
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling the physics and chemistry of binary and ternary praseodymium and cerium oxide systems
Gang
Niu
ab,
Marvin Hartwig
Zoellner
ab,
Thomas
Schroeder
*ab,
Andreas
Schaefer
c,
Jin-Hao
Jhang
c,
Volkmar
Zielasek
c,
Marcus
Bäumer
*c,
Henrik
Wilkens
d,
Joachim
Wollschläger
*d,
Reinhard
Olbrich
d,
Christian
Lammers
d and
Michael
Reichling
*d
aIHP, Im Technologiepark 25, 15236 Frankfurt(Oder), Germany. E-mail: schroeder@ihp-microelectronics.com
bBTU Cottbus-Senftenberg, Institute of Physics, Konrad-Zuse-Str. 1, 03046 Cottbus, Germany
cInstitute of Applied and Physical Chemistry, University of Bremen, Leobener Str. UFT, 28359 Bremen, Germany. E-mail: mbaeumer@uni-bremen.de
dFachbereich Physik, Universität Osnabrück, Barbarastr. 7, 49076 Osnabrück, Germany. E-mail: reichling@uos.de; jwollsch@uos.de
First published on 10th September 2015
Abstract
Rare earth praseodymium and cerium oxides have attracted intense research interest in the last few decades, due to their intriguing chemical and physical characteristics. An understanding of the correlation between structure and properties, in particular the surface chemistry, is urgently required for their application in microelectronics, catalysis, optics and other fields. Such an understanding is, however, hampered by the complexity of rare earth oxide materials and experimental methods for their characterisation. Here, we report recent progress in studying high-quality, single crystalline, praseodymium and cerium oxide films as well as ternary alloys grown on Si(111) substrates. Using these well-defined systems and based on a systematic multi-technique surface science approach, the corresponding physical and chemical properties, such as the surface structure, the surface morphology, the bulk–surface interaction and the oxygen storage/release capability, are explored in detail. We show that specifically the crystalline structure and the oxygen stoichiometry of the oxide thin films can be well controlled by the film preparation method. This work leads to a comprehensive understanding of the properties of rare earth oxides and highlights the applications of these versatile materials. Furthermore, methanol adsorption studies are performed on binary and ternary rare earth oxide thin films, demonstrating the feasibility of employing such systems for model catalytic studies. Specifically for ceria systems, we find considerable stability against normal environmental conditions so that they can be considered as a “materials bridge” between surface science models and real catalysts.
 The research team behind the cooperative work presented in this paper. From left to right: Jin-Hao Jhang, Sebastian Gevers, Henrik Wilkens, Volkmar Zielasek, Christian Lammers, Hans-Hermann Pieper, Reinhard Olbrich, Joachim Wollschläger, Michael Reichling, Marvin Hartwig Zoellner and Marcus Baumer. Missing from the photo: Gang Niu, Andreas Schaefer and Thomas Schroeder |
I. Introduction
In Earth's crust, a family of relatively abundant elements are the rare earth elements (REEs, 15 lanthanides La to Lu as well as Sc and Y) accounting for 0.01% of the mass of all elements. REEs are generally reactive with respect to oxygen in the ambient atmosphere so that rare earth oxides (REOs) are mostly stable compounds. In fact, REEs were discovered as oxides being extraordinarily stable, which leads to the “earth” part of their name: in early Greek “earth” is defined as materials that could not be changed further by heating. The chemical and physical properties of REEs are rather similar and hard to separate from each other, which results in the “rare” part of their name, referring to the difficulty in obtaining the pure element.1REOs exhibit a wide range of fascinating physical and chemical properties and attract intense scientific interest as well as interest with respect to many applications. An important field of application of REOs is heterogeneous catalysis. For example, each year millions of tons of REO raw materials (in which more than 95% is La2O3) are utilised as catalysts for cracking of crude petroleum. REOs can also catalyse many other organic reactions and they form important ingredients in automobile exhaust-gas conversion catalysts.2 In fact, REOs are considered as the most active and selective catalysts for oxidative coupling of methane to form higher hydrocarbon products, a significant process in natural gas processing.3 Furthermore, REOs are widely used for optical applications.4 For instance, a mixture of europium and yttrium oxides offers a brilliant-red phosphor.5 REOs are significant in the glass industry to polish or to dope glass; they are also used as luminescence materials,6 optical coatings7 and laser active materials.4 In the microelectronics domain, there has been burgeoning interest since the 1990s in nanometre scale thin films of REOs, initially fuelled by the exploration of alternative high dielectric constant (high-κ) gate insulator materials to maintain “Moore's law” predicting an exponential growth of the number of transistors on a chip via a continuous scaling of complementary metal–oxide–semiconductor (CMOS) devices.8 Thanks to their remarkable properties such as high-κ, a large band gap, a large band offset and high thermal stability in combination with Si, REOs were considered as promising high-κ dielectric candidates.8 Moreover, the capability of growing high quality, epitaxial, single crystalline REO films on Si with a thickness of up to several hundred nanometres opens a pathway for their applications in “More than Moore” technology, namely the integration of more functions on “frozen” Si CMOS chips without further scaling.9 In this context, REOs not only demonstrate diverse functions beyond conventional semiconductor functionality such as the use as diluted magnetic semiconductors.10 They can also be used as buffers for the integration of other functional layers such as superconductive,11 ferromagnetic12 and multiferroic13 oxides as well as semiconductors like Si,14 Ge15 and III–V16 grown on Si.
Considering that the structural and chemical properties of REOs can strongly influence their electrical and catalytic characteristics, it is necessary to understand the structure–property relationship to improve their performance in device applications. However, the complex features of REOs like their polymorphism and varying oxygen stoichiometry make the exploration of the structure–property relationship rather difficult. In this context, a surface science approach for studying single crystalline REO films turns out to be a valuable method, because (i) many REO-related scientific issues or applications are focused on the surface properties. (ii) By starting from the well-ordered, flat, two dimensional and stoichiometric single crystalline REO film, one can increase the complexity step by step to understand the structure–property relationship from simple to complex materials. (iii) One can control the surface properties by tailoring thin film properties like thickness and surface morphology as REOs generally possess a high oxygen mobility,4 leading to a strong coupling between the volume and surface properties of the thin film.
In this paper, we review recent progress in a detailed understanding of the chemistry and physics of two representative REOs, namely ceria and praseodymia. Cerium and praseodymium both form the tetravalent RE dioxides having a fluorite structure (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, N.225). Based on this basic structure, they form a multitude of other phases due to the reduction of the oxygen content by the formation of oxygen vacancies in the lattice. Oxygen vacancies can be considered as ions with double positive charge and are commonly represented as
m, N.225). Based on this basic structure, they form a multitude of other phases due to the reduction of the oxygen content by the formation of oxygen vacancies in the lattice. Oxygen vacancies can be considered as ions with double positive charge and are commonly represented as  according to the Kröger–Vink notation.17
according to the Kröger–Vink notation.17 formation is one of the most important factors determining the properties of oxides because it has a strong impact on the solid state physical properties and, accordingly, the performance in different applications of oxides. The increasing formation of
formation is one of the most important factors determining the properties of oxides because it has a strong impact on the solid state physical properties and, accordingly, the performance in different applications of oxides. The increasing formation of 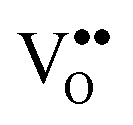 in CeO2 or PrO2 is associated with Ce4+ to Ce3+ and Pr4+ to Pr3+ reductions, respectively. Interestingly, Ce prefers the +4 valence while Pr favours the +3 valence in the oxides under commonly used preparation conditions, which is a result of the electrochemical reduction potential of Ce4+/Ce3+ (1.74 eV) being considerably smaller than that of Pr4+/Pr3+ (3.2 eV).18 Combining Ce and Pr oxides enables us to tailor the properties of oxides such as the oxygen mobility and the oxygen storage capacity. Therefore, we include two types of combined oxide systems in our study, namely CeO2/Pr2O3 bilayers and Ce1−xPrxO2−δ mixed oxide films.
in CeO2 or PrO2 is associated with Ce4+ to Ce3+ and Pr4+ to Pr3+ reductions, respectively. Interestingly, Ce prefers the +4 valence while Pr favours the +3 valence in the oxides under commonly used preparation conditions, which is a result of the electrochemical reduction potential of Ce4+/Ce3+ (1.74 eV) being considerably smaller than that of Pr4+/Pr3+ (3.2 eV).18 Combining Ce and Pr oxides enables us to tailor the properties of oxides such as the oxygen mobility and the oxygen storage capacity. Therefore, we include two types of combined oxide systems in our study, namely CeO2/Pr2O3 bilayers and Ce1−xPrxO2−δ mixed oxide films.
Fig. 1 shows the bulk phase diagrams of the Ce–O and Pr–O systems.19 Analysing the phase diagrams reveals general features that (i) almost all structures can be considered as quasi-cubic ones despite the existence of different non-stoichiometric phases, (ii) all structures are related to either a random or a periodic arrangement of  , (iii) the lattice constant increases as the oxygen content decreases because the cation radii increase with a lower oxidation state20,21 and (iv) the composition of the oxides strongly depends on thermodynamic and kinetic factors, i.e. the temperature as well as the physical state and preparation history.
, (iii) the lattice constant increases as the oxygen content decreases because the cation radii increase with a lower oxidation state20,21 and (iv) the composition of the oxides strongly depends on thermodynamic and kinetic factors, i.e. the temperature as well as the physical state and preparation history.
 | ||
| Fig. 1 Bulk phase diagrams for the systems Ce–O (left) and Pr–O (right). (Reproduced from ref. 19.) | ||
Rare earth oxides have been studied for a long time and the present work can build on a solid stock of fundamental knowledge on the physics and chemistry of these materials. Due to their outstanding role in chemical catalysis,22 the vast majority of publications in the field have been devoted to ceria where many fundamental questions related to surfaces and near-surface processes have been addressed. Related experimental and theory studies of recent years comprise the preparation of surfaces, their morphology and atomic structure,23–27 surface characterisation,28–34 surface defects with an emphasis on vacancies and their arrangement35–46 as well as investigations on adsorbates47–59 and metal atoms or clusters41,60–70 on ceria surfaces.
In model catalysis, the approach to grow a thin oxide film on a metal support has been widely employed to study the catalytic properties of REO systems.71 To model the REO systems by such thin film studies for gaining an in-depth understanding of the structure–reactivity correlation, high quality crystalline oxides are required. Several preparation methods have been studied, such as pulsed laser deposition (PLD),11 chemical vapour deposition (CVD),72 atomic layer deposition (ALD)73 and molecular beam epitaxy (MBE).74
The method of choice to grow REO films for the studies presented here is MBE on silicon substrates in combination with annealing in various environments including a vacuum, N2 or O2 atmosphere as well as oxygen-plasma-treatment. This preparation method has many advantages over other approaches, namely
• The method allows film growth on large samples (4 inch Si wafers) thus producing many samples with very similar properties from one batch suitable for surface science characterisation tools with sample stages typically 1 cm2 in size.
• The nature of the MBE technique provides good interface and stoichiometry control (including ternary alloys) as well as the possibility of in situ film growth monitoring by reflected high energy electron diffraction (RHEED) and X-ray photoemission spectroscopy (XPS), thus ensuring the realisation of high quality single crystalline films.
• Annealing in different atmospheres and in the ultra-high vacuum provides access to clean surfaces and various oxygen stoichiometries.
The produced film systems provide a multitude of research possibilities such as
• oxidation state and structural flexibility by exploiting the full range of PrOx and CeOx with x = 1.5–2,
• thickness dependence studies that help to clarify the role of oxygen exchange between the surface and bulk,
• stoichiometry studies with ternary systems,
• tailoring the morphology and nanostructure over a wide range of surface structures, and
• model system studies to explore new physical phenomena in materials like room temperature ferromagnetism.
It is important to note that this approach fundamentally differs from the growth of ultra-thin films on metal surfaces often used for surface science studies in this field.75–88 While basic atomic and defect structures of ultra-thin films can be assumed to be similar to surfaces of bulk oxide crystals, the ultra-thin film does not provide a significant reservoir of oxygen that is important for understanding surface processes depending on an interaction between the surface and volume. For this reason, in the context of the work reported here, mostly films of a thickness ranging from some tens to some hundreds of nm are used rather than films having a thickness of only a few atomic layers.
Single crystalline binary and ternary epitaxial ceria and praseodymia films are characterized by a multitude of techniques including high-resolution transmission electron microscopy (HRTEM), X-ray diffraction (XRD), synchrotron based grazing incidence X-ray diffraction (SR-GIXRD), X-ray specular reflectivity (XRR), synchrotron based X-ray absorption fine structure (SR-XAFS), X-ray photoelectron spectroscopy (XPS), Auger electron spectroscopy (AES), synchrotron radiation based photoelectron spectroscopy (SR-PES), spot profile analysis of low energy electron diffraction patterns (SPA-LEED), non-contact atomic force microscopy (NC-AFM), Kelvin probe force microscopy (KPFM) and temperature-programmed desorption (TPD) to explore the film bulk structure, interface and surface structures, surface morphology, local polarisation, adsorption properties and surface reactivity.
The paper is organized in sections, each covering a certain system or aspect of REO film growth or characterisation. In Section II we introduce the experimental details of film growth and post-deposition annealing processes. In Section III we present the results and discussion regarding the thin film structure (III.1) as well as the surface structure and morphology (III.2). Each of these sections is structured into sub-sections covering pure praseodymia and ceria phases, trivalent systems and phase transitions due to film reduction. Surface adsorption of methanol on pure and mixed systems in different reduction states is investigated with the results presented in Section IV. The main conclusions to be drawn from the entire work are summarized in Section V.
II. Experiment
Here, we focus on the film fabrication method and post-deposition annealing (PDA) processes. Information on the characterisation experiments can be found in the respective sections and references therein.II.1 Film growth
Before being introduced into the MBE growth chamber, boron-doped (1015 cm−3) p-type, 4-inch Si(111) substrates (ρ typically 10 Ω cm) are chemically cleaned using the following procedures to obtain an atomically smooth Si surface. The application of the standard Piranha process89 is followed by 30 min etching in 40% NH4F and finally 10 min rinsing in de-ionised water.90 Subsequently, the H-terminated Si(111) wafers are annealed in UHV for 5 min at 750 °C to prepare a clean (7 × 7) reconstructed Si(111) surface. The oxide thin film growth is carried out using an electron beam evaporation method. Highly purified oxide materials (Pr6O11 or CeO2) filled in carbon crucibles are bombarded by a high energy electron beam. Note that Pr6O11 is reduced to Pr2O3 in the crucible before evaporating. The growth of the oxides occurs at a rate of 0.05 to 0.1 nm s−1. Different from Si and Ge targets which melt under electron bombardment thus providing a stable evaporation rate, oxide materials in the crucible often do not melt resulting in an instable evaporation rate. The stability of the oxide growth rate is, therefore, controlled using a quartz crystal microbalance (QCM) sensor regulating the e-beam filament current for each electron gun controller. This also allows the measurement of the real time growth rate. The typical substrate temperature and oxygen partial pressure during the growth are 625 °C and 10−7 to 10−5 mbar, respectively. The film thickness is homogeneous over the whole 4-inch wafer and can be well controlled resulting in reproducible preparation results. For studies presented here, the film thickness is varied from 10 to 200 nm. For the preparation of ceria samples, a 1 nm thick Pr2O3 film is deposited on the Si substrate as a buffer layer before the growth of ceria to avoid amorphous SiO2 formation in the initial stages of epitaxial growth and to ensure single crystallinity of the ceria films.74 The mixed oxide films are prepared by co-evaporating the oxide materials in crucibles where the Ce/Pr ratio is tuned by adjusting the growth rate of each oxide.II.2 Post-deposition annealing
Different post-deposition annealing (PDA) processes are employed on the oxide/Si samples for different objectives, as illustrated by Fig. 2. Generally, post-deposition reduction processes are carried out on CeO2/Si(111) samples to obtain different CeOx phases while post-deposition oxidation processes are performed on Pr2O3/Si(111) samples to realise different PrOx phases. PDA treatments can be categorized as follows: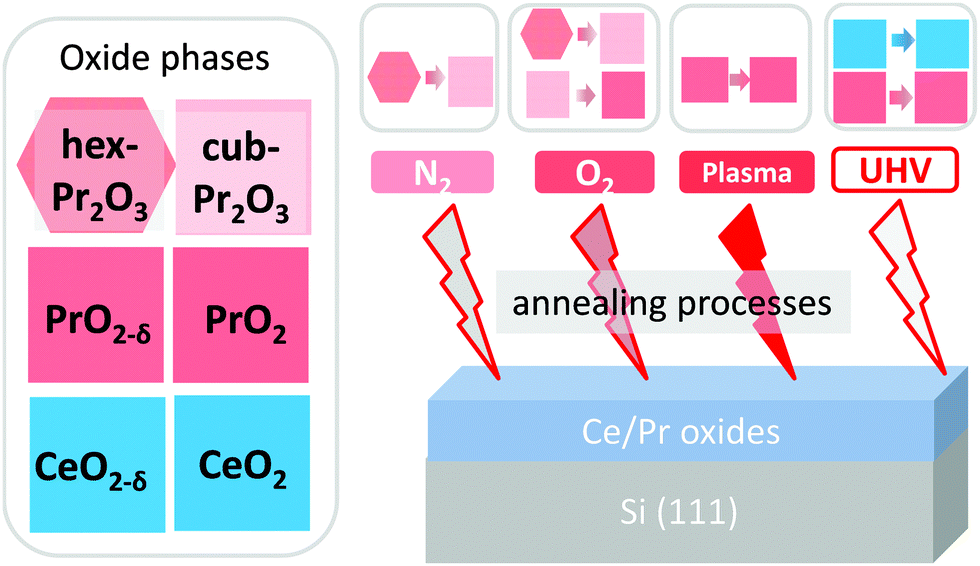 | ||
| Fig. 2 Illustration of effects of different PDA processes on the oxide phases. | ||
(1) N2-annealing. Annealing in a tubular furnace at 870 K under 1 bar N2 for 5 min. This method is used to induce a phase transition from the Pr2O3 hexagonal phase (hex-Pr2O3) to the cubic phase (cub-Pr2O3).91
(2) O2-annealing. Annealing in a tubular furnace at 670 K under 1 bar O2 for 30 min. This treatment can lead not only to a Pr2O3 hexagonal to cubic phase transition but also to a Pr2O3 to PrO2−δ phase transition.92
(3) Oxygen-plasma-treatment. A specially designed UHV-compatible, capacitively coupled radio frequency (RF) plasma source93 is used to fully oxidise defective PrO2 films.94,95 Here, the sample is inserted into the phase electrode of the capacitor and, thus, its surface is directly exposed to the oxygen-plasma (30 W power) for 30 to 60 min and under a continuous gas flow (15 sccm oxygen). The CeO2 films were also exposed to an oxygen plasma to clean their surfaces and to improve their crystalline structure. In this case, the plasma is generated by a microwave source (2.45 GHz, 360 W power, 80 sccm gas flow, 0.26 mbar gas pressure) and the sample is exposed for 15 min.
(4) UHV-annealing. Annealing under UHV conditions up to temperatures above 1270 K. This procedure is used to reduce CeO2 or PrO2 films.96,97
III. Thin film characterization
III.1 Thin film structure
A. hex-Pr2O3. The sesquioxide Pr2O3 can crystallise either in a hexagonal or a cubic phase. However, hex-Pr2O3(0001) growth is favoured in UHV on Si(111) due to the smaller lateral lattice mismatch of +0.5% compared to +2.7% mismatch for cub-Pr2O3(111).91 Specular XRD θ–2θ measurement on both hex-Pr2O3/Si(111) and cub-Pr2O3/Si(111) systems (obtained after the hex-to-cub transition due to PDA, see below) are shown in Fig. 3.74 Both, hex-Pr2O3 and cub-Pr2O3 films show diffraction peaks close to Si(hkl) (h = k = l = 1, 2, 3) reflections and hex-Pr2O3 exhibits additional peaks, e.g. hex-Pr2O3(0003) and hex-Pr2O3(0005). More detailed X-ray diffraction studies98,99 determine the epitaxial relationship of the hex-Pr2O3/Si system as hex-Pr2O3(0001); 〈−1100〉∥Si(111); 〈−1−12〉. As the interface structure and atomic bonding are of particular interest, Jeutter et al.100 analysed this system and concluded that the first layer on Si atoms is an oxygen layer, while the subsequent praseodymium layer prefers to be located above the silicon T4 site rather than the H3 site yielding the ACACAC… stacking sequence of the praseodymium atoms along the (0001) direction illustrated in Fig. 4(a). Initially, the hex-Pr2O3 film remains pseudomorphic and grows layer by layer. A relaxation process occurs when the thickness exceeds ∼12 nm and is accompanied by a roughening of the surface and a decrease of the domain size. Therefore, the growth follows the Stranski–Krastanov growth mode.89,101 Furthermore, an atomically sharp interface between Pr2O3 and Si is found, as shown in Fig. 4(b) by a cross-sectional HRTEM image of the hex-Pr2O3(0001)/Si(111) interface.102
 | ||
| Fig. 3 Specular XRD θ–2θ results for a 50 nm thick as-deposited hex-Pr2O3/Si(111) film (dotted line) and the cub-Pr2O3/Si(111) structure of the same film after annealing in oxygen (red solid line). Dashed lines indicate Si substrate bulk reflexions. (Reproduced from ref. 102.) | ||
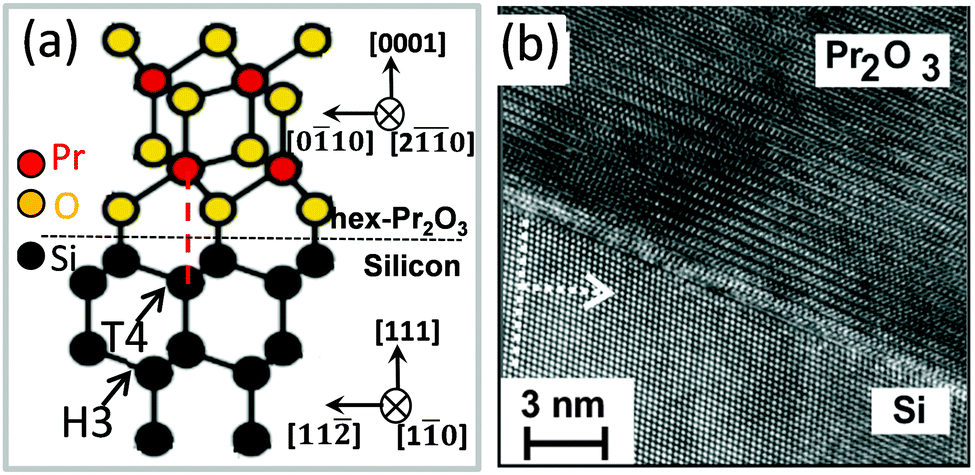 | ||
| Fig. 4 (a) Atomic model of the hex-Pr2O3(0001)/Si(111) interface. (b) Cross-sectional HRTEM image of the hex-Pr2O3(0001)/Si(111) interface. | ||
B. cub-Pr2O3. Because hex-Pr2O3(0001)/Si(111) thin films are metastable, it is possible to transform them by N2 or low pressure O2 annealing into twin-free, single crystalline type-B oriented cub-Pr2O3.74,91 The epitaxial relationship of the cub-Pr2O3/Si(111) system is studied in detail by XRD measurements and found to be cub-Pr2O3(111); 〈11−2〉∥Si(111); 〈−1−12〉.91,102 The phase transformation is accompanied by a change in the stacking sequence from ACACAC… to ABCABC… resulting in a type-B orientation. Fig. 5(a) shows the type-B cub-Pr2O3(111)/Si(111) structure model (upper panel) where praseodymium (red) oxide is connected to the Si(111) substrate through oxygen atoms (yellow). Note, that the first praseodymium layer is also above the silicon T4 site (cf.Fig. 4(a)). The bottom panel of Fig. 5(a) shows the registry of Pr and Si atoms in type A (left) and type B (right) interfaces. In a type A interface, Pr atoms from the first oxide layer are located above Si atoms from the second (111) double-layer of Si. In a type B interface, Pr atoms from the first oxide layer are located above Si atoms from the first (111) double-layer of Si. The results from ab initio calculations point to a stabilisation of the type-A/B stacking transition at the cub-Pr2O3(111)/Si(111) interface by electrostatic interactions.103 Moreover, an amorphous SiOx interfacial layer is observed at the cub-Pr2O3/Si interface by HRTEM as shown in Fig. 5(b). This interfacial layer is formed during the phase transformation annealing process due to the diffusion of oxygen through the Pr2O3 film to the Si surface, especially at elevated temperatures.102
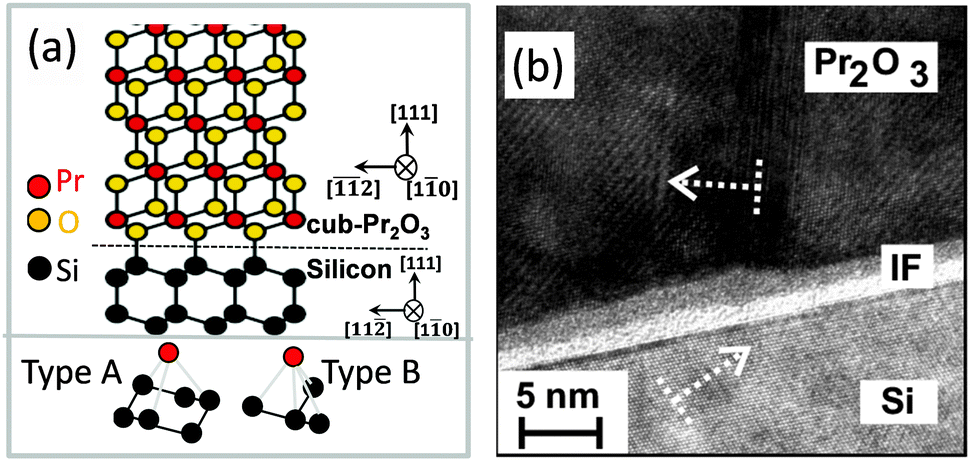 | ||
| Fig. 5 (a) Atomic model of the cub-Pr2O3(111)/Si(111) interface. (b) Cross-sectional HRTEM image of the cub-Pr2O3(0001)/Si(111) heterostructure. | ||
C. cub-PrO2−δ. It is well known that for bulk praseodymia, the β-phase Pr6O11 is stable under normal conditions104 while one has to increase both the oxygen pressure and the oxidation temperature to at least 1 bar O2 and above 300 °C, respectively, to form the PrO2 α-phase.105 Therefore, structural phase transformations due to the exposure of very thin hex-Pr2O3 films to 1 bar O2 for 30 min at a temperature of 400 °C have been studied by means of SR-GIXRD.92,106 In the following, XRD and GIXRD measurements are presented. h, k, and l values of the indexed peaks are chosen with respect to the bulk lattices of the corresponding material and will be referred to as (hkl)B, whereas labelling of the axes and rods corresponds to the hexagonal Si(111) lattice in surface coordinates (hkl)S. Fig. 6(a) shows specular out-of-plane XRD data (Θ–2Θ scans) obtained from hex-Pr2O3 films annealed at different temperatures. Obviously, the spectra obtained for films after annealing below 300 °C differ from those obtained after annealing at higher temperatures. The same result is obtained for GIXRD recording CTRs (crystal truncation rods) of higher diffraction order originating from the finite penetration depth of X-rays. We note here that CTRs are the scattering peaks arising between bulk Bragg reflections due to a sharp termination of the crystal lattice at the surface while the direction of CTRs is perpendicular to the surface and is sensitive to the surface/interface structure. An analysis of the Bragg peak positions shows that the films annealed at low temperatures keep the hexagonal structure while films annealed at higher temperatures are transformed into a cubic structure which is B oriented with respect to the Si(111) substrate. Therefore, as shown in Fig. 6(b), the ACACAC… stacking of the Pr sublattice of hex-Pr2O3 is transformed to the ABCABC… stacking of the Pr sublattice of PrO2 having the fluorite structure. In addition, the oxygen sublattice has to be rearranged. Especially, the alternating stacking of oxygen monolayers and bilayers between Pr layers for hex-Pr2O3 (denoted in Fig. 6(b) by b and c/a, respectively) is substituted by a homogeneous distribution of oxygen bilayers for PrO2. Similar to the cation stacking, the oxygen stacking follows the pattern bcabca (cf.Fig. 6(b)).
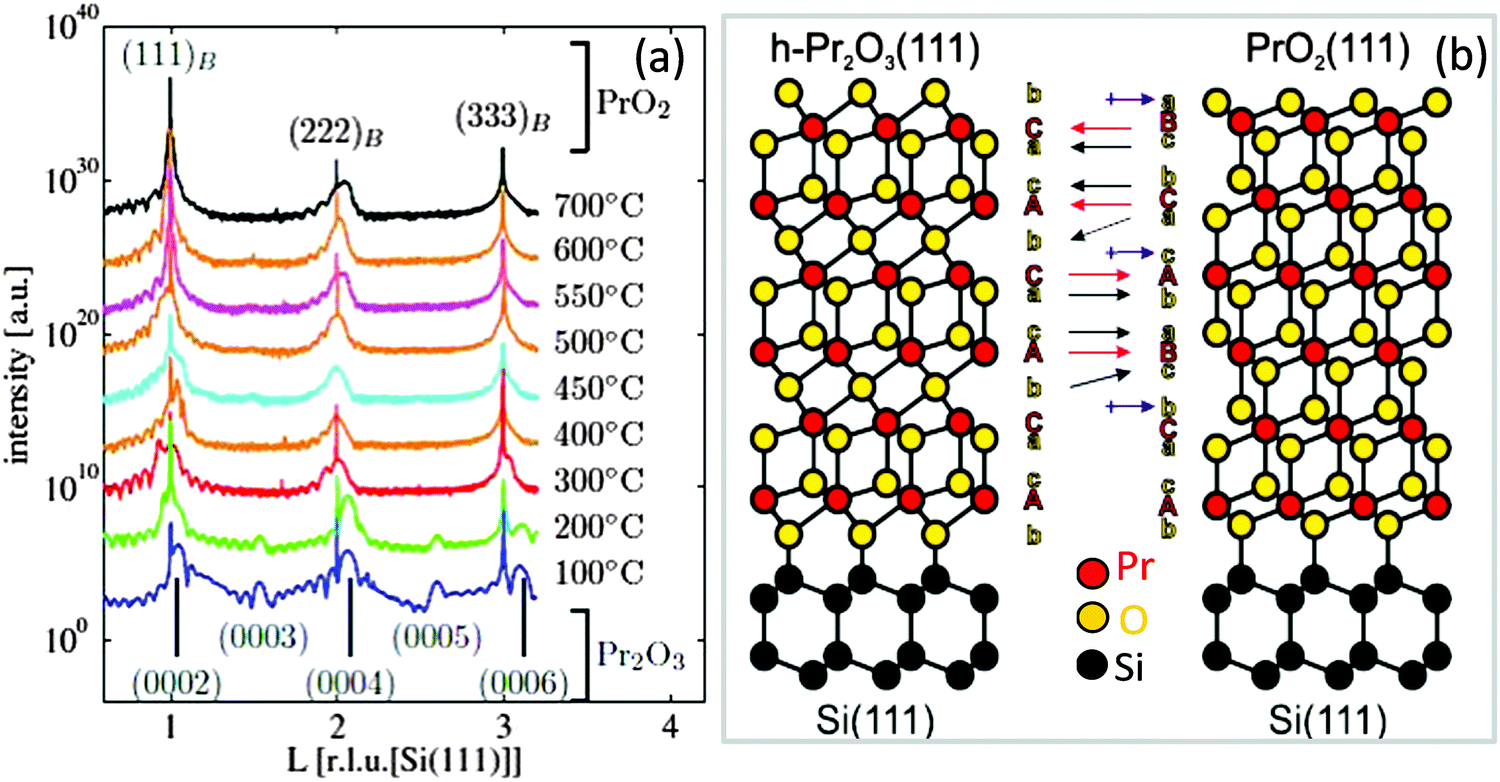 | ||
| Fig. 6 (a) Specular XRD θ–2θ measurements on a 5 nm thick hex-Pr2O3 film after annealing at different temperatures in a 1 bar O2 atmosphere. (b) Model proposed for the transition from the hex-Pr2O3 phase to the cub-Pr2O3 phase. (Reproduced from ref. 92.) | ||
A closer inspection of the specular Bragg peaks presented in Fig. 7(a) shows that a cubic praseodymia film does not exhibit one single phase but consists of two laterally separated phases with different (111) layer distances as sketched in Fig. 7(b). The layer distance of these phases decreases with increasing annealing temperature as expected for increasing oxygen load accompanied by a decreasing cation radius. In-plane scans (not presented) reveal that the lateral lattice constant of the higher oxidised film is pinned to the lateral lattice constant of hex-Pr2O3(0001). This effect has been attributed to the excellent lateral lattice matching between hex-Pr2O3(0001) and Si(111). Assuming that the praseodymia films are elastically deformed, one can calculate the cubic lattice constant of the undistorted praseodymia depending on the film stoichiometry.107 Taking into account the Poisson ratio of ν = 0.32 reported by Zaumseil et al.,108 one obtains that the inhomogeneously distributed phases of the cubic praseodymia film have Pr6O11 and PrO2−δ stoichiometry if the films are annealed at temperatures below 550 °C. After further annealing, the PrO2−δ phase is transformed into PrO2 while the coexisting Pr6O11 phase is transformed into PrO2−δ when annealing at temperatures above 600 °C. Therefore, after this treatment, the praseodymia film exhibits both laterally coexisting PrO2−δ and PrO2 phases. With respect to the Si(111) substrate, PrO2−δ as well as PrO2 are aligned by PrOx(111); 〈11−2〉 ||Si(111); 〈−1−12〉.
 | ||
| Fig. 7 (a) Close-up of three CTRs presented in Fig. 6(a): praseodymia Bragg peaks are split into two contributions. The Bragg positions are obtained from fitting: with two fitted sub-functions due to the coexistence of Pr6O11 (green line) and PrO2−δ (red line). (b) Model for the coexistence of Pr6O11 and PrO2−δ with a lateral phase separation (two column model). The layer distance in Pr6O11(111) is expanded compared to the one of PrO2−δ(111) due to the larger cation size for the lower (average) oxidation state. (Reproduced from ref. 92 and 106.) | ||
The results from XRR studies show that the previously sharp interface between the hex-Pr2O3(0001) film and the Si(111) substrate is dissolved and a praseodymium silicate with increasing thickness evolves after annealing at temperatures above 500 °C.91 The silicate interface layer consumes more and more of the praseodymia film with increasing annealing temperature. Therefore, oxidation by molecular oxygen has to be performed at temperatures below 500 °C to minimize the formation of interface silicate and alternative methods have to be applied to completely oxidise the praseodymia film.
D. PrO2. The oxidation process can be accelerated by exposing the film to an oxygen plasma. Therefore, cubic praseodymia films oxidized in molecular oxygen at annealing temperatures below 500 °C are exposed to a cold RF plasma94 to study this effect. Synchrotron based XRD studies shown in Fig. 8 demonstrate that the oxygen plasma treated praseodymia films exhibit two laterally coexisting phases of PrO2−δ and PrO2 stoichiometry. Taking into account the elastic deformation of the film due to lateral pinning at the interface (lateral lattice constant of hex-Pr2O3, cf. above) the extrapolated cubic lattice constants are 540.4 pm and 538.6 pm for PrO2−δ and PrO2, respectively. For comparison, a lattice constant of 539.2 pm has been reported for bulk PrO2 after annealing at 720 °C in 92 bar O2.109 Thus, a higher oxygen load is obtained by plasma oxidation compared to oxidation in molecular oxygen at high pressure. Furthermore, a detailed analysis of the XRD data within the kinematic diffraction theory shows that a very thin crystalline Pr6O11 layer (thickness less than 2 nm) is stabilized at the interface even when the residual film is completely oxidised.
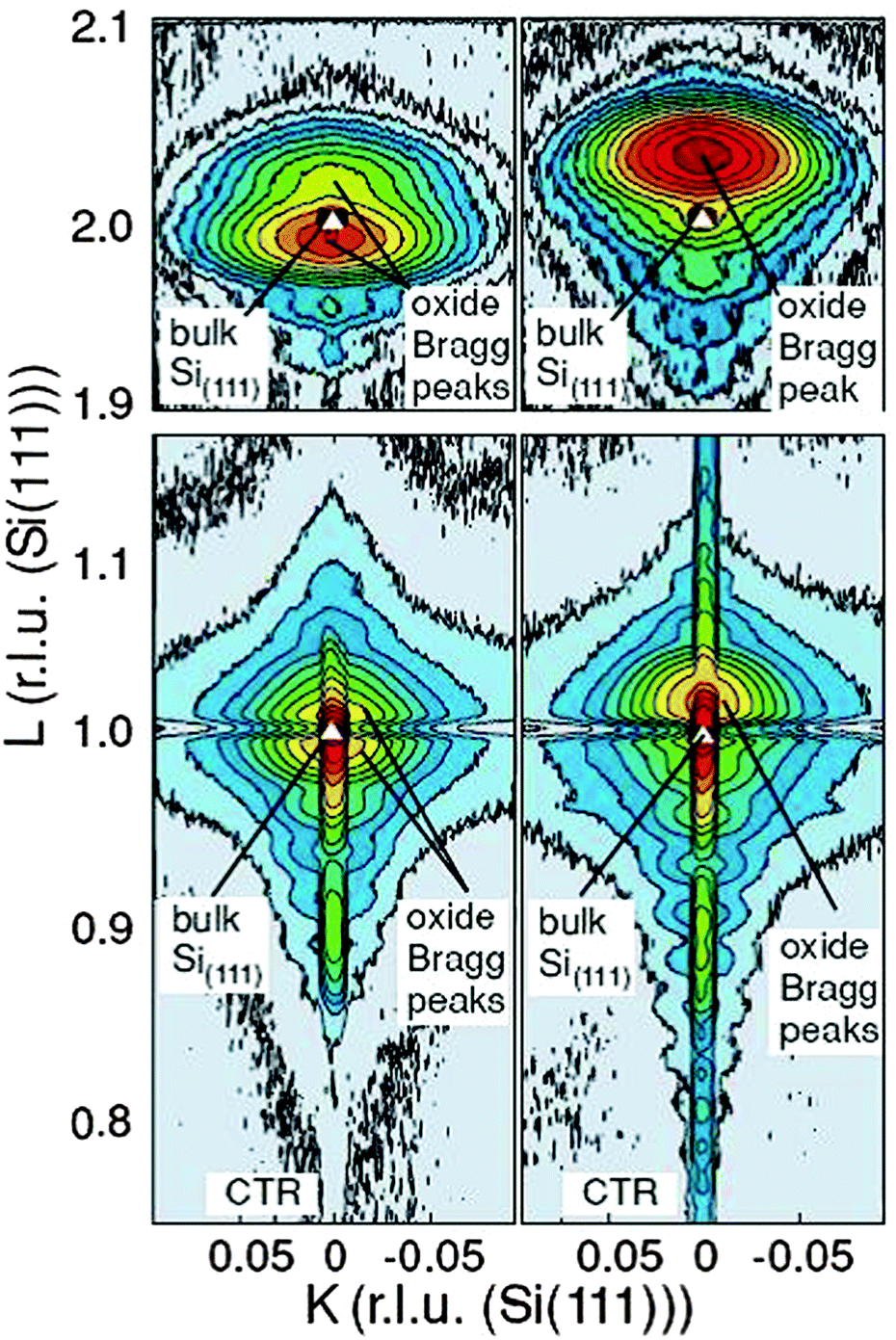 | ||
| Fig. 8 Reciprocal space maps close to the (111) Bragg peak (bottom) and (222) Bragg peak (top) of 15 nm thick praseodymia films investigated by SR-GIXRD. Left side: praseodymia film after oxidation in 1 atm oxygen. Right side: praseodymia film after oxygen plasma treatment. Clearly, the position of the praseodymia Bragg peaks shift due to the reduced lattice constant after the incorporation of oxygen. (Reproduced from ref. 95.) | ||
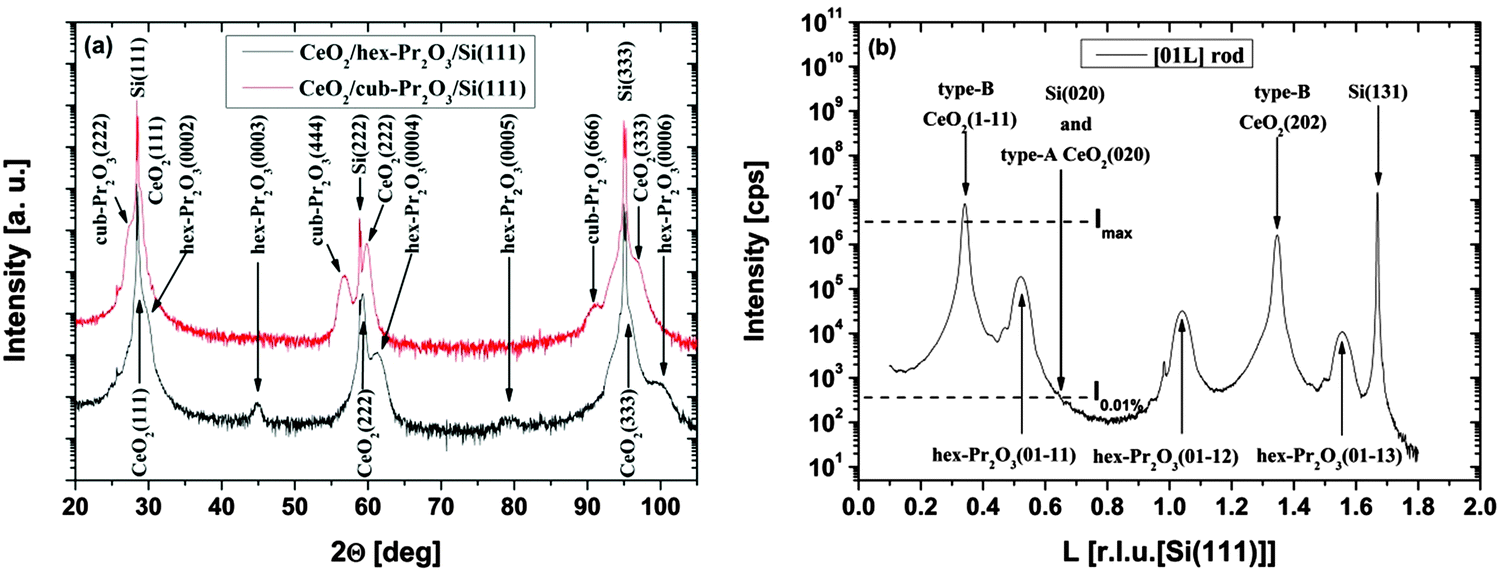 | ||
| Fig. 9 (a) Specular XRD θ–2θ measurements for a 38 nm thick CeO2/hex-Pr2O3/Si(111) and a 22 nm thick CeO2/cub-Pr2O3/Si(111) heterostructure. (b) SR-GIXRD scan along the [01l]S rod of the CeO2/hex-Pr2O3/Si(111) sample. (Reproduced from ref. 74.) | ||
As the type-B oriented growth of CeO2(111) on a hex-Pr2O3/Si(111) support appears to be surprising considering the stacking sequence transition from ACACAC… to ABCABC… at the CeO2(111)/hex-Pr2O3(0001) interface, ab initio calculations have been performed to identify the microscopic origin of this behaviour.26 The model of Fig. 10(a) shows that two facts are responsible for the preference of the type-B orientation. (i) The formation of a Pr4+ monolayer conserves the semiconducting character of the CeO2/Pr2O3 interface and leads to an electronic continuation. (ii) The oxygen sub-lattice transports a BACBAC stacking information and keeps the crystallographic continuation. In contrast, type-A oriented CeO2(111) would cause a stacking fault at the interface. Thus, type-B oriented CeO2(111) on hex-Pr2O3(0001)/Si(111) is favoured by 8 eV nm−2.74 HRTEM cross-sectional images shown in Fig. 10(b) and (c) exhibit the details of a CeO2 layer on hex-Pr2O3 and cub-Pr2O3 buffers, respectively. Notably, interfacial layers are formed even in a CeO2/hex-Pr2O3/Si heterostructure.
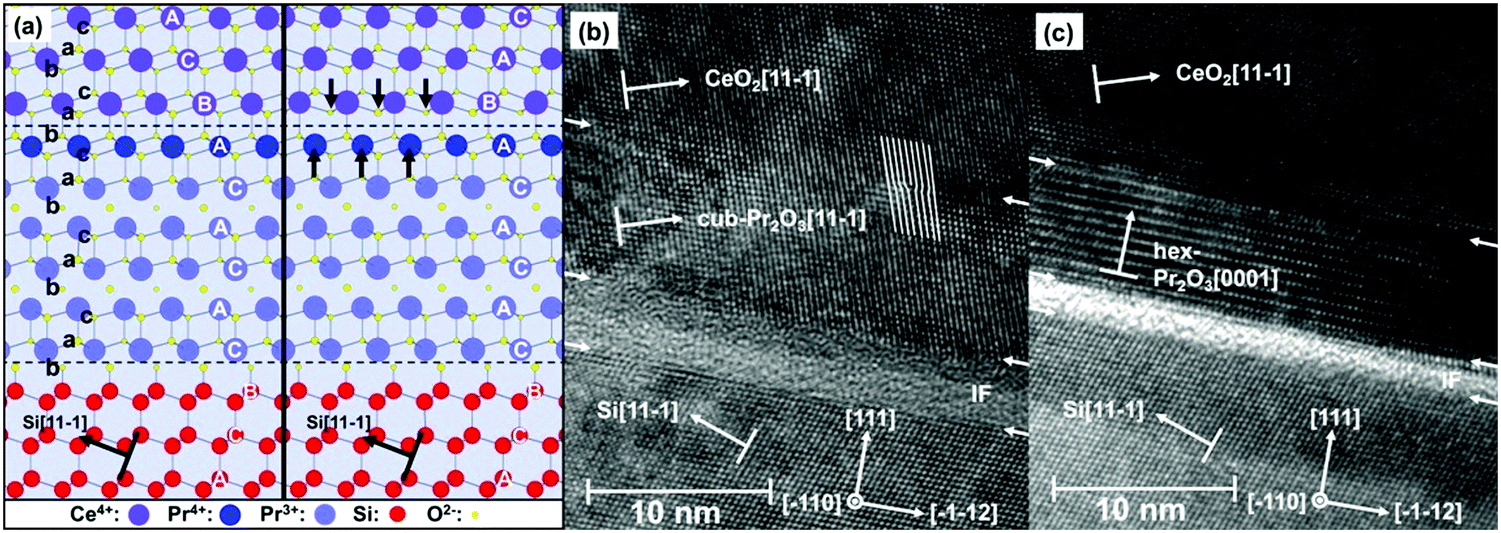 | ||
| Fig. 10 (a) Atomistic interface model for type-A and -B CeO2(111)/hex-Pr2O3(0001)/Si(111) layer stacking. (b) HRTEM image of the cub-Pr2O3(111)/Si(111) interface. (c) HRTEM image of the hex-Pr2O3(0001)/Si(111) interface. (Reproduced from ref. 26.) | ||
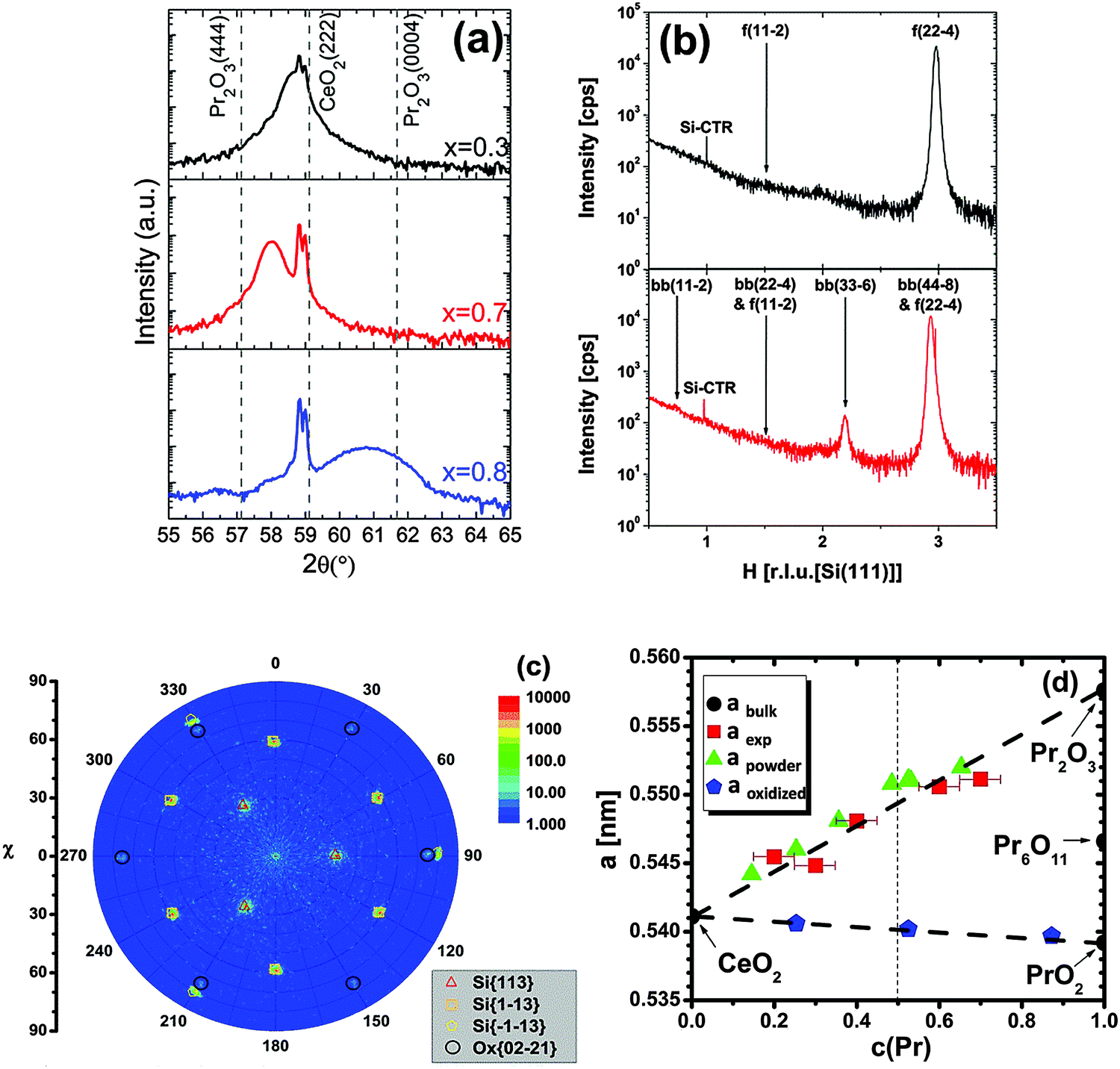 | ||
| Fig. 11 (a) Specular θ–2θ XRD measurements of 24 nm thick Ce1−xPrxO2−δ/Si(111) samples (x = 0.3, 0.7, 0.8). The dotted straight lines mark the Bragg peak positions of cubic Pr2O3(444), CeO2(222) and hexagonal Pr2O3(0004), respectively. (b) GIXRD scan along the [H00]S direction of Ce0.7Pr0.3O2−δ and Ce0.3Pr0.7O2−δ films (f: fluorite structure, bb: bixbyite structure). The Pr-rich sample exhibits an additional bixbyite phase as evident from the bb(33−6) peak. (c) Pole figure measurement of Ce0.2Pr0.8O2−δ(0001)/Si(111) measured at 2θ = 56.5 °. Black circles demonstrate the six-fold symmetry and thus a hexagonal crystal structure. (d) The bulk lattice parameter variation of the films as a function of the Pr concentration in the films. The reference from powder studies21,115 is given for comparison. (Reproduced from ref. 113.) | ||
We note here that due to the valence state difference between Pr3+ and Ce4+, the doping of Pr into the CeO2 lattice is an effective way to induce oxygen vacancies  in CeO2 films. It is well known that oxygen vacancies can strongly affect physical and chemical properties of oxides, such as the conductivity, optical and catalytic properties. For example, recently it has been reported that the room temperature ferromagnetism (RTFM) in diluted magnetic oxides is strongly related to oxygen vacancies118,119 and our Ce0.8Pr0.2O2−δ film shows strong magnetic properties.120 The oxidative reactivity as a function of the Ce–Pr stoichiometry within different gas ambiances like CO, H2O and O2 is introduced in Section III.2.6.
in CeO2 films. It is well known that oxygen vacancies can strongly affect physical and chemical properties of oxides, such as the conductivity, optical and catalytic properties. For example, recently it has been reported that the room temperature ferromagnetism (RTFM) in diluted magnetic oxides is strongly related to oxygen vacancies118,119 and our Ce0.8Pr0.2O2−δ film shows strong magnetic properties.120 The oxidative reactivity as a function of the Ce–Pr stoichiometry within different gas ambiances like CO, H2O and O2 is introduced in Section III.2.6.
III.2 Surface structure and morphology
The surface structure of praseodymia films as well as of ceria films has been studied by the high resolution diffraction of low energy electrons to obtain a better insight into their symmetry properties. Furthermore, the surface morphology has been studied analysing the profiles of diffraction peaks to explore the diversity of the film systems. | ||
| Fig. 12 2D high resolution electron diffraction patterns of praseodymia film surfaces; (a) for a 9 nm thick hex-Pr2O3(0001), (b) for a 9 nm thick cub-Pr2O3(111) and (c) for a 12 nm PrO2(111) film. (Reproduced from ref. 123 and 124.) | ||
For cubic Pr2O3, hydroxide formation is limited to the topmost layers of the film.95 If transported under ambient conditions, the majority of the bulk structure is preserved and only minor oxidation occurs as shown by XRD.124 It is further found that a clean single phase Pr2O3(111) film can be prepared by annealing the cub-Pr2O3(111) at temperatures above 300 °C under UHV conditions to remove hydroxides from the surface.124 Therefore, we assume that a hydroxide layer as well as the excess oxygen can be removed by annealing at higher temperatures and no capping layer is needed to transport cub-Pr2O3(111) film under ambient conditions. The SPA-LEED measurements of the cub-Pr2O3(111) film shown in Fig. 12(b) exhibit a hexagonal (4 × 4) diffraction pattern. This pattern is attributed to the bixbyite structure of (111) oriented Pr2O3 having a 16 times larger surface unit cell than hex-Pr2O3. In addition, a three-fold symmetry of the first order diffraction spot intensities clearly appears due to the ABCABC… stacking of the cubic structure.
Contamination free cub-PrO2(111) surfaces can be prepared by plasma oxidation of cubic PrO2−δ(111) films.94 SPA-LEED measurements of such films show a hexagonal (1 × 1) diffraction pattern due to the (111) orientation of the cubic fluorite type unit cell. As shown in Fig. 12(c), the intensity of the first order diffraction spots exhibit the same threefold symmetry based on the ABCABC… stacking as observed for the cub-Pr2O3(111) film.
 | ||
| Fig. 13 Cross-section of the central diffraction spot for a 9 nm thick hex-Pr2O3(0001) film for (a) anti-Bragg conditions (out-of-phase) and (b) Bragg conditions (in-phase). Two Lorentzian functions are necessary to describe the spot profile. (Reproduced from ref. 123.) | ||
 | ||
| Fig. 14 The FWHM of the central diffraction spot for a 9 nm thick hex-Pr2O3(0001) film oscillates with increasing scattering vector K⊥ perpendicular to the surface indicating single atomic step heights. By modelling the oscillation with a cosine function, the average step height d and terrace width can be determined. The additional constant offset of the FWHM is attributed to small grain sizes. (Reproduced from ref. 123.) | ||
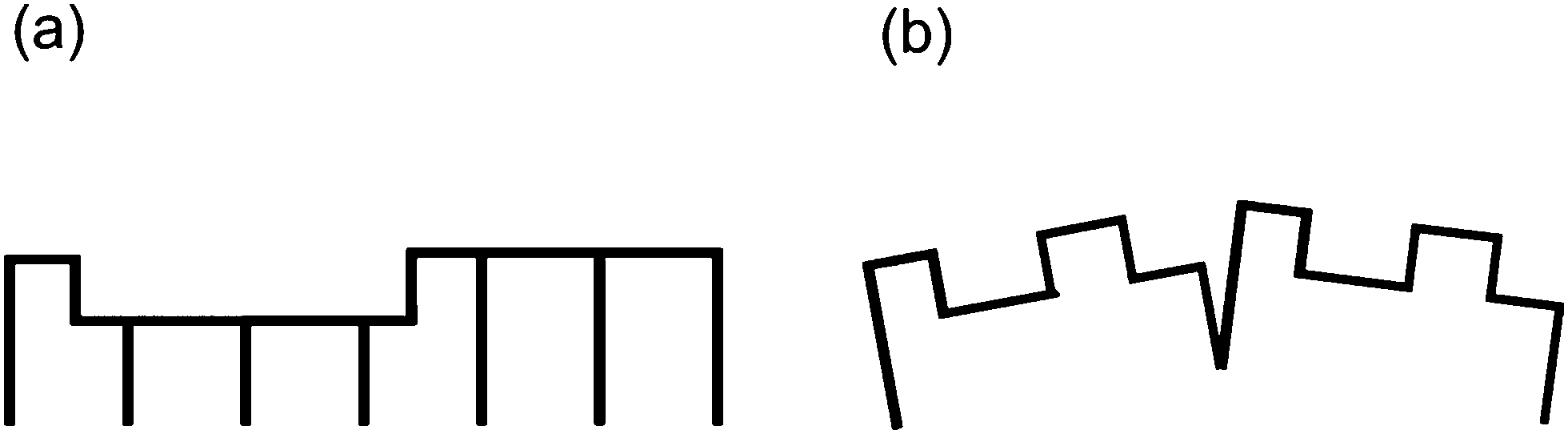 | ||
| Fig. 15 Schematic model of the hex-Pr2O3 (a) and cub-Pr2O3 (b) surface morphology. | ||
The SPA-LEED analysis of single phased cub-Pr2O3(111) also shows an oscillating FWHM of the specular diffraction peak due to single atomic steps at the surface as shown in Fig. 16. The average terrace size on cub-Pr2O3(111) is, however, significantly smaller than that for hex-Pr2O3(0001) and the density of atomic steps is significantly larger.95 Furthermore, in contrast to hex-Pr2O3(0001) films, a mosaic spread is concluded from an additional linear increase of the FWHM with increasing electron energy indicating a higher overall roughness of the surface. We assume that the surface roughening originates from strain effects induced during the hex-to-cub transition. The stress is caused by the lateral pinning of the oxide film to the lattice of hex-Pr2O3(0001) at the Si interface causing a strong vertical distortion of the cub-Pr2O3(111) film.92 Thus the film partly releases the high compressive stress via the formation of tilt mosaics as sketched in Fig. 15(b).
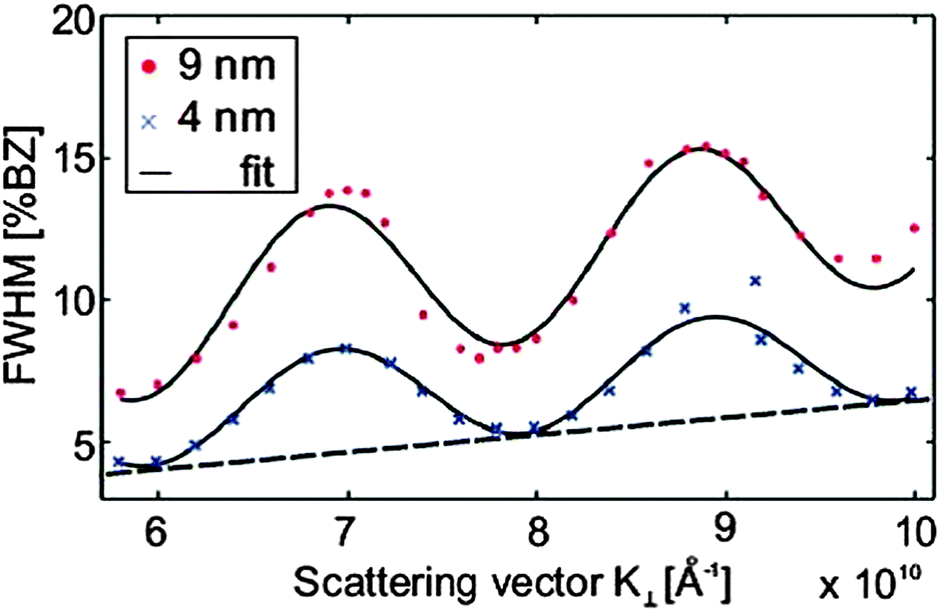 | ||
| Fig. 16 FWHM of the central diffraction spot against scattering vector K⊥ for cub-Pr2O3(111) films of different thicknesses. Similar to the hex-Pr2O3(0001) films, an oscillating behaviour due to single atomic steps is observed. The step height and terrace width can be determined by fitting a cosine function superimposed by a linear function to the data. The linear contribution is caused by the mosaic spread of the cub-Pr2O3(111) films. (Reproduced from ref. 124.) | ||
To confirm and elaborate on the spot profile analysis results, the surface morphology of cub-Pr2O3(0001) films as grown and after annealing is investigated by NC-AFM under UHV conditions125 allowing for direct imaging of surface topographic features and a detailed analysis of the origins of roughness. Experimental methods for sample preparation, NC-AFM imaging and the instrument used have been described in detail elsewhere.26,27,126,127Fig. 17 shows a comparison of imaging results for films with 10, 50, 100 and 200 nm thickness for surfaces prepared by post-deposition annealing for one hour at 830 K. This annealing temperature is chosen because it causes no significant change in the surface morphology compared to non-prepared samples but removes some contaminants present due to the transport of the sample under ambient conditions. The frame size of 250 nm × 250 nm allows for an identification of individual surface features but also for an assessment of the roughness at different scales and the overall homogeneity. The surface morphology significantly varies with film thickness. On the surface of the 10 nm film shown in Fig. 17(a), no well-shaped features are found and the height of the irregular structures present is in the range of 2 to 3 nm. The 50 nm film shown in Fig. 17(b) reveals more contiguous, slightly rounded features where some of them appear with a triangular shape. Height differences on the surface are larger compared to the 10 nm film, however, overall the surface is relatively flat. This can also be deduced for the roughness analysis shown in Fig. 17(e) where the 10 and 50 nm films appear with a rather narrow height distribution. For the 100 nm film shown in Fig. 17(c), the surface is dominated by roundish features with a typical size of 30 nm. In contrast to the thinner films, the surface is rough on a 10 nm lateral scale. With height differences amounting to several nanometres, this sample has a roughness that is by a factor of five larger than the roughness of the 50 nm film. The 200 nm film shown in Fig. 17(d), in contrast, reveals oriented triangular islands with rather straight edges where the vertices of the triangles point into [−1−12] directions. The surface roughness is comparable to the roughness of the 100 nm film. The trend in roughness can be explained by a phenomenon commonly referred to as kinetic roughening.128,129 The first layers of the film are grown in a two-dimensional mode resulting in a rather flat surface. Then the growth mode changes to a three-dimensional mode for increasing thickness where the growth of the next layers starts before the first layer is completed. This leads to the formation of islands and surface roughening. Note, however, that numerical values for the roughness of differently thick films cannot easily be compared with each other as the surface morphology changes with film thickness.
 | ||
| Fig. 17 (a) to (d) NC-AFM images representing the topography of cub-Pr2O3(0001) films with a thickness of (a) 10 nm, (b) 50 nm, (c) 100 nm and (d) 200 nm after post deposition annealing to 830 K. The in-plane crystallographic directions denoted in (a) are the same for all images. (e) The height distribution illustrates a significant difference in roughness between thinner and thicker films. The quantity plotted along the ordinate represents the number of image pixels from the 512 × 512 total pixels having a height within an interval of 0.33 nm around the value plotted along the abscissa. The width of the distributions are a measure for the RMS surface roughness. | ||
The results of a measurement on a larger scale after annealing the 200 nm film for one hour at 570 K are presented in Fig. 18(a). The image is dominated by triangular islands often having a 10 nm wide rim with an opening at one side as is best seen in the three-dimensional representation of an island shown in Fig. 18(b). Triangular islands have a side length of 50 to 60 nm and a height in the range of 6 to 10 nm where the side facets have a slope of approximately 30°. While we speculate that the rim formation is related to peculiarities of the growth in a fluorite type lattice,130 the formation of pyramids with a triangular base is discussed below in the context of ceria film growth in Section III.2.4. Attempts for obtaining a better ordered and cleaner film surface by annealing the film at higher temperature to allow for imaging with higher resolution fail as the film decomposes when annealed at temperatures higher than 850 K as demonstrated in Section III.2.5.1.
 | ||
| Fig. 18 (a) Topography of a 200 nm thick cub-Pr2O3(0001) film exhibiting a morphology dominated by triangular islands oriented in the [−1−12] direction. The orientation of the film is the same as in Fig. 17. (b) Three-dimensional representation of the island marked in (a) revealing the 10 nm wide rim with an opening terminating the island. (c) Photo of the as-prepared sample with the film exhibiting a red/violet colour. (d) Photo of the sample holder with the sample after annealing at a temperature above 850 K yielding a grey colour. (e) Topography of a 200 nm thick cub-Pr2O3(0001) film subjected to oxygen plasma treatment. (f) Photo of the sample holder with the sample after plasma treatment of the as-grown sample yielding a green colour. | ||
In summary, we note that the surface of the cub-Pr2O3(0001) film exhibits characteristic structural features where the morphology of the thickest films investigated is dominated by regular and oriented triangular islands with a surface appearing rather flat. Annealing at higher temperatures yields a cleaner surface and slight changes in detail; however, the nanoscale surface morphology does not change during annealing at temperatures up to 830 K defining the limit for the decomposition of the film. The as-grown film visually appears with different shadings of red and violet depending on film thickness and preparation details as shown in the example of Fig. 18(c). The colour changes to grey upon heating beyond the decomposition limit of 830 K as seen in Fig. 18(d). When the as-grown film is treated in an oxygen plasma as described in Section II.2, the surface morphology does not change (see Fig. 18(e)), however, the colour changes to green as shown in Fig. 18(f) and back to violet upon heating the plasma-treated sample to a temperature of 630 K (the results not shown).
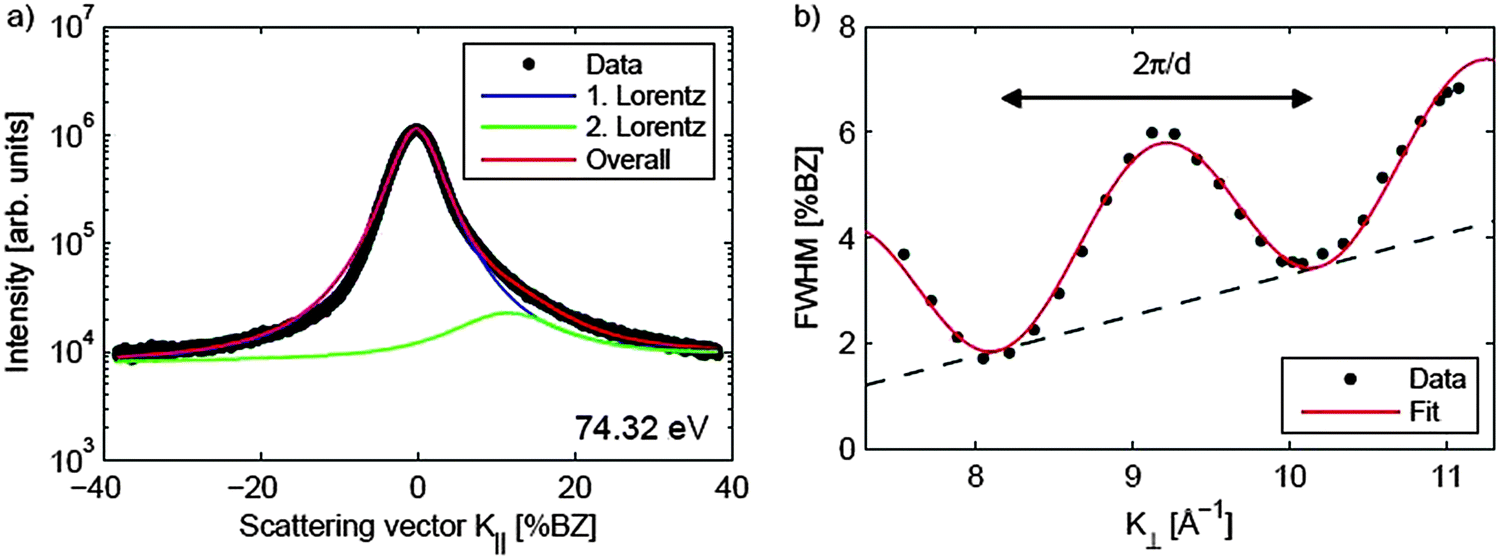 | ||
| Fig. 19 (a) Spot profile of the specular (00) diffraction spot of a 250 nm thick CeO2(111) film. The asymmetric peak shape is caused by surface facets, thus, two functions are needed to describe the spot profile. (b) FWHM of the central peak plotted against the scattering vector K⊥. Similar to observations for a praseodymia film, single atomic steps are present at the surface resulting in an oscillation of the FWHM. In addition, a linear increase of the FWHM due to mosaics is observed. (Reproduced from ref. 97.) | ||
To reveal the structure of the facets in detail, NC-AFM imaging is performed on surfaces of as-prepared and slightly heated cub-CeO2(111) samples131 using instruments and methods described earlier.23,26,27,127 Here, we discuss the morphology for the as-prepared and slightly heated 180 nm cub-CeO2(111) film that will further on be denoted as the low temperature morphology. In this temperature regime, post-deposition preparation does not affect the film surface structure significantly; however, the film is slightly reduced as will be discussed in Section III.2.5.2. The NC-AFM image shown in Fig. 20(a) represents a result for the as-prepared film while the results for a film heated to 510 K is shown in Fig. 20(b). Annealing the film to 510 K removes parts of the contamination layer on the sample surface, thus, allowing for imaging more structural details on the surface. The images exhibit a distinct surface structure consisting of large triangular based, oriented pyramidal islands that are 4 to 6 nm high and have an edge length of typically 100 nm. Close inspection reveals a terrace structure forming the pyramids as most clearly seen in Fig. 20(c). Pyramidal faces are composed of 3 to 5 nm wide (111) terraces separated by step edges with a height of 0.31 nm corresponding to the height of one O–Ce–O triple layer in agreement with the SPA-LEED results introduced above. The average slope of the pyramidal facets is determined by a cross-sectional analysis performed on several pyramids. The analysis yields different slopes for the bottom and top layers of the pyramids. For the bottom layers, we find a slope angle of 10° relative to the (111) surface plane which can be associated with a {332} facet while the top layers exhibit an angle of 5° relative to the (111) surface plane that can be associated with a {665} facet. Fig. 20(d) represents a side view model of the two facets yielding slope angles and terrace widths in good agreement with experimental observations. This analysis shows that the as-grown cub-CeO2(111) film provides a ceria surface exhibiting an exceptionally rich structure of step edges regularly arranged in terraced pyramids. Such a film could be most interesting for applications requiring a large number of regular steps with a height of one O–Ce–O triple layer.
 | ||
| Fig. 20 (a) to (c) NC-AFM images taken on a cub-CeO2(111) sample representing (a) the as-prepared ceria sample and (b and c) the sample heated to a temperature of 510 K. The terrace structure of the pyramid is clearly seen in (c). Bright spots preferentially attached to step edges are due to contaminants that could not be removed by the heat treatment. (d) Side view model for the bottom (typically 10) and top (typically 5) layers of the pyramids yielding nominal values for the terrace widths and slope angles. (Reproduced from ref. 131.) | ||
Beyond possible applications of the ceria film, it is interesting to discuss the growth mechanism yielding the pyramidal island structure. As a hypothesis, we anticipate that the nucleation of CeO2 is templated by the praseodymia buffer layer grown in a well-defined orientation and registry on the Si(111) substrate (see Section III.1.2). We further assume that the growth of the pyramidal structures is due to a kinetically limited process, similar to the one proposed for the homoepitaxial growth of platinum on Pt(111).132 As ceria crystallises in the fluorite structure, two different types of step edges, namely type-A and type-B, are relevant for a growth process on the (111) surface.23,133 The probability for particle attachment is higher at steps of type-A so that type-B steps grow faster and preferentially triangles are grown, although, the substrate exhibits a hexagonal surface symmetry.130 The pyramidal structures are commonly referred to as wedding cakes that are the result of a multilayer growth in the presence of a significant Ehrlich–Schwoebel barrier.134–136 Adatoms that adsorb during MBE on the top of a terrace diffuse to the borders if enough thermal excitation energy is available. In the absence of a step edge barrier, the adatoms would easily overcome the ascending step edge at the border and diffuse to a lower terrace finally resulting in the growth of a flat surface. However, if an Ehrlich–Schwoebel barrier is present and larger than the average diffusion energy, the adatoms cannot overcome step edges easily. While the probability for diffusion of adatoms to lower terraces is reduced, the probability for building a new layer by the remaining adatoms increases. This results in an enhanced intra-layer diffusion on the cost of inter-layer diffusion and the synchronous growth of structures on all terrace levels. The change in the slope between the bottom and top layers is attributed to a change in the Ehrlich–Schwoebel barrier during the growth process.
III.2.5.1 Reducing PrO2. The reduction behaviour of PrO2(111) films has been the subject of several detailed studies.94,96,107,137 As discussed in Section II.1, XPS measurements reveal that fully oxidised PrO2(111) films can only be achieved by oxygen plasma treatment.94 The Pr 3d spectra shown in Fig. 21 exhibit a complex peak structure based on hybridization between the Pr 4f states and the O 2p valence band. However, the amount of Pr4+ ions can be estimated by the relative intensity of the peak labelled as z (or z′). By thermally reducing the film, it can be observed that the intensity of the z (or z′) peak is lowered. Thus, the Pr4+ concentration is decreased revealing a homogeneous reduction of the film surface.
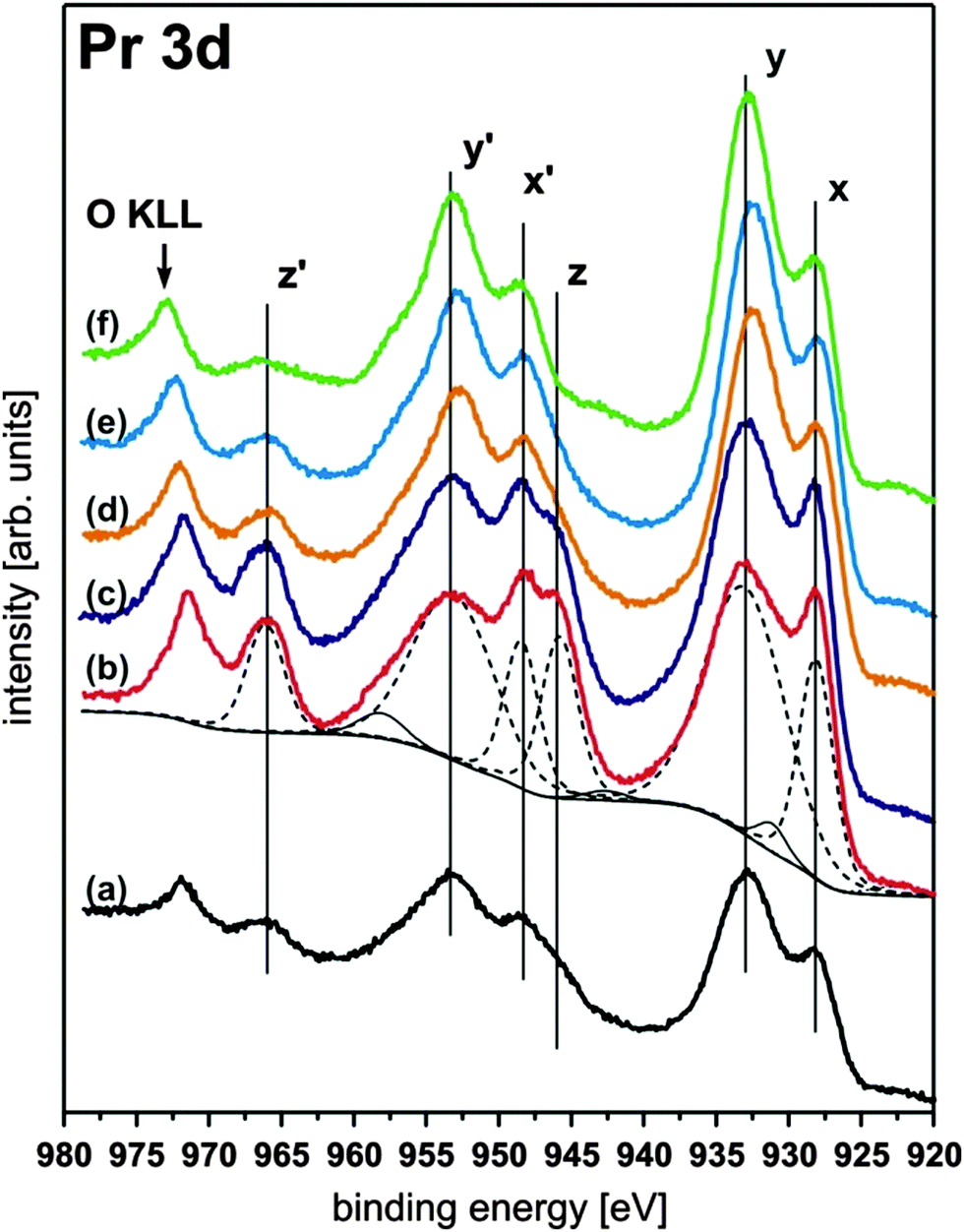 | ||
| Fig. 21 Pr 3d XP spectra of a 15 nm thick PrO2(111) film composed of three peaks (x, y, z) in each 3d5/2 and 3d3/2 component. The peaks labelled with z and z′ are directly related to the amount of Pr4+ ions present at the surface. Hence, the intensity of the z and z′ peaks drastically increases after the as-prepared PrO2−δ sample (a) is exposed to an oxygen plasma yielding a contamination free PrO2 surface (b). Subsequent annealing under UHV conditions (405 K (c), 580 K (d), 680 K (e), and 920 K (f)) leads to a reduction of the films as shown by the decreasing intensity of the z or z′ peak. (Reproduced from ref. 94.) | ||
The loss of oxygen has also been confirmed by TPD experiments. Characteristic peaks can be observed in the TPD spectra of molecular oxygen shown in Fig. 22 suggesting thermally induced phase transitions.94,107 However, the corresponding XRD analysis of the PrOx(222) Bragg peak reveals that these transitions result in coexisting oxide phases of different stoichiometries as evident from the appearance of coexisting praseodymia Bragg peaks in XRD measurements shown in Fig. 23.
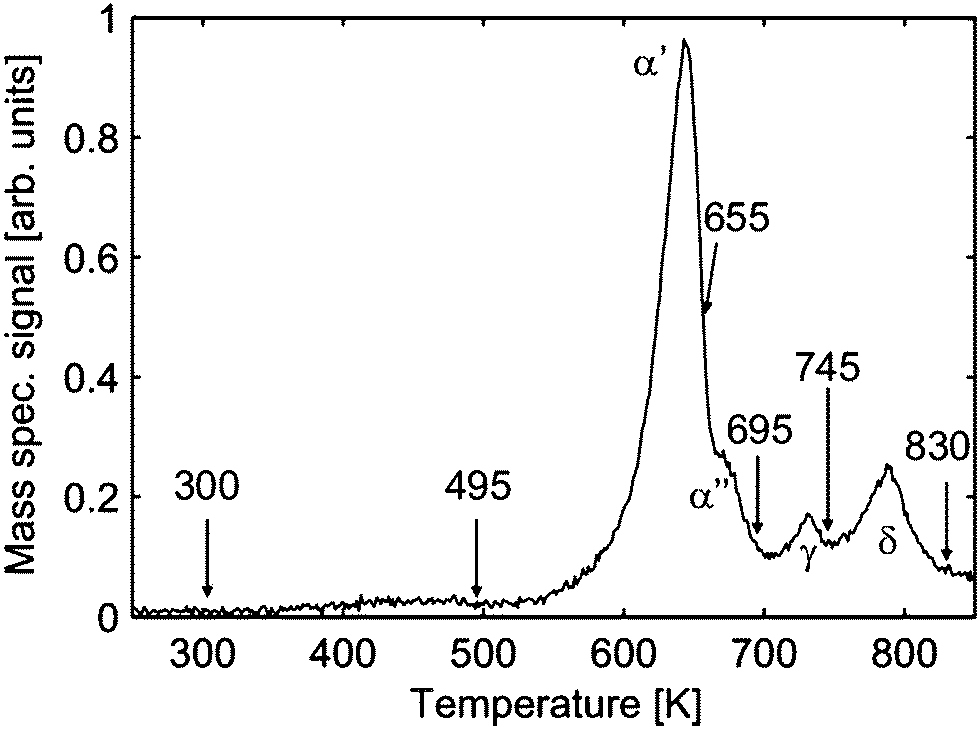 | ||
| Fig. 22 Temperature programmed desorption spectrum (heating rate 1 K s−1) of molecular oxygen for a 16 nm thick PrO2(111) film. After a certain characteristic desorption peak is observed the desorption is stopped (arrows) and the sample is immediately cooled to RT to perform GIXRD experiments. (Reproduced from ref. 107.) | ||
 | ||
| Fig. 23 XRD measurements of the region close to the (222)B Si Bragg peak for samples quenched at different temperatures. The oxide peak is described by two Lorentzian functions representing two laterally coexisting praseodymia phases. For the non-reduced films, an additional broad peak (dashed lines) is observed which can be attributed to a very thin Pr6O11(111) interface layer. (Reproduced from ref. 107.) | ||
It was further shown by out-of-plane GIXRD measurements that praseodymia films are pinned to the lateral lattice constant of hex-Pr2O3(0001) and, thus, tetragonally distorted during film growth.92 This growth induced exclusive pinning of the lateral lattice constant is breached during the reduction process. An explanation for the de-pinning is the transformation to an amorphous interface between the substrate and film at elevated temperatures.137 The stoichiometries of the coexisting oxide phases are determined by a calculation of their pseudo-cubic lattice constants from the in-plane and out-of-plane GIXRD data for the tetragonally distorted film with the results shown in Fig. 24.107 All intermediate phases of praseodymia are formed by vacancy generation in the oxygen sublattice of the fluorite structure of PrO2 and, thus, these vacancies result in an expansion of the cubic unit cell due to the decreased average oxidation state of praseodymium. Following Vegard's law,114 the film stoichiometry can be determined. Thus, the expansion of the re-calculated bulk lattice constant is directly proportional to the stoichiometry of the corresponding intermediate phase.
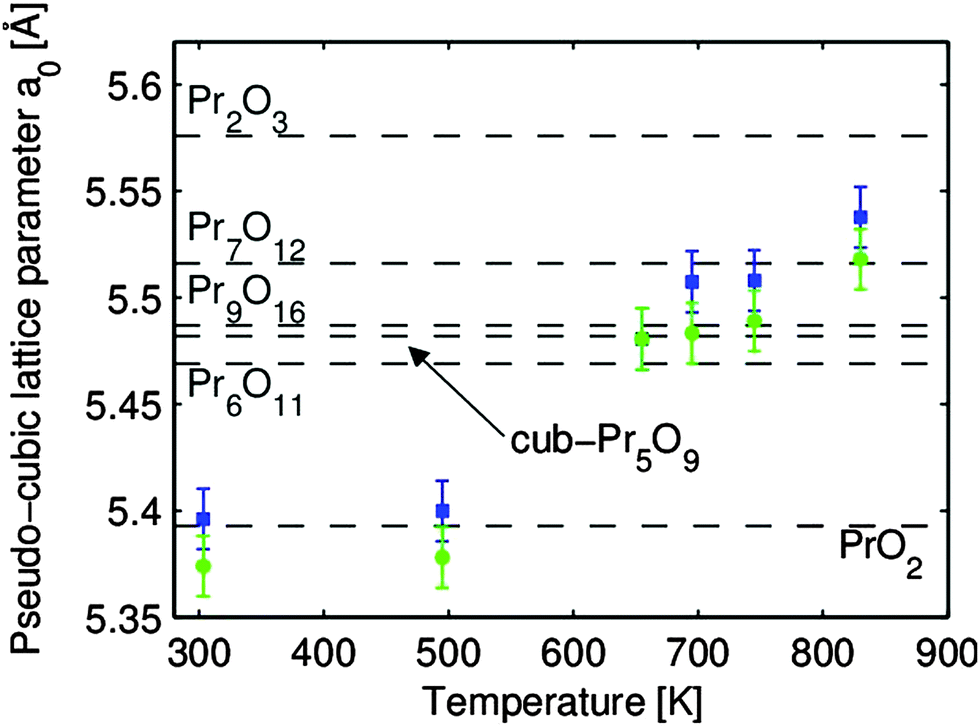 | ||
| Fig. 24 The pseudo-cubic lattice constants a0 of the two coexisting praseodymia phases related to thermal reduction. The lattice constants are determined under consideration of the elastic deformation of the film. The bulk values of the praseodymia phases stable at room temperature are marked by dashed lines. (Reproduced from ref. 107.) | ||
In summary, we find that PrO2 films are not homogenously reduced.96,107,137 Combining the results from the surface sensitive SPA-LEED measurements with an analysis of XRD results, a structure model for the reduction process is developed as depicted in Fig. 7. During the reduction, a mosaic spread is formed at the surface92,137 while the amorphisation of the interface layer underneath the coexisting oxide phases progresses during heat treatment.
III.2.5.2 Reducing CeO2. The reduction properties of CeO2(111) films show similarities but also significant differences in comparison to PrO2(111) films. For instance, after annealing at 600 °C, praseodymia films are fully reduced to cub-Pr2O3(111)124 while ceria films exhibit only slight oxygen loss at the surface as shown by XPS while the bulk does not change its stoichiometry as evident from the XRD results shown in Fig. 25 and 26, respectively.97 As a similarity we find that also ceria films like praseodymia films form coexisting phases when annealed under UHV conditions. After annealing at 650 °C, the Ce7O12 phase is stabilised which can also be detected at the surface of the film.138 The non-uniform reduction of ceria is concluded from the CeO2(111)B bulk diffraction peak that is present even after the formation of the Ce7O12 phase. As evident from Fig. 26, further annealing at 700 °C leads to the additional stabilisation of the Ce11O20 phase having a higher oxygen content than the coexisting Ce7O12 phase. Since no increase of the oxygen content is observed at the surface as evident from Fig. 25, we assume that Ce11O20 is formed close to the interface between the substrate and film due to oxygen diffusion towards the substrate. In contrast to praseodymia, single phased cub-Ce2O3(111) films cannot be prepared by annealing in UHV. The high temperatures needed to form cub-Ce2O3(111) for the entire film destroy the film by the formation of silicides and silicates.97,131 However, careful UHV annealing close to 880 °C forms cub-Ce2O3(111) in the near-surface region as proved by SPA-LEED measurements (not shown) and XPS results shown in Fig. 25.
 | ||
| Fig. 25 Ce 3d region measured by XPS for a 250 nm CeO2(111) film. The areas under the peaks are normalised to unity after a Shirley background is subtracted. The reference spectrum of CeO2 is measured after plasma cleaning of the samples and consists of three peaks (x, y, z) in each 3d5/2 and 3d3/2 component. The surface near the region is fully reduced to Ce2O3 with two peaks (u, v) in each 3d5/2 and 3d3/2 component after annealing at 890 °C. The Ce3+/Ce4+ ratio of the intermediate states is determined by fitting with a weighted linear combination of the CeO2 and Ce2O3 spectra. (Reproduced from ref. 97.) | ||
 | ||
| Fig. 26 Specular (00l) XRD scans close to the Si(111)B Bragg peak after annealing at temperatures up to 800 °C for 30 min. After annealing at 650 °C Ce7O12(111) Bragg peaks appear at lower l values than the CeO2(111) peak. A third peak attributed to Ce11O20(111) becomes visible at temperatures after annealing at 700 °C. (Reproduced from ref. 97.) | ||
As in the case of praseodymia (see Section III.2.5.1), the nanoscale surface structure of a ceria film is not influenced by its reduction. However, a detailed NC-AFM study reveals a dramatic change in surface morphology upon annealing the film.131 The as-prepared surface exhibits a metastable structure that is denoted as the low temperature morphology being stable against annealing at temperatures up to 850 K. A distinctly different morphology, referred to as the high temperature morphology, is found in the temperature range of 930 K to 1100 K where Ce7O12, Ce11O20 and Ce2O3 stoichiometries are present.
The basic surface structures of the as-prepared ceria surface exhibiting the low temperature morphology are pyramidal islands discussed in detail in Section III.2.4. The compilation of NC-AFM images of Fig. 27 taken with different resolution on films annealed at various temperatures shows the high degree of regularity in pyramid formation and their thermal stability. The only feature that significantly changes as a function of annealing temperature in the range from 510 K to 790 K is the formation of spikes rising to a height above the highest pyramids. These very sharp spikes grow mostly between the pyramids and have a width of less than 3 nm; the density and height of the spikes increases with annealing temperature, however, the spikes can be removed by annealing the film to 850 K, a temperature leaving the pyramids entirely unchanged. The origin of the spikes could not be clarified so far, however, we speculate that they result from contaminants in the film or in the praseodymia buffer layer segregating to the film surface.
 | ||
| Fig. 27 Selected NC-AFM images of 180 nm thick cub-CeO2(111) films representing the low temperature morphology. Films have been annealed to temperatures of (a) 300 K (as prepared sample), (b) 390 K, (c) 510 K and (d) to (f) 690 K, 790 K and 850 K, respectively. Images in rows (a)–(c) were taken under identical conditions with only the frame size varied. Dot-like bright features appearing in frames (c) to (e) are commented in the main text. (Reproduced from ref. 131.) | ||
The transformation of the ceria film into the high temperature morphology occurs for annealing at temperatures in the range of 930 K to 1100 K. The respective surface structure and that of preceding transition states are shown in Fig. 28(a) to (j). The onset of the transformation observed for annealing the sample at 930 K is shown in Fig. 28(a) to (c). The pyramidal structure is gradually dissolved into rounded, flat islands as relicts marking the position of former pyramidal islands (see triangles in Fig. 28(b)). The flat islands have a much larger contiguous surface area with (111) orientation and exhibit terraces with a width of 10 nm to 15 nm. While most terraces are separated by one triple-layer high step edges, the height of some step edges amounts to several triple-layers indicating that step-bunching occurs during the transformation of the islands.
 | ||
| Fig. 28 Selected NC-AFM and KPFM images of 180 nm thick cub-CeO2(111) films representative for the high temperature morphology and the decomposed film. Films were annealed to temperatures of 930 K (a) to (c), 1060 K (d) to (j) and 1150 K (k). The photos in (l) illustrate the change in optical properties of the films yielded by treatment at different temperatures. (Reproduced from ref. 131.) | ||
When annealing the film at 1060 K, the average extension of the terraces increases significantly and the transformation into the high temperature morphology is completed as shown in Fig. 28(d) to (j). Terraces formed during such preparations have a width of 50 nm to 60 nm that is significantly larger than the width of terraces on similarly prepared bulk single crystal surfaces. The final results are hexagonal pits and protrusions with mostly straight step edges so that the nanoscale morphology is very similar to the one found for the surface preparation of bulk single crystals.23,26,29 In contrast to similarly prepared surfaces of bulk ceria crystals developing perfect atomic regularity on terraces, surfaces of the film exhibit many atomic scale irregularities that are clearly seen in high resolution images compiled in Fig. 28(i) to (k). These irregularities yield a specifically strong local KPFM contrast as demonstrated in Fig. 28(f) indicating the presence of local charge or polarisation in or underneath the surface. The irregularities prevail even after the sample has been prepared with Ar+ ion sputter cycles and annealing at 1090 K. As the transformation into the high temperature morphology is accompanied by a strong reduction of the ceria film (see Section III.2.5.2), we speculate that the irregularities are the result of oxygen vacancy clusters appearing during reduction.
When annealing the ceria film at temperatures higher than 1100 K, the surface undergoes a major change. As seen in Fig. 28(k), large hexagonally shaped hillocks having a lateral extension of several hundreds of nanometres separated by 100 nm deep trenches are formed. This dramatic change in morphology is the result of a decomposition of the film related to the formation of silicate and silicide at the interface to the Si(111) substrate as outlined in the XPS and XRD parts of Section III.2.5.2.97 As for praseodymia, the different states of the films are reflected by their optical properties as illustrated by the photos shown in Fig. 28(l). The low temperature and high temperature states are characterised by shiny purple/blue and green/blue colours, respectively, while the decomposed film attains a dull grey colour.
In summary, the as-grown ceria films exhibit metastable surface structures in the form of terraced pyramids grown in the 〈111〉 direction with the base oriented in 〈−1−12〉 directions. In this low temperature morphology, the surface exhibits an exceptionally large number of edges and kink sites at step edges exclusively having a height of one O–Ce–O triple-layer. When the film is annealed to temperatures between 930 K and 1100 K, the pyramids undergo a phase transition into large terraces separated by steps with predominantly one O–Ce–O triple-layer height but also some higher steps. The properties of the film surface with this high temperature morphology resemble those of ceria crystals; however, the film exhibits exceptionally large terraces. Having distinctly different morphological characteristics, the film is in both states very well defined and ordered at the nanoscale. Provided it will be possible to remove the atomic scale irregularities by further efforts in a refined post-deposition treatment, for instance, involving a controlled re-oxidation of thermally treated films, the ceria film is a perfect model system for ceria surface studies but can also be used for applications requiring either a large number of edge and kink sites or large atomically flat terraces.
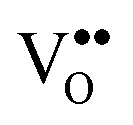 , lattice distortion and, thus, enhances the oxygen storage/release capacity and the ionic conductivity of ceria.110,139–142 Therefore, we investigate the reduction behaviour of the ternary single crystalline film Ce1−xPrxO2−δ as a function of the doping level x = 0.2, 0.4 and 0.6 by means of TPD, XRD, XPS and Raman spectroscopy.143
, lattice distortion and, thus, enhances the oxygen storage/release capacity and the ionic conductivity of ceria.110,139–142 Therefore, we investigate the reduction behaviour of the ternary single crystalline film Ce1−xPrxO2−δ as a function of the doping level x = 0.2, 0.4 and 0.6 by means of TPD, XRD, XPS and Raman spectroscopy.143
The oxygen release from in situ plasma-oxidised samples (“clean surfaces”) is investigated by TPD where the results (shown in Fig. 29(a)) reveal that as the Pr concentration x is increased, the samples release O2 at lower temperatures compared to bulk CeO2 films, what is attributed to the high oxygen exchange activity of the Pr4+/Pr3+ system. From the results shown in the inset of Fig. 29(a), we conclude that the oxygen storage effect is mainly based on the Pr4+/Pr3+ redox system as the ratio between the integrated O2 TPD signal and the film thickness IO2/t increases linearly with x. In contrast, ex situ atmosphere exposed samples (“hydrocarbon covered surfaces”) show inverse results, namely, the release of oxygen in the form of H2O, CO and CO2, where the desorption peak is shifted towards lower temperatures with decreasing x (i.e. more Ce in the mixed oxide) as demonstrated in Fig. 29(b). The fact that oxygen is not released as O2 could be attributed to a kinetically favoured reaction path of hydrocarbon oxidation instead of oxygen recombination. Moreover, ceria has a superior hydrocarbon oxidation capability, better than praseodymia, lowering the CO2 desorption temperature in Ce richer samples.144 However, no significant Ce4+/Ce3+ valence state change is found during the annealing in the temperature range from 200 °C to 300 °C as revealed by an XPS analysis.143 It is possible that an oxygen transport occurs to Ce3+ by the reduction of Pr4+, since the oxygen prefers to locate around Ce ions145 and, therefore, praseodymia serves as an oxygen reservoir in the mixed oxide.
 | ||
| Fig. 29 (a) TPD spectra of O2 for Ce1−xPrxO2−δ(111)/Si(111) samples treated in situ with oxygen plasma with the inset showing the IO2/t versus x plot. (b) TPD spectra of CO2 for the same samples treated ex situ with molecular oxygen. (c) Temperature dependent Δd222 of the same samples exposed to the ambient atmosphere with the inset showing Δd222(500 °C) as a function of x. (d) Model of the Ce1−xPrxO2−δ redox process. (Reproduced from ref. 143.) | ||
Complementary to the oxygen release, the reduction state of the sample with a hydrocarbon covered surface is revealed in an indirect way by measuring the temperature dependent lattice spacing variation Δd222 extracted from XRD measurements yielding the results shown in Fig. 29(c). The lattice spacing clearly increases with increasing temperature and this can be attributed to the Pr4+/Pr3+ transition because the ionic radius of Pr3+ is larger than that of Pr4+. The lattice spacing variation Δd222 (500 °C) increases linearly with x as shown in the inset of Fig. 29(c). This indicates that also for the mixed oxide with a hydrocarbon covered surface, the oxygen release is mainly triggered by the Pr4+/Pr3+ redox system.
Based on these results, a reduction model for Ce1−xPrxO2−δ has been developed that is schematically shown in Fig. 29(d). While the OSC is supported by the Pr4+/Pr3+ redox system (thick arrow in the purple region), the hydrocarbon oxidation capability originates from the Ce4+/Ce3+ redox system (thick arrow crossing the purple and yellow region). Since Ce favours the +4 valence state more than Pr, in the mixed oxide an exchange occurs by oxygen transport to Ce3+ (and mainly to the surface region) accompanied by the reduction of Pr4+. The oxygen mobility is provided by oxygen vacancies playing an important role for such kinds of oxidative processes.145
IV. Adsorption of methanol on CeOx, PrOx and Ce1−xPrxO2−δ films
Methanol oxidation has been well established as a test reaction for characterising the surface of metal oxide catalysts, allowing for a quantification and classification of active sites.146,147 Their chemical properties, rendering the oxide, for instance, an acid–base or a redox catalyst, can be deduced from the spectrum of surface species and products evolving after the exposure to methanol. Chemisorption of methanol on oxides is often observed to break the OH bond, generating methoxy species (CH3O)ads at the surface.148–151 While acidic sites lead to the formation of coupling products and at basic sites typically complete dehydrogenation of the surface methoxy is observed as indicated by CO or CO2 desorption, dehydrogenation is more selective on redox catalysts and, hence, formaldehyde (CH2O) desorbs as a major product at elevated temperatures.146,147Among the range of REOs, ceria has, so far, been the primary target of surface science studies on oxide-catalysed methanol oxidation. Besides ceria crystal surfaces152 also epitaxial cerium oxide thin films of various thicknesses on different substrates such as α-Al2O3, Y-stabilised zirconia, Nb-doped SrTiO3(100), Cu(111), and Ru(0001) have been studied as model systems for ceria catalysts.50,57,152–154 In the following, common findings of these studies as well as differences are pointed out.
Consistently, physisorption of methanol multilayers was found at temperatures below 140 K. Desorption of these multilayers and of a chemisorbed methanol monolayer is observed between 140 K and 200 K,50,154 leaving predominantly methoxy and OH groups on the surface.50,152,153 Their formation from methanol goes along with the reduction of Ce and, obviously, consumes oxygen from the ceria lattice.50 More dissociative methanol adsorption was found on reduced ceria films than on fully oxidised films.57,152,154 The species H2O, CH2O, H2, CO, and CO2 were the only products of methanol oxidation detected during TPD at elevated temperatures (T > 200 K).57,153,154 While on CeO2(111) predominantly H2O and CH2O are observed, indicating selective dehydrogenation of the methoxy, also CO and H2, i.e. products of complete dehydrogenation were found to desorb from partially reduced ceria surfaces.
Besides common trends, previous studies also found distinct differences between measurements revealing dependencies of methanol oxidation not only on the Ce valency but also on details of the cerium oxide film preparation and on the surface structure. Comparing 19 nm thick CeO2(100) films grown on Nb-doped SrTiO3(100) and 5 nm thick CeO2(111) films grown on Ru(0001), Mullins and co-workers found that the CeO2(100) surface, which presumably exhibits more under-coordinated Ce and O sites than CeO2(111), has a higher activity for methanol oxidation and shows a trend towards more extensive decomposition of methanol.57 Besides methoxy, which was the only prevalent carbonaceous species found by Mullins and co-workers in their studies,57,154 Matolín and co-workers also detected formate (CHOO−)ads as a resident on the surface of 1.5 nm thick CeO2(111) films grown on Cu(111),50 indicating, besides complete dehydrogenation, a second route of methanol oxidation on these films directly involving lattice oxygen.
In the present work, the reported dependencies of activity and selectivity of reactions on REO surface composition are tested by exposing epitaxial cerium oxide, praseodymium oxide, and mixed Ce0.4Pr0.6O2−δ films grown as detailed in Section III.1 to methanol. In contrast to previous work, where reduced cerium oxide surfaces were prepared by repeatedly exposing ceria films to methanol, which provides only limited control of the oxidation state, three well characterised cerium oxide film surface stoichiometries, namely CeO2, Ce7O12 and Ce2O3 are prepared by plasma and thermal treatments as described in Section III.2.5.2. Also for praseodymia, methanol oxidation is studied on the fully oxidised phase (PrO2), a partially reduced film with a stoichiometry close to Pr6O11, and a film fully reduced at the surface as described in Section III.2.5.1. The role of the rare earth valency for the selectivity of methanol dehydrogenation at the model REO catalysts is the focus of combined TPD and XPS experiments.
For TPD, a UHV chamber with a base pressure of 10−8 Pa is available that is equipped with a quadrupole mass spectrometer, LEED and an UHV-compatible RF plasma source for oxygen plasma treatment as described in Section II.2. SR-XPS experiments are performed in a separate UHV chamber at MAX IV laboratory at Beamline D1011 in Lund, Sweden. At both sites, samples from the same Si wafers, on which 15 nm thick ceria, 16 nm thick praseodymia and 23 nm thick mixed Ce0.4Pr0.6O2−δ films have been grown are used for parallel investigation. The REO films are initially fully oxidised by oxygen plasma treatment and then thermally reduced while the surface structure is monitored by LEED to observe, for instance, the characteristic pattern of the Ce7O11 phase.138
Prior to TPD measurements, the samples are exposed to 15 L methanol at a sample temperature of 105 K by a gas doser directed to the sample surface. The sample is then placed close to the aperture of a quartz cap surrounding the mass spectrometer so that only products desorbing from the sample surface are collected. During TPD, a heating rate of 1 K s−1 is maintained by an electronic controller. To detect the carbonaceous products CO, CH2O, and CO2, atomic masses 28, 29, and 44 are monitored as CHO is the most abundant fragment of CH2O after electron ionisation in the mass spectrometer. The number of recorded masses is restricted to maintain sufficiently fast data recording.
For SR-XPS experiments, samples are exposed to 15 L methanol at a sample temperature of 100 K to 120 K by backfilling the UHV chamber. The samples are then heated in intervals up to a maximum temperature that is step-wise increased in analogy to procedures described in ref. 50. After each heating interval with the maximum temperature maintained for 1 min, the sample is cooled down to ∼140 K and SR-XPS data are recorded. In this way, a series of XP spectra are obtained with each spectrum being representative of the resident surface species present at the respective maximum temperature.
IV.1 Methanol oxidation on CeOx
TPD experiments on ceria shown in Fig. 30 are referenced to previous work50,57,154 identifying the temperature range above 200 K as relevant in terms of detecting carbonaceous products of methanol oxidation. In accordance with this previous work, the spectral intensities of masses 28 and 29 observed at temperatures below 200 K for CeO2 and Ce7O12 can be interpreted as fragments of methanol155 desorbing from physisorbed multilayers or from a chemisorbed methanol layer leaving methoxy species at the surface. At elevated temperatures, no desorption of formaldehyde is detected from CeO2(111) films, however, significant amounts of CO and CO2 are produced, visible as broad desorption peaks centred at ∼600 K in Fig. 30 indicating complete oxidation of methanol at the surface. Formaldehyde desorption at ∼600 K as an indicator for selective dehydrogenation is only observed from the partially reduced surface Ce7O12 concomitantly with CO and CO2 desorption. While the CO signal comprises ionisation-induced fragments from CH2O and CO2, these contributions account for less than 50% of its intensity as a quantitative analysis of the desorption peak intensities at masses 28, 29, and 44 reveals. This implies that CO is also produced at the Ce7O12 surface.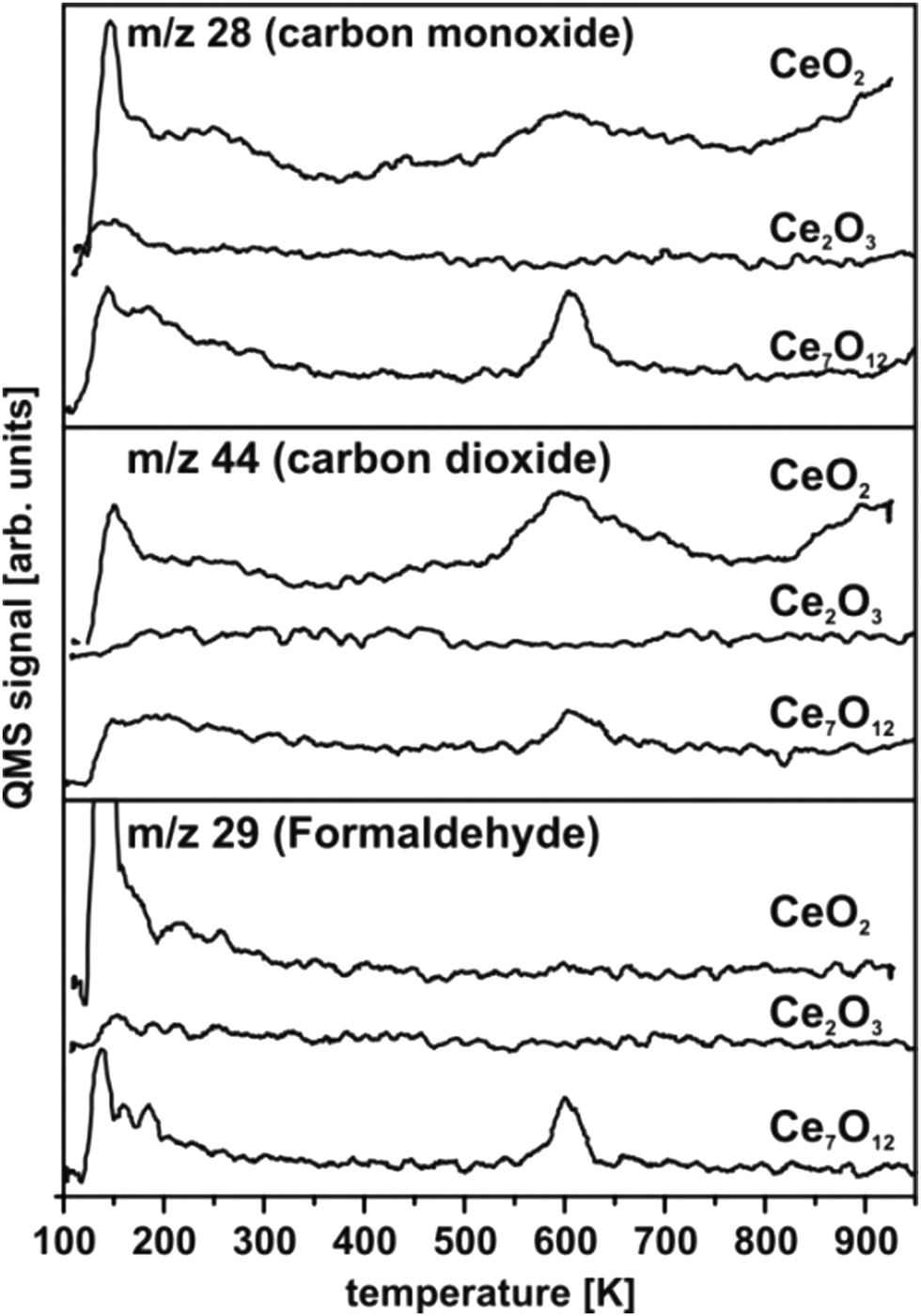 | ||
| Fig. 30 TPD spectra of methanol adsorbed on ceria surfaces of different oxidation states. Only the partially reduced Ce7O12 surface yields CH2O. On CeO2 total oxidation is observed while the Ce2O3 surface shows no reactivity towards methanol. | ||
After exposing the Ce2O3 surface to methanol at 105 K, TPD yields no evolution of products at all. Even below 200 K, no fragments of methanol desorption are detected. This puzzling result observed in repeated experiments indicates that methanol adsorption on the fully reduced cerium oxide film is negligible and, therefore, also its activity for methanol oxidation is negligible.
Methanol oxidation on ceria films does not match previously observed trends.57,152,154 Quite opposite, complete oxidation and significant activity is observed on fully oxidised CeO2(111) while the partially reduced surface with Ce7O12 stoichiometry tends to catalyse selective dehydrogenation of methanol. Furthermore, the present study does not indicate an increased amount of methanol adsorbed on reduced ceria as compared to CeO2. In fact, the fully reduced Ce2O3 surface exhibits the lowest sticking coefficient for methanol. Details of the surface morphology and defect structure, both depending, for instance, on substrate, growth conditions, and film thickness very likely play a prominent role for the different trends observed here and in previous studies.
IV.2 Methanol oxidation on PrOx
SR-XPS data for the C 1s binding energy region provide insight into carbonaceous species resident on the surface after methanol adsorption on PrO2 and Pr2O3 and subsequent heating with respective results shown in Fig. 31(a) and (b). The fully stoichiometric PrO2(111) surface exhibits a minor residual surface contamination by carbon (the “as-prepared” curve in Fig. 31(a)) and OH groups (the O 1s spectrum not shown), detected due to the high sensitivity of the SR-XPS experiment. No contamination is found on the fully reduced Pr2O3 surface. After methanol exposure at 100 K to 120 K, an intense C 1s peak at 287.7 eV observed for both, PrO2 and Pr2O3 (grey curves in Fig. 31), provides evidence for methanol at the surface, probably with multilayer coverage. The shift of this peak to ∼287.0 eV and the decrease of its intensity upon heating PrO2 and Pr2O3 up to 290 K and 300 K, respectively, indicate the desorption of methanol and the formation of methoxy species on the surface as previously reported for ceria.50,154 Besides methoxy, also formate species are formed on the surface between 200 K and 400 K as revealed by C 1s peaks at binding energies between 289 eV and 290 eV detected for both PrO2 and Pr2O3. The variation of the C 1s binding energy (289.0 eV on PrO2 and 289.7 eV on Pr2O3 at 400 K) corresponds to that observed for formate adsorption on fully oxidised and partially reduced ceria.156 The build-up of formate on the surface shows that, besides complete dehydrogenation, there is another route for methanol oxidation, as it has previously been reported for the CeO2/Cu(111) system.50 For PrO2, SR-XPS even reveals a third carbon species with a C 1s binding energy of 291.8 eV on the surface at 400 K, indicating carbonate (CO32−) formation on the cost of even more oxygen taken from the praseodymia lattice. Upon heating PrO2 above 400 K and Pr2O3 above 600 K, the C 1s peak intensities strongly decrease, implying that carbonaceous products desorb above these temperatures. | ||
| Fig. 31 C 1s core level spectra recorded at 410 eV photon energy after methanol adsorption and subsequent heating to the temperatures indicated for 16 nm thick PrO2 (a) and Pr2O3 (b) films as well as a 23 nm thick Ce4+0.4Pr3+0.6O1.7 (c) film after thermal reduction. Shown in addition: spectra of the REO film before methanol exposure denoted “as-prepared” and spectra recorded directly after methanol exposure before the initial heating interval shown as grey curves. | ||
The results from TPD after exposing PrO2 and films with stoichiometries close to Pr6O11 and to Pr2O3 to methanol at 105 K are shown in Fig. 32. Below 200 K, the detection of masses 28 and 29 indicates desorption of physisorbed and chemisorbed methanol in agreement with TPD and SR-XPS data for ceria. A strong CO2 desorption peak and concomitantly CO desorption are observed from PrO2 films at 480 K. Formaldehyde desorption is only minor, showing that complete oxidation of the methanol, obviously by consumption of lattice oxygen, is favoured. The grey curve in the top panel of Fig. 32 represents TPD of oxygen from an as-prepared PrO2(111) film, i.e. without surface methoxy species. From as-prepared PrO2, the desorption of lattice oxygen starts at ∼500 K, hence, surface methoxy promotes the release of lattice oxygen from PrO2, possibly via intermediate oxidation products such as surface formate and carbonate. From 500 K up to higher temperatures, the maxima of CO desorption from PrO2 correlate with the maxima of oxygen release from the as-prepared PrO2 film (grey curve), i.e. they coincide with REO reduction during phase transitions.
 | ||
| Fig. 32 TPD spectra of methanol adsorbed on surfaces of 16 nm thick praseodymia films in different oxidation states. A large CO2 and CO desorption signal is observed for PrO2 ∼20 K below the onset of lattice oxygen desorption from an as-prepared PrO2 film (grey spectrum, top panel). Pr2O3 only shows weak CO/CO2 signals as well as a weak CH2O signal (arrow, bottom panel). Pr6O11 is rather unreactive towards methanol adsorption up to 655 K. | ||
For a partially reduced film close to the Pr6O11 stoichiometry prepared by heating PrO2(111) up to 655 K, the detection of masses 28, 29, and 44 in TPD up to 400 K is ascribed to methanol desorption as discussed before and in accordance with XPS results (not shown), we observe a shift of the C1 s binding energy from 287.7 eV to 287.2 eV. No further product desorption is detected up to temperatures of 655 K. The increasing CO and CO2 desorption above this temperature indicates a complete oxidation of surface resident carbonaceous species by the lattice oxygen that is expected to be released when Pr6O11 is thermally reduced.
For the fully reduced Pr2O3 surface produced by heating a PrO2 film up to 830 K (SR-XPS results not shown exhibit Pr only in the oxidation state 3+), it should be noted that, opposite to the surface, the entire film may not be fully reduced.107 Up to 655 K, TPD results obtained after methanol exposure at 105 K are qualitatively similar to those of the partially reduced praseodymia surface. At 680 K, a weak formaldehyde desorption peak is observed and concomitantly CO and CO2 are produced. Increasing CO and CO2 desorption as the temperature is raised above 830 K is ascribed to a decomposition of the praseodymia film accompanied by further release of lattice oxygen.
As for ceria, it is the fully oxidised PrO2 film exhibiting a trend towards full oxidation of methanol while desorption of formaldehyde is observed only from the fully reduced surface, indicating selective dehydrogenation of adsorbed methoxy catalysed by Pr2O3. PrO2 and the partially reduced Pr6O11 film are not stable up to the desorption temperature of CH2O from Pr2O3, and the release of lattice oxygen below 680 K fully oxidises surface methoxy before formaldehyde can be formed. Of all ceria, praseodymia, and mixed oxide films studied, PrO2 shows the highest activity for methanol oxidation below 500 K.
IV.3 Methanol oxidation on mixed CePrOx
Partially reduced Ce0.4Pr0.6O2−δ films are prepared from the fully oxidised state (δ = 0) when heating up to ∼900 K. The LEED pattern recorded during heating indicates the presence of the ι-phase.138 SR-XPS of the reduced films shows Ce exclusively in the oxidation state 4+ and for Pr exclusively 3+ valency (data not shown), yielding the stoichiometry Ce0.4Pr0.6O1.7.TPD results shown in Fig. 33 obtained after exposing the Ce0.4Pr0.6O2 film to methanol are qualitatively similar to those obtained for PrO2. At 500 K, a pronounced CO2 desorption peak and concomitant CO desorption is observed, however, no formaldehyde, revealing full oxidation of adsorbed methanol. As for PrO2, the CO2 desorption peak is observed 20 K below the temperature at which the release of lattice oxygen starts from as-prepared Ce0.4Pr0.6O2, i.e. without surface methoxy species (the grey spectrum in the top panel of Fig. 33 shows the desorption of O2 from the as-prepared film). The CO2 desorption temperature is 20 K higher for Ce0.4Pr0.6O2 than for PrO2 (peak at 480 K), indicating that Ce4+ present in the mixed oxide film impedes the reduction of Pr4+ and the release of lattice oxygen.
 | ||
| Fig. 33 TPD spectra of methanol adsorbed on surfaces of 23 nm thick Ce0.4Pr0.6O2−δ films in different oxidation states. The partially reduced oxide shows selectivity towards CH2O formation. | ||
SR-XPS results of Ce0.4Pr0.6O1.7 shown in Fig. 31(c) reveal the prevalence of surface methoxy after the desorption of physisorbed methanol multilayers and the build-up of formate species at temperatures up to ∼500 K, as it is observed on the fully reduced praseodymia surface. TPD after exposing the Ce0.4Pr0.6O1.7 film to methanol yields spectra shown in Fig. 33 that are similar to those observed for the Ce7O12 phase. A formaldehyde desorption peak is observed at ∼580 K and concomitantly also CO and CO2 desorb as oxidation products.
In summary, praseodymia as well as ceria and mixed films exhibit a trend towards complete oxidation of methanol and, overall, a higher activity when they are fully oxidised. A selectivity towards partial dehydrogenation of surface methoxy formed by dissociative adsorption of methanol is observed for the partially reduced REO films. From the fully reduced Ce2O3 surface, no desorption of carbonaceous products is observed after exposure to CH3OH at 105 K and also the Pr2O3 surface shows a comparatively low activity for methanol oxidation. The partially reduced films provide the best balance between activity and the often desired catalytic selectivity towards partial dehydrogenation and formaldehyde production. Mixed ceria/praseodymia films show a potential for creating a catalyst with a good balance between the high oxygen mobility in PrOx and the high stability of CeOx.
V. Conclusions
Rare earth oxides are well established as materials for a large variety of technical applications with chemical catalysis being the most prominent. The desired functionality for specific applications is often created by using complex systems with tailored macro-, micro- and nanostructures and a suitable composition of elements forming binary, ternary or even quaternary rare earth oxide systems. Among the many base materials used, praseodymia and ceria are outstanding with respect to their oxygen release, storage and transfer capability based on their exceptional stability properties with respect to phase transitions involving oxygen transfer. In a fully reversible process controlled by sample temperature and environment, oxygen can be removed in large amounts from the fully oxidised REO2 state to form a variety of stable and atomically well-ordered intermediate oxide states until the fully reduced sesquioxide RE2O3 is reached while fully maintaining the structural integrity of the material throughout the series of phase transitions. While the oxygen transfer related phase diagrams for bulk samples of praseodymia and ceria have been explored in the past, in our work, we establish procedures for a controlled synthesis and post-deposition preparation of stable films to yield a system in any desired stoichiometry between the fully oxidised and fully reduced limiting phases. As a working platform well suitable for basic research model studies, we introduce the growth of rare earth oxides on Si(111) wafers by molecular beam epitaxy. High quality films can be grown as a binary oxide or a ternary oxide in the form of a mixed or layered material with a thickness ranging from a few to a few hundred nanometres. For ceria, we are able to very well define the surface morphology varied from a kinetically determined pyramidal nanostructure exhibiting a large number of steps and kinks at the atomic scale to a thermodynamically fully relaxed surface exhibiting large atomically flat terraces separated by a small number of single unit steps by a suitable post-deposition thermal treatment. Likewise, the stoichiometry of the surface can be tailored by thermal, gas exposure or plasma treatment, however, we find that for some intermediate states, a coexistence of phases is unavoidable. The film and surface structure can further be controlled by exploiting internal stress and relaxation effects induced by the lattice mismatch at the substrate–film interface, which is specifically interesting for tailoring ternary systems. Our studies clearly reveal the capabilities and limitations of the oxide films in terms of oxygen transport capability as well as thermal and environmental stability. While praseodymia clearly sets the benchmark in oxygen mobility, ceria is superior regarding thermal and environmental stability. The opposite electrochemical properties of the Ce4+/Ce3+ and Pr4+/Pr3+ redox systems on the one hand and the crystallographic similarities allowing the formation of truly mixed systems on the other hand allows tailoring rare earth oxide systems with most interesting properties. Thus, in their great variety of possible compositions, structures, stoichiometries and surface morphologies, the films are equally suitable for thin film model system studies and industrial applications where robust, bulk-like samples are required. The preparation of films by standard techniques used in the semiconductor industry greatly facilitates the creation of nanostructured, specifically functionalised and integrated systems by lithography, implantation and contacting techniques. By mastering the enormous complexity of advanced rare earth oxide systems and providing a facile route for production, the established thin film synthesis platform creates a great potential for expanding our understanding of rare earth oxides used in chemical catalysis,157–159 microelectronics, specifically resistive random-access memory devices,160,161 energy technologies,110,111,140,162 sensors163,164 and many other fields of application such as magneto-optic or spintronic devices based on room temperature ferromagnetism.118,120,165,166Acknowledgements
The authors gratefully acknowledge the funding of the project by the Deutsche Forschungsgemeinschaft via grants BA 1710/17-1, RE 1186/12-1, SCHR 1123/4-1, WO 533/16-1 and support from the COST Action CM1104. G. N. expresses his gratitude to the Alexander von Humboldt foundation for a post-doctoral fellowship. M.R. gratefully acknowledges stimulating discussions about film growth mechanisms with P. Maaß.References
- S. D. Barrett and S. S. Dhesi, The Structure of Rare-Earth Metal Surfaces, Imperial College Press, London, 2001 Search PubMed.
- K. C. Taylor, Catalysis – Science and Technology, Springer-Verlag, Berlin, 1984 Search PubMed.
- K. Otsuka, K. Jinno and A. Morikawa, J. Catal., 1986, 100, 353 CrossRef CAS.
- G. Adachi, N. Imanaka and Z. C. Kang, Binary Rare Earth Oxides, Springer, Dordrecht, 2004 Search PubMed.
- J. R. Jayaramaiah, B. N. Lakshminarasappa and B. M. Nagabhushana, Sens. Actuators, B, 2012, 173, 234 CrossRef CAS.
- S. S. Kim, J. H. Moon, B.-T. Lee, K.-S. Sohn, T. S. Kang and J. H. Je, Appl. Surf. Sci., 2004, 221, 231 CrossRef CAS.
- S. Thakur, N. K. Sahoo, M. Senthilkumar and R. B. Tokas, Opt. Mater., 2005, 27, 1402 CrossRef CAS.
- D. P. Norton, Mater. Sci. Eng., R, 2004, 43, 139 CrossRef.
- http://www.itrs.net .
- J. Stankiewicz, F. Villuendas and J. Bartolomé, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 235308 CrossRef.
- Y. A. Boikov, T. Claeson and D. Erts, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 11312 CrossRef CAS.
- F. Sánchez, R. Bachelet, P. d. Coux, B. Warot-Fonrose, V. Skumryev, L. Tarnawska, P. Zaumseil, T. Schroeder and J. Fontcuberta, Appl. Phys. Lett., 2011, 99, 211910 CrossRef.
- J. H. Choi, J. Y. Lee and Y. T. Kim, Appl. Phys. Lett., 2000, 77, 4028 CrossRef CAS.
- A. Fissel, R. Dargis, E. Bugiel, D. Schwendt, T. Wietler, J. Krügener, A. Laha and H. J. Osten, Thin Solid Films, 2010, 518, 2546 CrossRef CAS.
- N. A. Bojarczuk, M. Copel, S. Guha, V. Narayanan, E. J. Preisler, F. M. Ross and H. Shang, Appl. Phys. Lett., 2003, 83, 5443 CrossRef CAS.
- G. Saint-Girons, P. Regreny, L. Largeau and G. Patriarche, Appl. Phys. Lett., 2007, 91, 241912 CrossRef.
- F. A. Kröger and H. J. Vink, Solid State Phys., Academic Press, 1956, vol. 3, p. 307 Search PubMed.
- Z. L. Wang and Z. C. Kang, Functional and smart materials: structural evolution and structure analysis, Plenum Press, New York, 1998 Search PubMed.
- G. Adachi and N. Imanaka, Chem. Rev., 1998, 98, 1479 CrossRef CAS.
- H.-W. Chiang, R. N. Blumenthal and R. A. Fournelle, Solid State Ionics, 1993, 66, 85 CrossRef CAS.
- A. Trovarelli, Catalysis by ceria and related materials, Imperial College Press, London, 2002 Search PubMed.
- P. Fornasiero and A. Trovarelli, Catalysis by Ceria and Related Materials, Imperial College Press, London, 2014 Search PubMed.
- S. Torbrügge, M. Cranney and M. Reichling, Appl. Phys. Lett., 2008, 93, 073112 CrossRef.
- S. M. F. Shahed, Y. Sainoo and T. Komeda, Jpn. J. Appl. Phys., 2011, 50, 08LB05 CrossRef.
- N. Nilius, S. M. Kozlov, J. F. Jerratsch, M. Baron, X. Shao, F. Viñes, S. Shaikhutdinov, K. M. Neyman and H. J. Freund, ACS Nano, 2012, 6, 1126 CrossRef CAS PubMed.
- H. H. Pieper, C. Derks, M. H. Zoellner, R. Olbrich, L. Tröger, T. Schroeder, M. Neumann and M. Reichling, Phys. Chem. Chem. Phys., 2012, 14, 15361 RSC.
- H. H. Pieper, C. Lammers, L. Tröger, S. Bahr and M. Reichling, Rev. Sci. Instrum., 2012, 83, 055110 CrossRef CAS PubMed.
- S. Gritschneder, Y. Namai, Y. Iwasawa and M. Reichling, Nanotechnology, 2005, 16, S41 CrossRef CAS.
- S. Gritschneder and M. Reichling, Nanotechnology, 2007, 18, 044024 CrossRef.
- P. S. Bagus, C. J. Nelin, E. S. Ilton, M. Baron, H. Abbott, E. Primorac, H. Kuhlenbeck, S. Shaikhutdinov and H. J. Freund, Chem. Phys. Lett., 2010, 487, 237 CrossRef CAS.
- D. C. Grinter, R. Ithnin, C. L. Pang and G. Thornton, J. Phys. Chem. C, 2010, 114, 17036 CrossRef CAS.
- X. Shao, J. F. Jerratsch, N. Nilius and H. J. Freund, Phys. Chem. Chem. Phys., 2011, 13, 12646 RSC.
- J. I. Flege, B. Kaemena, A. Meyer, J. Falta, S. D. Senanayake, J. T. Sadowski, R. D. Eithiraj and E. E. Krasovskii, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 235428 CrossRef.
- D. C. Grinter, C. L. Pang, C. A. Muryn, F. Maccherozzi, S. Dhesi and G. Thornton, J. Electron. Spectrosc. Relat. Phenom., 2014, 195, 13 CrossRef CAS.
- S. Torbrügge, M. Reichling, A. Ishiyama, S. Morita and O. Custance, Phys. Rev. Lett., 2007, 99, 056101 CrossRef PubMed.
- M. V. Ganduglia-Pirovano, J. L. F. Da Silva and J. Sauer, Phys. Rev. Lett., 2009, 102, 026101 CrossRef PubMed.
- Z. X. Yang, X. H. Yu, Z. S. Lu, S. F. Li and K. Hermansson, Phys. Lett. A, 2009, 373, 2786 CrossRef CAS.
- C. J. Zhang, A. Michaelides, D. A. King and S. J. Jenkins, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 075433 CrossRef.
- J. F. Jerratsch, X. Shao, N. Nilius, H. J. Freund, C. Popa, M. V. Ganduglia-Pirovano, A. M. Burow and J. Sauer, Phys. Rev. Lett., 2011, 106, 246801 CrossRef PubMed.
- G. E. Murgida and M. V. Ganduglia-Pirovano, Phys. Rev. Lett., 2013, 110, 246101 CrossRef PubMed.
- Y. Pan, N. Nilius, H. J. Freund, J. Paier, C. Penschke and J. Sauer, Phys. Rev. Lett., 2013, 111, 206101 CrossRef PubMed.
- V. Stetsovych, F. Pagliuca, F. Dvořák, T. Duchoň, M. Vorokhta, M. Aulická, J. Lachnitt, S. Schernich, I. Matolínová, K. Veltruská, T. Skála, D. Mazur, J. Mysliveček, J. Libuda and V. Matolín, J. Phys. Chem. Lett., 2013, 4, 866 CrossRef CAS PubMed.
- Y. G. Wang, D. H. Mei, J. Li and R. Rousseau, J. Phys. Chem. C, 2013, 117, 23082 CrossRef CAS.
- T. Duchoň, F. Dvořák, M. Aulická, V. Stetsovych, M. Vorokhta, D. Mazur, K. Veltruská, T. Skála, J. Mysliveček, I. Matolínová and V. Matolín, J. Phys. Chem. C, 2014, 118, 357 CrossRef.
- S. M. Kozlov and K. M. Neyman, Phys. Chem. Chem. Phys., 2014, 16, 7823 RSC.
- J. Kullgren, M. J. Wolf, C. W. M. Castleton, P. Mitev, W. J. Briels and K. Hermansson, Phys. Rev. Lett., 2014, 112, 156102 CrossRef CAS PubMed.
- S. Gritschneder, Y. Iwasawa and M. Reichling, Nanotechnology, 2007, 18, 044025 CrossRef.
- M. B. Watkins, A. S. Foster and A. L. Shluger, J. Phys. Chem. C, 2007, 111, 15337 CrossRef CAS.
- M. Fronzi, S. Piccinin, B. Delley, E. Traversa and C. Stampfl, Phys. Chem. Chem. Phys., 2009, 11, 9188 RSC.
- V. Matolín, J. Libra, M. Škoda, N. Tsud, K. C. Prince and T. Skála, Surf. Sci., 2009, 603, 1087 CrossRef.
- C. Müller, K. Hermansson and B. Paulus, Chem. Phys., 2009, 362, 91 CrossRef.
- C. Müller, B. Paulus and K. Hermansson, Surf. Sci., 2009, 603, 2619 CrossRef.
- D. Fernández-Torre, K. Kośmider, J. Carrasco, M. V. Ganduglia-Pirovano and R. Pérez, J. Phys. Chem. C, 2012, 116, 13584 CrossRef.
- D. R. Mullins, P. M. Albrecht, T.-L. Chen, F. C. Calaza, M. D. Biegalski, H. M. Christen and S. H. Overbury, J. Phys. Chem. C, 2012, 116, 19419 CrossRef CAS.
- S. Torbrügge, O. Custance, S. Morita and M. Reichling, J. Phys.: Condens. Matter, 2012, 24, 084010 CrossRef PubMed.
- D. R. Mullins and P. M. Albrecht, J. Phys. Chem. C, 2013, 117, 14692 CrossRef CAS.
- P. M. Albrecht and D. R. Mullins, Langmuir, 2013, 29, 4559 CrossRef CAS PubMed.
- N. Tsud, R. G. Acres, M. Iakhnenko, D. Mazur, K. C. Prince and V. Matolín, J. Phys. Chem. B, 2013, 117, 9182 CrossRef CAS PubMed.
- D. Fernández-Torre, J. Carrasco, M. V. Ganduglia-Pirovano and R. Pérez, J. Chem. Phys., 2014, 141, 014703 CrossRef PubMed.
- J.-L. Lu, H.-J. Gao, S. Shaikhutdinov and H.-J. Freund, Catal. Lett., 2007, 114, 8 CrossRef CAS.
- J. A. Rodriguez, X. Wang, P. Liu, W. Wen, J. C. Hanson, J. Hrbek, M. Pérez and J. Evans, Top. Catal., 2007, 44, 73 CrossRef CAS.
- M. Baron, O. Bondarchuk, D. Stacchiola, S. Shaikhutdinov and H. J. Freund, J. Phys. Chem. C, 2009, 113, 6042 CrossRef CAS.
- N. J. Castellani, M. A. Branda, K. M. Neyman and F. Illas, J. Phys. Chem. C, 2009, 113, 4948 CrossRef CAS.
- N. C. Hernández, R. Grau-Crespo, N. H. de Leeuw and J. F. Sanz, Phys. Chem. Chem. Phys., 2009, 11, 5246 RSC.
- J. Majimel, M. Lamirand-Majimel, I. Moog, C. Feral-Martin and M. Tréguer-Delapierre, J. Phys. Chem. C, 2009, 113, 9275 CrossRef CAS.
- M. Škoda, M. Cabala, I. Matolínová, K. C. Prince, T. Skála, F. Šutara, K. Veltruská and V. Matolín, J. Chem. Phys., 2009, 130, 034703 CrossRef PubMed.
- C. J. Weststrate, R. Westerström, E. Lundgren, A. Mikkelsen and J. N. Andersen, J. Phys. Chem. C, 2009, 113, 724 CrossRef CAS.
- C. J. Zhang, A. Michaelides, D. A. King and S. J. Jenkins, J. Phys. Chem. C, 2009, 113, 6411 CrossRef CAS.
- A. Akita, S. Tanaka, K. Tanaka and M. Kohyama, Mater. Sci. Forum, 2010, 654, 2362 CrossRef.
- M. M. Branda, N. C. Hernández, J. F. Sanz and F. Illas, J. Phys. Chem. C, 2010, 114, 1934 CrossRef CAS.
- T. s. Duchoň, F. Dvořák, M. Aulická, V. Stetsovych, M. Vorokhta, D. Mazur, K. i. Veltruská, T. s. Skála, J. Mysliveček, I. Matolínová and V. Matolín, J. Phys. Chem. C, 2013, 118, 357 CrossRef.
- Z. Lu, R. S. Feigelson, R. K. Route, S. A. DiCarolis, R. Hiskes and R. D. Jacowitz, J. Cryst. Growth, 1993, 128, 788 CrossRef CAS.
- H. C. Aspinall, in Rare Earth Oxide Thin Films: Growth, Characterization, and Applications, ed. M. Fanciulli and G. Scarel, Springer Verlag, Berlin and Heidelberg, 2007, vol. 106, p. 53 Search PubMed.
- M. H. Zoellner, J. Dabrowski, P. Zaumseil, A. Giussani, M. A. Schubert, G. Lupina, H. Wilkens, J. Wollschläger, M. Reichling, M. Bäumer and T. Schroeder, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 035302 CrossRef.
- T. Staudt, Y. Lykhach, L. Hammer, M. A. Schneider, V. Matolín and J. Libuda, Surf. Sci., 2009, 603, 3382 CrossRef CAS.
- F. Dvořák, O. Stetsovych, M. Steger, E. Cherradi, I. Matolínová, N. Tsud, M. Škoda, T. Skála, J. Mysliveček and V. Matolín, J. Phys. Chem. C, 2011, 115, 7496 CrossRef.
- J. I. Flege, B. Kaemena, S. Gevers, F. Bertram, T. Wilkens, D. Bruns, J. Bätjer, T. Schmidt, J. Wollschläger and J. Falta, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 235418 CrossRef.
- P. Luches, F. Pagliuca and S. Valeri, J. Phys. Chem. C, 2011, 115, 10718 CrossRef CAS.
- K. Mašek, J. Beran and V. Matolín, Appl. Surf. Sci., 2012, 259, 34 CrossRef.
- S. D. Senanayake, J. T. Sadowski, J. Evans, S. Kundu, S. Agnoli, F. Yang, D. Stacchiola, J. I. Flege, J. Hrbek and J. A. Rodriguez, J. Phys. Chem. Lett., 2012, 3, 839 CrossRef CAS PubMed.
- O. Stetsovych, F. Dvořak, L. Szabová, S. Fabris, J. Mysliveček and V. Matolín, Phys. Rev. Lett., 2012, 109, 266102 CrossRef PubMed.
- L. Szabová, O. Stetsovych, F. Dvořák, M. F. Camellone, S. Fabris, J. Mysliveček and V. Matolín, J. Phys. Chem. C, 2012, 116, 6677 CrossRef.
- J. I. Flege, B. Kaemena, S. D. Senanayake, J. Höcker, J. T. Sadowski and J. Falta, Ultramicroscopy, 2013, 130, 87 CrossRef CAS PubMed.
- B. Kaemena, S. D. Senanayake, A. Meyer, J. T. Sadowski, J. Falta and J. I. Flege, J. Phys. Chem. C, 2013, 117, 221 CrossRef CAS.
- P. Luches, F. Pagliuca, S. Valeri and F. Boscherini, J. Phys. Chem. C, 2013, 117, 1030 CrossRef CAS.
- F. Pagliuca, P. Luches and S. Valeri, Surf. Sci., 2013, 607, 164 CrossRef CAS.
- J. I. Flege, B. Kaemena, J. Höcker, F. Bertram, J. Wollschläger, T. Schmidt and J. Falta, Appl. Phys. Lett., 2014, 104, 131604 CrossRef.
- J. I. Flege, B. Kaemena, T. Schmidt and J. Falta, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2014, 32, 03D124 Search PubMed.
- T. Schroeder, T. L. Lee, L. Libralesso, I. Joumard, J. Zegenhagen, P. Zaumseil, C. Wenger, G. Lupina, G. Lippert, J. Dabrowski and H. J. Mussig, J. Appl. Phys., 2005, 97, 074906 CrossRef.
- A. Schaefer, T. Schroeder, G. Lupina, Y. Borchert, J. Dabrowski, C. Wenger and M. Bäumer, Surf. Sci., 2007, 601, 1473 CrossRef CAS.
- J. P. Liu, P. Zaumseil, E. Bugiel and H. J. Osten, Appl. Phys. Lett., 2001, 79, 671 CrossRef CAS.
- T. Weisemoeller, F. Bertram, S. Gevers, A. Greuling, C. Deiter, H. Tobergte, M. Neumann, J. Wollschläger, A. Giussani and T. Schroeder, J. Appl. Phys., 2009, 105, 124108 CrossRef.
- B. Gehl, U. Leist, V. Aleksandrovic, P. Nickut, V. Zielasek, H. Weller, K. Al-Shamery and M. Bäumer, Rev. Sci. Instrum., 2006, 77, 083902 CrossRef.
- A. Schaefer, S. Gevers, V. Zielasek, T. Schroeder, J. Falta, J. Wollschläger and M. Bäumer, J. Chem. Phys., 2011, 134, 054701 CrossRef CAS PubMed.
- S. Gevers, T. Weisemoeller, A. Schaefer, V. Zielasek, M. Bäumer and J. Wollschläger, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 193408 CrossRef.
- S. Gevers, T. Weisemoeller, B. Zimmermann, F. Bertram, C. Deiter and J. Wollschlager, J. Phys.: Condens. Matter, 2009, 21, 8 CrossRef PubMed.
- H. Wilkens, O. Schuckmann, R. Oelke, S. Gevers, M. Reichling, A. Schaefer, M. Bäumer, M. H. Zoellner, G. Niu, T. Schroeder and J. Wollschläger, Phys. Chem. Chem. Phys., 2013, 15, 18589 RSC.
- E. J. Tarsa, J. S. Speck and M. Robinson, Appl. Phys. Lett., 1993, 63, 539 CrossRef CAS.
- H. J. Osten, J. P. Liu, E. Bugiel, H. J. Mussig and P. Zaumseil, Mater. Sci. Eng., B, 2001, 87, 297 CrossRef.
- N. M. Jeutter, W. Moritz, A. Sidorenko and A. Stierle, Appl. Phys. Lett., 2007, 90, 062906 CrossRef.
- A. Schaefer, V. Zielasek, T. Schmidt, A. Sandell, M. Schowalter, O. Seifarth, L. E. Walle, C. Schulz, J. Wollschläger, T. Schroeder, A. Rosenauer, J. Falta and M. Bäumer, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 045414 CrossRef.
- T. Schroeder, P. Zaumseil, G. Weidner, W. Ch, J. Dabrowski, H. J. Müssig and P. Storck, J. Appl. Phys., 2006, 99, 014101 CrossRef.
- T. Schroeder, P. Zaumseil, O. Seifarth, A. Giussani, H. J. Müssig, P. Storck, D. Geiger, H. Lichte and J. Dabrowski, New J. Phys., 2008, 10, 113004 CrossRef.
- L. Eyring, in Handbook of the Physics and Chemistry of Rare Earths, ed. K. A. J. Gschneidner and L. Eyring, North-Holland, Amsterdam, 1979, ch. 337 Search PubMed.
- B. G. Hyde, E. E. Garver, U. E. Kuntz and L. Eyring, J. Phys. Chem., 1965, 69, 1667 CrossRef CAS.
- T. Weisemoeller, C. Deiter, F. Bertram, S. Gevers, A. Giussani, P. Zaumseil, T. Schroeder and J. Wollschläger, Appl. Phys. Lett., 2008, 93, 032905 CrossRef.
- H. Wilkens, S. Gevers, S. Röhe, A. Schaefer, M. Bäumer, M. H. Zoellner, T. Schroeder and J. Wollschläger, J. Phys. Chem. C, 2014, 118, 3056 CrossRef CAS.
- P. Zaumseil and T. Schroeder, J. Phys. D: Appl. Phys., 2011, 44, 055403 CrossRef.
- C. L. Sieglaff and L. Eyring, J. Am. Chem. Soc., 1957, 79, 3024 CrossRef CAS.
- S. D. Park, J. M. Vohs and R. J. Gorte, Nature, 2000, 404, 265 CrossRef CAS PubMed.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345 CrossRef CAS PubMed.
- M. A. Henderson, C. L. Perkins, M. H. Engelhard, S. Thevuthasan and C. H. F. Peden, Surf. Sci., 2003, 526, 1 CrossRef CAS.
- M. H. Zoellner, P. Zaumseil, H. Wilkens, S. Gevers, J. Wollschläger, M. Bäumer, Y.-H. Xie, G. Niu and T. Schroeder, J. Cryst. Growth, 2012, 355, 159 CrossRef CAS.
- L. Vegard, Z. Phys., 1921, 5, 17 CrossRef CAS.
- J. D. McCullough, J. Am. Chem. Soc., 1950, 72, 1386 CrossRef CAS.
- G. Niu, P. Zaumseil, M. A. Schubert, M. H. Zoellner, J. Dabrowski and T. Schroeder, Appl. Phys. Lett., 2013, 102, 011906 CrossRef.
- G. Niu, M. A. Schubert, F. d'Acapito, M. H. Zoellner, T. Schroeder and F. Boscherini, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 116, 123515 Search PubMed.
- J. M. D. Coey, M. Venkatesan and C. B. Fitzgerald, Nat. Mater., 2005, 4, 173 CrossRef CAS PubMed.
- N. Paunović, Z. Dohčević-Mitrović, R. Scurtu, S. Aškrabić, M. Prekajski, B. Matović and Z. V. Popović, Nanoscale, 2012, 4, 5469 RSC.
- G. Niu, E. Hildebrandt, M. A. Schubert, F. Boscherini, M. H. Zoellner, L. Alff, D. Walczyk, P. Zaumseil, I. Costina, H. Wilkens and T. Schroeder, ACS Appl. Mater. Interfaces, 2014, 6, 17496 Search PubMed.
- J. M. Haschke and L. Eyring, Inorg. Chem., 1971, 10, 2267 CrossRef.
- Z. Kang and L. Eyring, J. Solid State Chem., 1988, 75, 60 CrossRef CAS.
- H. Wilkens, J. Rodewald, S. Gevers, M. H. Zoellner, T. Schroeder and J. Wollschläger, J. Phys. D: Appl. Phys., 2013, 46, 285306 CrossRef.
- S. Gevers, T. Weisemoeller, B. Zimmermann, C. Deiter and J. Wollschläger, Phys. Status Solidi C, 2010, 7, 292 CrossRef CAS.
- J. V. Lauritsen and M. Reichling, J. Phys.: Condens. Matter, 2010, 22, 263001 CrossRef CAS PubMed.
- S. Gritschneder and M. Reichling, J. Phys. Chem. C, 2008, 112, 2045 CrossRef CAS.
- S. Torbrügge, J. Lübbe, L. Tröger, M. Cranney, T. Eguchi, Y. Hasegawa and M. Reichling, Rev. Sci. Instrum., 2008, 79, 083701 CrossRef PubMed.
- A. Zangwill, J. Cryst. Growth, 1996, 163, 8 CrossRef CAS.
- R. Cuerno, M. Castro, J. Muñoz-García, R. Gago and L. Vázquez, Eur. Phys. J.: Spec. Top., 2007, 146, 427 Search PubMed.
- M. Körner, F. Loske, M. Einax, A. Kühnle, M. Reichling and P. Maass, Phys. Rev. Lett., 2011, 107, 016101 CrossRef PubMed.
- R. Olbrich, H. H. Pieper, R. Oelke, H. Wilkens, J. Wollschläger, M. H. Zoellner, T. Schroeder and M. Reichling, Appl. Phys. Lett., 2014, 104, 081910 CrossRef.
- M. Kalff, P. Šmilauer, G. Comsa and T. Michely, Surf. Sci., 1999, 426, L447 CrossRef CAS.
- H. H. Pieper, C. Barth and M. Reichling, Appl. Phys. Lett., 2012, 101, 051601 CrossRef.
- G. Ehrlich and F. G. Hudda, J. Chem. Phys., 1966, 44, 1036 Search PubMed.
- J. Krug, Phys. A, 2002, 313, 47 CrossRef CAS.
- I. V. Markov, Crystal growth for beginners: Fundamentals of nucleation, crystal growth and epitaxy, World Scientific, 2008 Search PubMed.
- S. Gevers, T. Weisemoeller, D. Bruns, A. Giussani, T. Schroeder and J. Wollschläger, J. Phys.: Condens. Matter, 2011, 23, 115904 CrossRef CAS PubMed.
- H. Wilkens, O. Schuckmann, R. Oelke, S. Gevers, A. Schaefer, M. Bäumer, M. H. Zoellner, T. Schroeder and J. Wollschläger, Appl. Phys. Lett., 2013, 102, 111602 CrossRef.
- J. B. Goodenough, Nature, 2000, 404, 821 CrossRef CAS PubMed.
- T. Hibino, A. Hashimoto, T. Inoue, J.-i. Tokuno, S.-i. Yoshida and M. Sano, Science, 2000, 288, 2031 CrossRef CAS PubMed.
- M. Nakayama and M. Martin, Phys. Chem. Chem. Phys., 2009, 11, 3241 RSC.
- S. Y. Chen, R. J. Chen, W. Lee, C. L. Dong and A. Gloter, Phys. Chem. Chem. Phys., 2014, 16, 3274 RSC.
- M. H. Zoellner, G. Niu, J.-H. Jhang, A. Schaefer, P. Zaumseil, M. Bäumer and T. Schroeder, J. Phys. Chem. C, 2013, 117, 24851 CrossRef CAS.
- M. P. Rosynek, Catal. Rev.: Sci. Eng., 1977, 16, 111 CrossRef CAS.
- Y. Tang, H. Zhang, L. Cui, C. Ouyang, S. Shi, W. Tang, H. Li, J.-S. Lee and L. Chen, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125104 CrossRef.
- J. M. Tatibouët, Appl. Catal., A, 1997, 148, 213 CrossRef.
- M. Badlani and I. E. Wachs, Catal. Lett., 2001, 75, 137 CrossRef CAS.
- C. T. Au, W. Hirsch and W. Hirschwald, Surf. Sci., 1989, 221, 113 CrossRef CAS.
- C. Houtman and M. A. Barteau, Langmuir, 1990, 6, 1558 CrossRef CAS.
- M. C. Wu, C. A. Estrada, J. S. Corneille and D. W. Goodman, J. Chem. Phys., 1992, 96, 3892 CrossRef CAS.
- P. A. Dilara and J. M. Vohs, Surf. Sci., 1994, 321, 8 CrossRef CAS.
- R. M. Ferrizz, G. S. Wong, T. Egami and J. M. Vohs, Langmuir, 2001, 17, 2464 CrossRef CAS.
- A. Siokou and R. M. Nix, J. Phys. Chem. B, 1999, 103, 6984 CrossRef CAS.
- D. R. Mullins, M. D. Robbins and J. Zhou, Surf. Sci., 2006, 600, 1547 CrossRef CAS.
- J. H. Beynon, A. E. Fontaine and G. R. Lester, Int. J. Mass Spectrom. Ion Phys., 1968, 1, 1 CrossRef CAS.
- S. D. Senanayake and D. R. Mullins, J. Phys. Chem. C, 2008, 112, 9744 CrossRef CAS.
- Y. He, B. Yang and G. Cheng, Catal. Today, 2004, 98, 595 CrossRef CAS.
- R. Juarez, P. Concepcion, A. Corma and H. Garcia, Chem. Commun., 2010, 46, 4181 RSC.
- Y. Sun, Q. Liu, S. Gao, H. Cheng, F. Lei, Z. Sun, Y. Jiang, H. Su, S. Wei and Y. Xie, Nat. Commun., 2013, 4, 2899 Search PubMed.
- A. Younis, D. Chu, I. Mihail and S. Li, ACS Appl. Mater. Interfaces, 2013, 5, 9429 Search PubMed.
- J. Zhang, H. Zhao, F. Wei, M. Yang, Z. Yang, Q. Chen and J. Chen, Phys. Status Solidi RRL, 2014, 8, 95 CrossRef CAS.
- A. J. Jacobson, Chem. Mater., 2009, 22, 660 CrossRef.
- N. Izu, W. Shin, N. Murayama and S. Kanzaki, Sens. Actuators, B, 2002, 87, 95 CrossRef CAS.
- L. Liao, H. X. Mai, Q. Yuan, H. B. Lu, J. C. Li, C. Liu, C. H. Yan, Z. X. Shen and T. Yu, J. Phys. Chem. C, 2008, 112, 9061 CrossRef CAS.
- V. Fernandes, P. Schio, A. J. A. de Oliveira, W. A. Ortiz, P. Fichtner, L. Amaral, I. L. Graff, J. Varalda, N. Mattoso, W. H. Schreiner and D. H. Mosca, J. Phys.: Condens. Matter, 2010, 22, 216004 CrossRef CAS PubMed.
- M. C. Prestgard, G. Siegel, Q. Ma and A. Tiwari, Appl. Phys. Lett., 2013, 103, 102409 CrossRef.
| This journal is © the Owner Societies 2015 |