
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of electron correlation on the adsorption of hydrogen fluoride and water on magnesium fluoride surfaces
Elisavet
Kanaki
*a,
Giuseppe
Sansone
b,
Lorenzo
Maschio
b and
Beate
Paulus
a
aFreie Universität Berlin, Institut für Chemie und Biochemie, Takustraße 3, 14195 Berlin, Germany. E-mail: kanaki@chemie.fu-berlin.de
bUniversity of Torino, Department of Chemistry, Theoretical Chemistry Group, Via P. Giuria 5, 10125 Torino, Italy
First published on 29th June 2015
Abstract
We have performed periodic density functional and periodic local MP2 calculations for the adsorption of hydrogen fluoride and water on the four low index surfaces (001), (100), (101) and (110) of magnesium fluoride. While the adsorption of HF is described well using B3LYP, MP2 is required for a good description of the adsorption of H2O. Post-optimization dispersion corrections of B3LYP are found to consistently overestimate the adsorption energy. The coordination of surface cations, the presence of hydroxyls on the surface, as well as the coverage appear to play an equally important role in the adsorption.
1 Introduction
Catalysis research plays a substantial role in green chemistry by improving the efficiency of large scale synthetic processes. A good catalyst is expected to be efficient, low-cost and environmentally friendly. Nanoscopic sol–gel synthesized MgF2 has recently found applications in various catalytic processes1,2 and at the same time fulfils these requirements; its synthesis in a one-pot reaction allows for the modification of the nanomaterial's surface composition through variation of the synthetic conditions.3 Indeed, in a recent study it was shown, by means of first principles thermodynamics, that temperature and pressure during synthesis strongly influence the shape of MgF2 crystallites leading to the exposure of different surfaces.4Different surfaces are expected to have distinct catalytic properties: based on the adsorption energy of CO and its vibrational frequency shift upon adsorption on MgF2, the Lewis acidity of coordinatevely unsaturated cations on MgF2 surfaces was shown to vary significantly for different terminations.5 However, the evaluation of the reactivity is by no means trivial. Although the vibrational frequency shift of CO is generally regarded as a measure for the Lewis acidity of surface sites, Huesges et al.5 observed a discrepancy in the trends of adsorption energies and vibrational frequency shifts, which in turn show no agreement with the order of stability of the surfaces as deduced from the surface energies. The observed discrepancies are not necessarily surprising. Numerous factors are involved in adsorption processes, e.g. the Lewis acidity of the cationic surface sites, the electronegativity of surface anions, the tendency of surfaces and adsorbates to form hydrogen bonds and the topology of the surface.
The focus of this work is to compare the performance of different computational methods in the description of adsorption and at the same time to investigate which factors contribute to the stabilization of adsorption structures. To this end, we have studied the adsorption of two probe molecules, H2O and HF, on MgF2 with different terminations and at different coverages: we have considered clean and partially hydroxylated terminations of the low index surfaces (001), (100), (101) and (110) of MgF2 in rutile-type structure, at full, half and 25% coverages. The structures obtained from the geometry optimizations are presented in Section 3.1. They are used for the calculation of the adsorption energy using different methods, the performance of which is discussed in Section 3.2 for selected structures. In Sections 4.1 and 4.2 we present the full series of adsorption energies for HF on clean surfaces and for H2O on clean and partially hydroxylated surfaces. Finally, in Section 4.3 we discuss the interpretation of adsorption energy with respect to catalytic activity.
2 Computational details
We have performed periodic density functional theory calculations using B3LYP,6,7 and in one case for testing using PBE8 and B3PW,6,9 as implemented in the CRYSTAL09 code.10,11 Mg and F are described with basis sets of triple-zeta quality12,13 previously optimized for MgF2, while O and H with basis sets from a previous study on AlF3 adsorption systems.14 From the full relaxation of the bulk, we obtained lattice parameters a = 4.6672 Å, b = 3.0829 Å and x(F) = 0.304 which result in a volume cell 3% larger than the experimental value.15 Bulk relaxation using B3LYP-D2 on the other hand underestimates the volume of unit cell by 2% giving a = 4.5969 Å, b = 3.0348 Å and x(F) = 0.302. B3LYP is shown to yield satisfactory results for the bulk properties of MgF2 compared to other functionals.5 Stoichiometric symmetric slabs with a thickness of 12 formula units show a good convergence of the surface energy4,5 and are used for modelling the surfaces. For the optimization of the slabs, lattice constants are fixed at the values previously optimized using B3LYP and the nuclear coordinates of all layers are relaxed.The total energy convergence criterion is set to 10−8 Ha. The accuracy of the Coulomb overlap, Coulomb penetration and exchange overlap is set to 10−6, and of the first and second pseudo-overlap to 10−12 and 10−24, respectively. The eigenvectors are calculated for a Pack–Monkhorst grid of 8 k-points in each periodic lattice direction of the irreducible Brillouin zone. The density matrix is calculated on a 16 × 16 k-point Gilat net.
Grimme's scheme for long range dispersion contributions is applied on selected optimized structures using version 3.1 of the DFT-D3 package.16,17 The D3 correction is applied using Becke–Johnson damping,18 while all other parameters are adopted as proposed in ref. 17; it is compared to results obtained using D2.16 D*, a modification of the Grimme scheme for solids proposed by Civalleri et al.,19 is also tested for selected structures. To overcome the overestimation of dispersion contribution by D2, Civalleri et al. modified the atomic van der Waals radii in the damping function obtaining excellent results for cohesive energies of crystals.20
Second-order Møller-Plesset (MP2) corrections on the optimized structures at full and half coverage have been computed using the periodic Local MP2 (LMP2) method as implemented in the Cryscor code.21,22 The Wannier functions (WFs) of H2O excitation domains are constructed taking into account only its own atoms, while for fluorines of the slabs the domains include the fluorines and their first magnesium neighbours. In cases of hydroxylated slabs, the WF centred on the oxygen atoms of the slab domains includes the oxygen itself, the nearest hydrogen and the first neighbouring magnesium atoms. Bielectronic integrals are evaluated up to a WF–WF distance of 9 Å using different fast integral evaluation techniques: for nearest integrals the density fitting procedure is employed23 in their direct space formulation24 using a valence triple-zeta level auxiliary basis with mixed Poisson- and Gaussian-type functions. The other integrals are calculated via multipolar expansion up to hexadecapoles.
In selected cases the dual basis set (d.b.) option is tested.25,26 Extended basis sets are necessary for the correction of the virtual manifold in LMP2 but diffuse functions can cause problems of linear dependencies when employed in periodic calculations. Using the d.b. option, we perform the Hartree–Fock calculation with a suitable basis set, which is afterwards appropriately extended to solve the LMP2 equations.22 Where the d.b. option is used, the Hartree–Fock part is performed with the basis set described previously. For the subsequent LMP2 calculation the basis set is extended by one polarized function with an exponent equal to half of the exponent of the initial basis set's last polarized function. The underlying Hartree–Fock calculations are performed with convergence criteria for the Coulomb overlap, Coulomb penetration, and exchange overlap set to 10−8 and for the first and second pseudo-overlap set to 10−20 and 10−80, respectively.
The adsorption energy Eads of HF and H2O on magnesium fluoride surfaces has been corrected for basis set superposition according to the counterpoise scheme27 and relaxation effects:
| Eads = E(s + ads) − E(s + gads) − E(ads + gs) + ΔErel(s) + nΔErel(ads) | (1) |
ΔErel = Eads![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) str − Erlxdstr str − Erlxdstr | (2) |
3 Models and methods
3.1 Adsorption structures of H2O and HF
A variety of initial structures was tested for each adsorption system, in search for different minima. In all but one cases only one stable adsorption structure was found. Adsorption structures of H2O and HF on clean MgF2 surfaces are presented in Fig. 1. The orientation of the adsorbates is in all cases such, that the electronegative atom X (oxygen or fluorine) is located above the coordinatively unsaturated magnesium. At the same time, the hydrogens of the adsorbates coordinate with one of the surface fluorines, so that the molecules “lie” on the surface. The distance Mg–X is about 2.1 Å independent of the adsorbate and the surface, but the distances between the adsorbate's hydrogen and its nearest surface fluorine are for HF 1.4–1.5 Å, whereas for H2O 1.6–1.8 Å. The arrangement of H2O on the surfaces is such, that one hydrogen points towards a surface fluorine, with the exception of surface (001). There, both hydrogens form hydrogen bonds with the surface. The orientation of the adsorbates to each other strongly depends on the surface structure. For example, as a consequence of the “staggered” surface cations on (101), adsorbates build an alternating arrangement (Fig. 1(c) and (g)), while adsorbates on the other surfaces are parallel to each other. No change is observed in the orientation of the adsorbates on clean surfaces with changing coverage. The surface structures remain practically unaffected by the presence of the adsorbates. Only for surface (110) we observe a slight distortion of the outer layer, which takes a zig-zag form. In all cases, the Mulliken population analysis shows a small population transfer from the surface to the adsorbate. Depending on the structure, the population increase on the adsorbate varies between 0.03–0.09 for H2O and 0.07–0.14 for HF.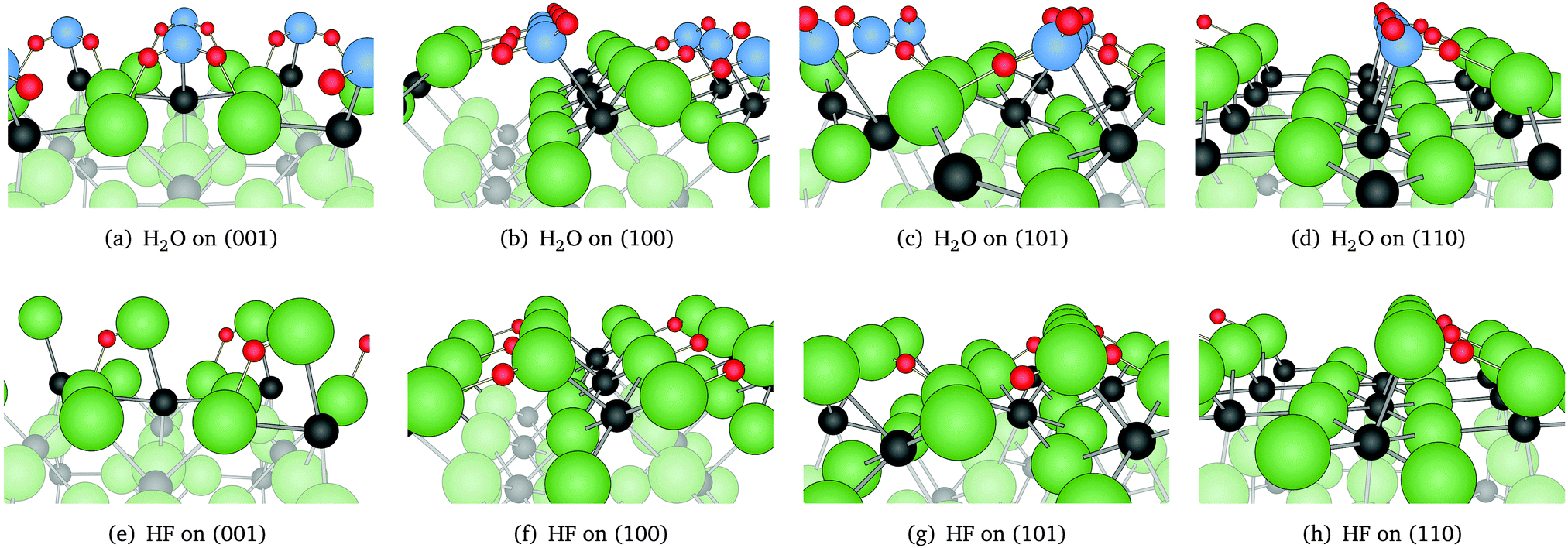 | ||
| Fig. 1 Adsorption structures of H2O and HF on low index MgF2 surfaces, full coverage, obtained using B3LYP. Fluorine atoms are represented as green, magnesium as black, hydrogen as red and oxygen as blue spheres. Surface atoms are highlighted. | ||
In addition to clean surfaces, H2O adsorption was modelled on partially hydroxylated MgF2 surfaces. The adsorption structures are shown in Fig. 2. On (001) and (101), which contain both fluorine and hydroxyl, H2O forms preferably a hydrogen bond to the oxygen of the hydroxyl group. As a result, the symmetric adsorption structure on (001) shown in Fig. 1(a) is distorted, with a short hydrogen bond to the oxygen and a longer one to the fluorine (Fig. 2(a)). Only one hydrogen bond with the hydroxy group is formed on (101).
 | ||
| Fig. 2 Adsorption structures of H2O on partially hydroxylated MgF2 surfaces obtained using B3LYP. Fluorine atoms are represented as green, magnesium as black and hydrogen as red and oxygen as blue spheres. Surface atoms are highlighted. | ||
The effect of hydroxylation on the adsorption structure of H2O is prominent on surface (100). In contrast to the structure in Fig. 1(b), molecules of a full H2O layer on the hydroxylated (100) coordinate to the hydrogen of the surface hydroxyls instead of the coordinatively unsaturated cations, while forming at the same time a hydrogen bond with the surface fluorine. At half coverage, H2O molecules coordinate to the cation as on clean surfaces, but form two hydrogen bonds with the surface hydroxyls as shown in Fig. 2(d).
The orientation of H2O at full coverage on surface (110) does not change when the surface is hydroxylated. However, of the two hydroxylation patterns investigated (Fig. 2(e) and (f)), the latter demonstrates two possible adsorption patterns of H2O at half coverage, presented in Fig. 2(g) and (h). One of them is the orientation of H2O in a manner analogous to the full layer of H2O. The other adsorption structure is similar to the one observed on surface (100) at half coverage, with the oxygen-end of H2O coordinating to the hydroxyl on the surface instead of the magnesium, while the hydrogens coordinate to the two surface fluorines. The structure shown in Fig. 2(h) is the only one, for which the Mulliken analysis shows a population shift from the molecule to the surface. We have observed a population decrease of 0.14 for 50%, (110)-OHb (ii), whereas an increase between 0.02−0.30 for all other structures of H2O adsorbed on partially hydroxylated MgF2 surfaces. The surface structures remain – as in the case of clean surfaces – largely unaffected by the presence of the adsorbates. Only the surface hydroxyls are shifted towards the H2O molecules. The results presented in the following sections for the adsorption energies refer to these structures, which were obtained from the B3LYP optimization.
3.2 Comparison of methods
In the following, adsorption energies are reported as negative values to illustrate the stabilizing nature of the interaction. The comparison of adsorption energies refers to the absolute values of the energies, so that an energy value of e.g. −0.50 eV is regarded as larger than the one of −0.30 eV.We started by calculating the adsorption energy of HF and H2O on three surfaces at full coverage, with the structures optimized using B3LYP. These results were combined with D2-, D3- and D*-corrections. For the structures obtained using B3LYP, the adsorption energy was also calculated at the Hartree–Fock and LMP2 level. LMP2 has been shown to yield excellent results for adsorption systems on MgF2 in comparison to CCSD(T) results,28 hence it is used in this study as a reference for the performance of DFT. The comparison of the results, shown in Table 1, reveals qualitative differences in the description of HF and H2O adsorption.
| Method | E ads(HF) [eV] | E ads(H2O) [eV] | ||||
|---|---|---|---|---|---|---|
| (100) | (101) | (110) | (100) | (101) | (110) | |
| Hartree–Fock | −0.54 | −0.72 | −0.78 | −0.62 | −0.68 | −0.78 |
| B3LYP | −0.67 | −0.83 | −0.89 | −0.67 | −0.74 | −0.83 |
| B3LYP-D2 | −0.97 | −1.11 | −1.21 | −1.03 | −1.09 | −1.20 |
| B3LYP-D3 | −0.90 | −1.04 | −1.13 | −0.98 | −1.06 | −1.16 |
| B3LYP-D* | −0.90 | −1.03 | −1.12 | −0.93 | −0.99 | −1.06 |
| LMP2 | −0.67 | −0.81 | −0.89 | −0.74 | −0.80 | −0.90 |
| LMP2-d.b. | −0.70 | −0.85 | −0.92 | −0.75 | −0.83 | −0.92 |
The discrepancy between B3LYP and LMP2 is less than 3% for adsorption energies of HF, yielding a 10–20% stronger adsorption than Hartree–Fock. Dispersion corrections overestimate the adsorption energy by up to 30% compared to LMP2, with D3 and D* performing somewhat better than D2.
Electron correlation effects are not described as well using B3LYP in the case of H2O adsorption. Here, LMP2 predicts up to 10% stronger adsorption than B3LYP, adding up to 17% to Hartree–Fock results. D* performs better than D3, unlike the case of HF adsorption where there is nearly no difference between the two corrections. Dispersion corrected B3LYP again overestimates the adsorption energies. The use of dual basis set for better description of the virtual space (LMP2-d.b in Table 1) adds less than 5% to the LMP2 results.
As a complementary test for the performance of functionals and dispersion corrections, we calculated the adsorption energy of H2O on (110) at full coverage using PBE and B3PW, for the previously optimized structure. The results are presented in Table 2. As indicated by the difference of only 1% between B3LYP and B3PW, the adsorption energy is not sensitive to the different correlation description in the two functionals. The calculation using PBE, on the other hand, results in an adsorption energy higher than the two hybrid functionals and considerably closer to LMP2. The D2 and D3 corrections on PBE add a contribution of 61% and 48% to the uncorrected result, respectively, overestimating in both cases the adsorption energy. A D2 parametrization is not available for B3PW, but the D3 correction adds a dispersion contribution of −690 meV, which corresponds to 84% of the uncorrected adsorption energy and a 60% overestimation compared to the LMP2 energy.
| Functional | DFT | DFT-D2 | DFT-D3 |
|---|---|---|---|
| B3LYP | −0.83 | −1.20 | −1.17 |
| B3PW | −0.82 | — | −1.51 |
| PBE | −0.87 | −1.40 | −1.29 |
4 Results
4.1 Adsorption of HF
It was shown in the previous section that the adsorption of HF on MgF2 surfaces is described satisfactorily at the B3LYP level. We present in Table 3 the complete series of adsorption energies for HF on surfaces (001), (100), (101) and (110). We have calculated adsorption structures for three coverages, each optimized using B3LYP, to examine the influence of HF–HF interaction on the adsorption patterns and energies. Changes in coverage do not affect the molecule's arrangement on the adsorption sites.| Coverage (%) | (001) | (100) | (101) | (110) |
|---|---|---|---|---|
| 100 | −0.58 | −0.67 | −0.83 | −0.89 |
| 50 | −0.60 | −0.69 | −0.88 | −0.86 |
| 25 | −0.59 | −0.63 | −0.86 | −0.83 |
On the other hand, the energy differences between coverages vary from 3% to 10% depending on the arrangement of the adsorption sites on the surface. Adsorption energies on surface (001) are least affected, presumably due to the square unit cell, which favours the uniform placement of adsorbates on the surface (Fig. 1(e)). The unit cell of surface (100), on the other hand, is oblong, resulting in a considerably denser placement of adsorption sites in one periodic direction than the other. This offers an explanation for the behaviour of the adsorption energy with decreasing coverage on (100). Full coverage is shown in Fig. 1(f). Half coverage is achieved by the removal of every other HF in the direction of the narrow side of the cell, eliminating the destabilizing interaction between parallel HF molecules. At 25% coverage, the neighbouring molecules in the other direction are also removed. The resulting adsorption energy is lower than that at half coverage, indicating a stabilizing effect by the HF molecules in a row. A similar trend occurs for surface (101), where an increase of 6% in the absolute value of the adsorption energy is observed upon decreasing the coverage from 100% to 50%. For 25% coverage, the adsorption of HF on (101) is slightly lower than that at 50%. In contrast to the other surfaces the strongest adsorption of HF on (110) is observed at 100%, decreasing with falling coverage.
4.2 Adsorption of H2O
The preliminary calculations in Section 3.2 indicated that B3LYP, even combined with dispersion correction, does not adequately describe the adsorption of H2O. For that reason we performed additional Hartree-Fock and LMP2 calculations to explicitly include correlation effects for the adsorption of H2O on further clean and partially hydroxylated MgF2 surfaces.The adsorption energies of H2O on clean MgF2 surfaces are listed in Table 4. All three methods capture the same trends in the adsorption energy for the variation of termination and coverage. The strongest adsorption is observed on surface (001), followed by (110) and (101), while the weakest adsorption occurs on (100). Variations in coverage barely influence the adsorption energy on (001), whereas on (100) and (110) the adsorption becomes stronger with lowering coverage. H2O adsorption on (101) is the weakest for half coverage, followed by 100%, while the strongest adsorption is calculated for 25% coverage.
| Surface | Coverage (%) | B3LYP | B3LYP + D2 | B3LYP + D3 | Hartree–Fock | LMP2 |
|---|---|---|---|---|---|---|
| (001) | 100 | −1.01 | −1.30 | −1.28 | −0.92 | −1.01 |
| 50 | −1.01 | −1.28 | −1.26 | −0.93 | −1.03 | |
| 25 | −1.00 | −1.27 | −1.25 | −0.93 | — | |
| (100) | 100 | −0.67 | −1.03 | −0.98 | −0.62 | −0.74 |
| 50 | −0.73 | −1.03 | −1.01 | −0.68 | −0.80 | |
| 25 | −0.72 | −1.01 | −0.99 | −0.70 | — | |
| (101) | 100 | −0.75 | −1.09 | −1.06 | −0.68 | −0.80 |
| 50 | −0.53 | −0.74 | −0.74 | −0.49 | −0.57 | |
| 25 | −0.85 | −1.11 | −1.10 | −0.79 | −0.89 | |
| (110) | 100 | −0.83 | −1.20 | −1.16 | −0.78 | −0.90 |
| 50 | −0.89 | −1.20 | −1.17 | −0.85 | −0.96 | |
| 25 | −0.89 | −1.20 | −1.17 | −0.88 | — | |
Generally, LMP2 adds 10–17% to the Hartree–Fock results. Depending on the structure, B3LYP underestimates the adsorption energy by up to 10% – giving similar energies to Hartree–Fock, while D2 and D3 corrections overestimate the adsorption energy on clean surfaces by up to 30% for all terminations and coverages, compared to LMP2 calculations. The invariance of the dispersion energy with respect to coverage is a strong indication that adsorbate–adsorbate interactions have little or no contribution in the dispersion correction.
Turning to hydroxylated surfaces, we observe for the first time a discrepancy in the description of correlation with B3LYP and LMP2 (Table 5). Hartree–Fock results are in most cases approximately 20% lower than LMP2. The performance of B3LYP is in general satisfactory, giving in some cases the same results as LMP2 but ranging up to a 10% deviation. However, there are a few notable exceptions, which need to be discussed in detail.
| Surface | Coverage (%) | B3LYP | B3LYP + D2 | B3LYP + D3 | Hartree–Fock | LMP2 |
|---|---|---|---|---|---|---|
| (001)-OH | 100 | −1.15 | −1.50 | −1.45 | −0.67 | −0.83 |
| 50 | −0.93 | −1.27 | −1.24 | −0.78 | −0.94 | |
| (100)-OH | 100 | −0.32 | −0.55 | −0.56 | −0.12 | −0.55 |
| 50 | −0.77 | −1.19 | −1.14 | −0.56 | −0.85 | |
| (101)-OH | 100 | — | — | — | — | — |
| 50 | −0.95 | −1.26 | −1.25 | −0.80 | −0.98 | |
| (110)-OHa | 100 | −0.89 | −1.31 | −1.25 | −0.78 | −0.99 |
| (110)-OHb | 100 | −0.73 | −1.11 | −1.07 | −0.66 | −0.81 |
| 50 (i) | −0.78 | −1.11 | −1.08 | −0.69 | −0.84 | |
| 50 (ii) | −0.52 | −0.75 | −0.74 | −0.35 | −0.52 | |
The first case concerns the adsorption of H2O at full coverage on (001)-OH. The B3LYP adsorption energy is remarkably higher than the LMP2 energy. While the Hartree–Fock adsorption energy is 19% lower than LMP2, calculations using B3LYP give an adsorption energy higher by 39% than LMP2. This behaviour is not observed at half coverage on the same surface, where B3LYP and LMP2 result in almost the same adsorption energy. It resembles, however, the previously discussed case of the non-hydroxylated (001), where B3LYP and LMP2 give the same adsorption energy. The small contribution of electron correlation to the adsorption of H2O on (001)-OH shows that the binding is dominated by Pauli repulsion and H-bonds instead of dispersive contributions. As a consequence, the standard B3LYP functional with 30% exchange contribution underestimates the Pauli repulsion and yields therefore overbinding.
Surface (100)-OH stands out as well: the adsorption energy calculated using LMP2 is more than three times higher than that calculated using Hartree–Fock for full coverage and 52% higher for half coverage. B3LYP also fails to describe the adsorption at full coverage, showing a deviation of 42% from the LMP2 energy, but shows the expected performance for half coverage. The failure of Hartree–Fock can be understood by looking at the structures in Fig. 2(c) and (d). The contribution of dispersion on the adsorption structure at 100% is expected to be large, since it is entirely due to hydrogen bonding, not only between surfaces and adsorbates but also among H2O molecules forming chains. The adsorbate–adsorbate stabilization at full coverage on (100)-OH is essential. This is illustrated by the fact that in the absence of this stabilization at half coverage, the adsorption structure is completely different. Adsorption at half coverage includes a large contribution from hydrogen bonds between H2O and surface hydroxyls, but no obvious adsorbate–adsorbate interaction. As a result, dispersion effects have a smaller contribution to the adsorption, so B3LYP approaches the performance of MP2.
An adsorption structure entirely based on hydrogen bonds between surfaces and adsorbates is (ii) on (110)-OHb, shown in Fig. 2(h). In line with the observations on (100)-OH, to which the structure shows a great similarity, we find that Hartree–Fock fails to describe the interactions adequately, giving an adsorption energy 32% lower than LMP2. In this case B3LYP gives the same adsorption energy as LMP2 as expected, considering the absence of significant adsorbate–adsorbate interactions.
4.3 Reactivity of the surfaces
The adsorption energy of probe molecules on a surface can be an indicator of the reactivity of a surface. Another indicator is the surface energy: an unstable surface is expected to be more reactive. The vibrational frequency shift of CO is also used both in theory and in experiments as a measure for Lewis acidity. In Table 6, we have summarized these quantities for the clean MgF2 surfaces. To minimize the contribution of adsorbate–adsorbate interaction we have used the values for 25% coverage. Comparing the data in Table 6, the lack of a consistent trend is striking. Similarities could be identified when comparing individual series, e.g. Eads(HF) and Δν(CO), or Eads(H2O) and Eads(CO), however, a generalization based on these similarities should be avoided. The adsorption structure and strength appear to be specific to the combinations of surface termination, adsorbates and coverage. This type of behaviour is desired for the catalytic application of sol–gel synthesized MgF2. It supports the hypothesis that crystallites of different shapes have different catalytic properties and opens up the way towards tailor-made selective catalysts.5 Conclusions
We have investigated in this work the adsorption of HF and H2O on clean and partially hydroxylated MgF2 surfaces. We have employed LMP2, Hartree–Fock and B3LYP to calculate adsorption energies for these systems. LMP2 was tested in previous work28 and showed performance as good as CCSD(T). Based on its reliability, we have used LMP2 in this work as a measure for the performance of B3LYP and post-optimization dispersion corrections (D2 and D3). D2 and D3 have in all cases overestimated the adsorption energies compared to LMP2.The description of HF adsorption is rather uncomplicated, so that the use of B3LYP is adequate for a good description. In the case of H2O, however, B3LYP underestimates the adsorption energies. The description of H2O adsorption on hydroxylated surfaces is even more demanding due to the variety of factors playing a role in the interactions. Here, B3LYP fails to catch the trends in the adsorption energies, because it does not describe dispersion effects well. As a consequence, particularly in cases of strong adsorbate–adsorbate interactions or hydrogen bonded structures B3LYP underestimates the adsorption energy. The use of LMP2 is essential for reliable calculations on H2O adsorption.
The comparison of adsorption energies and structures of different probe molecules on the surfaces and terminations studied reveals a selectivity in the surface activity of MgF2. These results on the adsorption behaviour provide evidence that support the potential application of MgF2-based materials in catalysis.
Acknowledgements
We gratefully acknowledge Lukas Hammerschmidt for his contribution to this work and the fruitful discussions. Also, we thank Deutsche Forschungsgemeinschaft (DFG) for the financial support of this study through the graduate school ‘Fluorine as a key element’, and the ZEDAT at the Freie Universität Berlin as well as HLRN (North-German Supercomputing Alliance) for the computational time.References
- E. Kemnitz, S. Wuttke and S. M. Coman, Eur. J. Inorg. Chem., 2011, 4773–4794 CrossRef CAS PubMed.
- H. A. Prescott, Z.-J. Li, E. Kemnitz, J. Deutsch and H. Lieske, J. Mater. Chem., 2005, 15, 4616–4628 RSC.
- G. Scholz, D. Heidemann and E. Kemnitz, Z. Anorg. Allg. Chem., 2013, 639, 694–701 CrossRef CAS PubMed.
- E. Kanaki, S. Gohr, C. Müller and B. Paulus, Surf. Sci., 2015, 632, 158–163 CrossRef CAS PubMed.
- Z. Huesges, C. Müller, B. Paulus, C. Hough, N. Harrison and E. Kemnitz, Surf. Sci., 2013, 609, 73–77 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- R. Dovesi, R. Orlando, B. Civalleri, C. Roetti, V. R. Saunders and C. M. Zicovich-Wilson, Z. Kristallogr., 2005, 220, 571–573 CrossRef CAS.
- R. Dovesi, V. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. Harrison, I. Bush, P. D'Arco and M. Llunell, CRYSTAL09 User's Manual, University of Torino, Torino, 2010 Search PubMed.
- L. Valenzano, Y. Noël, R. Orlando, C. Zicovich-Wilson, M. Ferrero and R. Dovesi, Theor. Chem. Acc., 2007, 117, 991–1000 CrossRef CAS.
- M. Catti, A. Pavese, R. Dovesi, C. Roetti and M. Causà, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 3509–3517 CrossRef CAS.
- C. L. Bailey, S. Mukhopadhyay, A. Wander, B. G. Searle and N. M. Harrison, J. Phys. Chem. C, 2009, 113, 4976–4983 CAS.
- J. Haines, J. M. Léger, F. Gorelli, D. D. Klug, J. S. Tse and Z. Q. Li, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 134110 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 123, 154101 CrossRef PubMed.
- B. Civalleri, C. M. Zicovich-Wilson, L. Valenzano and P. Ugliengo, CrystEngComm, 2008, 10, 405–410 RSC.
- B. Civalleri, L. Maschio, P. Ugliengo and C. M. Zicovich-Wilson, Phys. Chem. Chem. Phys., 2010, 12, 6382–6386 RSC.
- C. Pisani, M. Schütz, S. Casassa, D. Usvyat, L. Maschio, M. Lorenz and A. Erba, Phys. Chem. Chem. Phys., 2012, 14, 7615–7628 RSC.
- A. Erba and M. Halo, CRYSCOR09 User's Manual, Università degli Studi di Torino, Torino - Italy, 2009 Search PubMed.
- L. Maschio and D. Usvyat, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 73102 CrossRef.
- M. Schütz, D. Usvyat, M. Lorenz, C. Pisani, L. Maschio, S. Casassa and M. Halo, Accurate Condensed-Phase Quantum Chemistry, CRC press, 2010, ch. 2, pp. 29–56 Search PubMed.
- W. Liang and M. Head-Gordon, J. Phys. Chem. A, 2004, 108, 3206–3210 CrossRef CAS.
- D. Usvyat, L. Maschio, C. Pisani and M. Schütz, Z. Phys. Chem., 2010, 224, 441–454 CrossRef CAS.
- S. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS PubMed.
- L. Hammerschmidt, C. Müller and B. Paulus, J. Chem. Phys., 2012, 136, 124117 CrossRef PubMed.
| This journal is © the Owner Societies 2015 |