
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparing the electronic relaxation dynamics of aniline and d7-aniline following excitation at 272–238 nm†
Oliver M.
Kirkby
a,
Matthieu
Sala
b,
Garikoitz
Balerdi
c,
Rebeca
de Nalda
d,
Luis
Bañares
c,
Stéphane
Guérin
b and
Helen H.
Fielding
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: h.h.fielding@ucl.ac.uk
bLaboratoire Interdisciplinaire Carnot de Bourgogne UMR 6303 CNRS, Université Bourgogne Franche-Comté, 9 Av. A. Savary, BP 47870, F-21078 Dijon Cedex, France
cDepartamento de Química Física I (Unidad Asociada I + D + i al CSIC), Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain
dInstituto de Química Física Rocasolano, CSIC, C/Serrano 119, 28006 Madrid, Spain
First published on 14th May 2015
Abstract
Femtosecond time-resolved photoelectron spectroscopy experiments have been used to compare the electronic relaxation dynamics of aniline and d7-aniline following photoexcitation in the range 272–238 nm. Together with the results of recent theoretical investigations of the potential energy landscape [M. Sala, O. M. Kirkby, S. Guérin and H. H. Fielding, Phys. Chem. Chem. Phys., 2014, 16, 3122], these experiments allow us to resolve a number of unanswered questions surrounding the nonradiative relaxation mechanism. We find that tunnelling does not play a role in the electronic relaxation dynamics, which is surprising given that tunnelling plays an important role in the electronic relaxation of isoelectronic phenol and in pyrrole. We confirm the existence of two time constants associated with dynamics on the 11πσ* surface that we attribute to relaxation through a conical intersection between the 11πσ* and 11ππ* states and motion on the 11πσ* surface. We also present what we believe is the first report of an experimental signature of a 3-state conical intersection involving the 21ππ*, 11πσ* and 11ππ* states.
1 Introduction
There is continuing interest in the role of 1πσ* states in the photochemistry of small aromatic molecules containing OH and NH groups.1,2 These states are characterised by dissociative potential energy curves along O–H or N–H stretching coordinates and have been shown to provide efficient electronic relaxation pathways to conical intersections with the electronic ground state. Consequently, these dissociative states play an important role in protecting biological molecules from harmful photochemical reactions.3–5Aniline (C6H5NH2) is a structural motif found in the purine nucleotides, adenine and guanine, and in the pyrimidine nucleotide, cytosine. The UV absorption spectrum of aniline is dominated by two bands centered around 282 nm and 230 nm, corresponding to π* ← π transitions to the first two 1ππ* states, labelled 11ππ* and 21ππ* (Fig. 1).6–11 The first 1πσ* state, labelled 11πσ*, lies between the 11ππ* and 21ππ* states.7 It is composed of N-centered π3s and πσ* configurations, arising from 3s ← π and σ* ← π transitions, respectively.12,13 The 11πσ* state has π3s character in the Franck–Condon region but becomes dissociative along the N–H stretch coordinate and forms conical intersections with the 11ππ* state and ground electronic state at modest N–H bond-lengths. It has also been proposed that the two 3p Rydberg states may lie between the 11ππ* and 21ππ* states.14,15
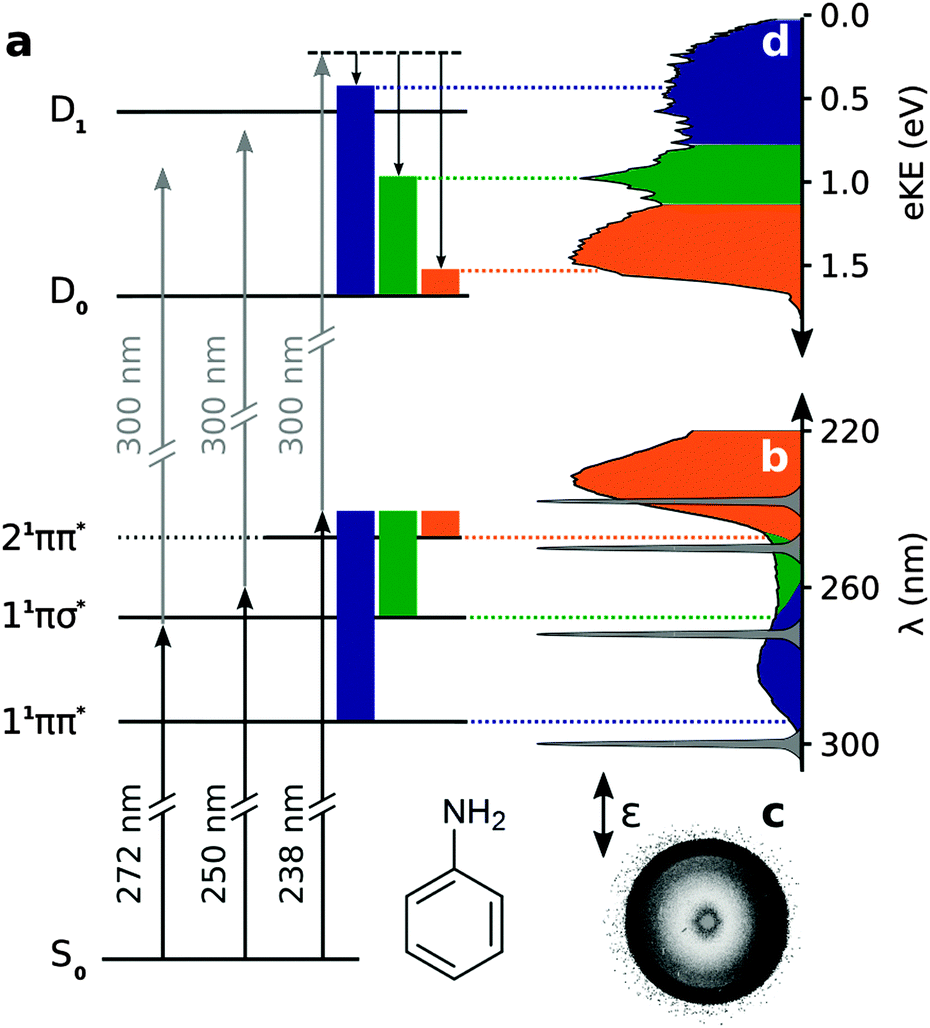 | ||
| Fig. 1 (a) Schematic excitation scheme in aniline. Solid coloured blocks represent vibrational energy in the excited electronic states of aniline and the equivalent vibrational energy in the ground electronic state of the cation, illustrating the propensity for Δv = 0 upon photoionisation. (b) Gas-phase absorption spectrum of aniline and spectral profiles of pump and probe laser pulses. Representative photoelectron image (c) and spectrum (d) of aniline collected with pump–probe delay t = −13 fs, using a 238 nm pump and 300 nm probe. The double-headed arrow shows the direction of the electric field vector of the pump and probe photons. | ||
There have been a number of recent experimental studies of the photochemistry and photophysics of isolated aniline molecules in vacuo using H-atom (Rydberg) photofragment translational spectroscopy,16 femtosecond pump–probe photoionisation spectroscopy,17 femtosecond pump–probe velocity map imaging,18 and time-resolved photoelectron imaging spectroscopy.19–22 There have also been a number of recent theoretical investigations of the potential energy landscape and relaxation pathways following photoexcitation to the first few singlet excited states.14,18,23 However, there is not yet a consensus on the electronic relaxation mechanism.
Questions that remain are: (1) Is there evidence to support the suggestion that following excitation close to the 11πσ* origin, tunnelling occurs through the barrier on the 11πσ* potential surface?16,18,21 (2) Following excitation above the 11πσ* origin, is the relaxation pathway 11πσ* → 11ππ*19,20 or 11ππ* → 11πσ*?16–18 (3) Is there evidence to support the suggestion that non-radiative decay from the 21ππ* state to the ground-state passes through a three-state conical intersection involving the 21ππ*, 11πσ* and 11ππ* states23 or a 21ππ*/11ππ* conical intersection18 or involves the 3p Rydberg states?14
These questions motivated us to revisit the nonradiative relaxation dynamics of aniline and, in this paper, we present the results of new femtosecond time-resolved photoelectron spectroscopy experiments comparing the relaxation dynamics of aniline and deuterated aniline (d7-aniline) following photoexcitation in the range 272–238 nm, from just below the 11πσ* origin up to the 21ππ* state.
2 Methods
The experimental setup has been described elsewhere.19,20,24–26 Briefly, aniline (Sigma-Aldrich, >99%) and d7-aniline (98%, Sigma-Aldrich) were introduced into the velocity map imaging spectrometer by passing 800 mbar helium carrier gas through the liquid samples and expanding through a 50 μm nozzle. After collimation by a 1 mm skimmer, the molecular beam was intersected by femtosecond pump (272–238 nm) and probe (300 nm) laser pulses generated by frequency upconverting the outputs of two optical parametric amplifiers pumped by a commercial amplified Ti:sapphire femtosecond laser system. Pump–probe cross-correlation full-width half-maximum (FWHM) measurements were in the range 180–225 fs. The pump and probe fluxes were attenuated to ∼1 μJ per pulse to minimise multiphoton processes and to keep the photoelectron count-rates below 20 photoelectrons per pulse, thus avoiding space-charge effects. The probe wavelength was selected to access as much of the ionisation continuum as possible whilst keeping 11ππ* ← S0 absorption to a minimum (Fig. 1). Multiphoton ionisation time-of-flight mass spectra were recorded to ensure minimal cluster formation and fragmentation of the parent molecules before collecting photoelectron images.For each excitation wavelength, a set of approximately 18 photoelectron images with pump–probe delays in the range −0.2 ps to 1 ps were recorded, together with the total integrated photoelectron signal for pump–probe delays from −0.5 ps to 100 ps (272 nm) and −0.5 ps to 10 ps (250 nm and 238 nm). Photoelectron velocity distributions were recovered from the raw photoelectron images using the pBasex image inversion algorithm27 and the energy scale was calibrated by recording the 2 + 1 resonance-enhanced multiphoton ionisation spectrum of Xe at 249.6 nm.28 The resolution was ∼3.5%.
To extract decay times from a set of time-resolved photoelectron spectra, the total integrated areas of the photoelectron spectra recorded at each pump–probe delay were scaled to the total integrated photoelectron signal intensity and then integrated portions of the set of spectra were fit to sums of exponentially decaying profiles convoluted with a Gaussian cross-correlation function representing the cross-correlation of the pump and probe laser pulses g(t),19,20
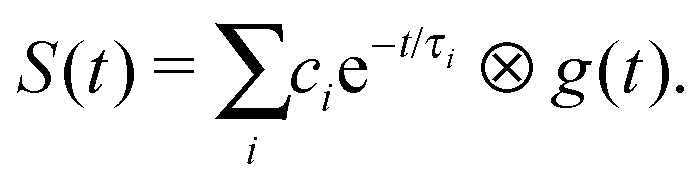 | (1) |
Compared with our previous work,19,20 our data analysis software has been rewritten and is now based on a least-squares fit using the Levenberg–Marquardt optimisation algorithm. We also increased the number of images collected for pump–probe delays between ±100 fs to improve confidence in our determination of t = 0 and the cross-correlation from our fitting procedure.
3 Results
Fig. 1(d) shows a representative photoelectron spectrum recorded at −13 fs pump–probe delay following photoexcitation at 238 nm in aniline. The propensity for vibrational energy to be conserved during photoionisation allows us to assign the asymmetric feature at high eKE to ionisation from the 21ππ* state and the symmetric feature at low eKE to ionisation from the 11ππ* state (Fig. 1).19,20 This Δv = 0 propensity also allows us to assign the narrow peak at 1 eV to ionisation from the 11πσ* state and its anisotropic photoelectron angular distribution (maximum β ≈ 0.8) is consistent with ionisation from a π3s configuration.Time-resolved photoelecron spectra are shown in Fig. 2. We do not observe any variation in anisotropy parameter with pump–probe delay for aniline or d7-aniline, in agreement with all recent time-resolved photoelectron imaging experiments with aniline.19–22 For each photoexcitation wavelength, the integrated areas of the photoelectron spectra have been scaled to the total integrated photoelectron signals at the corresponding pump–probe delays and plotted as a contour map. Notably, the time-resolved photoelectron spectra are very similar for aniline and d7-aniline. The symmetric feature at low eKE, corresponding to ionisation from the 11ππ* state, is observed in all the contour maps and appears to have a lifetime >1 ps. The sharp feature around 1 eV, corresponding to ionisation from the 11πσ* state, is observed in the 250 nm and 238 nm contour maps and appears to have a lifetime ≳100 fs. The asymmetric feature corresponding to ionisation from the 21ππ* state is observed in the 238 nm contour maps and appears to decay very rapidly, on a timescale <100 fs.
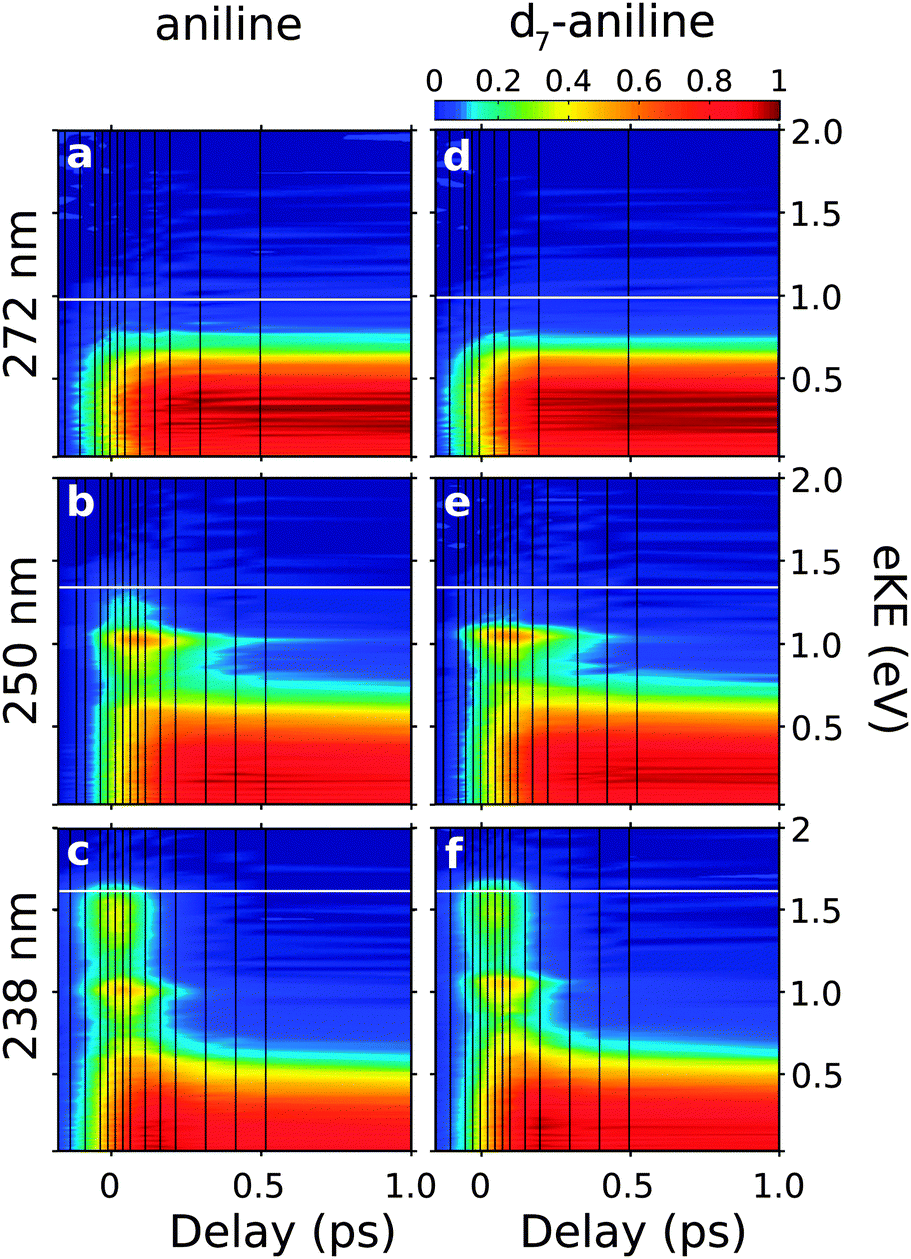 | ||
| Fig. 2 Contour plots showing experimental time-resolved photoelectron spectra for aniline (a–c) and d7-aniline (d–f), following excitation just below the 11πσ* origin at 272 nm (4.56 eV), above the 11πσ* origin at 250 nm (4.96 eV) and to the 21ππ* state at 238 nm (5.21 eV). Individual plots were normalised to their maximum photoelectron signals. The shading was smoothed using bilinear interpolation and the vertical black lines mark the pump–probe delays at which photoelectron spectra were recorded. Horizontal white lines mark the maximum eKE possible from 1 + 1′ ionisation, calculated using the central wavelengths of the pump and probe laser pulses and the adiabatic ionisation potential of 7.72 eV.29 | ||
The observed dynamics were fit to eqn (1) and the lifetimes obtained are presented in Table 1. Only one lifetime was required to fit the 272 nm data sets, three lifetimes were required to fit the 250 nm data sets and four lifetimes were required to fit the 238 nm data sets.
The time-resolved photoelectron spectra were then fit to sums of the exponential decays listed in Table 1 convoluted with g(t),
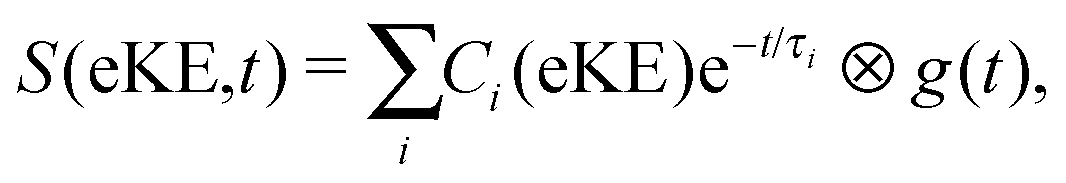 | (2) |
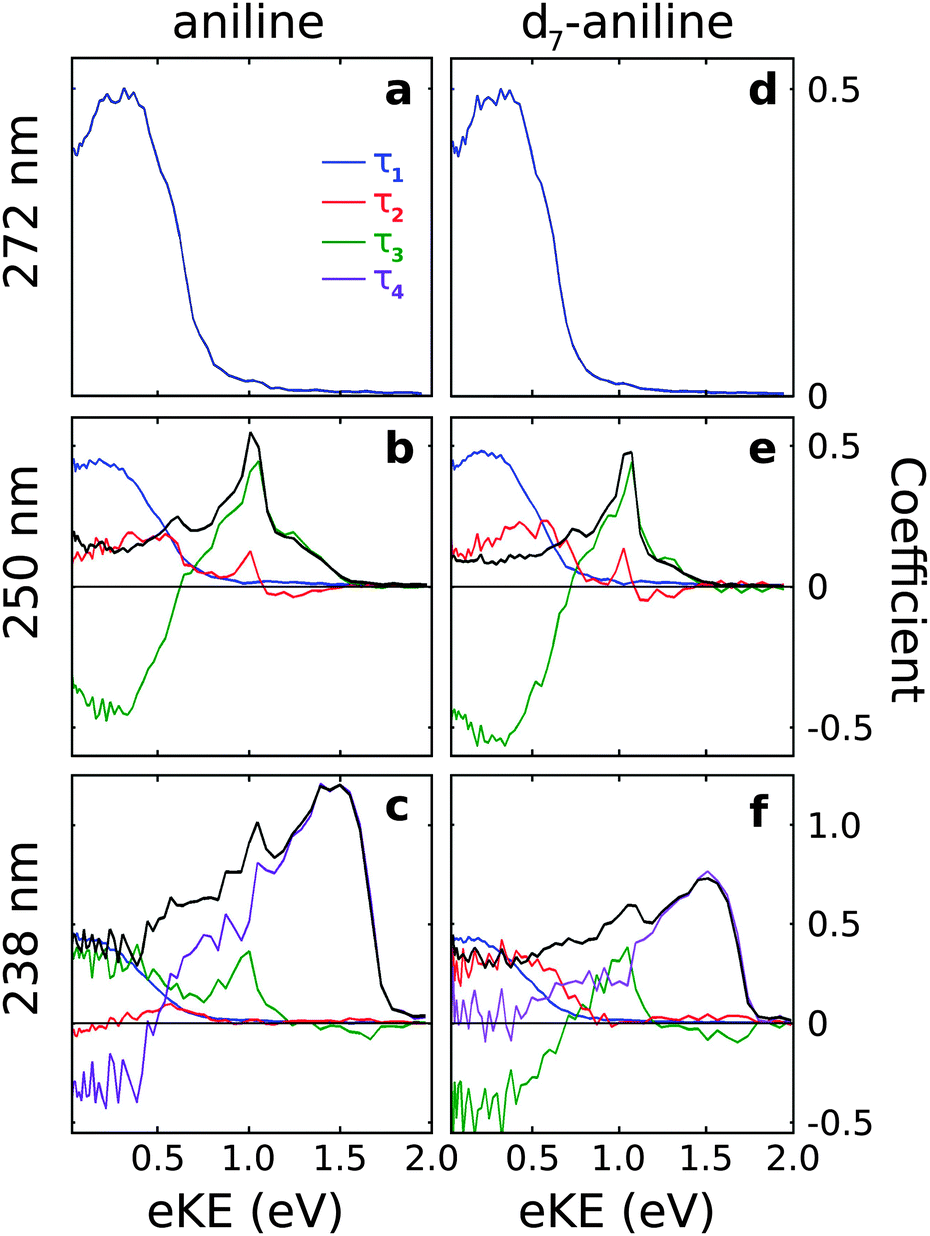 | ||
| Fig. 3 Spectral components of Ci(eKE) extracted from the time-resolved photoelectron spectra (Fig. 2) using the decay times listed in Table 1 and eqn (2). The sum of Ci(eKE) (black lines) represents the photoelectron spectrum of the initially excited state. | ||
For all photoexcitation energies, the spectra associated with the τ1 lifetime are centered around 0.2 eV with positive amplitude components, indicating a decay of population from the 11ππ* state. Following photoexcitation at 250 nm, the spectra associated with the τ2 lifetime extends from 0 eV to ≳1 eV with positive amplitude components, indicating a decay of population on a timescale τ2. In contrast, the spectra obtained for the τ3 lifetime have sharp features around 1 eV with positive amplitude components and broad features centered around 0.2 eV with negative amplitude components. This suggests an evolution along the excited potential energy surface from the 11πσ* state to the 11ππ* state with timescale τ3.
Following photoexcitation of d7-aniline at 238 nm, the spectra associated with the τ2 and τ3 lifetimes are similar to those observed at 250 nm, although the spectrum associated with the τ2 timescale no longer has significant amplitude at 1 eV. The spectrum associated with the τ4 timescale has a broad, asymmetric feature at high eKE with positive amplitude components, indicating a decay of population from the 21ππ* state with timescale τ4. The absence of negative amplitude components prevents us identifying the fate of this population but it does not rule out the possibility of energy flow into 11πσ* or 11ππ* states because the photoelectron spectra of these components overlap and the decay associated spectra are the sum of positive and negative amplitude components. In contrast, for aniline, the spectrum associated with the τ2 timescale is much less intense than that for d7-aniline, the spectrum associated with τ3 only has positive amplitude components, both at low eKE and around 1 eV, and the spectrum associated with the τ4 timescale has positive amplitude components at high eKE and negative amplitude components at low eKE, suggesting an evolution on the excited potential energy surface from the 21ππ* state into the 11ππ* state.
4 Discussion
To assist with the interpretation of the photoelectron spectra, we begin with a summary of the results of our recent theoretical investigation of the relaxation pathways following photoexcitation to the first few singlet excited states of aniline in Fig. 4.23 On the 11ππ* potential energy surface (blue lines in Fig. 4), we found a prefulvene-like minimum energy conical intersection (MECI) connecting the 11ππ* state with the ground state in which the carbon-atom carrying the amino group is distorted out-of-plane. On the 11πσ* potential energy surface (green lines in Fig. 4), we found a MECI connecting the 11πσ* state and the 11ππ* state close to the local minimum on the 11πσ* surface, suggesting that population on the 11πσ* surface could relax through this MECI to the 11ππ* state and to the dissociative component of the 11πσ* state. On the 21ππ* state (orange lines in Fig. 4) we found evidence for a barrierless pathway from the Franck–Condon geometry to the ground state through a three-state conical intersection involving the 21ππ*, 11ππ* and 11πσ* states.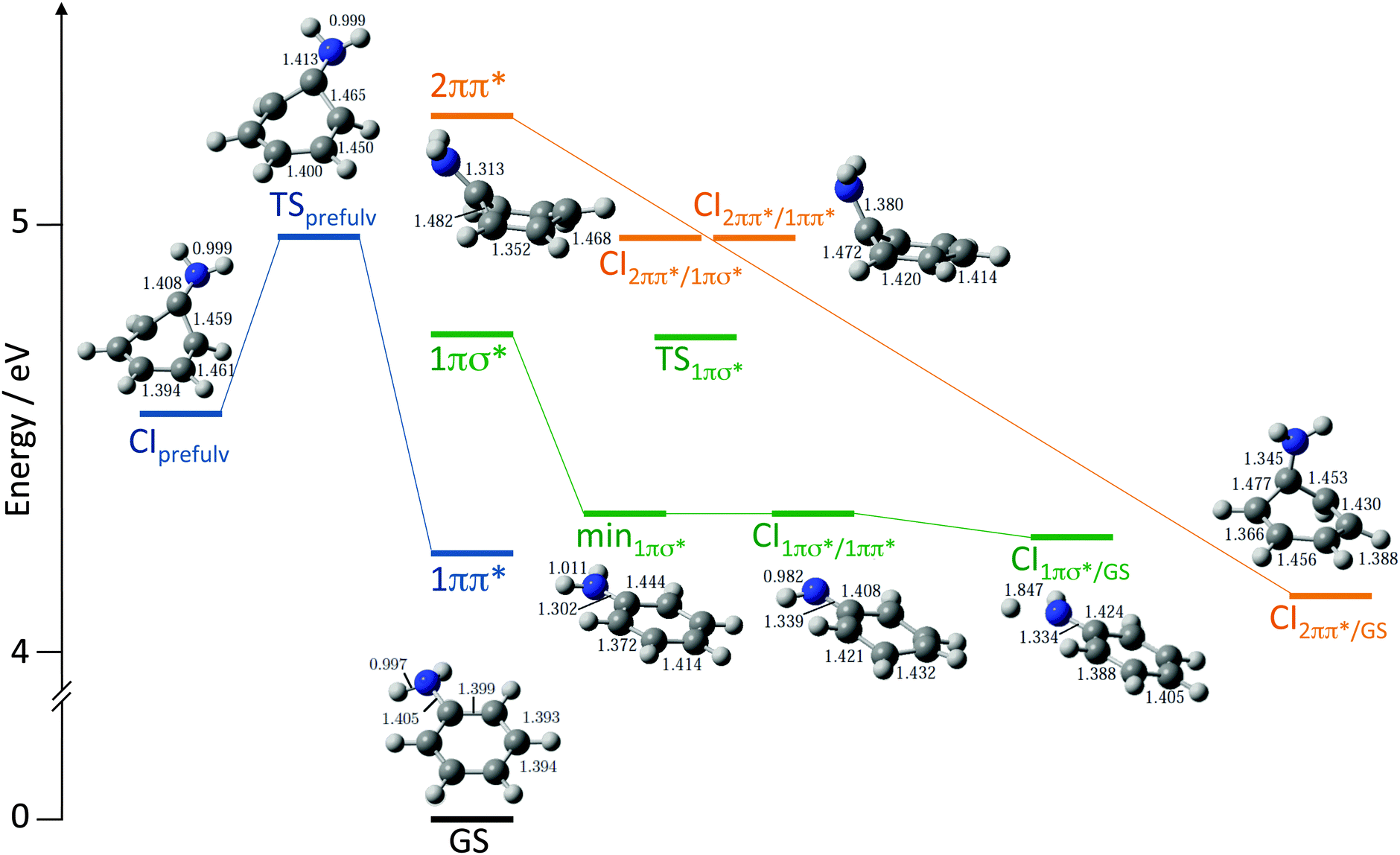 | ||
| Fig. 4 Schematic diagram illustrating key features on the potential energy landscape and relaxation pathways following photoexcitation to the first few singlet excited states of aniline.23 The vertical excitation energies of the 11ππ*, 11πσ* and 21ππ* levels, calculated at the XMCQDPT2 level of theory, are plotted vertically above the minimum energy of the ground electronic state (GS). XMCQDPT2 energies of key transition states (TS), minima (min) and conical intersections (CI) on the 11ππ* (blue), 11πσ* (green) and 21ππ* surfaces (orange) are presented together with their corresponding CASSCF geometries. | ||
The experiments presented in this paper confirm our earlier observation that four time constants are required to characterise the photochemistry of aniline following photoexcitation in the range 272–238 nm,19,20 which is consistent with the existence of at least four relaxation pathways.
The longest time constant τ1 corresponds to the decay of population from the 11ππ* state. It has been suggested that following excitation close to the 11πσ* origin, population on the 11ππ* state may tunnel through the barrier formed by the crossing between the 11ππ* and 11πσ* states onto the dissociative part of the 11πσ* surface,16,18,21 as has been observed in phenol.12,31–34 Tunnelling is sensitive to the mass of the particle involved and therefore the time constant for a relaxation mechanism involving tunnelling of one of the H-atoms of the amino group would be expected to be significantly longer for d7-aniline than aniline. However, τ1 is only a factor of two longer for d7-aniline (Table 1), which is not enough to be attributed to quantum mechanical tunnelling although it can be accounted for by differences in the vibration frequencies between aniline and d7-aniline. Thus, we do not find any evidence to support the suggestion that tunnelling occurs through the barrier formed by the crossing between the 11ππ* and 11πσ* states in aniline, which is surprising given that such tunnelling has been observed in isoelectronic phenol. We therefore interpret τ1 as the lifetime for intramolecular vibrational redistribution (IVR), intersystem crossing (ISC)11 or fluorescence10 following excitation close to the 11πσ* origin.
Following excitation with wavelengths ≲250 nm, the prefulvene MECI connecting the 11ππ* state with the ground state is accessible (Fig. 4). Thus, following excitation above the barrier between the Franck–Condon region and this MECI, it is likely that τ1 corresponds to the lifetime for IC through the prefulvene MECI. The decrease in lifetime with increase in photoexcitation energy is consistent with the presence of a barrier.
Curiously, whereas we only need one time constant to fit the aniline and d7-aniline time-resolved photoelectron spectra recorded following photoexcitation at 272 nm and photoionisation at 300 nm (Fig. 3a and d), Thompson and coworkers found that an additional time constant of 640 fs was required to fit the time-resolved photoelectron spectra of aniline they recorded following excitation at 273 nm and photoionisation at 305 nm.21 They attributed the additional time constant to IVR on the 11ππ* surface. We cannot explain the difference between the two experiments as it seems unlikely that an IVR promoting mode is accessible in the 11ππ* state at 273 nm photoexcitation in aniline but not at 272 nm in either aniline or d7-aniline.
The τ3 time constant corresponds to a decay of population from the 11πσ* state and an increase of population in the 11ππ* state, following photoexcitation at 250 nm (Fig. 3b and e). This supports our earlier proposal that following photoexcitation above the 11πσ* origin, the 11πσ* state is excited directly and relaxes through the MECI connecting the 11πσ* and 11ππ* states close to the local minimum on the 11πσ* surface (Fig. 4).19,20,23 The MECI funnels population to the 11ππ* surface and also to the dissociative component of the 11πσ* state, which has been monitored directly in femtosecond pump–probe velocity map imaging experiments.18 We do not observe any population flow from the 11ππ* state to the 11πσ* state, so it seems unlikely that direct excitation of the 11ππ* state is followed by relaxation through the MECI to the dissociative component of the 11πσ* state, as suggested by King and coworkers16 and Roberts and coworkers,18 although we cannot rule out the possibility that there is a small contribution from such a relaxation mechanism that is not revealed by our fitting procedure. We note that a very recent study of the dynamics of aniline reported by Thompson and coworkers supports our conclusions.22
In aniline, the τ4 time constant corresponds to a rapid (<50 fs) flow of population from the 21ππ* state to the 11ππ* state and there is a decay of population from both the 11πσ* and 11ππ* states with a τ3 time constant (Fig. 3c). In d7-aniline, the τ4 time constant corresponds to a rapid (<50 fs) decay of population from the 21ππ* state out of the observation window of our experiment, and a flow of population from the 11πσ* state to the 11ππ* state with a τ3 time constant (Fig. 3f). The rapid flow of population from the 21ππ* state following excitation at 238 nm is consistent with our proposal that population decays along a barrierless path to the ground-state, through a CI between the 21ππ* state and the ground-state.19,20 It is also worth noting that similar ring-puckering relaxation pathways have been observed in other simple aromatic molecules such as pyrrole,35 and the purine DNA bases adenine36,37 and guanine.38 Moreover, the boat conformation of the phenyl ring at the CI between the 21ππ* state and the ground-state (Fig. 4) resembles the Dewar form of benzene, which is known to be populated following photoexcitation of the 21ππ* state of benzene.39,40 However, our observation of population in the 11πσ* and 11ππ* states at short times is consistent with passage through a three-state CI involving the 21ππ*, 11ππ* and 11πσ* states. It is worth noting that it is unlikely that there is significant direct population of 11πσ* and 11ππ* states at 238 nm. Thus, our new experiments support our earlier proposal that following excitation of the 21ππ* state the majority of the excited state population is transferred directly back to the ground state,19,20 but it seems likely that some population is also transferred to the 11ππ* and 11πσ* states at a three-state CI involving the 21ππ*, 11ππ* and 11πσ* states. From the 11πσ* and 11ππ* states, the molecule can then relax directly to the Franck–Condon geometry from where it can then relax through the prefulvene MECI connecting the 11ππ* state with the ground state (τ1) or through the MECI connecting the 11πσ* and 11ππ* states close to the local minimum on the 11πσ* (τ3).
The τ2 time-constant has been another subject of debate. Its broad decay associated spectrum originally led us to assign it to dynamics on the 11πσ* surface, possibly trapping,19,20 and Thompson and coworkers to assign it to IVR on the 11ππ* surface.21 The values of τ2 (Table 1) are similar to timescales that have been attributed to ultrafast IVR processes on the 11ππ* surfaces of phenol, catechol, resorcinol, and hydroquinone.41 However, we do not observe any dynamics with a τ2 time-constant following excitation below the 11πσ* origin, so we believe this time-constant must correspond to dynamics on the 11πσ* surface. Interestingly, Domcke and coworkers also found two time constants associated with the 11πσ* surface of pyrrole which, like aniline, has a quasi-bound well close to the Franck–Condon region and a dissociative potential curve along the N–H stretching coordinate.42 Their wave packet calculations revealed time constants of ∼10 fs and several hundreds of femtoseconds, corresponding to direct decay over the barrier and tunnelling through the barrier, respectively. However, τ2 has similar values for aniline and d7-aniline following photoexcitation at 250 nm (Table 1) and similar values over a range of excitation energies (Table 1 and ref. 20), which rules out tunnelling. Thus, we still conclude that there are two time constants associated with dynamics on the 11πσ* surface in aniline: τ3 associated with flow of population through the CI between the 11πσ* and 11ππ* states and τ2 associated with motion on the 11πσ* surface, although we concede that IVR may be a better description of the dynamics with time constant τ2. Quantum dynamics calculations may help gain further insight into the dynamics on the 11πσ* surface.
5 Conclusions
New femtosecond time-resolved photoelectron spectroscopy experiments following photoexcitation of aniline and d7-aniline from below the 11πσ* origin to the 21ππ* state have resolved a number of unanswered questions surrounding various electronic relaxation mechanisms.(1) We did not find evidence to support the suggestion that following excitation close to the 11πσ* origin, tunnelling occurs through the barrier on the 11πσ* potential surface.16,18,21 We attribute the dynamics following excitation below the 11πσ* origin in aniline and d7-aniline to IVR on the 11ππ* surface, ISC or fluorescence. Following photoexcitation above the barrier between the Franck–Condon geometry and the prefulvene MECI connecting the 11ππ* state with the ground state, we attribute the dynamics on the 11ππ* surface to relaxation through the prefulvene MECI.
(2) We observe clear evidence to support the conclusions of our earlier experiments that following photoexcitation above the 11πσ* origin, the 11πσ* state is excited directly and relaxes through the MECI connecting the 11πσ* and 11ππ* states close to the local minimum on the 11πσ* surface.19,20,23 Although we do not observe population flow from the 11ππ* state to the 11πσ* state, it is possible that there could be a small contribution from direct excitation of the 11ππ* state followed by relaxation through the MECI to the dissociative component of the 11πσ* state. We conclude that there is also a second time constant associated with dynamics on the 11πσ* surface.
(3) We find compelling evidence to support our proposal that non-radiative decay from the 21ππ* state involves a barrierless pathway from the Franck–Condon region to a CI between the 21ππ* state and the ground-state that passes through a three-state conical intersection involving the 21ππ*, 11πσ* and 11ππ* states.23
In summary, this work demonstrates that tunnelling does not play a role in the electronic relaxation dynamics of aniline following photoexcitation in the range 272–238 nm, which is surprising given that tunnelling has been found to play an important role in the electronic relaxation of the isoelectronic molecule phenol and in pyrrole. To the best of our knowledge, this work also provides the first experimental signature of a 3-state conical intersection. Detailed gas-phase experiments such as this, combined with the results of detailed theoretical studies, are essential in forming a good starting point for understanding the relaxation mechanisms of these biologically relevant chromophores in realistic environments.
Acknowledgements
This work was supported by the EPSRC, European Marie Curie Initial Training Network Grant No. GA-ITN-214962-FASTQUAST, Conseil Regional de Bourgogne and the Spanish Ministry of Economy and Competitiveness (MINECO) through grant CTQ2012-37404-C02-01. The authors are grateful to David Townsend (Herriot-Watt) for valuable discussions.References
- M. N. R. Ashfold, G. A. King, D. Murdock, M. G. D. Nix, T. A. A. Oliver and A. G. Sage, Phys. Chem. Chem. Phys., 2010, 12, 1218–1238 RSC.
- G. M. Roberts and V. G. Stavros, in The role of * states in the photochemistry of heteroaromatic biomolecules and their subunits: insights from gas-phase femtosecond spectroscopy, ed. R. de Nalda and L. Banares, Springer-Verlag, Berlin, 2014, vol. 107, pp. 119–143 Search PubMed.
- C. T. Middleton, K. de La Harpe, C. Su, Y. K. Law, C. E. Crespo-Hernández and B. Kohler, Annu. Rev. Phys. Chem., 2009, 60, 217–239 CrossRef CAS PubMed.
- T. Gustavsson, R. Improta and D. Markovitsi, J. Phys. Chem. Lett., 2010, 1, 2025–2030 CrossRef CAS.
- C. C.-W. Cheng, C. Ma, C. T.-L. Chan, K. Y.-F. Ho and W.-M. Kwok, Photochem. Photobiol. Sci., 2013, 12, 1351–1365 CAS.
- X. Song, M. Yang, E. R. Davidson and J. P. Reilly, J. Chem. Phys., 1993, 99, 3224 CrossRef CAS PubMed.
- T. Ebata, C. Minejima and N. Mikami, J. Phys. Chem. A, 2002, 106, 11070–11074 CrossRef CAS.
- E. R. T. Kerstel, M. Becucci, G. Pietraperzia and E. Castellucci, Chem. Phys., 1995, 199, 263–273 CrossRef CAS.
- K. Kimura, H. Tsubomura and S. Nagakura, Bull. Chem. Soc. Jpn., 1964, 37, 1336–1346 CrossRef CAS.
- R. Scheps, D. Florida and S. A. Rice, J. Chem. Phys., 1974, 61, 1730–1747 CrossRef CAS PubMed.
- B. Kim, C. P. Schick and P. M. Weber, J. Chem. Phys., 1995, 103, 6903–6913 CrossRef CAS PubMed.
- A. L. Sobolewski, W. Domcke, C. Dedonder-Lardeux and C. Jouvet, Phys. Chem. Chem. Phys., 2002, 4, 1093–1100 RSC.
- H. Reisler and A. I. Krylov, Int. Rev. Phys. Chem., 2009, 28, 267–308 CrossRef CAS PubMed.
- F. Wang, S. P. Neville, R. Wang and G. A. Worth, J. Phys. Chem. A, 2013, 117, 7298–7307 CrossRef CAS PubMed.
- B. N. Rajasekhar, A. Veeraiah, K. Sunanda and B. N. Jagatap, J. Chem. Phys., 2013, 139, 064303 CrossRef CAS PubMed.
- G. A. King, T. A. A. Oliver and M. N. R. Ashfold, J. Chem. Phys., 2010, 132, 214307 CrossRef PubMed.
- R. Montero, Á. P. Conde, V. Ovejas, R. Martínez, F. Castaño and A. Longarte, J. Chem. Phys., 2011, 135, 054308 CrossRef PubMed.
- G. M. Roberts, C. A. Williams, J. D. Young, S. Ullrich, M. J. Paterson and V. G. Stavros, J. Am. Chem. Soc., 2012, 134, 12578–12589 CrossRef CAS PubMed.
- R. Spesyvtsev, O. M. Kirkby, M. Vacher and H. H. Fielding, Phys. Chem. Chem. Phys., 2012, 14, 9942–9947 RSC.
- R. Spesyvtsev, O. M. Kirkby and H. H. Fielding, Faraday Discuss., 2012, 157, 165–179 RSC.
- J. O. F. Thompson, R. A. Livingstone and D. Townsend, J. Chem. Phys., 2013, 139, 034316 CrossRef PubMed.
- J. O. F. Thompson, L. Saalbach, S. W. Crane, M. J. Paterson and D. Townsend, J. Chem. Phys., 2015, 142, 114309 CrossRef PubMed.
- M. Sala, O. M. Kirkby, S. Guérin and H. H. Fielding, Phys. Chem. Chem. Phys., 2014, 16, 3122–3133 RSC.
- D. S. N. Parker, R. S. Minns, T. J. Penfold, G. A. Worth and H. H. Fielding, Chem. Phys. Lett., 2009, 469, 43–47 CrossRef CAS PubMed.
- R. S. Minns, D. S. N. Parker, T. J. Penfold, G. A. Worth and H. H. Fielding, Phys. Chem. Chem. Phys., 2010, 12, 15607–15615 RSC.
- A. D. G. Nunn, R. S. Minns, R. Spesyvtsev, M. J. Bearpark, M. A. Robb and H. H. Fielding, Phys. Chem. Chem. Phys., 2010, 12, 15751–15759 RSC.
- G. A. Garcia, L. Nahon and I. Powis, Rev. Sci. Instrum., 2004, 75, 4989–4996 CrossRef CAS PubMed.
- J. E. Hansen and W. Persson, Phys. Scr., 1987, 36, 602–643 CrossRef CAS.
- M. A. Smith, J. W. Hager and S. C. Wallace, J. Chem. Phys., 1984, 80, 3097–3105 CrossRef CAS PubMed.
- This lifetime was difficult to fit due to its small amplitude (Fig. 3c) but excluding it resulted in a poor fit. The value of 450 fs was selected to give a reasonable fit to the experimental data.
- G. A. Pino, A. N. Oldani, E. Marceca, M. Fujii, S. I. Ishiuchi, M. Miyazaki, M. Broquier, C. Dedonder and C. Jouvet, J. Chem. Phys., 2010, 133, 124313 CrossRef CAS PubMed.
- R. N. Dixon, T. A. A. Oliver and M. N. R. Ashfold, J. Chem. Phys., 2011, 134, 194303 CrossRef PubMed.
- H. An and K. K. Baeck, J. Phys. Chem. A, 2011, 115, 13309–13315 CrossRef CAS PubMed.
- G. M. Roberts, A. S. Chatterley, J. D. Young and V. G. Stavros, J. Phys. Chem. Lett., 2012, 3, 348–352 CrossRef CAS.
- M. Barbatti, M. Vazdar, A. J. A. Aquino, M. Eckert-Maksić and H. Lischka, J. Chem. Phys., 2006, 125, 164323 CrossRef PubMed.
- L. Blancafort, J. Am. Chem. Soc., 2006, 128, 210–219 CrossRef CAS PubMed.
- S. Perun, A. L. Sobolewski and W. Domcke, J. Am. Chem. Soc., 2005, 127, 6257–6265 CrossRef CAS PubMed.
- L. Serrano-Andrés, M. Merchán and A. C. Borin, J. Am. Chem. Soc., 2008, 130, 2473–2484 CrossRef PubMed.
- H. R. Ward and J. S. Wishnok, J. Am. Chem. Soc., 1968, 90, 5353–5357 CrossRef CAS.
- A. L. Sobolewski, J. Chem. Phys., 1990, 93, 6433–6439 CrossRef CAS PubMed.
- R. A. Livingstone, J. O. F. Thompson, M. Iljina, R. J. Donaldson, B. J. Sussman, M. J. Paterson and D. Townsend, J. Chem. Phys., 2012, 137, 184304 CrossRef PubMed.
- V. Vallet, Z. Lan, S. Mahapatra, A. L. Sobolewski and W. Domcke, J. Chem. Phys., 2005, 123, 144307 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Time constant fits and decay associated spectra residuals. See DOI: 10.1039/c5cp01883h |
| This journal is © the Owner Societies 2015 |