
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Laser-driven electron dynamics for circular dichroism in mass spectrometry: from one-photon excitations to multiphoton ionization
Dominik
Kröner
Chemistry Department – Theoretical Chemistry, Universität Potsdam, Karl-Liebknecht-Straße 24-25, D-14476 Potsdam, Germany. E-mail: Dominik.Kroener@uni-potsdam.de
First published on 30th June 2015
Abstract
The distinction of enantiomers is a key aspect of chemical analysis. In mass spectrometry the distinction of enantiomers has been achieved by ionizing the sample with circularly polarized laser pulses and comparing the ion yields for light of opposite handedness. While resonant excitation conditions are expected to be most efficient, they are not required for the detection of a circular dichroism (CD) in the ion yield. However, the prediction of the size and sign of the circular dichroism becomes challenging if non-resonant multiphoton excitations are used to ionize the sample. Employing femtosecond laser pulses to drive electron wavepacket dynamics based on ab initio calculations, we attempt to reveal underlying mechanisms that determine the CD under non-resonant excitation conditions. Simulations were done for (R)-1,2-propylene oxide, using time-dependent configuration interaction singles with perturbative doubles (TD-CIS(D)) and the aug-cc-pVTZ basis set. Interactions between the electric field and the electric dipole and quadrupole as well as between the magnetic field and the magnetic dipole were explicitly accounted for. The ion yield was determined by treating states above the ionization potential as either stationary or non-stationary with energy-dependent lifetimes based on an approved heuristic approach. The observed population dynamics do not allow for a simple interpretation, because of highly non-linear interactions. Still, the various transition pathways are governed by resonant enantiospecific n-photon excitation, with preferably high transition dipole moments, which eventually dominate the CD in the ionized population.
1 Introduction
Mass spectrometry (MS) is a widely used and exceptionally sensitive analytic technique. While the obtained fragmentation pattern carries structural information, the distinction of isomers can become challenging, such that MS is often combined with other analytical, e.g. chromatographic, techniques or applied in a tandem setup of sequential mass analyzers. However, shaped femtosecond laser pulses, optimized in computer assisted feedback loops,1,2 may be used to identify constitutional isomers and even quantify their ratio in a mixture.3 When it comes to enantiomers, i.e. non-superposable molecules of the same constitution and related by reflection, the fragmentation pattern cannot simply be consulted to distinguish them. If, however, circular polarized nanosecond pulses are used for the ionization of the molecules, a difference in the ion yield is found, depending on the handedness of the light and the sample.4,5 Accordingly, a circular dichroism in the ion yield can be determined by comparing the ion yields of two measurements with opposite circular polarization, similar to the experimental proof of enantioselective laser pulse control proposed by us a few years before.6,7Circular dichroism in ion yields can also be detected employing shaped circular polarized femtosecond (fs) laser pulses.8 Despite the advances, the optimal conditions and laser pulse parameters for the maximum chiral distinction of enantiomers in MS remain unknown. While resonance enhanced multiphoton ionization (REMPI) is usually employed, Horsch et al. measured the circular dichroism (CD) in ion yields for (R)-propylene oxide after non-resonant multiphoton ionization with fs-laser pulses.9 Different laser frequencies were tested which were significantly lower than the transition frequency to the first electronic exited state. Measurements of the ion yields with respect to the laser pulse energy indicated three- to seven-photon processes for the different excitation wavelengths. Consequently, m-photon transitions composed of a resonant n-photon excitation to an electronic excited states followed by an (m–n)-photon transition to the ionization continuum were assumed. However, the detected CD in the ion yields did not correspond to the CD (or anisotropy factor) in one-photon absorption at the respective n-photon frequency. A fully satisfactory assignment of the excitation wavelengths to resonant n-photon transitions did, hence, not succeed, leaving the explanation for the origin of the detected CD in the ion yield incomplete.
For (R)-3-methylcyclopentanone we have proven that the CD in the ion yield after REMPI can be predicted by laser-driven quantum electron dynamics based on ab initio calculations, namely time-dependent configuration interaction singles with perturbative doubles (TD-CIS(D)).10,11 In particular, the impact of the laser pulse duration was successfully reproduced by our approach, allowing for an explanation of the observed decrease of the CD for very short laser pulses.10 The requirement was a multi-state system based on ab initio data with electric and magnetic dipole transition elements between every electronic state to allow for a dynamic polarization of the molecule with the laser. Although our approach allowed for the investigation of the CD as a function of various laser pulse parameters so far,11 it required an initial resonant transition to a specific electronic excited state.
In order to simulate the experiments by Horsch et al.,9,12 applying non-resonant multiphoton ionization, we, hence, have to extend our model by several aspects. We will demonstrate ways for the calculation of the CD based on stationary or non-stationary electronic states above the ionization potential. Moreover, electric quadrupole interactions will be introduced into the interaction Hamiltonian, and their contribution to resonant and non-resonant multiphoton excitations will be studied. Based on these extensions we are able to clarify which (enantiospecific) transitions are responsible for the CD in the ion yield after non-resonant laser pulse ionization.
Note that, our approach differs in several ways from the one of Ma and Salam, who studied the related problem of optimal selective excitation of enantiomers in a racemic mixture by circular polarized laser pulses.13 Their investigations were based on the analytical description of a two-level model system, for which resonant and off-resonant one-photon absorptions, mediated by electric and magnetic dipole interactions, were compared showing that off-resonant conditions significantly decreased the enantioselectivity.
The paper is organized as follows: Section 2 describes the model system, the applied quantum chemical methods and the extensions of the laser-driven many electron electron dynamics for the simulation of the circular dichroism in non-resonant multiphoton excitations. In Section 3 the results of the ab initio calculations as well as of the quantum dynamics are presented and discussed. Conclusions are given in Section 4. Appendices A to E give more insight into the quality of the ab initio results, and more detailed explanations for the more complex findings, partly based on a two-level model system.
2 Model and theory
2.1 Model system
The molecule investigated in mass spectrometry is 1,2-propylene oxide,9 also known as epoxypropane. As in the experiment, we focus on the (R)-enantiomer, which will be abbreviated by (R)-PO in the following. The minimum energy geometry was obtained from MP2 (2nd order Møller–Plesset perturbation theory) using the triple-zeta augmented correlation-consistent basis set aug-cc-pVTZ.14–16 The optimized structure is shown in Fig. 1. Characteristic geometric parameters are given in Table 6 in Appendix B. Simulated infrared (IR) and vibrational CD (VCD) are presented in Appendix C. The agreement with available experimental data is good. | ||
| Fig. 1 Minimum energy geometry of (R)-propylene oxide, MP2/aug-cc-pVTZ. | ||
Next, vertical excitation energies to the first n = 149 singlet electronic excited states were computed for the minimum energy geometry with CIS(D) (configuration interaction singles with perturbative doubles correction) for the aug-cc-pVTZ basis set.17 The computed energies are later used for the electron wavepacket dynamics, see Section 2.2, and to simulate UV and electronic CD (ECD) spectra for comparison with experimental spectra. For the ECD spectrum the rotatory strengths Rj for the transition from the electronic ground state S0 to the jth electronic excited state Sj are needed additionally. The rotatory strength is defined as
 | (1) |
 and
and  are the electric and magnetic dipole operators for masses mk with charges qk at positions
are the electric and magnetic dipole operators for masses mk with charges qk at positions ![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) k and with momenta
k and with momenta  (note that, the nuclei are fixed). The required electric and magnetic transition dipole moments between electronic ground and excited states are provided by the CIS calculations. The Gaussian broadened UV and ECD spectra are calculated as described in ref. 11.
(note that, the nuclei are fixed). The required electric and magnetic transition dipole moments between electronic ground and excited states are provided by the CIS calculations. The Gaussian broadened UV and ECD spectra are calculated as described in ref. 11.
For the quantum electron dynamics electric,  , and magnetic transition dipole moments,
, and magnetic transition dipole moments,  , between every pair of electronic states i and j have to be considered.18 They are also provided by the CIS calculations. Moreover, permanent electric dipole (and quadrupole) moments of each electronic state,
, between every pair of electronic states i and j have to be considered.18 They are also provided by the CIS calculations. Moreover, permanent electric dipole (and quadrupole) moments of each electronic state, ![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) ii, were obtained from additional CIS calculations based on the generalized electronic density.19 Note that all
ii, were obtained from additional CIS calculations based on the generalized electronic density.19 Note that all ![[m with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_006d_20d1.gif) ii are zero, and ij = −ji = ji* due to the (angular) momentum operator in
ii are zero, and ij = −ji = ji* due to the (angular) momentum operator in  . As the electric transition quadrupole moments are often in the same order of magnitude as the magnetic transition dipole moments, they are considered as well in the wavepacket dynamics. Additional CIS calculations based on the transition density matrices between every electronic state i and j yielded all required electric transition quadrupole moment tensors
. As the electric transition quadrupole moments are often in the same order of magnitude as the magnetic transition dipole moments, they are considered as well in the wavepacket dynamics. Additional CIS calculations based on the transition density matrices between every electronic state i and j yielded all required electric transition quadrupole moment tensors  , where
, where  is the αβ-Cartesian component (α, β∈ {x, y, z}) of the electric quadrupole operator.18 Since the lower order moment of the molecule, i.e. the electric dipole is non-zero, the electric quadrupole moment depends on the choice of the origin of the coordinate system. Moreover, an origin-dependence exists for the magnetic transition dipole moments, too. The center of charge was chosen as the origin. All ab initio calculation were done with the Gaussian09 program package.20
is the αβ-Cartesian component (α, β∈ {x, y, z}) of the electric quadrupole operator.18 Since the lower order moment of the molecule, i.e. the electric dipole is non-zero, the electric quadrupole moment depends on the choice of the origin of the coordinate system. Moreover, an origin-dependence exists for the magnetic transition dipole moments, too. The center of charge was chosen as the origin. All ab initio calculation were done with the Gaussian09 program package.20
2.2 Electron wavepacket dynamics
The electron dynamics are simulated in the state representation, using the time-dependent configuration interaction singles method, with perturbative doubles (TD-CIS(D)) treatment for the energies of the electronic states, as described elsewhere.21,22 In total n = 150 electronic states were used if not stated otherwise. The propagation was carried out employing the Runge–Kutta method of fourth order with a time step of Δt = 2.5 attoseconds, see Table 1.| Parameter | Value |
|---|---|
| n | 150 |
| Δt [as] | 2.5 |
| N | 144 |
| d [a0] | 100 |
| t p [fs] | 200 |
| fwhmI [fs] | 73 |
| ω 02 [eV ℏ−1] | 7.01 |

|
0.1 |
| Ī [GW cm−2] | 0.50 |
| γ K | 1150 |
| ω 02/2 [eV ℏ−1] | 3.50 |

|
1.0 |
| Ī [GW cm−2] | 50 |
| γ K | 57.4 |
| ω 02/4 [eV ℏ−1] | 1.75 |

|
10 |
| Ī [TW cm−2] | 5.0 |
| γ K | 2.87 |
The interaction with the laser is expressed as:23,24
 | (2) |
 , where the wavevector
, where the wavevector  for the field propagating in z-direction (
for the field propagating in z-direction ( is the unit vector in z-direction), see below. In contrast to ref. 11 we now include electric quadrupole interactions in addition to the magnetic dipole interactions. The electric field of the laser is given by:
is the unit vector in z-direction), see below. In contrast to ref. 11 we now include electric quadrupole interactions in addition to the magnetic dipole interactions. The electric field of the laser is given by: | (3) |
 and the frequency ω. The pulse shape is formed by
and the frequency ω. The pulse shape is formed by  for 0 ≤ t ≤ tp and
for 0 ≤ t ≤ tp and  , where the pulse duration tp = 2fwhm (full width at half maximum of the field). For better comparison with experimental conditions, the fwhm of the intensity may be calculated by:
, where the pulse duration tp = 2fwhm (full width at half maximum of the field). For better comparison with experimental conditions, the fwhm of the intensity may be calculated by:  . The polarization is given by
. The polarization is given by  , where the phase δ may be chosen
, where the phase δ may be chosen  for left-(LCP) and
for left-(LCP) and  for right circular polarization (RCP). The magnetic field is defined accordingly using
for right circular polarization (RCP). The magnetic field is defined accordingly using  .
.
The field amplitude  was chosen such that the impact on the system is not too strong (for reasons discussed below), while the amount of transferred population is still significantly above the numerical error, to allow for a reliable analysis of the results. Typical laser pulse parameters for one-, two- and four-photon transitions are listed in Table 1. The mean intensity (averaged over tp) is calculated as
was chosen such that the impact on the system is not too strong (for reasons discussed below), while the amount of transferred population is still significantly above the numerical error, to allow for a reliable analysis of the results. Typical laser pulse parameters for one-, two- and four-photon transitions are listed in Table 1. The mean intensity (averaged over tp) is calculated as  . The Keldysh parameter – defined as
. The Keldysh parameter – defined as  with
with  being the ponderomotive energy and the ionization potential IP = 10.22 eV25 – determines the transition between two regimes of nonlinear ionization:26 multiphoton ionization (MPI) is to occur if γK > 1, while tunneling ionization is dominant for γ ≪ 1. For all laser pulses investigate here the Keldysh parameter is well above one, see Table 1.
being the ponderomotive energy and the ionization potential IP = 10.22 eV25 – determines the transition between two regimes of nonlinear ionization:26 multiphoton ionization (MPI) is to occur if γK > 1, while tunneling ionization is dominant for γ ≪ 1. For all laser pulses investigate here the Keldysh parameter is well above one, see Table 1.
For (resonant) transitions to one specific electronic excited state Sj, we may define the circular dichroism (CD) via the population of the excited state after laser excitation:
 | (4) |
In our simulations the laser field interacts with the projections of ij and ij onto the x–y-plane for a given orientation of the molecule. In experiment the molecules are, however, randomly oriented. Therefore, propagations are performed for different orientations of the molecule and the results are averaged over all orientations. For it the molecule, i.e. all its (transition) dipole/quadrupole moments, is rotated around the laboratory fixed coordinate axes using Euler angles ϕ (∈[0, 2π]) and θ (∈[0, π]):
 | (5) |
 is the rotation matrix for a rotation about the k-axis by ξ degrees. A third rotation about the z-axis was omitted, because it has no influence on the laser-molecule interactions for the employed circularly polarized pulses propagating in z-direction and where 1/ω ≪ tp. The rotation angles are given by ϕ = i·Δξ and θ = j·Δξ for i = 0, 1,…, M − 1 or
is the rotation matrix for a rotation about the k-axis by ξ degrees. A third rotation about the z-axis was omitted, because it has no influence on the laser-molecule interactions for the employed circularly polarized pulses propagating in z-direction and where 1/ω ≪ tp. The rotation angles are given by ϕ = i·Δξ and θ = j·Δξ for i = 0, 1,…, M − 1 or  , where
, where  . The number of considered orientations
. The number of considered orientations  was set to 144 (Δξ = 22.5°), if not stated otherwise. For resonant transitions to Sj very small changes were found in the CD[Sj] when increasing the number of orientations. For non-resonant excitations where practically every transition moment element may play a role, this number has to be considered a compromise between an acceptable computational effort and a reasonable degree of convergence. The few performed test calculations with a higher number of orientations did not result in important changes.
was set to 144 (Δξ = 22.5°), if not stated otherwise. For resonant transitions to Sj very small changes were found in the CD[Sj] when increasing the number of orientations. For non-resonant excitations where practically every transition moment element may play a role, this number has to be considered a compromise between an acceptable computational effort and a reasonable degree of convergence. The few performed test calculations with a higher number of orientations did not result in important changes.
All CD values are calculated from rotationally averaged final populations (unless noted otherwise) which are obtained by:27
 | (6) |
 .
.
Note that, one can always (unless the vectors are collinear) find orientations for which the projections of ij and ij onto the xy-plane result in parallel or anti-parallel projection vectors, causing a sign change in the calculated CD-value. Therefore, rotational averaging is mandatory to balance these sign chances in the CD-values by those of all the other orientations. In addition, for a resonant excitation to an excited state Sj, it is convenient to initially orient the molecule such that its interaction with the light is maximized. This is accomplished by rotating the molecule beforehand such that the corresponding electric transition dipole moment, μ0j, becomes parallel to the x-axis, and the magnetic transition dipole moment, m0j, is in the xy-plane. That way we ensure that the target state is highly populated allowing for a reliable result for the CD[Sj]. Needless to say that this approach cannot uniquely be applied for non-resonant excitations with no specific target state. Moreover, a preferred orientation of the molecule could actually overestimate one specific transition moment unless rotational averaging over a huge number of orientations is carried out. As we are first of all interested in observing circular dichroism we chose an initial orientation which is optimal for a resonant transition to S2, because it is the state with the highest anisotropy factor within the experimental investigated range of excitations energies (6.8–9 eV), see Section 3.
2.3 Model for ionization
In experiment the ion yield is measured. Therefore, a determination of the CD is required which is more comparable to the experiment than the one used in eqn (4), in particular if the laser excitation is non-resonant. Here the challenge is not only to correctly describe the free electron, but also the transition from bound states to continuum states. The required transition matrix elements, however, cannot be obtained from our wavefunction calculations, i.e. by optimizing the neutral and cationic state, as the number of electrons differs. In addition, in the dynamics a large number of electronic eigenstates also above the IP is required to cover a maximal portion of the dynamic polarizability of the molecule.28 Hence, electronic states above the IP, as obtained from the ab initio calculations, cannot simply be omitted or exchanged by a continuum. Consequently, the population of these, quasi-ionized, states above the IP can be used to quantify the extent of ionization.Two models for the detection of the ionization yield are used in this work. Both rely on the sum of the population of all eigenstates above the IP, obtained from the total population remaining below the IP by:
 | (7) |
The quasi ionized population in eqn (7) can be used to calculate the CD[>IP] after the interaction with the laser pulse:
 | (8) |
Therefore, in order to reduce the effects of enantiospecific excitations to “ionized” states and population back-transfer, we can alternatively make the electronic states above the IP (11.81 eV) non-stationary by replacing their real eigenenergies, En (≥IP), by complex ones: En − iℏγn/2. Afterward the molecular Hamiltonian will not be Hermitian any more and, as such, the time evolution will not be unitary any more, i.e. the population of these states will decay irreversibly. The are many ways, how the decay rates γn may be chosen. In a heuristic model that proved to be suitable for the description of ionization in TD-CIS calculations the rates were determined by:28
 | (9) |
 , where εr is the energy of the orbital from which the electron escapes, and d is the escape distance. The rates are weighted by the probability of the excitation which is given by the absolute squared value of the corresponding CI-coefficients, Dra,n, for an electron excitation from the occupied orbital a to the virtual orbital r. Hence, the lifetime of the states above the IP are given by
, where εr is the energy of the orbital from which the electron escapes, and d is the escape distance. The rates are weighted by the probability of the excitation which is given by the absolute squared value of the corresponding CI-coefficients, Dra,n, for an electron excitation from the occupied orbital a to the virtual orbital r. Hence, the lifetime of the states above the IP are given by  ; those below the IP are infinite. According to eqn (9), the higher the orbital energy of a virtual orbital the lower the lifetime of an electronic state which is dominated by this orbital. Orbital energies and CI-coefficients are obtained from the CIS calculations.18 The parameter d may be used to scale the rates. In contrast to studies by others,28 we wish to adjust the rates such that a meaningful comparability to the CD[>IP] is ensured, but preventing the return of population to states below the IP. However, too high rates may suppress transient population of states above the IP which has an impact on the CD as well, see Discussion in Section 3.4. Therefore, a rather high value of 100 a0 was chosen for d.
; those below the IP are infinite. According to eqn (9), the higher the orbital energy of a virtual orbital the lower the lifetime of an electronic state which is dominated by this orbital. Orbital energies and CI-coefficients are obtained from the CIS calculations.18 The parameter d may be used to scale the rates. In contrast to studies by others,28 we wish to adjust the rates such that a meaningful comparability to the CD[>IP] is ensured, but preventing the return of population to states below the IP. However, too high rates may suppress transient population of states above the IP which has an impact on the CD as well, see Discussion in Section 3.4. Therefore, a rather high value of 100 a0 was chosen for d.
Worth mentioning are a similar approaches to model ionization in TD-CIS which are based on complex absorbing potentials (CAP) with space-grid or atom-centered basis functions, see e.g.ref. 30 and 31.
The CD in case of non-stationary states above the IP is still calculated using eqn (7) and (8), but will be denoted CDion to tell it apart from the CD[>IP]. Note that for the CDion, however, P[S>IP] covers the decayed as well as the remaining population above the IP (if existing). In both approaches, P[S>IP] may be considered a lower limit for the “ionized” population, as a large number of states are missing to the (quasi) continuous basis set limit.
3 Results
3.1 UV and ECD spectra
The simulated UV and ECD spectra are shown in Fig. 2. The spectra are based on the vertical excitation energies, oscillator strengths or rotatory strengths, see eqn (1), calculated with CIS(D)/aug-cc-pVTZ. Selected values are given in Table 2. At first glance three main peaks are found between 185 nm (6.7 eV) and 140 nm (8.9 eV) in agreement with experimental observations.32 Some of the bands result however from several electronic states, some of which are close in energy, with strongly varying oscillator strengths, see Table 2. This and the fact that the gas phase spectrum shows a fine structure attributable to vibrational modes (which are not considered in our model) make a straight comparison with experiment rather complicated. | ||
Fig. 2 Simulated UV (top) and ECD (bottom) spectra of of (R)-propylene oxide, as obtained from CIS(D)/aug-cc-pVTZ. Gaussian broadening with 500 cm−1 . Oscillator strengths or rotatory strengths are plotted as stick spectra. Spectra are shown up to ionization potential of 10.22 eV.25 Inset show the ECD spectrum with a Gaussian broadening of 1000 cm−1 for an easier comparison with the experimental gas phase spectrum, see text and ref. 32 and 33. . Oscillator strengths or rotatory strengths are plotted as stick spectra. Spectra are shown up to ionization potential of 10.22 eV.25 Inset show the ECD spectrum with a Gaussian broadening of 1000 cm−1 for an easier comparison with the experimental gas phase spectrum, see text and ref. 32 and 33. | ||
| Sj | E j [eV] | λ j [nm] | f osc j | R j | Main character |
|---|---|---|---|---|---|
| S1 | 6.95 | 178 | 0.0462 | −17.0 | n → Ryd (3s) |
| S2 | 7.01 | 177 | 0.0083 | 12.3 | n → Ryd (3s') |
| S3 | 7.10 | 175 | 0.0066 | 4.16 | n → Ryd (3sp) |
| S4 | 7.29 | 170 | 0.0403 | 7.37 | n → Ryd (3p) |
| S5 | 7.72 | 161 | 0.0031 | −0.521 | σ → Ryd (3s) |
| S6 | 8.01 | 155 | 0.0407 | −16.3 | σ → Ryd (3sp) |
| S7 | 8.14 | 152 | 0.0259 | 5.53 | n → Ryd (3p′,3s′′) |
| S8 | 8.16 | 152 | 0.0263 | −6.28 | σ → Ryd (3s′) |
| S9 | 8.19 | 151 | 0.0227 | 14.1 | n → Ryd (3p′′) |
| S10 | 8.30 | 149 | 0.0032 | −3.08 | n → Ryd (3s′′′) |
| S11 | 8.35 | 149 | 0.0154 | −13.9 | σ → Ryd (3p) |
| S12 | 8.50 | 146 | 0.0201 | 3.26 | n → Ryd (3p′,3s′′) |
Although agreement with experiment is found for the most part, our results deviate from experimental one-photon absorption spectra as well as from theoretical data based on coupled cluster (CC) or symmetry-adapted cluster-CI (SAC-CI) calculations in some parts.34,35 These deviations are not surprising given the quality of the CIS(D) method and the fact that most excited states are of Rydberg character which is difficult to capture entirely. The simulated UV and ECD spectra are analysed in more detail in Appendix A. The analysis shows that CIS(D) provides a reasonable description of the electronic structure, in particular for the low lying electronic excited states. Since we wish to employ time-dependent CIS to reveal the fundamental steps of the electron dynamics, a perfect agreement with the experiment is, however, not required as the laser pulse parameter can easily be adapted to the calculated electronic structure of the system. For the following many electron dynamics the electronic structure is a good compromise between accuracy and computational effort.
3.2 One-photon excitations
The electron wavepacket dynamics allow comparison of resonant one- with two- or higher multi-photon excitations to a target state Sj, for which the CD[Sj] can easily be determine according to eqn (4). For this purpose it is important to understand which types of interactions mediate these transitions. Beforehand, a suitable target state for the analysis has to be identified. Here it is instructive to learn about the correlation between the CD[Sj] and the rotatory strength or the anisotropy factor.
mediate these transitions. Beforehand, a suitable target state for the analysis has to be identified. Here it is instructive to learn about the correlation between the CD[Sj] and the rotatory strength or the anisotropy factor.
In a resonant one-photon transition, the difference in the population, ΔP[Sj] = PLCP[Sj] − PRCP[Sj], of the target state Sj is determined by the lengths of the corresponding electric and magnetic transition dipole vectors, |0j| and |j0|, as well as the angle  between them. These values are listed in Table 3 for a selection of electronic states. Note that the angle θ is origin-dependent, as shown e.g. for computed VCD spectra.36
between them. These values are listed in Table 3 for a selection of electronic states. Note that the angle θ is origin-dependent, as shown e.g. for computed VCD spectra.36
0j and ![[Fraktur I]](https://www.rsc.org/images/entities/char_e186.gif) (j0), their ratio, the angle θ between them, and the calculated anisotropy factor g (CIS/aug-cc-pVTZ). Values for the S0 → S1 transition in (R)-3-methylcyclopentanone are given for comparison11
(j0), their ratio, the angle θ between them, and the calculated anisotropy factor g (CIS/aug-cc-pVTZ). Values for the S0 → S1 transition in (R)-3-methylcyclopentanone are given for comparison11
| Sj | |0j| [e a0] |
|j0| [e ℏ me−1] |
|
θ [°] | g [a0Ehℏ−1] |
|---|---|---|---|---|---|
| S1 | 0.45 | 0.19 | 0.41 | 116 | −0.72 |
| S2 | 0.19 | 0.14 | 0.76 | 11 | 3.0 |
| S3 | 0.17 | 0.35 | 2.0 | 82 | 1.2 |
| S4 | 0.41 | 0.085 | 0.21 | 63 | 0.38 |
| S5 | 0.12 | 0.27 | 2.3 | 92 | −0.31 |
| S6 | 0.42 | 0.13 | 0.32 | 128 | −0.77 |
| S7 | 0.31 | 0.049 | 0.16 | 40 | 0.47 |
| S8 | 0.34 | 0.17 | 0.49 | 104 | −0.47 |
| S9 | 0.29 | 0.14 | 0.47 | 42 | 1.4 |
| S10 | 0.11 | 0.22 | 2.0 | 105 | −2.0 |
| S11 | 0.26 | 0.19 | 0.73 | 128 | −1.8 |
| S12 | 0.28 | 0.16 | 0.57 | 81 | 0.36 |
| S1 (3MCP) | 0.029 | 0.54 | 19 | 11 | 74 |

In a two-level system, ΔP of the target state maximizes with increasing size of the electric or magnetic transition dipole moments and for angles θ close to 0° or 180°, as e.g. shown by Ma and Salam.37 Naturally, this seems to perfectly correspond to the rotatory strength Rj = |0j||j0|cos(θ), see eqn (1). While the rotational strength is determined by the product of |0j| and |j0|, we observe a significant CD[Sj] if |j0| is large with respect to |0j|. Thus, a high  ratio is desired for a good chiral distinction in our simulations. Calculated values are given in Table 3. Please, recall that the CD[Sj] is the weighted difference in the populations, see eqn (4); a high population of the target state (mediated by 0j) is not required for a high absolute CD value. Therefore, although the absolute value of the rotational strength for the S0 → S1 excitation is quite high (R1 = −17.0 × 10−40 cgs), see Table 2, we find a
ratio is desired for a good chiral distinction in our simulations. Calculated values are given in Table 3. Please, recall that the CD[Sj] is the weighted difference in the populations, see eqn (4); a high population of the target state (mediated by 0j) is not required for a high absolute CD value. Therefore, although the absolute value of the rotational strength for the S0 → S1 excitation is quite high (R1 = −17.0 × 10−40 cgs), see Table 2, we find a  ratio below one (0.41 a0Eh ℏ−1), see Table 3. For the S0 → S1 excitation of the previously investigated (R)-3-methylcyclopentanone (3MCP) the |j0|/|0j| ratio is more than forty times higher (19 a0Eh ℏ−1), although the rotational strength is more than two times lower in magnitude (R0 = 7.25 × 10−40 cgs).11 Therefore, the absolute CD[S1] value is expected to be much larger for (R)-3MCP than for (R)-PO. The reason is that the S0 → S1 excitation in 3MCP is characterized by a n → π*-transition in the carbonyl group, which is typically almost electric dipole forbidden, but strongly magnetic dipole allowed.
ratio below one (0.41 a0Eh ℏ−1), see Table 3. For the S0 → S1 excitation of the previously investigated (R)-3-methylcyclopentanone (3MCP) the |j0|/|0j| ratio is more than forty times higher (19 a0Eh ℏ−1), although the rotational strength is more than two times lower in magnitude (R0 = 7.25 × 10−40 cgs).11 Therefore, the absolute CD[S1] value is expected to be much larger for (R)-3MCP than for (R)-PO. The reason is that the S0 → S1 excitation in 3MCP is characterized by a n → π*-transition in the carbonyl group, which is typically almost electric dipole forbidden, but strongly magnetic dipole allowed.
The  ratio may be connected to the anisotropy factor g, which is the ratio of the CD to the absorption in absorption spectroscopy, i.e. the difference of the extinction coefficients for LCP and RCP (Δε = εLCP − εRCP), to the absorption:
ratio may be connected to the anisotropy factor g, which is the ratio of the CD to the absorption in absorption spectroscopy, i.e. the difference of the extinction coefficients for LCP and RCP (Δε = εLCP − εRCP), to the absorption:  . The factor may be approximated by
. The factor may be approximated by  , with the electric dipole strength Dj = 0jj0 = |0j|2, if the shapes for the CD signal and the absorption signals are the same as well as the permittivity is set to one.38 Both assumptions are reasonable, as we consider only vertical excitations in our model, and treat single molecules in vacuum. Values of g are listed in Table 3. A comparison of the g-factors for the S0 → S1 transitions in (R)-3MCP and (R)-PO supports our observations that the corresponding absolute CD[S1] values differ by about two orders of magnitude when using the same laser parameters (results not shown).
, with the electric dipole strength Dj = 0jj0 = |0j|2, if the shapes for the CD signal and the absorption signals are the same as well as the permittivity is set to one.38 Both assumptions are reasonable, as we consider only vertical excitations in our model, and treat single molecules in vacuum. Values of g are listed in Table 3. A comparison of the g-factors for the S0 → S1 transitions in (R)-3MCP and (R)-PO supports our observations that the corresponding absolute CD[S1] values differ by about two orders of magnitude when using the same laser parameters (results not shown).
Employing TD-CIS(D), with the parameters listed in Table 1, we obtain CD[Sj] values for one-photon excitations in (R)-PO to states S1, S2 and S3 of −0.52%, 2.2% and 0.87%, respectively. Note that, the CD[Sj] ratios almost perfectly match the ratios of the corresponding anisotropy factors, see Table 3. Moreover the values are about 137.036 times smaller than the listed g-values, because the laser interaction with the magnetic dipole is  times smaller than with the electric one, see Section 2.2. Still, the anisotropy factor is not the perfect quantity to directly compare to the circular dichroism obtained after one-photon excitation, because the absolute value of CD[Sj] decreases with increasing field strengths eventually ruining the almost perfect agreement with g, see Appendix D for more details. Only for very low field strength (weak field limit) the correlation between CD[Sj] and the anisotropy factor is good.
times smaller than with the electric one, see Section 2.2. Still, the anisotropy factor is not the perfect quantity to directly compare to the circular dichroism obtained after one-photon excitation, because the absolute value of CD[Sj] decreases with increasing field strengths eventually ruining the almost perfect agreement with g, see Appendix D for more details. Only for very low field strength (weak field limit) the correlation between CD[Sj] and the anisotropy factor is good.
In order to compare resonant one- and multi-photon excitations in the following, we will use the S0 → S2 transitions in PO, because the |g|-factor or CD[Sj] is the highest of the low lying states.
In Table 4 we study the influence of the electric quadrupole contributions on the one-photon (ω02) CD[S2] value. If electric quadrupole interactions are included, the result will not significantly change. Very small deviations (less than 1% of the CD[S2] value) are due to the fact that perfect numerical averaging cannot be achieved here. Apparently, the CD[Sj] in one-photon excitations depends only on the electric and magnetic dipole contributions. Analytically the contribution of the electric quadrupole vanishes entirely upon rotational averaging.23 For oriented molecules the electric quadrupole does, however, add to the CD-value (not shown).
 or electric quadrupole
or electric quadrupole  or both interactions for an ensemble of rotationally averaged (R)-PO molecules. Laser pulse parameters are given in Table 1
or both interactions for an ensemble of rotationally averaged (R)-PO molecules. Laser pulse parameters are given in Table 1
| CD[S2] [%] | ω 02 | ω 02/2 | ω 02/4 |
|---|---|---|---|

|
2.2 | −1.0 | 0.40 |

|
0.0 | −0.13 | −0.078 |

|
2.2 | −1.2 | 0.32 |
In the next section, we will compare the one-photon excitation to two- and four-photon excitations.
3.3 Multiphoton excitations
Although (R)-PO is presumably non-resonantly excited before it ionizes, the slopes of the experimental parent ion yields as a function of the laser pulse energy indicate approximately three-, five- and seven-photon processes for the three different excitation wavelengths.9 Due to the manifold of electronic states, in particular when approaching the ionization potential, such resonant multiphoton excitations, i.e. where the laser frequency (within its spectral width) is an integer multiple of the respective transition frequency, are indeed possible. Therefore, for a start the CD[S2] of two- and four-photon resonant S0 → S2 excitations are evaluated in the following. The role of the electric quadrupole moment is in particular of interest.In a two-photon transition the amount population transferred to the target state is significantly smaller than in the respective one-photon transition for a given field strength. The same is true when going to the four-photon excitation. In order to transfer enough population to S2 for a reliable determination of the CD[S2], the field strength  is increased by a factor of ten for each doubling of the number of photons. In addition, the central laser frequency ω is half or a quarter of ω02. The used parameters are listed in Table 1.
is increased by a factor of ten for each doubling of the number of photons. In addition, the central laser frequency ω is half or a quarter of ω02. The used parameters are listed in Table 1.
Table 4 summarizes the results on (one-,) two- and four-photon S0 → S2 transitions obtained from electron wavepacket dynamics. In case of the two-photon excitation the contribution of the electric quadrupole moment does not vanish upon orientation averaging and is, hence, not negligible any more. This is consistent with conclusions drawn from analytical expressions for absorption rates of elliptically polarized light, derived from time-dependent perturbation theory.23 Moreover, the absolute value of the two-photon CD[S2] is smaller than of the one-photon one; in addition, the sign has changed. This is surprising, because for a two-level system (n = 2) of the states S0 and S2 (using the same pulse parameters), the two-photon CD[S2] equals + 1.9% instead of −1.2%. This is a clear indication for interaction with other electronic states. At tp/2, i.e. when the maximum field strength is reached, population is indeed distributed among states up to S63. However, at the end of the pulse (tp) only states S0 to S3 remain populated below the IP, with populations in S1 and S3 being negligible compared to the target states S2. Apparently S1 and S3 are energetically close enough (60 to 90 meV), see Table 2, to partly compete with S2 due to spectral width of the (73 fs) pulse of approximately 30 meV (3.295 ℏ per fwhmI). Note also that the electric transition dipole moment to S1 is more than twice the in magnitude than one to S2. However, the interactions with the two neighboring states of S2 seem not to be enough to be alone responsible for the significant change of the CD[S2] when all 150 electronic states are included. Seemingly, transient interactions with higher states contribute to the observed CD.
In case of the four-photon excitation, the absolute value of the CD[S2] is even smaller than in the two-photon case. The reason seems to be again transient interactions, now with an even larger number of electronic states due to the lower frequency and the higher field amplitude. For a two-level system of S0 and S2 (using the same pulse parameters), we actually obtain 1.8%. Moreover, the fraction of the CD[S2] value caused by electric quadrupole interactions is relatively larger for the four-photon transition (∼24%) than in the case of the two-photon transition (∼11%). We also note that the sign caused by the quadrupole interactions does not have to be the same as for the electric/magnetic dipole interactions only. For a correct description of the CD in multiphoton excitations the electric quadrupole contribution can, therefore, not be omitted. It is important to note that, in particular, the difference of the permanent electric dipole moments, ![[d with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0064_20d1.gif) j0 = jj − 00, as well as the difference between the permanent electric quadrupole moments,
j0 = jj − 00, as well as the difference between the permanent electric quadrupole moments,  , should not be zero for a multiphoton excitation.23 In fact, for the limiting case of an “achiral” transition, i.e. Rj = gj = 0, j0 and
, should not be zero for a multiphoton excitation.23 In fact, for the limiting case of an “achiral” transition, i.e. Rj = gj = 0, j0 and  can still cause a non-zero CD[S2], as shown in Appendix E for a two-level model system.
can still cause a non-zero CD[S2], as shown in Appendix E for a two-level model system.
Note that some population remains in states above the IP after the laser is off. This can be used to detect the CD[>IP] in the following. In the next section we wish to determine the CD in the “ionized” population rather than in a target state.
3.4 Ionization
Since in the experiment the ion yield is detected, one of the two approaches for analyzing the “ionized” population (see Section 2.3) has to be applied if non-resonant conditions are used. In order to estimate the performance of the two models, they are tested for one-, two- and four-photon S0 → S2 excitations, see Table 5. We realize that the CD[>IP] qualitatively reproduces the results of the CD[S2], but slightly differs quantitatively. In particular for ω02/2 and ω02/4 differences are not surprising, because these frequencies allow resonances to several states below the IP which can alter the CD detected above the IP.| CD[S2] [%] | CD[>IP] [%] | CDion [%] | |
|---|---|---|---|
| ω 02 | 2.2 | 2.0 | 1.4 (1 GV m−1) |
| ω 02/2 | −1.2 | −0.51 | 0.22 (5 GV m−1) |
| ω 02/4 | 0.32 | 0.19 | −0.16 (20 GV m−1) |
Evidently, the result for the resonant one-photon excitation supports our previous assumptions that the detection of the CD in the population of the target state is a valid approximation for the CD in the ion yield (or at least for the “ionized” population). The small difference of the CD[>IP] with respect to the CD[S2] could be caused by transient population transferred back to states below the IP, due to the limited number of 150 states, or simply by resonant enantiospecific transitions from S2 to states above the IP with an opposite CD effect. Indeed, for the S2 → S128 transition with E128 − E2 = (7.01 − 14.01) eV = 7.00 eV, a negative g-value can be computed. Still, the state S128 is not the most populated state above the IP though. This is state S129 at 14.03 eV due to the higher S2 → S129 dipole strength compared to the S2 → S128 transition. And for S2 → S129 we calculated a positive g-value. These findings indicate that transitions to the states above the IP may have a impact on the CD[>IP]. Yet, the effect seems to be small and should, for all we know, not appear in the ionization step.
The CDion results tell a somewhat different story: while for the one-photon S0 → S2 excitation the CDion is still close to the CD[>IP] and CD[S2] values, there are significant deviations for the two- and four-photon transitions. Although the CDion is for all three frequencies in the same order of magnitude as the CD[>IP], the absolute values are all smaller and, in particular, differ in their sign for the two- and four-photon excitations, see Table 5. There are several possible reasons for these discrepancies.
First of all, the chosen escape distance of d = 100 a0 is relatively large (compared to values used by others28), and results in rather low decay rates ranging from 0.19/fs to 0.29/fs for electronic states above the IP (11.81 eV). For comparison, the laser frequency for the resonant S0 → S2 excitation is with 11/fs about fifty times higher. The large d-value was, however, necessary as small values cause the |CDion| value to diminish. Apparently, interim interactions even with states above the IP are important at these laser pulse conditions, such that any transient population is quasi instantaneously destroyed during the action of the field if the decay rates are too high. Second, the field strengths had to be further increased, as otherwise the CDion values came out two to up to three orders of magnitude smaller than the corresponding CD[>IP] values. This may again be attributed to the weakened interactions with the decaying states above the IP. However, high field strengths usually result in less enantiospecific transitions, see Appendix D and ref. 11, and may eventually amplify the transfer of population back to lower states, in particular, for low decay rates.
Therefore, we cannot tell for sure whether CD[>IP] or CDion is the better description for the CD in the ion yield. It is possible that 150 states are still too few to obtain reliable values for CDion, or that there are actually too many states above the IP which are accessible by enantiospecific transitions altering the CD[>IP] too much. A higher number of states is, however, beyond the scope of this study. Because the CD[>IP] produces values which are closer to the model we used in the previous sections, i.e. it seems to better reflect the CD of the states below the IP, we will stick with the CD[>IP] to study the CD as a function of frequency in the next section.
3.5 Frequency dependence
Fig. 3 shows the CD[>IP] as a function of the excitation wavelength. Except for the wavelength, the pulse parameters are the same as employed for the four-photon excitations, see Table 1, because we study wavelengths between 700 and 930 nm to cover the range used in experiment.9 As our simulations are based on ab initio data which most certainly differ from the real energetics of the molecule for high electronic states, we do not expect to perfectly reproduce the experiment, but to learn more about what determines the CD in the ion yield. | ||
| Fig. 3 CD[>IP] of rotationally averaged (R)-PO molecules for a selection of excitation wavelengths, as obtained from TD-CIS(D). Other laser pulse parameters are the same as for the four-photon excitation, see Table 1. | ||
The wavelength dependent CD[>IP] values are roughly one order of magnitude smaller than the CD in ion yields found in experiment, which are (0.4 ± 0.5)% at 738 nm, (2.2 ± 0.9)% at 810 nm and (1.9 ± 1.1)% for 878 nm.9 Furthermore, we find wavelengths that cause positive CD values and those which cause negative ones. In the experiment only positive values were found for the three investigated wavelengths. Apparently, the CD[<IP] reacts very sensitively to the wavelength, as there are strong changes within a few ten nanometers for some cases. If this sensitivity is caused by achieving resonance with a single (or very few) electric excited states of (R)-PO, then a population analysis at the middle of the laser pulse (tp/2) and at the end of the laser pulse (tp) should reveal it. This is particularly of interest, as measurements of the ion yield with respect to the laser pulse energy indicate that three- to seven-photon processes take place for the three investigated wavelengths.9 We picked the highest CD[>IP] value (+0.44%) at 704 nm (1.76 eV) and the lowest (−0.34%) at 918 nm (1.35 eV), as well as the prominent peak (+0.29%) at 892 nm (1.39 eV) for further analysis:
For 1.76 eV (704 nm) we find that the full manifold of electronic states are populated at tp/2. Except for S0, S1 is the most populated one below the IP at that moment (followed by S17 and others), although its energy is further off resonance than the energy of S2 or even S3. Here the magnitude of the electric dipole moment seems to be more important than the resonance condition, as it is for S1 much larger than for S2 or S3, see Table 3. At the end of the laser pulse (tp) states (S0,) S2 and S3 are the most populated states below the IP, which can be interpreted by an initial four-photon transition (4 × 1.76 eV = 7.04 eV) to S2 and S3, followed by e.g. a three-photon transition to the ionization continuum (7.04 eV + 3 × 1.76 eV > 11.81 eV). The CD[S2] value for the four-photon transition to S2 is positive and about two times larger in magnitude compared to the corresponding negative value for S3 (not shown). Although this is consistent with the positive sign of CD[>IP] found at 704 nm, it does not explain why the CD[>IP] is significantly larger (0.44%) than for the exact four-photon excitation to S2 (0.19%), see Table 5. However, many more states are additionally populated. For instance, we cannot ignore the fact that states S38 to S41 energetically lie (10.46–10.72 eV) well within the range of a six-photon excitation (6 × 1.76 eV = 10.56 eV). In addition, there are more states involved which cannot all easily be attributed to an excitation by a integer number of photons. Thus we can conclude that complex excitation pathways take place.
For 1.35 eV (918 nm) and 1.39 eV (892 nm) similar observations are made as before: At tp/2 mainly those states with highest absolute electronic transition dipole moments are populated, although many of them are not in resonance, i.e. the molecule is significantly polarized during interaction with the laser pulse. At tp many states below the IP remain populated and offer clues to which states the electronic wavepacket has passed. Those states with rather high populations may indeed be attributed to resonant multiphoton transitions from the electronic ground state. It is these transitions which dominate the CD[>IP] with their dipole and (electric) quadrupole moments. Therefore, we did not find any indication of a single dominant multiphoton transition to or via one particular electronic state. In addition, the observed CD[>IP] could not be derived from the one-photon g-values of the dominant transitions.
Although our analyses indicate that resonant multiphoton excitations via several specific electronic states determine the CD[>IP], the process is still more complex, as we only analysed the most prominent excitation pathways. Actually many more states are involved. One must also keep in mind that in our model even the transitions from intermediate states below the IP to those above the IP may be enantiospecific, altering the population difference built up in the previous transition.
We also wish to stress again that one-photon g-values did not prove to be suitable for the analysis, as they did not always agree with the CD-value of a multiphoton transition to the same excited state, see also Section 3.3. One-photon anisotropy factors are, however, in many cases the only available data for a prediction.9 Therefore, a prediction of the CD in the ion yield after non-resonant multiphoton ionization remains challenging.
4 Conclusions
In this paper we extended our methodology to calculate the CD in ion yields via laser driven electron wavepacket dynamics by several aspects. Instead of resonant one-photon excitations, also resonant two- and four-photon as well as non-resonant multiphoton excitations were treated. Due to the two- and higher photon transitions we introduced electric quadrupole interactions. As for non-resonant excitations no target electronic state can uniquely be determined, the CD had to be calculated from the population of electronic states above the ionization potential whose lifetimes were either infinite or finite. These extensions allowed for the simulation of the experimental CD in ion yields as a function of the fs-laser pulse frequency for the non-resonant multiphoton ionization of (R)-propylene oxide.Several findings were reported. For one-photon transitions is was shown that the rotatory strength is not a good quantity for estimating the distinction of enantiomers in the ion yield. In fact, the anisotropy factor is more reliable in this sense, because it accounts for the fact that the CD value is normalized by the sum of populations (or ion yields). However, these findings apply strictly speaking only for a two-level system in the weak field limit. For increasing field strengths, the CD value decreases, namely the faster the higher the respective electric transition dipole moments. While for one-photon transitions the electric quadrupole interactions do not contribute to the CD at all, they become of importance for multiphoton excitations, being even capable of changing the sign of the CD-value.
For non-resonant excitations the CD is, in general, determined by non-linear interactions with the manifold of electronic states which require permanent and transition matrix elements for and between every electronic state. However, electric (transition) dipole moments are usually significantly larger than their magnetic dipole and electric quadrupole analogs. Therefore, they seem to determine which states are polarized most during laser interactions. Then, multiphoton resonance condition determined by the laser frequency mainly control which states remain populated at the end. Eventually the strongest transitions seem to dominate the observed CD. However, since a large variety of transitions take place and none of them are resonant one-photon excitations, the prediction of the CD in ion yield only based on one-photon anisotropy factors appears to be practically impossible. This confirms experimental observations, where the CD in the ion yield after an assumed n-photon excitation with a wavelength of nλ could not be assigned to the CD in one-photon absorption at wavelength λ.9
Conditions that ensure a resonant one-photon excitation from the start (preferably to a state with a high absolute anisotropy factor) seem, hence, the best way to distinguish enantiomers in ion yields on the first glance. Still, for one-photon transitions with small magnetic dipole transition moments, two-photon transitions might be an alternative if certain multipole moments (inter alia permanent electric dipole and quadrupole moments) support a good chiral distinction.29 Finally, a subsequent enantiospecific excitation of the parent ion after ionization might also result in higher CD values in the ion yields of fragments preferably formed from the electronically excited parent ion.12,29
Some of the extensions of our methodology have proven to be limited. In electron wavepacket dynamics we found that electronic states above the ionization potential have an impact on the CD due to transient interactions with these states, i.e. they allow for additional polarization of the molecule by the field. However, these electronic states are not a perfect description of the continuum states of the free electron. Yet, the approach of bound stationary states above the IP seems to work rather well, while rendering these states non-stationary significantly reduced the response of the electronic structure to the field. But it is also clear that simply adding a manifold of quasi-continuum states will not automatically improve the model in terms of the calculation of the CD, in particular, as transition matrix elements are not available by first principle calculations. Although one can always increase the number of electronic states to the basis set limit of the ab initio calculations, if the computational effort can be handled, the awkward character of the quasi-ionized highly excited electronic states will remain. An alternative ansatz could be to apply hybrid basis sets, composed of local Gaussian-type and plane wave basis functions, for the ab initio calculations. If this will improve the description of the CD ion yield remains to be shown.
Despite the incomplete description of the continuum, the presented methodology is applicable to any molecule for which excited state energies and transitions moments are available. They may be obtained by any quantum chemical method, for instance, by density functional theory in case of larger biomolecules.
Appendix A: detailed analysis of UV and ECD spectra
The simulated UV and ECD spectra are shown in Fig. 2, the corresponding data is given in Table 2. The positions of the three main peaks (7.0, 7.3 and around 8.2 eV) of our simulated UV spectrum are all slightly too low in energy compared to the three band maxima found in experiment (7.1, 7.7 and 8.4 eV33). These deviations are even more pronounced for CC2 (6.52, 6.92, 6.97 and 7.13 eV) on a similar level of theory, but decrease when advancing to CCSD (7.31, 7.55, 7.75 and 7.95 eV) or SAC-CI (7.01, 7.71 and 8.44 eV).34,35 In accordance with these theoretical investigations all excited states within this energy range are mainly characterized by transitions into Rydberg-like orbitals starting either from HOMO−1, the non-bonding orbital at the oxygen n(O), or the HOMO, a σ-type MO localized along the bonds of the oxirane ring, see Table 2. A clear characterization of the various Rydberg-like orbitals was, however, in many cases very difficult, as s- and p-character often mix. Still, for the two energetically lowest states with high oscillator strength (S1, S4) we can assign n(O) → 3s Rydberg and n(O) → 3p Rydberg transitions in agreement with others.33,35 However, their energies (6.95 and 7.29 eV) are by about 0.2–0.4 eV too low compared to the maxima of the vibrationally broadened bands found in experiment (7.1 and 7.7 eV). Moreover, the states S2, S3 lie so close to S1 such that they cannot clearly be attributed to two of the three expected n(O) → 3p Rydberg excitations.For the third broad band that spans from 144 to 157 nm, see Fig. 2, the excited state S11 may be picked as reference, as it agrees very well in excitation energy (8.35 eV, 149 nm) and the nature of the transition (σ → 3p Rydberg) to experimental (8.4 eV) and theoretical findings of others (8.44 eV).33,35 We note, however, that S6 is actually the third lowest state of high oscillator strength, but which is again about 0.4 eV too low in energy (8.01 eV) in comparison to the gas phase UV spectrum (8.4 eV).33 The character of this transition is dominated by a σ → 3s′′ Rydberg transition, where the Rydberg orbitals also feature a larger amount of diffuse p-type basis functions. Moreover, the following excited states lie well within in the energy range of the broad experimental band around 8.2–8.8 eV.
The characteristics of the simulated ECD spectrum resemble those of the experimental spectrum,32,33 in particular if a larger broadening is used, see inset of Fig. 2. Theory and experiment differ, however, with regard to the details: While the first excited state, S1, has a large negative rotatory strength (R1 = −17 × 10−40 cgs), the second one, S2, which is very close in energy (+0.06 eV), has a large positive one (R2 = +12 × 10−40 cgs). This state did not make a strong appearance in the UV spectrum, because its oscillator strength is very low. It significantly reduces the peak height of the first band in the ECD spectrum though, causing only a fair agreement with the particular deep (negative) peak found in experiment. Still sign and size of R1 agree well with the experimental one (−13 × 10−40 cgs)33 and even very well with those of CCSD calculations (−17 × 10−40 cgs).34 The state S4 has a rotatory strength of +7 × 10−40 cgs which is again close to the experiment (+6 × 10−40 cgs) and well within the range of other methods, e.g. +5 × 10−40 for SAC-CI.35 The states between S1 and S4 contribute to the positive band around 171 nm, see inset of Fig. 2. The third broad peak of the UV spectrum basically appears in the ECD spectrum as three peaks of alternating signs, or two peaks of negative sign (at 148 and 156 nm) if a larger broadening is allowed, see inset of Fig. 2. The gas phase ECD spectrum shows indeed a very broad band of very low negative height with a vivid fine structure.32,33 We find seven excited states (S6 to S12) with rotatory strengths of different sizes and signs, which are reached by excitations from either the n(O) or σ-MO to a variety of Rydberg orbitals of s- and p-type or combinations of them. As such a one-to-one comparison to the experiment is impossible, in particular as we cannot distinguish between electronic states and vibrational modes. The general negative character of this band is reproduced for the most part, although the rotatory strength is on average too large in magnitude compared to the experiment (−4 × 10−40 cgs).33 But even SAC-CI, which was shown to provide a good theoretical description for the first two spectral bands, overestimates the size of the rotatory strengths for the state that forms the third band (−10 × 10−40 cgs).35
Appendix B: optimized minimum geometry of (R)-PO
Table 6 lists a selection of bond lengths, angles and dihedrals of the minimum energy geometry obtained from a MP2/aug-cc-pVTZ optimization. Note that, MP2 calculations at 6-311++G(2d,2p) level of theory or with the density functional B3LYP, using either basis set, yield values which differ very little (less than one percent) to none from the ones listed in Table 6. Good agreement to the equilibrium structures calculated by similar and more sophisticated methods is found as well.39| Parameter | Size | Ref. 39 |
|---|---|---|
| R(C1–O) | 1.44 Å | 1.43 Å |
| R(C2–O) | 1.44 Å | 1.43 Å |
| R(C1–C2) | 1.46 Å | 1.46 Å |
| R(C1–C3H3) | 1.50 Å | 1.50 Å |
| α(C1–O–C2) | 61.1° | 61.3° |
| δ(O–C1–C2–C3) | 103.4° | — |
Appendix C: IR and VCD spectra of (R)-PO
IR and VCD spectra are obtained from a normal mode analysis of the minimum energy geometry at B3LYP/aug-cc-pVTZ level of theory. The molar absorption coefficient, ε, and its difference between left and right circularly polarized light, Δε, are calculated as described in the supporting information of ref. 11. The simulated spectra (unscaled harmonic frequencies) are shown in Fig. 4 using a Lorentzian broadening of 15 cm−1. Overall a good qualitative agreement to the experimental IR and VCD spectra as well as to first principles calculations is found.39–41 Merely, the relative intensities of some of the CH-stretching modes, see peaks around 3100 cm−1, are not perfectly reproduced and they are slightly blue-shifted with respect to the experiment. | ||
| Fig. 4 Simulated IR (top) and VCD (bottom) of (R)-propylene oxide, B3LYP/aug-cc-pVTZ (unscaled), Lorentzian broadening of 15 cm−1 (≙HWHM). | ||
Appendix D: correlation between CD[Sj] and gj
In Fig. 5 (top) the CD[Sj] is plotted as a function of the field strength for selected electronic excited states of (R)-PO. In each case the molecule was kept in its optimal initial orientation, as described in Section 2.3, to allow for a best possible comparison with the anisotropy factor g. The CD[Sj] was calculated for a single orientation after resonant excitation with ω = (Ej − E0)/ℏ. The laser pulse duration tp was 200 fs in each case.
for selected electronic excited states of (R)-PO. In each case the molecule was kept in its optimal initial orientation, as described in Section 2.3, to allow for a best possible comparison with the anisotropy factor g. The CD[Sj] was calculated for a single orientation after resonant excitation with ω = (Ej − E0)/ℏ. The laser pulse duration tp was 200 fs in each case.
 | ||
Fig. 5 CD[Sj] (top) and the absolute value of the relative change of CD[Sj] (bottom) as a function of the laser field strength  for selected electronic excited states of (R)-PO. Right ordinate (top): CD[Sj] scaled by the speed of light (c) for comparison with the anisotropy factor g. Data for the S1 state of (R)-3MCP is given for comparison, see circles and dashed lines. for selected electronic excited states of (R)-PO. Right ordinate (top): CD[Sj] scaled by the speed of light (c) for comparison with the anisotropy factor g. Data for the S1 state of (R)-3MCP is given for comparison, see circles and dashed lines. | ||
As can be seen on the right ordinate of the graph, the CD[Sj] values at very low field strengths, namely  , correspond very well to the anisotropy factor gj of the respective state, cf.Table 3. With increasing field strength, however, the CD values deviate from their respective g-values and approach zero. That is, because the sum of populations
, correspond very well to the anisotropy factor gj of the respective state, cf.Table 3. With increasing field strength, however, the CD values deviate from their respective g-values and approach zero. That is, because the sum of populations  , see eqn (4), increases much faster the difference ΔP (data not shown). In addition, ΔP will even decrease once most of the population has been transferred to the target state, see ref. 11 for more details.
, see eqn (4), increases much faster the difference ΔP (data not shown). In addition, ΔP will even decrease once most of the population has been transferred to the target state, see ref. 11 for more details.
In Fig. 5 (bottom) the relative change of the CD with respect to  , more precisely
, more precisely  , is plotted as a function of
, is plotted as a function of  . The functional form may be approximated by a quadratic function. The largest change is observed for S1: at
. The functional form may be approximated by a quadratic function. The largest change is observed for S1: at  the CD has lost 60% of its initial value, i.e. the chiral distinction has significantly decreased. The relative change of the CD with
the CD has lost 60% of its initial value, i.e. the chiral distinction has significantly decreased. The relative change of the CD with  is dominated by the electric transition dipole moment |0j|, see Table 3. The higher the |0j| the larger the relative change of the CD with the laser field strength. Therefore, the anisotropy factor is only a good measure for the CD if field intensity and the electric transition dipole moments in question are rather low. A comparison with the S1 state of (R)-3MCP confirms that its large ratio
is dominated by the electric transition dipole moment |0j|, see Table 3. The higher the |0j| the larger the relative change of the CD with the laser field strength. Therefore, the anisotropy factor is only a good measure for the CD if field intensity and the electric transition dipole moments in question are rather low. A comparison with the S1 state of (R)-3MCP confirms that its large ratio  of 19 a0Ehℏ−1, see Table 3, ensures a high stability of the CD[S1] with respect to
of 19 a0Ehℏ−1, see Table 3, ensures a high stability of the CD[S1] with respect to  , see dashed line in Fig. 5 (bottom).
, see dashed line in Fig. 5 (bottom).
Appendix E: non-zero CD[1] for an “achiral” two-level system
While in one-photon excitations the electric quadrupole moment contributions to the circular dichroism vanish for randomly oriented molecules, they play a role in two-photon (or higher) excitations.23 Although these contributions are usually small, they can become decisive, in particular if the one-photon CD is very small or even zero. The later is shown in the following for a two-level model system of states 0 and 1 and parameters given in Table 7. The electric and magnetic transition dipole moments are chosen such that the one-photon CD[1] is zero, i.e. the01 is perpendicular to {01} making the system quasi achiral, see Table 7.
| Parameter | Value |
|---|---|
| E 1 − E0 [eV] | 1 |
| μ x 01 [e a0] | 1 |
| ℑ{my01} [e ℏ me−1] | 1 |
| |00| [e a0] |
1 |
| Q yz 01 [e a02] | −54.4 |
| Q xy 00 [e a02] | −54.4 |
| ω [eV ℏ−1] | 0.5 |

|
5.0 |
| t p [fs] | 200 |
For a two-photon excitation (ω = 0.5 eV ℏ−1), the CD[1] remains zero (i.e. does not change), as long as = 11 − 00 is zero and electric quadrupole interactions are entirely ignored  , see case (a) in Table 8. A two-photon CD[1] unequal zero is, however, obtained, if ≠
, see case (a) in Table 8. A two-photon CD[1] unequal zero is, however, obtained, if ≠ ![[0 with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/char_0030_20d1.gif) and as long as is not perpendicular to either 01 or {01}, see case (b) in Table 8. This even holds if quadrupole contributions are excluded. Moreover, if is perpendicular to 01 and {01} (μ00z ≠ 0, i.e. μ00x = μ00y = 0), the electric quadrupole interactions allow for a nonzero two-photon CD[1], as long as either
and as long as is not perpendicular to either 01 or {01}, see case (b) in Table 8. This even holds if quadrupole contributions are excluded. Moreover, if is perpendicular to 01 and {01} (μ00z ≠ 0, i.e. μ00x = μ00y = 0), the electric quadrupole interactions allow for a nonzero two-photon CD[1], as long as either  (case (c) in Table 8) or
(case (c) in Table 8) or  (case (d) in Table 8) is nonzero. Therefore, permanent electric dipole moments as well as permanent and transition electric quadrupole moments can result in a nonzero two-photon CD, even if the respective one-photon CD is zero. These findings agree with predictions made by Meath and Power based on analytical expressions for Einstein B-coefficients.23
(case (d) in Table 8) is nonzero. Therefore, permanent electric dipole moments as well as permanent and transition electric quadrupole moments can result in a nonzero two-photon CD, even if the respective one-photon CD is zero. These findings agree with predictions made by Meath and Power based on analytical expressions for Einstein B-coefficients.23
| Case | Interactions | CD[1] [%] |
|---|---|---|
| (a) |

|
0.00 |
| (b) |

|
0.35 |
| (c) |

|
0.35 |
| (d) |

|
−0.54 |
Acknowledgements
The author would like to thank Karl-Michael Weitzel (Marburg), Tillmann Klamroth and Peter Saalfrank for helpful discussions. Financial support by the German Research Foundation (project KR 2942/2) is gratefully acknowledged.References
- R. Judson and H. Rabitz, Phys. Rev. Lett., 1992, 68, 1500–1503 CrossRef CAS PubMed.
- A. Assion, T. Baumert, M. Bergt, T. Brixner, B. Kiefer, V. Seyfried, M. Strehle and G. Gerber, Science, 1998, 282, 919–922 CrossRef CAS PubMed.
- J. Dela Cruz, V. Lozovoy and M. Dantus, J. Phys. Chem. A, 2005, 109, 8447–8450 CrossRef CAS PubMed.
- U. Boesl von Grafenstein and A. Bornschlegl, ChemPhysChem, 2006, 7, 2085–2087 CrossRef PubMed.
- R. Li, R. Sullivan, W. Al-Basheer, R. M. Pagni and R. N. Compton, J. Chem. Phys., 2006, 125, 144304 CrossRef CAS PubMed.
- D. Kröner, M. F. Shibl and L. González, Chem. Phys. Lett., 2003, 372, 242–248 CrossRef.
- D. Kröner and L. González, Phys. Chem. Chem. Phys., 2003, 5, 3933–3942 RSC.
- H. G. Breunig, G. Urbasch, P. Horsch, J. Cordes, U. Koert and K.-M. Weitzel, ChemPhysChem, 2009, 10, 1199–1202 CrossRef CAS PubMed.
- P. Horsch, G. Urbasch and K.-M. Weitzel, Z. Phys. Chem., 2011, 225, 587–594 CrossRef CAS.
- P. Horsch, G. Urbasch, K.-M. Weitzel and D. Kröner, Phys. Chem. Chem. Phys., 2011, 13, 2378–2386 RSC.
- D. Kröner, J. Phys. Chem. A, 2011, 115, 14510–14518 CrossRef PubMed.
- P. Horsch, G. Urbasch and K.-M. Weitzel, Chirality, 2012, 24, 684–690 CrossRef CAS PubMed.
- Y. Ma and A. Salam, Chem. Phys., 2006, 324, 367–375 CrossRef CAS.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- R. A. Kendall, T. H. Dunning Jr. and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- M. Head-Gordon, R. J. Rico, M. Oumi and T. J. Lee, Chem. Phys. Lett., 1994, 219, 21–29 CrossRef CAS.
- Gaussian 09 (Rev. A.02) prints out the magnetic transition dipole matrix elements as
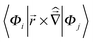 , i.e. the values have to be multiplied by 1/2 to obtain the employed ij in atomic units (e ℏ me−1). Electric transition quadrupole moments
, i.e. the values have to be multiplied by 1/2 to obtain the employed ij in atomic units (e ℏ me−1). Electric transition quadrupole moments 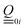 between ground and electronic excited state i, obtained from the transition density, have to be divided by
between ground and electronic excited state i, obtained from the transition density, have to be divided by 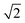 . CI-coefficients Dra are multiplied by
. CI-coefficients Dra are multiplied by 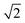 to preserve a norm of one (
to preserve a norm of one (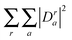 should be one).
should be one). - K. B. Wiberg, C. M. Hadad, T. J. LePage, C. M. Breneman and M. J. Frisch, J. Phys. Chem., 1992, 96, 671–679 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- T. Klamroth, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 245421 CrossRef.
- P. Krause and T. Klamroth, J. Chem. Phys., 2008, 128, 234307 CrossRef PubMed.
- W. J. Meath and E. A. Power, J. Mod. Opt., 1989, 36, 977–1002 CrossRef CAS.
- W. J. Meath and A. E. Kondo, J. Mol. Struct.: THEOCHEM, 1991, 232, 23–42 CrossRef.
- K. Watanabe, T. Nakayama and J. Mottl, J. Quant. Spectrosc. Radiat. Transfer, 1962, 2, 369–382 CrossRef.
- L. Keldysh, Sov. Phys. JETP, 1965, 20, 1307–1314 Search PubMed.
- E. A. Power and T. Thirunamachandran, J. Chem. Phys., 1974, 60, 3695–3701 CrossRef CAS.
- S. Klinkusch, P. Saalfrank and T. Klamroth, J. Chem. Phys., 2009, 131, 114304 CrossRef PubMed.
- C. Logé and U. Boesl, Phys. Chem. Chem. Phys., 2012, 14, 11981–11989 RSC.
- L. Greenman, P. J. Ho, E. Karmarchik, D. A. Mazzoni and R. Santra, Phys. Rev. A: At., Mol., Opt. Phys., 2010, 82, 023406 CrossRef.
- P. Krause, J. A. Sonk and H. B. Schlegel, J. Chem. Phys., 2014, 140, 174113 CrossRef PubMed.
- M. Carnell, S. D. Peyerimhoff, A. Breest, K. H. Gödderz, P. Ochmann and J. Hormes, Chem. Phys. Lett., 1991, 180, 477–481 CrossRef CAS.
- A. Breest, P. Ochmann, F. Pulm, K. H. Gödderz, M. Carnell and J. Hormes, Mol. Phys., 1994, 82, 539–551 CrossRef CAS.
- J. Kongsted, T. B. Pedersen, M. Strange, A. Osted, A. E. Hansen, K. V. Mikkelsen, F. Pawlowski, P. Jørgensen and C. Hättig, Chem. Phys. Lett., 2005, 401, 385–392 CrossRef CAS.
- T. Miyahara, J. Hasegawa and H. Nakatsuji, Bull. Chem. Soc. Jpn., 2009, 82, 1215–1226 CrossRef CAS.
- S. Góbi and G. Magyarfalvi, Phys. Chem. Chem. Phys., 2011, 13, 16130–16133 RSC.
- Y. Ma and A. Salam, Chem. Phys. Lett., 2006, 431, 247–252 CrossRef CAS.
- J. A. Schellman, Chem. Rev., 1975, 75, 323–331 CrossRef CAS.
- V. Barone, M. Biczysko, J. Bloino and C. Puzzarini, J. Chem. Phys., 2014, 141, 034107 CrossRef PubMed.
- P. Chu, F. Guenther, G. Rhoderick and W. Lafferty, in NIST Chemistry WebBook, NIST Standard Reference Database Number 79: Quantitative Infrared Database, ed. P. Linstrom and W. Mallard, National Institute of Standards and Technology, Gaithersburg, MD, 20899, 2014 Search PubMed.
- P. L. Polavarapu, B. A. Hess Jr. and L. J. Schaad, J. Chem. Phys., 1985, 82, 1705–1710 CrossRef CAS.





| This journal is © the Owner Societies 2015 |