypeptide on silver and its influence on the release of silver was investigated, Fig. 7, and compared with findings for BSA, Fig. 6. Shifts in Δf and ΔD upon injection of the PGA-containing (1 g L−1) NaNO3 solution were less than 2 Hz and approximately 0.6 × 10−6, respectively. These small changes in frequency and energy dissipation indicate a negligible adsorption of the negatively charged PGA onto the similarly charged silver surface. Observed changes in frequency may also be attributed to changes in viscosity and density of the bulk solution in the presence of PGA compared with the non-polypeptide containing solution. Literature findings show that the adsorption of polypeptides on charged substrates predominantly is governed by electrostatic interactions.51,52 In neutral solution, PGA is highly ionized (>90%), which supports a strong electrostatic repulsion between COO−1 groups and the silver surface.53 PGA molecules in solution close to the surface will hence experience an electrostatic repulsion from the negatively charged silver surface and only a small shift in Δf. The shifts of both Δf and ΔD regress upon rinsing with non-PGA containing NaNO3. Δf regresses to its original level after the second rinsing cycle (t2). The small shift in Δf is hence most probably caused by the difference of solution properties of the NaNO3 solution with and without PGA. Even though some PGA adsorption does occur, the adsorbed mass is small and the adsorption reversible.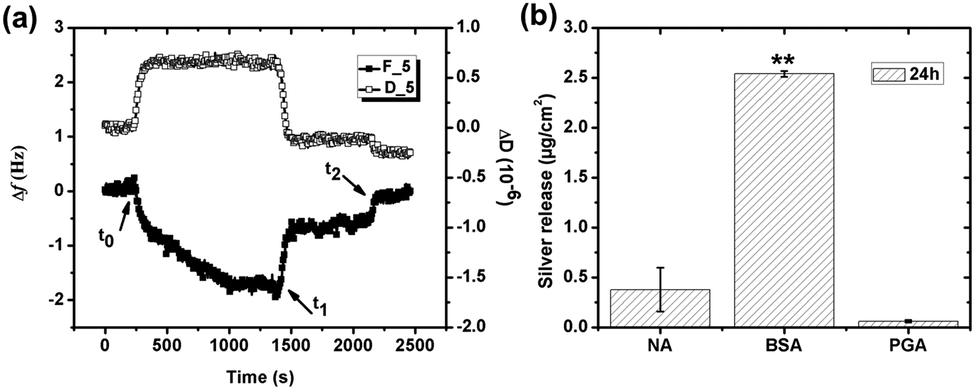 | ||
| Fig. 7 (a) Change in frequency (Δf) (filled squares) and dissipation (ΔD) (unfilled squares) versus time during adsorption of PGA (1 g L−1) on silver (pH 7 and 25 °C) monitored with QCM-D. The time points, t0, t1, t2 correspond to PGA injection, rinsing, and continued rinsing, respectively. Overtone 5 was randomly selected to illustrate the results. (b) Amount of released silver from silver surfaces exposed to different solutions: NaNO3 (NA), NaNO3 with BSA (1 g L−1), or NaNO3 with PGA (1 g L−1) for 24 h at pH 7 and 25 °C measured by means of GF-AAS. Asterisks indicate significant differences in released silver in protein or polypeptide solutions compared with non-protein/polypeptide containing NaNO3 solutions, as calculated by a student t-test, p < 0.01 (**). | ||
Generated results show that the negatively charged BSA successfully adsorbs onto the negatively charged silver surface while the negatively charged PGA molecule does not. A protein molecule surface is heterogeneous with hydrophobic and hydrophilic patches of positive and negative charge, dipoles and with hydrogen-bonded surface groups.18 Approximately 11% of the exposed surface of BSA is, according to the literature rich in glutamic acid,54 and even though BSA is net negatively charged at a neutral pH, approximately 17% of the surface is still positively charged via amino acids such as lysine and tyrosine. Attractive electrostatic interactions of positively charged regions of the BSA molecule can hence contribute to the overall interaction with the negatively charged silver surface, i.e. gain entropy due to release of water molecules and counter ions and conformational changes of the protein molecule. In contrast, the PGA molecule only consists of glutamic acid, and is more than 90% ionized with negative charges, which hinder its adsorption onto the silver surface.
Amounts of released silver from massive silver are presented in Fig. 7b after 24 h in the different solutions and immersion conditions, NaNO3 (NA), NaNO3 + BSA (1 g L−1) or NaNO3 + PGA (1 g L−1). Significantly more silver is released in solutions containing BSA (average released amount of 2.54 μg cm−2) compared with corresponding released amounts in solutions with PGA, levels that are not statistically significantly different from released amounts in non-protein/polypeptide containing NaNO3 solutions (<0.4 μg cm−2). The presence of PGA in solution hence does not influence the release of silver into solution at given conditions.
3.4 Surface complexation between BSA and silver followed by the detachment of BSA is the most likely mechanism for the enhanced release of silver in BSA containing solutions of pH neutral conditions
As illustrated above, the presence of BSA in solution enhances the release of silver from silver surfaces at given pH neutral conditions, Fig. 6. This is consistent with previous findings on e.g. chromium metal19 and stainless steel,19,20 but contradictory to findings for titanium55 and iron.20 This study shows that the surface is fully covered by a side-on monolayer at a BSA concentration of 1 g L−1, cf. Section 3.1. From the Boltzmann distribution it is evident that the proton concentration outside a charged surface is different compared to the bulk concentration. This may result in a lower local pH close to the surface. If the adsorption of counter ions from the 0.15 M NaNO3 solution is not taken into account before BSA adsorption, the local pH at surface would only be reduced to 6.5 for a zeta potential of −30 mV at pH 7. Surface adsorption of the net negatively charged BSA onto silver induces an enrichment of counter ions such as H+ and Na+ at the interface between the surface and the protein layer, a process that further may lower the surface pH. Such a reduction in local pH may enhance the release of silver. Due to charge regulation during surface adsorption of BSA, the apparent interfacial potential is most probably not influenced by the density of adsorbed proteins,43 and a negative interfacial potential is hence expected at the protein solution interface. As a consequence, electrostatic attraction of released silver will take place during its transport through the protein layer and enable its release into solution.In order to study this in more detail, the silver release process was investigated by consecutive exposure of massive silver coupons into sequences of BSA and/or BSA-free NaNO3-solutions, Fig. 8. As previously observed, the amount of released silver is enhanced by the presence of BSA, Fig. 8a and b. The consecutive exposure of the silver coupons to solutions with and without proteins mimics the adsorption and rinsing steps of the QCM-D/ellipsometry investigation, Fig. 8c and d. These sequences clearly show that more silver is released in solutions containing BSA compared with the non-protein containing NaNO3 solutions even though BSA is irreversible adsorbed as a fully surface-covered monolayer, and elucidate the importance of proteins in bulk solution for the silver release process.
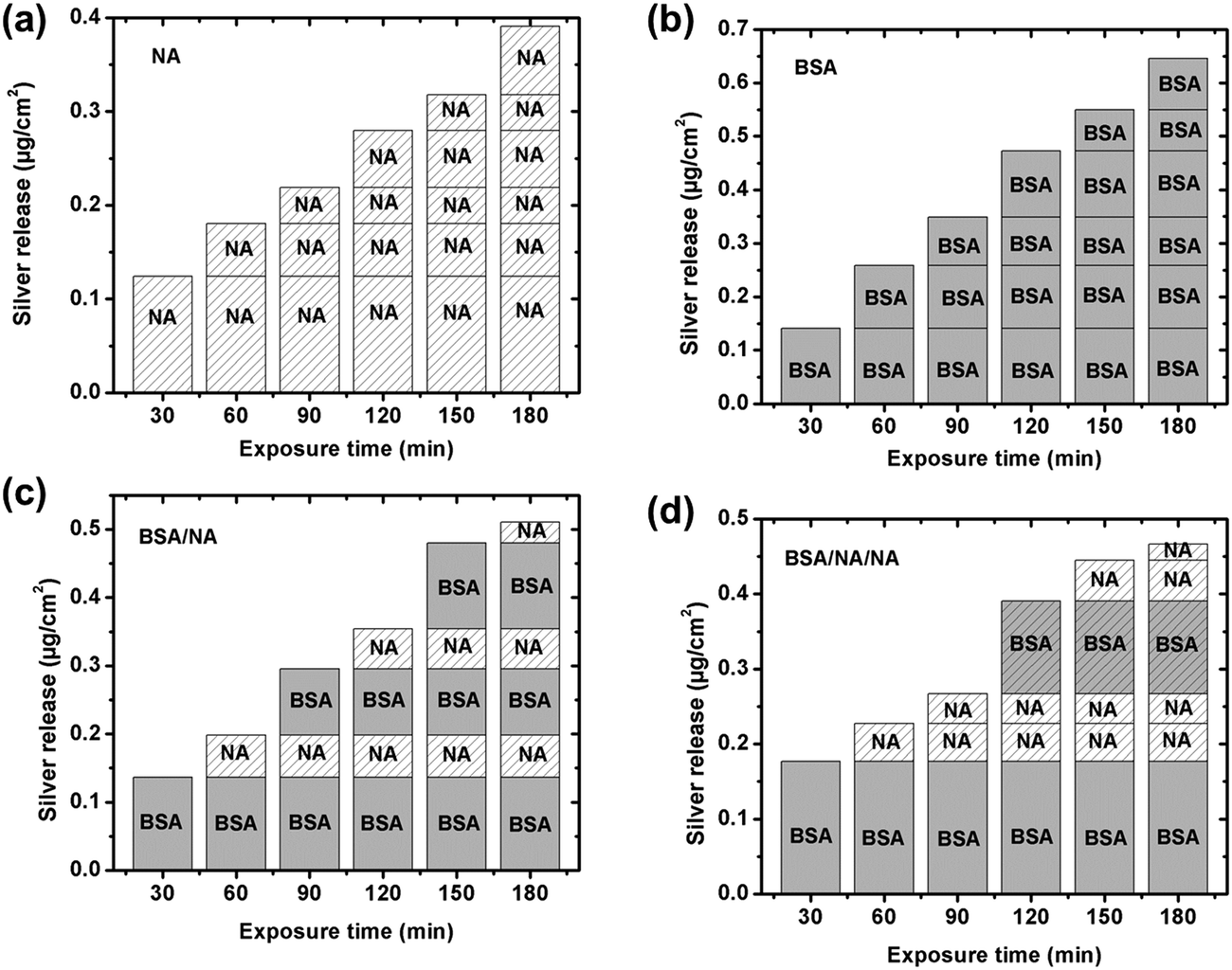 | ||
| Fig. 8 Total amount of released silver per surface area from massive silver coupons with time, consecutively immersed in different solutions determined by means of GF-AAS. Each step involves a 30 min immersion period before each coupon is transferred to the next “fresh” solution. Four different sequences were investigated; (a) 0.15 M NaNO3 (NA): NA–NA–NA–NA–NA–NA, (b) 1 g L−1 BSA in 0.15 M NaNO3 (BSA): BSA–BSA–BSA–BSA–BSA–BSA, (c) BSA/NA: BSA–NA–BSA–NA–BSA–NA, and (d) BSA/NA/NA: BSA–NA–NA–BSA–NA–NA. Average values are presented based on triplicate measurements for each immersion step with blank values subtracted if >LOD. | ||
Together with the direct release of silver ions through voids in the hydrated BSA layer, the most probable mechanism behind the enhancement of released silver from massive silver surfaces in the presence of BSA is most likely its complexation with silver surface and, the dynamic equilibrium obtained between surface adsorbed BSA and BSA molecules in the bulk solution. Based on the Pearson's classification (the HSAB principle), the Ag(I) ion is a “soft” cation that prefers to bind with soft ligands that are relatively polarizable.56 The softest ligands of the functional groups of BSA are sulfur groups. BSA consists in total of 583 amino acid residues that contain 17 disulfide bonds (S–S) in 34 cysteine residues, a free thiol group (R–S–H) in a cysteine residue, R–S–R thiols in 5 methionine residues, and the free Cys-34 sulfhydryl that has been identified as a binding site for silver ions.23 Literature findings show that eight disulfide bonds in the BSA molecule are distributed in the same plane.57 Since at least one cysteine residue for each disulfide pair on this plane has a large area (>18 Å2) on the surface, it favors surface adsorption via disulfide bonds. It is hence reasonable to assume that disulfide bonds in BSA are prone to bind to the silver surface, e.g. via cysteine residues, and form BSA–silver surface complexes. The carboxylic functional groups of the PGA molecule prefer, according to HSAB principle, to complex with hard acids such as Fe(III) and Cr(III), rather than to the soft acid Ag(I). This would explain the lack of significant surface adsorption of PGA onto silver, Fig. 7a. Protein adsorption equilibrium has previously been proven as a dynamic process in which adsorption and desorption of proteins at the surface take place with equal rates.18,58,59 With the exchange of proteins on the surface follows the release of complexed silver into solution, which in turn induces more silver release from the surface. The absence of BSA in solution disables its exchange with the surface59 and the concomitant detachment of silver–protein complexes from the surface. Such conditions reduce the released amounts of silver from the surface, despite the presence of an adsorbed protein layer, Fig. 8c and d.
The influence of BSA in solution on the extent of released silver was investigated for different concentrations (0.01, 0.1 and 1 g L−1), Fig. 9. No significant differences in released amounts of silver compared with the non-protein containing solution were observed for the lowest BSA concentrations (0.01 and 0.1 g L−1). A significant effect was observed for the highest concentration (1 g L−1). According to the QCM-D measurements, the sensed mass gradually increased with increasing BSA concentration (from 0.001 to 1 g L−1, Fig. 3), and the layer was more rigid at the lowest BSA concentrations compared with the higher concentration. This indicates a higher extent of spreading of adsorbed BSA molecules at lower concentrations, and hence an increased number of protein segments exposed at the surface, effects that will influence the exchange rate between adsorbed proteins and proteins in solution.18 Previous findings show that the volume fraction of voids within a HSA layer on mica increases with the layer thickness, which is indicative of reduced conformational changes with increased HSA concentration.43 This will most probably also influence the exchange rate of adsorbed proteins with proteins in solution, processes that in turn are facilitated by an increased concentration of proteins in solution due to a higher flux towards the surface.59 The extent of spreading increases as the time to fill the surface increases. Results of this study show a rapid adsorption of BSA in solutions of high protein concentration (1 g BSA per L, Fig. 3c). This means that the time for the relaxation of the protein is short and, hence, that the area of exposed BSA segments at the surface becomes small, which is beneficial for its detachment from the surface. Increased amounts of metals in solutions of increased concentration of BSA (≥1 g L−1) have also been observed for stainless steels.20 Higher concentrations of BSA in solution further enhance the release of metals, which is related to surface complexation, local defects in the surface oxide induced by complexation, and/or local lowering of the surface pH. In this study, the adsorbed BSA undergoes the repulsion between adsorbed proteins molecules and the repulsion between adsorbed proteins and protein in solution. With increase of BSA concentration, the amount of adsorbed BSA and BSA in solution is increased as a result the repulsion to adsorbed BSA might be increased. Hence, the most likely explanation for the observed enhancement of silver release with increasing concentration of BSA in solution is an increased exchange of BSA complexed with surface and BSA molecules in solution. However, some release of silver from the surface due to induced surface defects and lowering of local pH cannot be completely excluded. A model to depict the adsorbed BSA layer and its role in the enhancement of silver release is schematically compiled in Fig. 10.
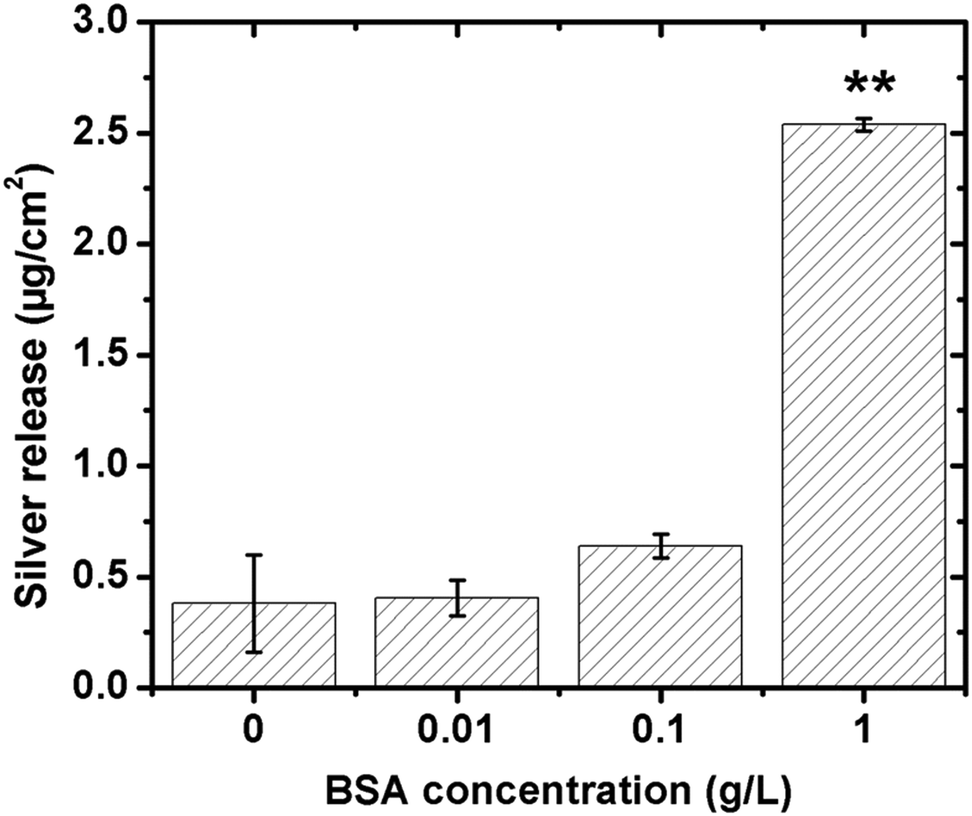 | ||
| Fig. 9 Released amounts of silver per surface area after 24 h of exposure in 0.15 M NaNO3-solutions (pH 7, 25 °C) of increased BSA concentration (0, 0.01, 0.1 and 1 g L−1) determined by means of GF-AAS. Asterisks indicate significant enhancement of the released silver in the BSA solution compared with the non-protein containing NaNO3 solution, as calculated by a student t-test, p < 0.01 (**). | ||
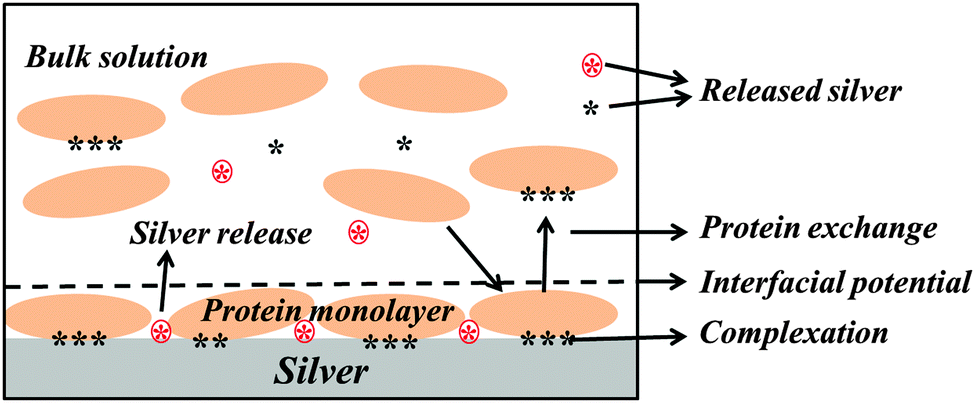 | ||
| Fig. 10 A schematic model that proposes the possible mechanisms behind the enhancement of silver release from silver surfaces in the presence of BSA in pH neutral aqueous solutions. Red stars in a circle represent silver ions that are directly released from the silver surface and black stars represent silver ions that are released from the surface due to complexation with BSA. | ||
4. Conclusions
Protein adsorption and its influence on the release of silver from silver surfaces have been investigated in 0.15 M NaNO3 with and without BSA at pH 7. The negatively charged BSA protein adsorbs on silver, an adsorption that increases with protein concentration, while the similarly charged PGA polypeptide, investigated for comparison, scarcely and reversibly adsorbs. Differences in adsorption mechanisms are associated with the intrinsic properties and structures of the protein and polypeptide molecules.The present study shows no significant differences in released amounts of silver in NaNO3 solutions containing BSA for protein concentrations ≤0.1 g L−1, compared with non-protein containing NaNO3 solutions. Exposures in NaNO3 solutions with 1 g L−1 BSA significantly (7-fold after 24 h) enhance the released amount of silver, whereas no effects are seen for solutions containing 1 g L−1 PGA. The effect of BSA is attributed to the structure of surface adsorbed BSA and the presence of BSA molecules in solution. It also indicates that the macromolecules need to be physically adsorbed onto the surface and not only be present in the proximity of the surface to influence the silver release. The adsorption of a monolayer of BSA with a “side-on” orientation and with some molecules adsorbed at a certain angle from the surface, gives rise to low conformational changes that promote exchange with protein molecules in solution. These processes trigger the detachment of silver–BSA surface complexes and enhance the release of silver at given conditions.
The property of the adsorbed protein layer has a significant impact on the silver release process. In the case of LSZ, the formation of a bilayer structure on the silver surface efficiently hinders the release of silver.22 This can be explained by the interfacial potential of the adsorbed protein layer. As positively charged LSZ adsorbs on the surface, the release of silver will, due to electrostatic repulsion between the positively charged silver ions and the net positively charged outermost layer at the LSZ/solution interface, be reduced during transport through the layer. For negatively charged BSA, this will not be the case since the interfacial potential is anticipated to be net negative. This may result in electrostatic attraction that facilitates transport of released silver trough the layer and enable silver ions to be released into the surrounding solution.
It was also evident that the amount of released silver was significantly enhanced by the presence of BSA in solution. The proposed mechanism explaining this is related to the formation and exchange of BSA–silver complexes at the surface and with BSA molecules in solution that will detach from the surface and increase the amount of silver in solution. However, some release of silver due to induced surface defects and a local lowering in surface pH cannot be completely rolled out.
Acknowledgements
The Chinese Scholarship Council (CSC) is gratefully acknowledged for the financial support of Xin Wang. Financial support from the Swedish National Research Council (VR) is highly acknowledged. Dr Yolanda Hedberg and Dr Jonas Hedberg are both acknowledged for invaluable help to analyze and evaluate the amount of silver release by means of GF-AAS. Dr Lubica Macakova is acknowledged for valuable discussions and help with ellipsometry measurements and data analysis.References
- E. Sheehan, J. McKenna, D. Dowling, D. McCormack and J. M. Fitzpatrick, J. Bone Jt. Surg. Br., 2003, 85-B(SUPP I), 3 Search PubMed.
- N. Alt and G. Gosheger, Eur. Musculoskeletal Rev., 2011, 6, 166–168 Search PubMed.
- H. H. Lara, E. N. Garza-Trevino, L. Ixtepan-Turrent and D. K. Singh, J. Nanobiotechnol., 2011, 9, 30 CrossRef CAS PubMed.
- D. J. Leaper, Int. Wound J., 2006, 3, 282–294 CrossRef PubMed.
- W. Stewart and W. S. Halstead, JAMA, J. Am. Med. Assoc., 1913, 60, 1119–1126 CrossRef.
- J. A. Spadaro, S. E. Chase and D. A. Webster, J. Biomed. Mater. Res., 1986, 20, 565–577 CrossRef CAS PubMed.
- M. A. Wassall, M. Santin, C. Isalberti, M. Cannas and S. P. Denyer, J. Biomed. Mater. Res., 1997, 36, 325–330 CrossRef CAS.
- J. Hardes, C. von Eiff, A. Streitbuerger, M. Balke, T. Budny, M. P. Henrichs, G. Hauschild and H. Ahrens, J. Surg. Oncol., 2010, 101, 389–395 Search PubMed.
- I. Sondi and B. Salopek-Sondi, J. Colloid Interface Sci., 2004, 275, 177–182 CrossRef CAS PubMed.
- W. K. Jung, H. C. Koo, K. W. Kim, S. Shin, S. H. Kim and Y. H. Park, Appl. Environ. Microbiol., 2008, 74, 2171–2178 CrossRef CAS PubMed.
- R. L. Zapata-Sirvent and J. F. Hansbrough, J. Burn Care Rehabil., 1993, 14, 132–140 CrossRef CAS PubMed.
- I. L. Steffensen, O. J. Mesna, E. Andruchow, E. Namork, K. Hylland and R. A. Andersen, Gen. Pharmacol., 1994, 25, 1621–1633 CrossRef CAS.
- M. Trop, M. Novak, S. Rodl, B. Hellbom, W. Kroell and W. Goessler, J. Trauma, 2006, 60, 648–652 CrossRef PubMed.
- E. Sudmann, H. Vik, M. Rait, K. Todnem, K. J. Andersen, K. Julsham, O. Flesland and J. Rungby, Med. Prog. Technol., 1994, 20, 179–184 CAS.
- C. N. Lok, C. M. Ho, R. Chen, Q. Y. He, W. Y. Yu, H. Sun, P. K. Tam, J. F. Chiu and C. M. Che, JBIC, J. Biol. Inorg. Chem., 2007, 12, 527–534 CrossRef CAS PubMed.
- S. Y. Liau, D. C. Read, W. J. Pugh, J. R. Furr and A. D. Russell, Lett. Appl. Microbiol., 1997, 25, 279–283 CAS.
- S. Omanovic and S. G. Roscoe, Langmuir, 1999, 15, 8315–8321 CrossRef CAS.
- W. Norde, Colloids Surf., B, 2008, 61, 1–9 CrossRef CAS PubMed.
- M. Lundin, Y. Hedberg, T. Jiang, G. Herting, X. Wang, E. Thormann, E. Blomberg and I. Odnevall Wallinder, J. Colloid Interface Sci., 2012, 366, 155–164 CrossRef CAS PubMed.
- Y. Hedberg, X. Wang, J. Hedberg, M. Lundin, E. Blomberg and I. Odnevall Wallinder, J. Mater. Sci.: Mater. Med., 2013, 24, 1015–1033 CrossRef CAS PubMed.
- H. L. Karlsson, P. Cronholm, Y. Hedberg, M. Tornberg, L. De Battice, S. Svedhem and I. Odnevall Wallinder, Toxicology, 2013, 313, 59–69 CrossRef CAS PubMed.
- X. Wang, G. Herting, I. Odnevall Wallinder and E. Blomberg, Langmuir, 2014, 30, 13877–13889 CrossRef CAS PubMed.
- D. C. Carter and J. X. Ho, Adv. Protein Chem., 1994, 45, 153–203 CrossRef CAS.
- P. Rondeau and E. Bourdon, Biochimie, 2011, 93, 645–658 CrossRef CAS PubMed.
- T. Peters Jr, in Adv. Protein Chem., ed. J. T. E. C. B. Anfinsen and M. R. Frederic, Academic Press, 1985, vol. 37, pp. 161–245 Search PubMed.
- L. Porcar, A. Codoner, C. M. Gomez, C. Abad and A. Campos, Eur. Polym. J., 2004, 40, 819–828 CrossRef PubMed.
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 CrossRef CAS.
- J. A. De Feijter, J. Benjamins and F. A. Veer, Biopolymers, 1978, 17, 1759–1772 CrossRef CAS PubMed.
- J. Wen, T. Arakawa and J. S. Philo, Anal. Biochem., 1996, 240, 155–166 CrossRef CAS PubMed.
- J. Vörös, Biophys. J., 2004, 87, 553–561 CrossRef PubMed.
- F. P. Mertens, P. Theroux and R. C. Plumb, J. Opt. Soc. Am., 1963, 53, 788–793 CrossRef.
- L. E. Bailey, D. Kambhampati, K. K. Kanazawa, W. Knoll and C. W. Frank, Langmuir, 2002, 18, 479–489 CrossRef CAS.
- M. C. Dixon, J. Biomol. Tech., 2008, 19, 151–158 Search PubMed.
- T. J. Halthur and U. M. Elofsson, Langmuir, 2004, 20, 1739–1745 CrossRef CAS.
- J. Vogelsang and J. Hädrich, Accredit. Qual. Assur., 1998, 3, 242–255 CrossRef.
- I. Reviakine, D. Johannsmann and R. P. Richter, Anal. Chem., 2011, 83, 8838–8848 CrossRef CAS PubMed.
- H. Stadler, M. Mondon and C. Ziegler, Anal. Bioanal. Chem., 2003, 375, 53–61 CAS.
- M. J. Desroches, N. Chaudhary and S. Omanovic, Biomacromolecules, 2007, 8, 2836–2844 CrossRef CAS PubMed.
- D. R. Jackson, S. Omanovic and S. G. Roscoe, Langmuir, 2000, 16, 5449–5457 CrossRef CAS.
- D. F. Evans and H. Wennerström, The Colloidal Domain: Where Physics, Chemistry, Biology, and Technology Meet, 2nd edn, 1999 Search PubMed.
- J. E. Kinsella and D. M. Whitehead, Adv. Food Nutr. Res., 1989, 33, 343–438 CAS.
- E. Podstawka, Y. Ozaki and L. M. Proniewicz, Appl. Spectrosc., 2004, 58, 1147–1156 CrossRef CAS PubMed.
- E. Blomberg, P. M. Claesson and R. D. Tilton, J. Colloid Interface Sci., 1994, 166, 427–436 CrossRef CAS.
- M. Lundin, T. Sandberg, K. D. Caldwell and E. Blomberg, J. Colloid Interface Sci., 2009, 336, 30–39 CrossRef CAS PubMed.
- D. Johannsmann, Phys. Chem. Chem. Phys., 2008, 10, 4516–4534 RSC.
- J. Andersson, K. N. Ekdahl, J. D. Lambris and B. Nilsson, Biomaterials, 2005, 26, 1477–1485 CrossRef CAS PubMed.
- F. Höök, J. Vörös, M. Rodahl, R. Kurrat, P. Böni, J. J. Ramsden, M. Textor, N. D. Spencer, P. Tengvall, J. Gold and B. Kasemo, Colloids Surf., B, 2002, 24, 155–170 CrossRef.
- K. Rezwan, L. P. Meier, M. Rezwan, J. Vörös, M. Textor and L. J. Gauckler, Langmuir, 2004, 20, 10055–10061 CrossRef CAS PubMed.
- J. Hedberg, M. Lundin, T. Lowe, E. Blomberg, S. Wold and I. O. Wallinder, J. Colloid Interface Sci., 2012, 369, 193–201 CrossRef CAS PubMed.
- A. W. Czanderna, J. Phys. Chem., 1964, 68, 2765–2771 CrossRef CAS.
- N. G. Hoogeveen, M. A. Cohen Stuart and G. J. Fleer, J. Colloid Interface Sci., 1996, 182, 133–145 CrossRef CAS.
- T. J. Halthur and U. M. Elofsson, Langmuir, 2004, 20, 1739–1745 CrossRef CAS.
- S. Ozeki, K. Ohtani, T. Sato, Y. Oowaki and C. Morita, in Fundamentals of Adsorption, ed. M. D. LeVan, Kluwer Academic Publishers, 1996 Search PubMed.
- C. H. Schein, Nat. Biotechnol., 1990, 8, 308–317 CrossRef CAS.
- S. Takemoto, M. Hattori, M. Yoshinari, E. Kawada and Y. Oda, Biomaterials, 2005, 26, 829–837 CrossRef CAS PubMed.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- L. M. Wang, J. Y. Li, J. Pan, X. M. Jiang, Y. L. Ji, Y. F. Li, Y. Qu, Y. L. Zhao, X. C. Wu and C. Y. Chen, J. Am. Chem. Soc., 2013, 135, 17359–17368 CrossRef CAS PubMed.
- I. Lundström and H. Elwing, J. Colloid Interface Sci., 1990, 136, 68–84 CrossRef.
- J. L. Brash and Q. M. Samak, J. Colloid Interface Sci., 1978, 65, 495–504 CrossRef CAS.
| This journal is © the Owner Societies 2015 |