
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic and magnetic properties of DUT-8(Ni)
Kai
Trepte
*a,
Sebastian
Schwalbe
b and
Gotthard
Seifert
a
aTechnische Universität Dresden, Theoretical Chemistry, Germany. E-mail: kai.trepte@chemie.tu-dresden.de
bTU Bergakademie Freiberg, Institute for Theoretical Physics, Germany
First published on 27th May 2015
Abstract
First principles calculations using density functional theory (DFT) have been performed to investigate the electronic and magnetic properties of DUT-8(Ni) (DUT – Dresden University of Technology). This flexible metal–organic framework (MOF) exists in two crystalline forms: DUT-8(Ni)open and DUT-8(Ni)closed. To identify the energetically favoured magnetic ordering, the density of states (DOS) and the energy difference between a low-spin (LS) and a high-spin (HS) coupling ΔELS–HS for those crystalline structures have been computed. Calculations on supercells have been carried out to include a variety of different magnetic couplings beyond a single unit cell. Several molecular model systems have been employed to further investigate the magnetic behaviour by introducing a diversity of chemical environments to the magnetic centers. The magnetic ground state of both crystalline structures has been found to be the low-spin state (S = 0). This low-spin ordering can be seen in the DOS as well as from ΔELS–HS calculations. Additionally, the calculations on the supercells confirm that the local character of the ordering (i.e. within the Ni dimers) is the most favoured one. However, the model systems indicate a change from the low-spin (S = 0) to a high-spin (S ≠ 0) ordering by introducing certain alterations into the chemical environment. Such alterations could be incorporated into the crystalline systems which should lead to similar results.
1 Introduction
Metal–organic frameworks (MOFs) gained increasing interest since their initial description1,2 due to their high porosity, high surface areas and absorption behaviour.3–5 Such compounds consist of certain metal centers (metal) within their so-called secondary building unit which are connected via specific organic linkers (organic) to build a crystalline network (framework). Inside MOFs, there can be pores of different sizes. Those pores enable the MOF to be utilized for absorption due to their size and resulting large surface area. Several subclasses of frameworks, including so-called paddle wheels, can nowadays be synthesized leading to a variety of different types of MOFs. Some MOFs are flexible,6 meaning that several stable crystalline structures can be obtained by absorption and desorption. From the diversity of available metal centers, open shell transition metals can introduce a magnetic ground state. In general, the main research on MOFs is concerned with their absorption behaviour and how to increase their surface areas and porosities. On the other hand, the magnetism in MOFs has not been studied yet, but is thought to extend the already wide field of applications for MOFs. Furthermore there is a connection to the widely studied field of binuclear complexes,7–11 which are the carriers of the magnetic interaction within DUT-8(Ni) and other MOFs. A special case are Ni(II) dimer systems, which have been studied in the last few years.7,8 Within Ni(II) dimer complexes, each Ni can be viewed as a d8-Ni(II). As a consequence the highest possible spin is SHS = 2 while the lowest spin is SLS = 0. In all discussions low-spin accounts for an antiparallel alignment of the spins at the magnetic centers while high-spin stands for a parallel alignment. However, the magnetic ground state in DUT-8(Ni)12,13 (Fig. 1), with one Ni dimer per magnetic unit, is still unclear.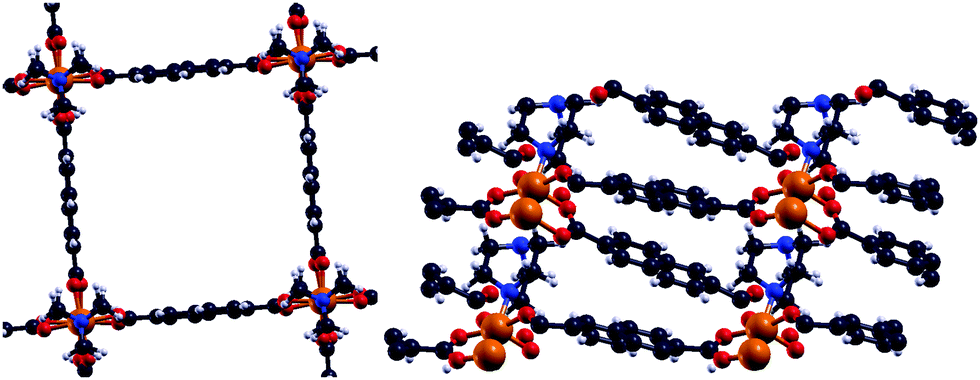 | ||
Fig. 1 Comparison of the tetragonal DUT-8(Ni)open12 (along ![[c with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0063_20d1.gif) ) and the triclinic DUT-8(Ni)closed13 (along ) and the triclinic DUT-8(Ni)closed13 (along ![[b with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0062_20d1.gif) ) (four Ni dimers are displayed in each structure for comparison). ) (four Ni dimers are displayed in each structure for comparison). | ||
DUT-8(Ni) is flexible corresponding to two crystalline structures with so-called paddle wheels (DUT-8(Ni)open/DUT-8(Ni)closed), with a ratio of the unit cell volumes of VDUT-8(Ni)open ≈ 2.6·VDUT-8(Ni)closed. The Ni atoms in these structures are coordinated by four oxygens of the organic linkers (2,6-naphthalenedicarboxylate C12O4H6),12 which connect the magnetic units in two dimensions to form the paddle wheels. The paddle wheels are connected via dabco units (1,4-diazabicyclo[2·2·2]octane C6N2H12).12 This leads to an additional coordination of the Ni by a nitrogen atom in the three-dimensional periodic structure of DUT-8(Ni). In connection to the structural flexibility a very interesting question arises concerning the magnetic properties of such structures.
The main question to answer in the first place is whether the structural change, which occurs due to absorption into the DUT-8(Ni)closed to gain DUT-8(Ni)open, alters the magnetic ground state. Such an alteration could be used as an additional measure of absorption and might even be sensitive to different absorbents, as it might lead to different strengths of the coupling. To gain insight into the magnetic behaviour of this compound, DFT calculations were performed. The magnetic properties are discussed on the basis of the calculated energy difference between a low-spin and a high-spin state ΔELS–HS, which describes the favoured magnetic ordering qualitatively as well as quantitatively.
For further evaluation of the strength of the magnetic interaction the Heisenberg–Dirac–Van Vleck model is applied. This model uses a Hamiltonian which describes the magnetic interaction between spins with the so-called coupling constant J. The energy values as well as the resulting magnetizations from spin-polarized DFT calculations allow the computation of this coupling constant. It scales the energy differences to the respective total spins of the high-spin state and the low-spin state (see eqn (2)). The sign of J is given by the favoured magnetic ordering (positive for high-spin and negative for low-spin) while its absolute value is a measure for the strength of such ordering.
2 Methodology
2.1 DFT calculations
All calculations have been carried out using the DFT14,15 based program package QUANTUM ESPRESSO (QE).16 It uses a pseudopotential approach with a plane wave17 basis set.The projector-augmented wave (PAW)18 method with a GGA-PBE19 exchange–correlation functional was used ([element].pbe-(n)-kjpaw_psl.0.1.UPF, for details see http://.quantum-espresso.org/pseudopotentials/). All calculations were performed spin-polarized. The van der Waals (vdW) interaction by Grimme20 has been used to validate the results of the crystalline structures. The results of the QE calculations have been compared with calculations using the GPAW21 program package. This was done to compare the results obtained with different basis sets (plane wave vs. local basis). GPAW is a real-space-grid DFT code based on the projector-augmented wave (PAW) method. Pseudo wavefunctions can be described in different ways (http://https://wiki.fysik.dtu.dk/gpaw/algorithms.html#algorithms), of which the LCAO ansatz22 was used. All LCAO calculations are spin-polarized and were performed using a double-ζ basis set with one polarization function. The set of k-points and the exchange–correlation functional are the same like in the QE calculations.
The application of the GGA-PBE approach is justified by its robustness (see below), its computational efficiency and the minimum number of required parameters. These are advantages compared to hybrid functionals (e.g. B3LYP) or DFT + U, although such approaches can correct the problem of too much delocalization of d-electrons in certain ways. Other methods, e.g. CI or CASSCF, are prohibited concerning the computational effort for large systems like MOFs. Within the research of binuclear complexes,9–11 different approaches to determine the magnetic properties of small molecules have been carried out. Such approaches include wave function based methods (e.g. CI9 or CASSCF10) and recently also DFT.11 The B3LYP hybrid functional with different amounts of exact exchange has been compared to DDCI23 (difference dedicated configuration interaction) and to a PBE functional.11 Calculations of the coupling constant show similar results for all methodologies. Especially the results obtained with B3LYP and PBE agree even quantitatively. Considering these results and taking into account the size of DUT-8(Ni), it is reasonable to apply the GGA-PBE as a good compromise between the computational effort and accuracy. Thus all calculations in the present work were performed within the GGA-PBE approximation.
The Brillouin-zone was sampled with a 3 × 3 × 3 Monkhorst–Pack grid (14 k-points per spin orientation). The kinetic energy cutoff is 90 Ry for the crystalline systems and 150 Ry for the model systems. Full geometry optimizations with and without the inclusion of the vdW interaction for the crystalline structures were performed. The optimizations were carried out on a 2 × 2 × 2 Monkhorst–Pack grid where convergence was achieved once all forces are smaller than 10−3 Ry/a0 ≈ 0.025 eV Å−1 (a0 is the Bohr radius).
It has to be considered that within the DUT-8(Ni)open unit cell the interaction between the Ni dimers is negligible (see Section 3.2). Thus the energy difference between a low-spin and a high-spin coupling ΔELS–HS has to be divided by 2. This scales ΔELS–HS,DUT-8(Ni)open to one Ni dimer. With that it can be compared to ΔELS–HS,DUT-8(Ni)closed.
For the calculation of DUT-8(Ni)open, no absorbents have been taken into account. Thus a comparison of the structures as shown in Fig. 1 was performed. However no major changes in the magnetic behaviour are expected once certain substances are absorbed (as long as those substances do not carry some spin polarization on their own).
2.2 Heisenberg–Dirac–Van Vleck model
A description of the magnetic behaviour, as introduced by the coupling of local spins at different magnetic centers, is given by the Heisenberg–Dirac–Van Vleck (HDVV) Hamiltonian24–26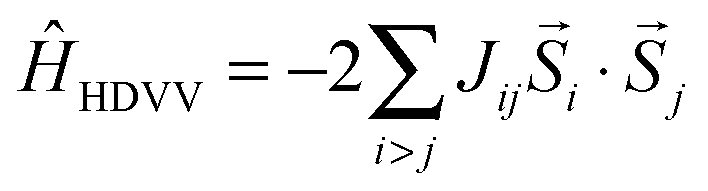 | (1) |
![[S with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0053_20d1.gif) i and j are spin operators of the magnetic centers i and j. A high-spin state, meaning a parallel alignment of the spins, is indicated by a positive value of the coupling constant Jij while a negative value refers to a low-spin state and an antiparallel alignment. The coupling constant for dimers (i = 1 and j = 2) can be related to the total energies and the total spins of two magnetic orderings
i and j are spin operators of the magnetic centers i and j. A high-spin state, meaning a parallel alignment of the spins, is indicated by a positive value of the coupling constant Jij while a negative value refers to a low-spin state and an antiparallel alignment. The coupling constant for dimers (i = 1 and j = 2) can be related to the total energies and the total spins of two magnetic orderings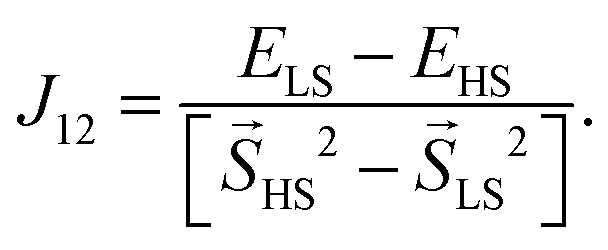 | (2) |
i and j at each centers do not have to be taken into account. The denominator in eqn (2) becomes six for a Ni dimer (SHS = 2, SLS = 0), two for a Cu dimer (SHS = 1, SLS = 0) and three for a mixed Ni–Cu dimer (SHS = 3/2, SLS = 1/2). This consideration leads to the values given in Table 3.
2.3 Structures and models
The tetragonal unit cell (a = b ≈ 18.679 Å and c ≈ 9.613 Å) for DUT-8(Ni)open contains 132 atoms and two Ni dimers. The unit cell of DUT-8(Ni)closed contains 66 atoms and only one Ni dimer. It is described by a ≈ 6.947 Å, b ≈ 8.180 Å, c ≈ 12.172 Å and α ≈ 91.23°, β ≈ 103.87°, γ ≈ 104.55°.For further analysis of the magnetic interaction, calculations on supercells (Fig. 2) were performed. The supercells allow the study of different magnetic coupling configurations (Fig. 3). Thus the magnetic interaction beyond a single unit cell is analyzed, as an evaluation of such couplings is not possible with single unit cells. The energetically most favoured interaction will be either within the magnetic dimer units (local interaction) or between them (global interaction). The DUT-8(Ni)open supercell (Fig. 2a) has been constructed by replicating two unit cells in the direction. For a comparable size of the DUT-8(Ni)closed supercell (Fig. 2b), four unit cells were taken into account, two in the ![[a with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0061_20d1.gif) and two in direction each. This leads to 264 atoms and four magnetic dimer units (I, II, III and IV) for each supercell. There is a major difference between the supercells of DUT-8(Ni)open and DUT-8(Ni)closed. This difference is connected with the organic linkers. In the DUT-8(Ni)closed supercell they are only introduced by the periodic boundary conditions. As the magnetic interaction along the organic linkers is small (see Section 3.2), an explicit consideration in the DUT-8(Ni)closed supercell is not mandatory. On the other hand those linkers are contained in the DUT-8(Ni)open supercell according to the elementary cell of DUT-8(Ni)open.
and two in direction each. This leads to 264 atoms and four magnetic dimer units (I, II, III and IV) for each supercell. There is a major difference between the supercells of DUT-8(Ni)open and DUT-8(Ni)closed. This difference is connected with the organic linkers. In the DUT-8(Ni)closed supercell they are only introduced by the periodic boundary conditions. As the magnetic interaction along the organic linkers is small (see Section 3.2), an explicit consideration in the DUT-8(Ni)closed supercell is not mandatory. On the other hand those linkers are contained in the DUT-8(Ni)open supercell according to the elementary cell of DUT-8(Ni)open.
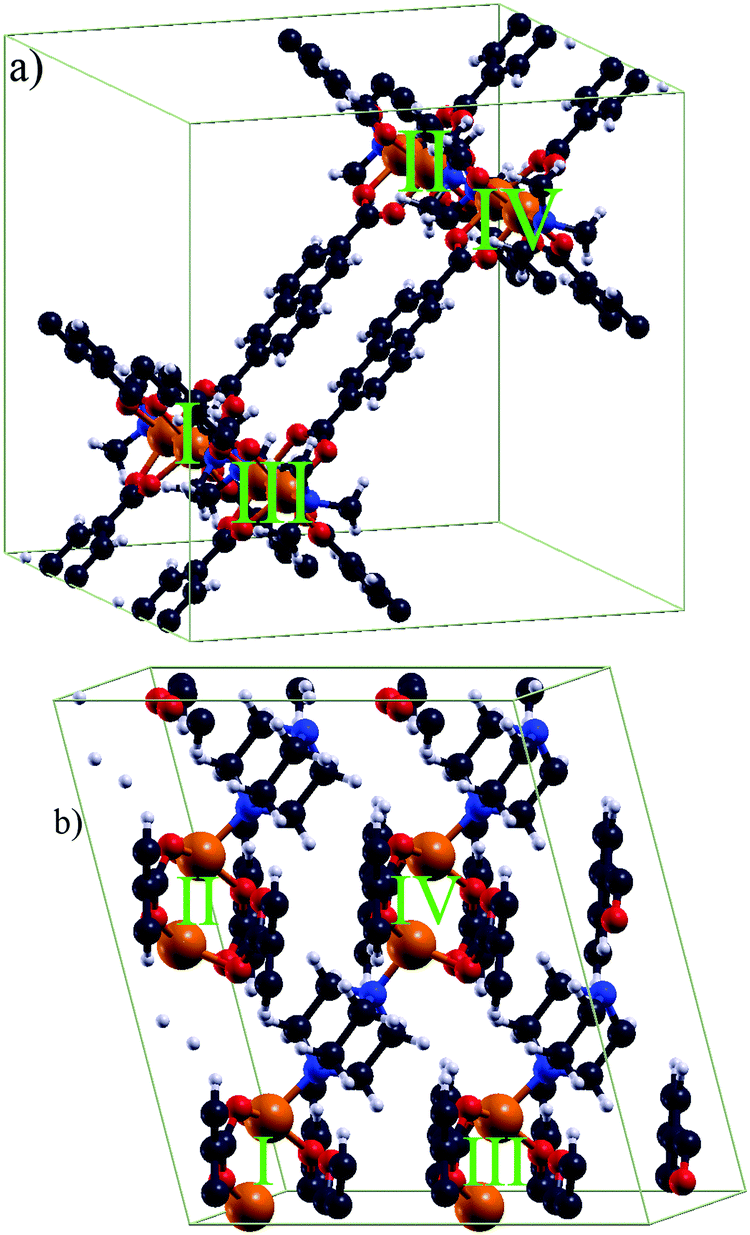 | ||
| Fig. 2 The used supercells (264 atoms) for (a) DUT-8(Ni)open and (b) DUT-8(Ni)closed, respectively, to investigate global magnetic orderings (see Fig. 3). The Ni pairs as magnetic dimer units are highlighted. | ||
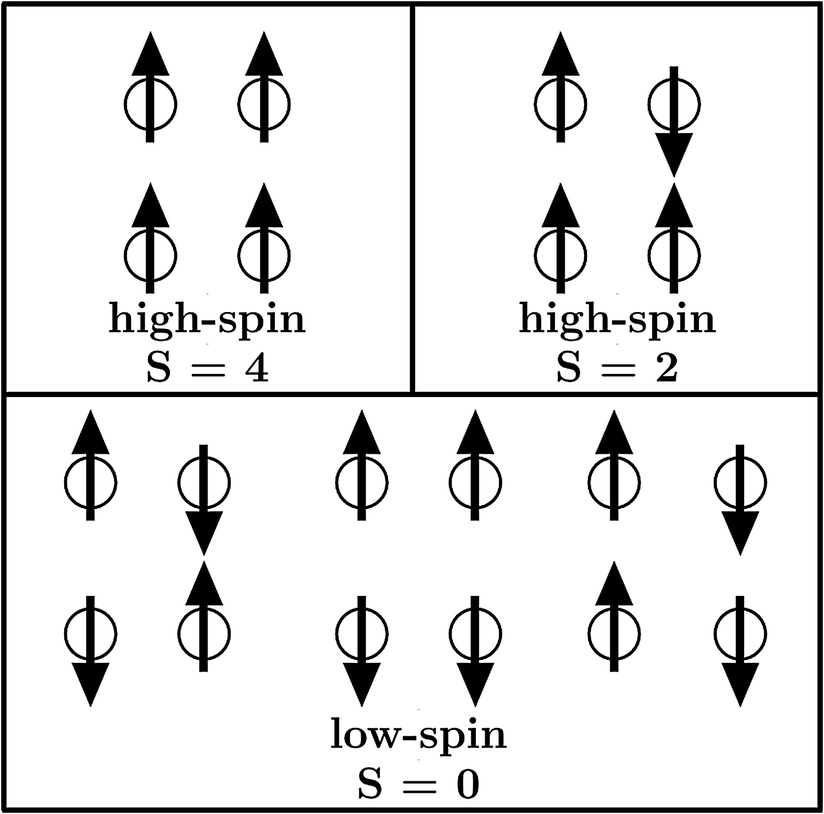 | ||
| Fig. 3 Possible couplings within the supercells leading to different global magnetic orderings. The local interaction at each magnetic dimer unit, indicated by spheres, is assumed to be high-spin. | ||
It can be shown, e.g. for the MOF HKUST-1,27 that the magnetic interaction can be described by molecular model systems. For an in-depth insight into the magnetic interaction within DUT-8(Ni), several molecular model systems were generated (Fig. 4 and Table 1). This allows to analyze possible alterations of the magnetic ground state which are related to changes in the chemical environment of the magnetic centers rather than the structural change in the periodic systems. Thus several ways to influence the magnetic properties have been employed. For the initial models M1 (Ni2(HCOO)4(NH3)2, Fig. 4) the coordinates of the atoms inside the magnetic unit within the crystalline systems were used. To achieve charge neutrality the carbon of the organic linkers as well as the nitrogen of the dabco units were terminated with hydrogen. These initial structures were modified (Table 1) to study the influence of the local environment of the Ni atoms on the magnetic properties. The modifications on the chemical environment include the addition of phenyl groups, the removal of NH3 groups and the replacement of NH3 groups with some other functional groups. Additionally, the magnetic centers themselves have been exchanged (Cu instead of Ni) without altering the chemical environment. The influence on the magnetic behaviour caused by such alterations was investigated. The initial models (M1) contain 26 atoms while all other models (Table 1) incorporate between 18 and 26 atoms. For the molecular model systems the unit cells have been chosen to impose a vacuum of 10 Å in each direction (x,y,z) to avoid interactions between neighbouring molecules.
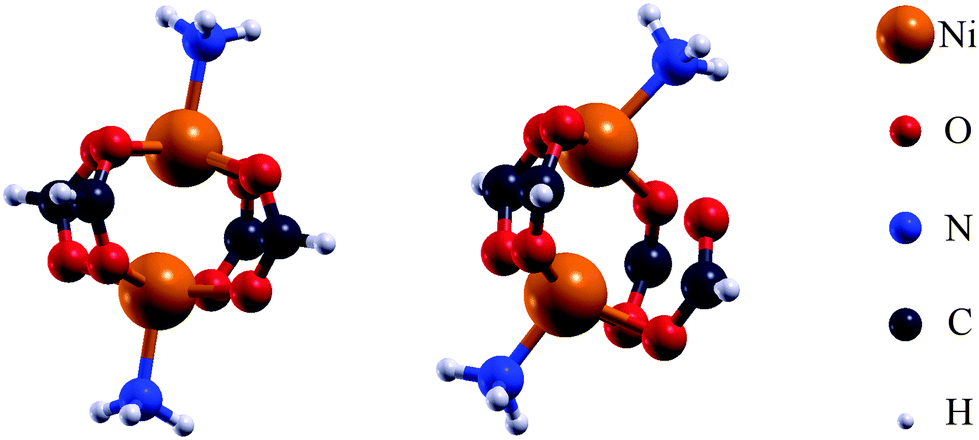 | ||
| Fig. 4 Structures of the initial model systems (M1) for DUT-8(Ni)open and DUT-8(Ni)closed used for further investigations. | ||
| Model system | Description | |||
|---|---|---|---|---|
| Unit per atom | Unit per atom | |||
| M2 | Include benzene | C–H | to | C–Ph |
| M3 | Remove NH3,1 | Only first NH3 | to | Vacuum |
| M4 | Remove NH3,2 | Only second NH3 | to | Vacuum |
| M5 | Remove NH3,1&2 | Both NH3 | to | Vacuum |
| M6 | Cu for Ni1 | Only first Ni | to | Cu |
| M7 | Cu for Ni2 | Only second Ni | to | Cu |
| M8 | Cu for Ni1&2 | Both Ni | to | Cu |
| M9 | Water | NH3 | to | OH2 |
| M10 | Carbon dioxide | NH3 | to | OCO |
| M11 | Saturated cyano | NH3 | to | NCH |
3 Results
3.1 Crystalline systems
The density of states (DOS) for DUT-8(Ni)open and DUT-8(Ni)closed around the Fermi energy EF is shown in Fig. 5. The electronic densities (n) have been decomposed into spin up (↑) and spin down (↓) contributions to get a first insight into the spin polarization (n↑−n↓) and the magnetic behaviour. The partial DOS (PDOS) for all Ni d-states with the respective spins are also shown next to the total DOS. Fig. 5(a), representing DUT-8(Ni)open, shows a very similar contribution of the ↑ and ↓ densities over the given energy range (E = EF ± 4 eV). This can be attributed to the symmetry of DUT-8(Ni)open. The PDOS for the Ni d-states shows that one Ni per dimer (dimer 1: Ni1,2 and dimer 2: Ni3,4) carries one specific spin (↑) while the respective other Ni carries the opposite spin (↓) at the same energy. Therefore a low-spin state is expected to be favoured. The resulting coupling constant J in such a case is negative. Further the contribution to the states around the Fermi level EF does not exclusively come from the Ni d-states. The biggest contribution next to such states are the oxygen p-states (not shown in Fig. 5). Such contributions have been found in the electronic structure of e.g. MOF-528 as well.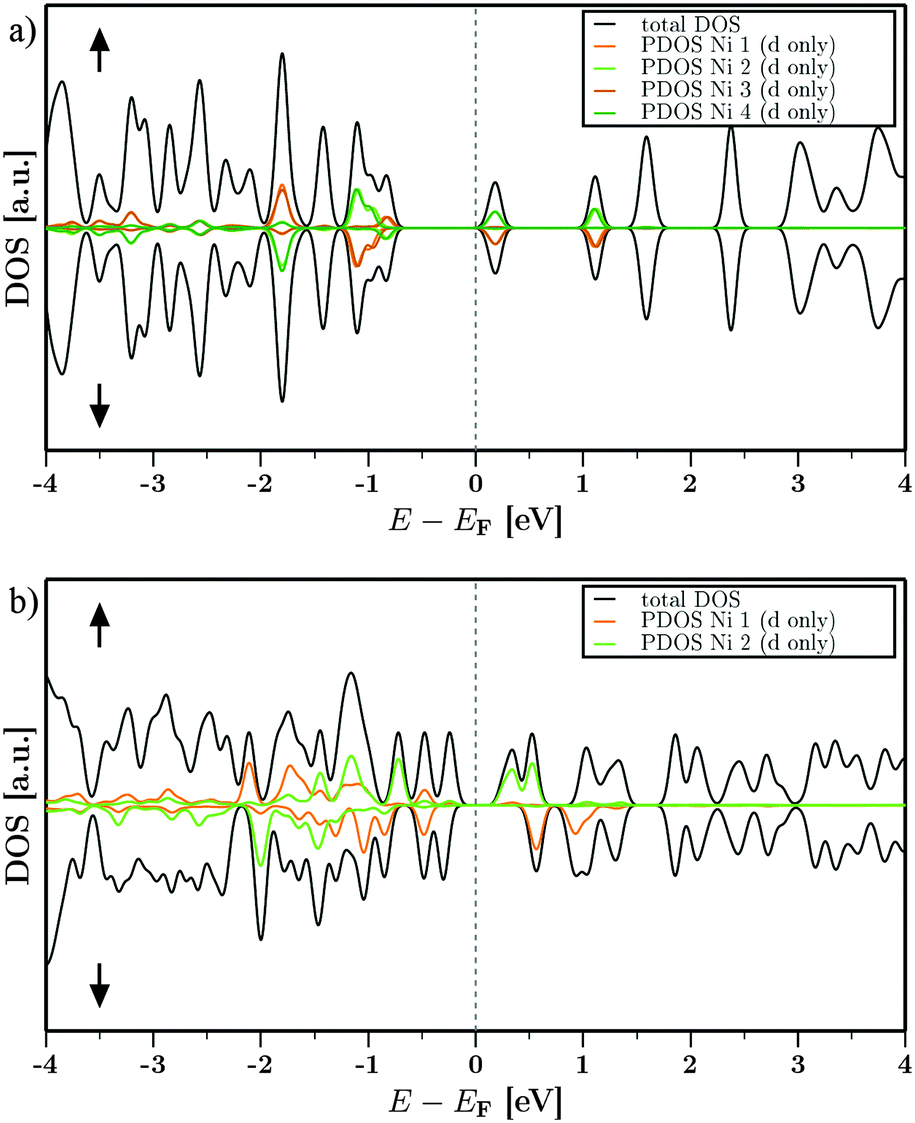 | ||
| Fig. 5 Density of states, splitted into spin up (↑) and spin down (↓) densities, for (a) DUT-8(Ni)open and (b) DUT-8(Ni)closed. States with energies around the Fermi level E = EF ± 4 eV are displayed. | ||
The contribution of the different spin densities for DUT-8(Ni)closed is not symmetric anymore (Fig. 5b). However, one of the Ni contributes mainly to one of the spin species while the other magnetic center contributes to the other one (Ni1↓, Ni2↑). The spins at the two magnetic centers are aligned antiparallel and the overall magnetic ordering should be the low-spin state as well.
The calculated energy differences between a low-spin and a high-spin coupled system for the two crystalline system give a qualitative as well as quantitative insight into the magnetic ordering (the results for DUT-8(Ni)open are adjusted, meaning that the given ΔELS–HS has been divided by 2 in order to account to one Ni dimer and to be comparable to DUT-8(Ni)closed). The calculations give
| ΔELS–HS,DUT-8(Ni)open,QE = −199 meV |
| ΔELS–HS,DUT-8(Ni)closed,QE = −81 meV |
| ΔELS–HS,DUT-8(Ni)open,GPAW = −206 meV |
| ΔELS–HS,DUT-8(Ni)closed,GPAW = −77 meV. |
| JDUT-8(Ni)open,QE = −267 cm−1 |
| JDUT-8(Ni)open,GPAW = −277 cm−1 |
| JDUT-8(Ni)closed,QE = −108 cm−1 |
| JDUT-8(Ni)closed,GPAW = −103 cm−1. |
ΔEDUT-8(Ni)open,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) optimized,novdW = −340 meV optimized,novdW = −340 meV |
| ΔEDUT-8(Ni)open,optimized,withvdW = −339 meV |
| ΔEDUT-8(Ni)closed,optimized,novdW = −183 meV |
| ΔEDUT-8(Ni)closed,optimized,withvdW = −194 meV. |
3.2 Supercell calculations
As magnetic interactions extend beyond elementary cells, supercells were generated. This allows the calculation of different magnetic couplings between several elementary cells. Further a comparison of global magnetic interactions and local ones can be carried out. A variety of different interactions which cannot be realized within elementary cells can be studied as well. As mentioned earlier, the organic linkers within the DUT-8(Ni)closed supercell are only implemented by the periodic boundary conditions. To verify this statement the interaction along the organic linkers in DUT-8(Ni)open has been analyzed. Calculating the energy difference between a low-spin and a respective high-spin ordering along the linkers gives ΔELS→HS ≈ 0.04 meV per unit cell. Thus the interaction along the organic linkers is small and does not have to be taken explicitly into account in the DUT-8(Ni)closed supercell. In comparison to the DUT-8(Ni)open supercell, this allows the study of the interaction of one magnetic dimer unit coupled antiparallelly to all others (second last entry in Table 2).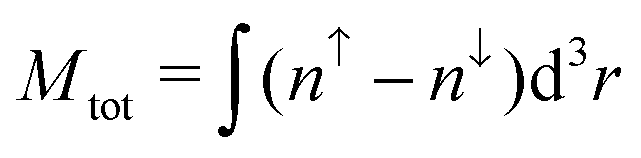 is the total magnetization of the system,
is the total magnetization of the system, 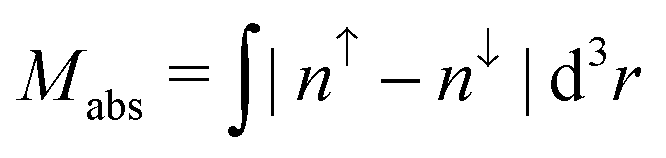 is the corresponding absolute magnetization and ΔELS(local) = ELS(local) − Eref is the energy difference between the local low-spin and the considered magnetic ordering
is the corresponding absolute magnetization and ΔELS(local) = ELS(local) − Eref is the energy difference between the local low-spin and the considered magnetic ordering
| I | II | III | IV | M tot [μB per cell] | M abs [μB per cell] | ΔELS(local) [meV per dimer] |
|---|---|---|---|---|---|---|
| DUT-8(Ni)open | ||||||
| ↑↓ | ↑↓ | ↑↓ | ↑↓ | 0.00 | 14.93 | 0.00 |
| ↑↑ | ↓↓ | ↓↓ | ↑↑ | 0.00 | 16.38 | −190.35 |
| ↑↑ | ↑↑ | ↓↓ | ↓↓ | 0.00 | 16.35 | −190.37 |
| ↑↑ | ↑↑ | ↑↑ | ↑↑ | 16.00 | 16.52 | −198.93 |
| DUT-8(Ni)closed | ||||||
| ↑↓ | ↑↓ | ↑↓ | ↑↓ | 0.00 | 14.80 | 0.00 |
| ↑↑ | ↓↓ | ↓↓ | ↑↑ | 0.00 | 16.63 | −80.75 |
| ↑↑ | ↑↑ | ↓↓ | ↓↓ | 0.00 | 16.36 | −58.09 |
| ↑↑ | ↓↓ | ↑↑ | ↓↓ | 0.00 | 16.34 | −58.06 |
| ↓↓ | ↓↓ | ↑↑ | ↓↓ | 8.00 | 16.49 | −69.41 |
| ↑↑ | ↑↑ | ↑↑ | ↑↑ | 16.00 | 16.62 | −80.71 |
Several magnetic orderings were introduced to the Ni dimers within the supercells and the resulting energies were compared to figure out which magnetic ordering is the energetically most favoured one (Table 2).
First the results indicate that the local low-spin ordering is the energetically most favoured coupling of all possible interactions. This confirms the results achieved by the calculations on single unit cells. Second they provide the information that for local high-spin behaviour, every global low-spin ordering shows lower energies than any global high-spin ordering. The only exception occurs for a high-spin coupling along the dabco linker units (the second coupling for DUT-8(Ni)closed in Table 2). There, a very small energy difference to the global high-spin case (↑↑↑↑) is found. An explanation is the interaction between Ni and N, which has a major influence on the magnetic behaviour (see Section 3.3). For the other global low-spin couplings similar energies are found where the coupling along the dabco units is always low-spin. Thus the interactions along the dabco units determine which coupling is favoured. A local high-spin state can be gained by altering the chemical environment of the magnetic centers, as shown in the next section.
3.3 Molecular models
To further study the magnetic interaction between the Ni atoms in DUT-8(Ni), several molecular model systems were generated. With such models, the chemical environment of the magnetic centers can be modified. Additionally, the magnetic centers themselves were exchanged (Table 1).The used molecular models (M1) (Fig. 4) show a similar behaviour considering the ΔELS–HS in comparison to their respective crystal structures (Table 3). These systems were modified to study influences of different chemical environments as well as an exchange of magnetic centers. If such alterations would be implemented into the crystalline structures, a similar magnetic behaviour is expected. The inclusion of the phenyl groups (M2), which describe the system with organic linkers, shows basically no change in the magnetic ground state and can be neglected for all other models.
| Model | DUT-8(Ni)open | DUT-8(Ni)closed | ||
|---|---|---|---|---|
| ΔE [meV] | J [cm−1] | ΔE [meV] | J [cm−1] | |
| M1 | −202.5 | −272.2 | −52.2 | −70.2 |
| M2 | −189.6 | −254.8 | −43.8 | −58.9 |
| M3 | 214.0 | 863.2 | 151.0 | 605.4 |
| M4 | 217.5 | 876.8 | −4.2 | −5.7 |
| M5 | 402.9 | 1624.6 | 193.4 | 780.0 |
| M6 | −92.2 | −247.9 | 0.7 | 1.8 |
| M7 | −90.2 | −242.5 | −38.9 | −104.6 |
| M8 | −128.4 | −517.6 | −17.2 | −69.5 |
| M9 | 67.8 | 273.7 | 21.4 | 86.3 |
| M10 | 301.9 | 1216.0 | 211.5 | 852.8 |
| M11 | 346.3 | 1396.5 | 242.2 | 976.8 |
The removal of the NH3 groups (M3, M4) is interesting in that it leads to a high-spin state due to the missing interaction between the N to one of the Ni. An increase in the coupling strength can be observed when removing both NH3 groups. Without the N–Ni interaction, the electron density of the Ni ‘relaxes’ towards to the other Ni, which changes the interaction between them. The resulting ground state has a SHS = 1 total spin. An overlap of the electron densities of the Ni due to the mentioned relaxation is assumed. Thus there are two electrons in between the Ni with a low-spin coupling (↑↓) while the outer electrons couple high-spin (↑↑). This is confirmed by calculations of the electron localization functions (ELFs) revealing paired electrons in between the Ni. As there are no paired electrons in the M1 models, the previous assumption is validated. Transferring the alterations to the crystalline systems can be thought of as a removal of a dabco unit which separates the paddle wheels within the DUT-8(Ni).12 When synthesizing this MOF, a reduced amount of dabco units should lead to a high-spin state while for a higher amount of such linker groups, a low-spin state should be present.
A different magnetic behaviour is expected when exchanging Ni with Cu (M6, M7, M8), considering the differences of their electronic configuration ([Ar]4s23d8 to [Ar]3d104s1). For a Ni dimer, there are two unpaired d-electrons per Ni while there is only one unpaired s-electron each in the case of Cu. Thus in Ni two 3d-states with one electron each can couple while on the other hand in Cu the magnetic interaction occurs between only one electron per atom. The high-spin energies within the models correspond to a SHS,Ni = 2 and a SHS,Cu = 1 ground state, respectively. As known from ref. 29, the magnetic ground state in Cu complexes usually results in a low-spin state (S = 0) with a rather small ΔELS–HS (at around 100 meV). This is confirmed by the calculations of the M6, M7 and M8 models. The results show that the favoured ground state for both Ni and Cu is the low-spin state. The energy difference between the low-spin state and the high-spin state ΔELS–HS for Ni is larger than for Cu. An explanation is the larger number of electrons which contribute to the magnetic interaction for Ni. On the other hand the ΔELS–HS in the Cu dimer has been found to be higher than in the mixed Ni–Cu dimer models. This can be explained by the magnetic configurations which have to be considered in such systems. Ni provides two unpaired electrons while Cu provides only one unpaired electron. A spin of SHS = 3/2 is obtained by the high-spin state while the low-spin state shows a SLS = 1/2. Unlike in the Cu dimer system, there are always two parallelly aligned electrons from Ni which interact with either another parallelly aligned (↑↑↑) or an antiparallelly aligned electron (↑↑↓) from Cu. The separation between the high-spin state and the low-spin state is not as distinct as in the Cu dimer, where the electrons either align parallelly (↑↑) or antiparallelly (↑↓). All those alterations can be related to a metal exchange in the crystalline systems while leaving the rest of the framework unchanged.
Further alterations were implemented to the M1 models by exchanging both NH3 with some other functional groups, such as H2O, CO2 and HCN (M9, M10, M11). In analogy to changing ligands in Cu dimers30 the expectations arise that major changes in the magnetic ground state occur and that the Ni–N interaction plays an important role for the magnetic interaction. For M9, M10 and M11 these expectations are met. In those models, the high-spin state is favoured and obtains a SHS = 1. For the interpretation again an ELF analysis can be used. In each of them, there is a clear indication of a bond between the two Ni, thus two spins are always ↑↓. In M9 the Ni is coordinated by yet another oxygen instead of a N. While ΔELS–HS is small in comparison to the other two systems, the magnetic ground state already changes to a high-spin state. This indicates that the Ni–N interaction is vital for the magnetism in the systems. For M10 the energy difference increases by about a factor of 5 compared to M9 due to the stronger interaction of the oxygen with its attached C. In M9, the hydrogens obtain single bonds to the oxygen. In M10 there is a double bond to C, thus redistributing the valence electrons of oxygen. In M11 a further increase in ΔELS–HS is observed, even though the N is neither removed nor exchanged. However, in the M1 models the N forms three bonds to H, thus obtaining a sp3-hybridisation. For M11 one gets N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H corresponding to a sp-hybridisation at the carbon atom which leads to a higher separation between the low-spin state and the high-spin state.
C–H corresponding to a sp-hybridisation at the carbon atom which leads to a higher separation between the low-spin state and the high-spin state.
All discussions so far were carried out for the DUT-8(Ni)open models. In the corresponding DUT-8(Ni)closed models a similar magnetic behaviour is found (Table 3). However, the quantitative values for ΔELS–HS are smaller. This can be attributed to the deviation in the structural conformation between DUT-8(Ni)open and DUT-8(Ni)closed. The biggest differences are observed for the models M4 and M6, where the DUT-8(Ni)closed models show a very small energy difference between the high-spin state and the low-spin state. This can be explained by the Ni–N distances. For DUT-8(Ni)open one has dNi1&2–N ≈ 2.11 Å for both Ni while the distances for DUT-8(Ni)closed are dNi1–N ≈ 1.91 Å and dNi2–N ≈ 2.09 Å, respectively. Thus the interaction between Ni1 and the neighbouring N is stronger than the respective interaction for Ni2. As a consequence the Ni–Ni interaction is weaker than for the DUT-8(Ni)open models which leads to smaller ΔELS–HS. Especially for DUT-8(Ni)closed (M4 and M6 models) the resulting values for ΔELS–HS become too small to be evaluated. This shows again the importance of the Ni–N interaction on the magnetic ground state.
To simulate the removal of a dabco linker unit within the crystalline structures another model system is introduced. The system consists of two M3 molecules which are facing each other at the free Ni sites. The results of this approach are displayed in Fig. 6. Only the DUT-8(Ni)open models were taken into account. Similar effects should be observed for the respective DUT-8(Ni)closed models. Starting from a distance between the free Ni sites of dNi–Ni = 2.3 Å (binding situation), it has been steadily increased by 0.1 Å steps to dNi–Ni = 9.3 Å (two isolated systems). The corresponding distance in the crystals with a dabco unit is dcrystal ≈ 6.8 Å. Trends of the energy depending on the distance were investigated while two different magnetizations were imposed to the systems. As shown earlier, the magnetic ground state for M3 has been found to be a high-spin state. Thus the two M3 molecules were generated with a local high-spin state, while the global coupling of the system was chosen to be either a high-spin (↑↑↑↑) or a low-spin one (↑↑↓↓). First, the global interaction has basically no influence on the resulting total energy. Thus, the local interaction is of major importance and only one graph is displayed in Fig. 6. Second, a shallow energy minimum is found at a distance of about 2.9 Å. Whether this minimum can be realized in the crystalline structures is however questionable. Another observation from Fig. 6 is that for a Ni–Ni distance lower than 2.5 Å the system becomes strongly unfavourable due to the formation of a bond between the two free Ni sites. This distance can be related to the metallic, crystalline fcc Ni phase, where a lattice constant of a = 3.524 Å31 is found. Thus the Ni–Ni distance accounts to 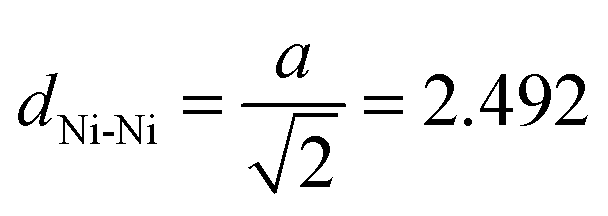 Å, in agreement with the trend seen in Fig. 6.
Å, in agreement with the trend seen in Fig. 6.
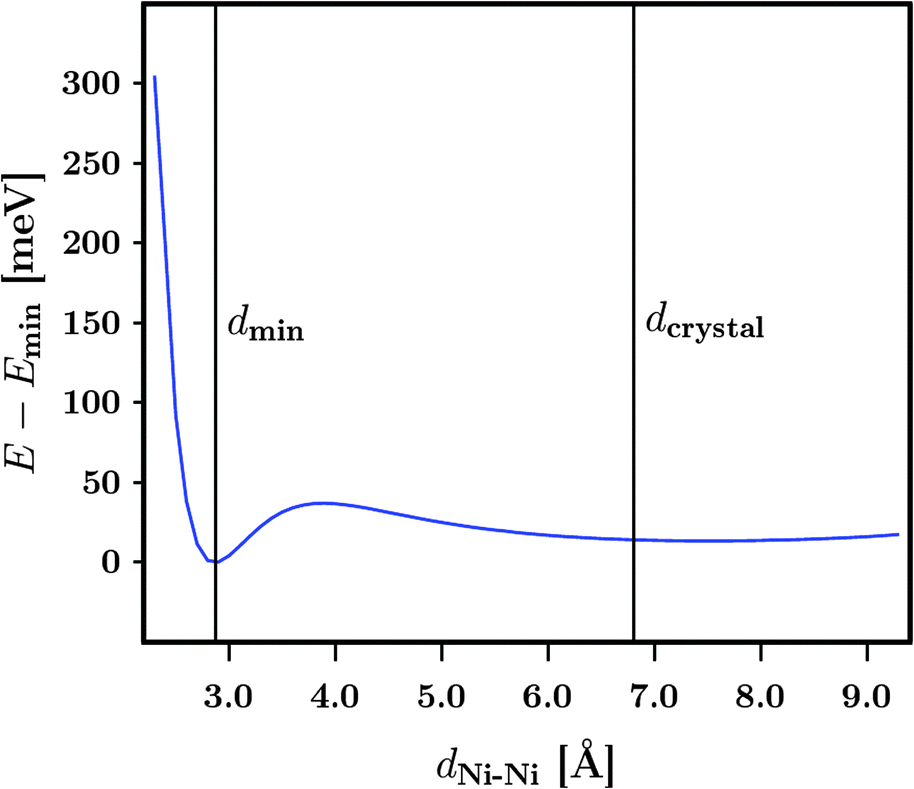 | ||
| Fig. 6 Relative energies of two DUT-8(Ni)open M3 molecules for an increasing distance dNi–Ni between them. The free Ni sites are facing each other. Emin is the energy minimum for the imposed magnetic ordering. | ||
4 Conclusions
The electronic and magnetic properties of the flexible MOF DUT-8(Ni) have been investigated by means of DFT. For a qualitative and quantitative analysis of the magnetic behaviour the energy difference between a low-spin and a high-spin coupling has been calculated. The DFT results were used as parameters for the calculation of the coupling constant as derived from the Heisenberg–Dirac–Van Vleck Hamiltonian. It has been found that the Ni–N interaction is most important for the magnetism within the systems. In both crystalline structures (DUT-8(Ni)open/DUT-8(Ni)closed) a low-spin ordering (S = 0) has been found to be the most favoured one. This result is confirmed by the supercell calculations, where a variety of different couplings between multiple unit cells has been analyzed, as well as from calculations including the vdW interaction. Several molecular model systems suggest a possible switch of the magnetic ground state to a high-spin character (S ≠ 0) of the coupling by certain changes in the structure. Especially the removal of the NH3 groups can be related to the removal of a dabco linker unit within the crystalline systems. All alterations can and should be introduced to the periodic systems both theoretically as well as experimentally to find out whether the modifications influence the magnetic ground state as indicated by the model systems.Acknowledgements
The authors want to thank the group of theoretical chemistry in Dresden for fruitful discussions, especially Diana Tranca and Igor Baburin. Thanks goes to Jens Kortus for cooperation and the ZIH in Dresden for computational time and support. This work is supported by funding of the Excellence Initiative by the German Federal and State Governments (Institutional Strategy, measure “support the best”).References
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1989, 111, 5962–5964 CrossRef CAS.
- H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2003, 427, 523–527 CrossRef PubMed.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valenta, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC.
- G. Ferey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC.
- A. Worsztynowicz, S. M. Kaczmarek, M. Bosacka, V. Mody and R. S. Czernuszewicz, Rev. Adv. Mater. Sci., 2007, 14, 24–32 CAS.
- U. Kortz, Y. P. Jeannin, A. Teze, G. Herve and S. Isber, Inorg. Chem., 1999, 38, 3670–3675 CrossRef CAS PubMed.
- P. de Loth, P. Cassoux, J. P. Daudey and J. P. Malrieu, J. Am. Chem. Soc., 1981, 103, 4007–4016 CrossRef CAS.
- C. Wang, K. Fink and V. Staemmler, Chem. Phys., 1995, 192, 25–35 CrossRef CAS.
- E. Coulaud, N. Guihery, J. P. Malrieu, D. Hagebaum-Reignier, D. Siri and N. Ferre, J. Chem. Phys., 2012, 137, 114106 CrossRef PubMed.
- N. Klein, C. Herzog, M. Sabo, I. Senkovska, J. Getzschmann, S. Paasch, M. R. Lohe, E. Brunner and S. Kaskel, Phys. Chem. Chem. Phys., 2010, 12, 11778–11784 RSC.
- H. C. Hoffmann, B. Assfour, F. Epperlein, N. Klein, S. Paasch, I. Senkovska, S. Kaskel, G. Seifert and E. Brunner, J. Am. Chem. Soc., 2011, 133, 8681–8690 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. Seitsonen, A. Smogunov, P. Umari and R. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- F. Bloch, Z. Phys., 1929, 52, 555–600 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- J. J. Mortensen, L. B. Hansen and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 1–11 CrossRef.
- A. H. Larsen, M. Vanin, J. J. Mortensen, K. S. Thygesen and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 195112 CrossRef.
- J. Miralles, O. Castell, R. Caballol and J. P. Malrieu, Chem. Phys., 1993, 172, 33–43 CrossRef CAS.
- W. Heisenberg, Z. Phys., 1926, 38, 411–426 CrossRef.
- P. A. M. Dirac, Proc. R. Soc. London, Ser. A, 1926, 112, 661–677 CrossRef CAS.
- J. H. Van Vleck, The theory of electric and magnetic susceptibilities, Clarendon Press, Oxford, 1932 Search PubMed.
- P. St. Petkow, G. N. Vayssilov, J. Liu, O. Shekhah, Y. Wang, C. Wöll and T. Heine, ChemPhysChem, 2012, 13, 2025–2029 CrossRef PubMed.
- L.-M. Yang, P. Vajeeston, P. Ravindran, H. Fjellvag and M. Tilset, Inorg. Chem., 2010, 49, 10283–10290 CrossRef CAS PubMed.
- O. Kahn, Molecular magnetism, Wiley, 1993 Search PubMed.
- E. Kokot and R. L. Martin, Inorg. Chem., 1964, 3, 1306–1312 CrossRef CAS.
- W. B. Pearson, A handbook of lattice spacings and structures of metals and alloys, Pergamon Press, London, 1958 Search PubMed.
| This journal is © the Owner Societies 2015 |