
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DFT-based Green's function pathways model for prediction of bridge-mediated electronic coupling†
Laura
Berstis
and
Kim K.
Baldridge
*
University of Zürich, Winterthurerstrasse 190, Zürich, CH-8057, Switzerland. E-mail: kimb@oci.uzh.ch
First published on 20th April 2015
Abstract
A density functional theory-based Green's function pathway model is developed enabling further advancements towards the long-standing challenge of accurate yet inexpensive prediction of electron transfer rate. Electronic coupling predictions are demonstrated to within 0.1 eV of experiment for organic and biological systems of moderately large size, with modest computational expense. Benchmarking and comparisons are made across density functional type, basis set extent, and orbital localization scheme. The resulting framework is shown to be flexible and to offer quantitative prediction of both electronic coupling and tunneling pathways in covalently bound non-adiabatic donor–bridge–acceptor (D–B–A) systems. A new localized molecular orbital Green's function pathway method (LMO-GFM) adaptation enables intuitive understanding of electron tunneling in terms of through-bond and through-space interactions.
Introduction
Considerable efforts in the last decade have been extended towards achieving accurate predictions of electronic coupling in bridge-mediated non-adiabatic electron transfer (ET) reactions. Of central interest are models based on quantum mechanical (QM) and density functional theory (DFT) schemes.1–10 For bridge-mediated ET reactions, tunneling can take place over large bridge (B) distances between donor (D) and acceptor (A), as a cooperativity of through-bond (TB) and/or through-space (TS) interactions (Fig. 1).‡ In this weak coupling regime, the rate of ET can be described by the Golden rule,11–14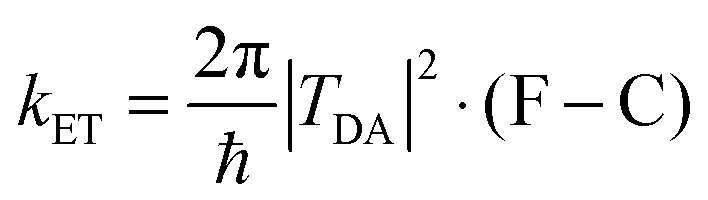 | (1) |
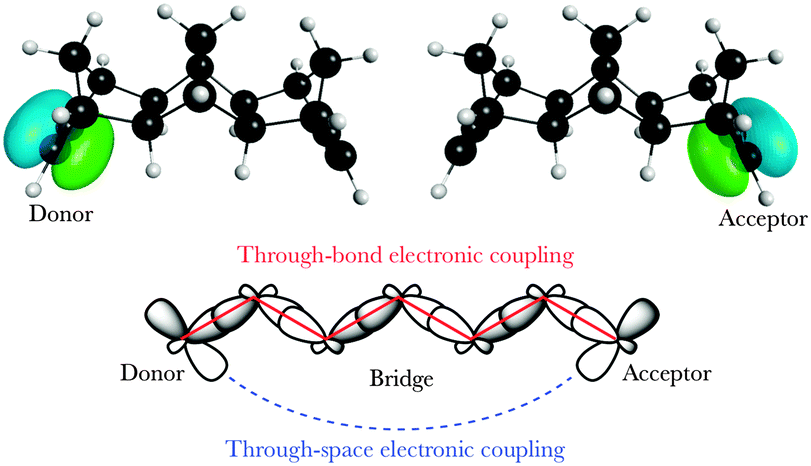 | ||
| Fig. 1 Degenerate localized π D/A orbitals on a model D–B–A system, with TB and TS interactions mediating electronic coupling. | ||
In this non-adiabatic regime, the reaction is controlled by two key factors. First, the Franck–Condon factor (F–C) is the nuclear factor that determines the thermally averaged density of states for configurations that have the reactant (transfer electron in the donor) and product (transfer electron in the acceptor) electronic states in resonance, enabling electron tunneling between them. The second factor is the electronic coupling between D and A states, TDA, which determines the tunneling frequency between D and A. In perturbation theory, kET is expressed as the product of the square of TDA and the probability of D and A forming a resonant activated complex.15,16 Perturbation theory requires that the electronic frequency, TDA/ℏ, be small compared to that of the relevant nuclear motion, to satisfy the non-adiabatic limit.6,17,18
An inherent challenge for quantitative theoretical predictions of reaction rate is high sensitivity to the accuracy of the electronic coupling, such that at ambient temperature, even a 0.1 eV error can lead to large error in rate prediction due to the square dependency. The electronic coupling is dependent on the mechanistic details through the bridge, which relates to the length and geometry of the bridge.19 As such, detailed understanding of TB and TS interactions and their influences on the electron tunneling pathways and transfer integrals aids in the understanding of the mechanism. Accurate computational prediction therefore requires methodology that can appropriately model the physics, particularly within the molecular context where D/A are covalently bound to the bridge.
In previous work, the pathways model was invoked to address questions of how tunneling through protein medium occurs.4,5,7,8,20 The methodology combined computationally inexpensive semi-empirical methods with molecular dynamics, to address important issues of dynamic effects and multiple pathway tubes in large protein structures. In the present effort, we return primarily to ET reactions dominated by a single pathway tube, but now further develop the models to improve quantitative TDA predictions with QM and density functional theory (DFT) methods, with an emphasis on accuracy as a function of wavefunction type or density functional, basis set, and localization scheme. In particular, a desirable goal is to enable prediction of electronic coupling to an error of less than 0.1 eV with respect to experiment. All implementations have been carried out in the computational chemistry suite, GAMESS,21 an open-source QM software. Results demonstrate predictive strengths of the method, as well as suggesting avenues of further development.
Theoretical methods for determination of electronic coupling and pathways in D–B–A systems
Several recent and extensive works have provided valuable perspectives of various theoretical approaches for determination of electronic coupling elements.1,5,6,9,10,22–30 Rather than repeat theoretical developments well discussed in the literature, we highlight components relevant to establishing reliable TDA predictions with Green's function methodologies.Effective Hamiltonian methodologies
Using a Löwdin partitioning scheme,10,24,25 the full D–B–A system is partitioned into two states, D/A and B, thereby mapping a high dimension QM eigenvalue problem onto an equivalent lower dimension problem with Hamiltonian,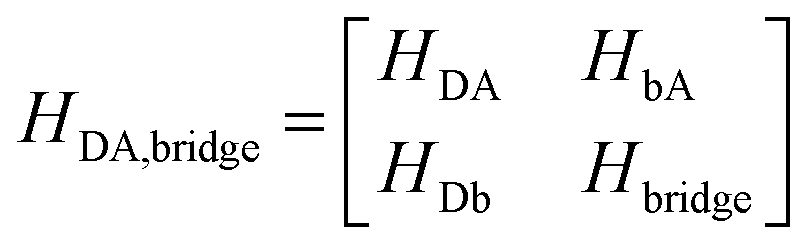 | (2) |
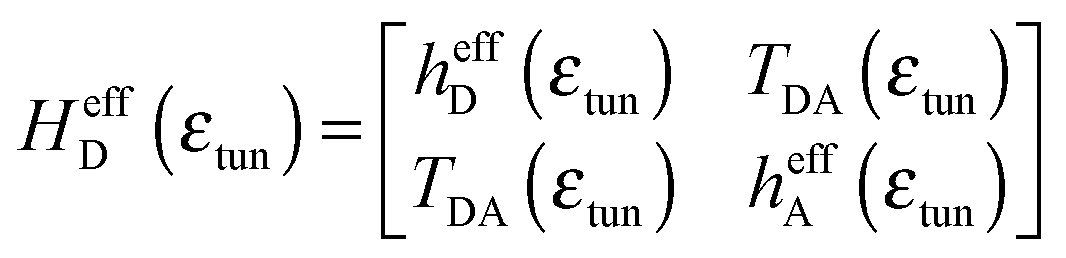 | (3) |
 | (4) |
 | (5) |
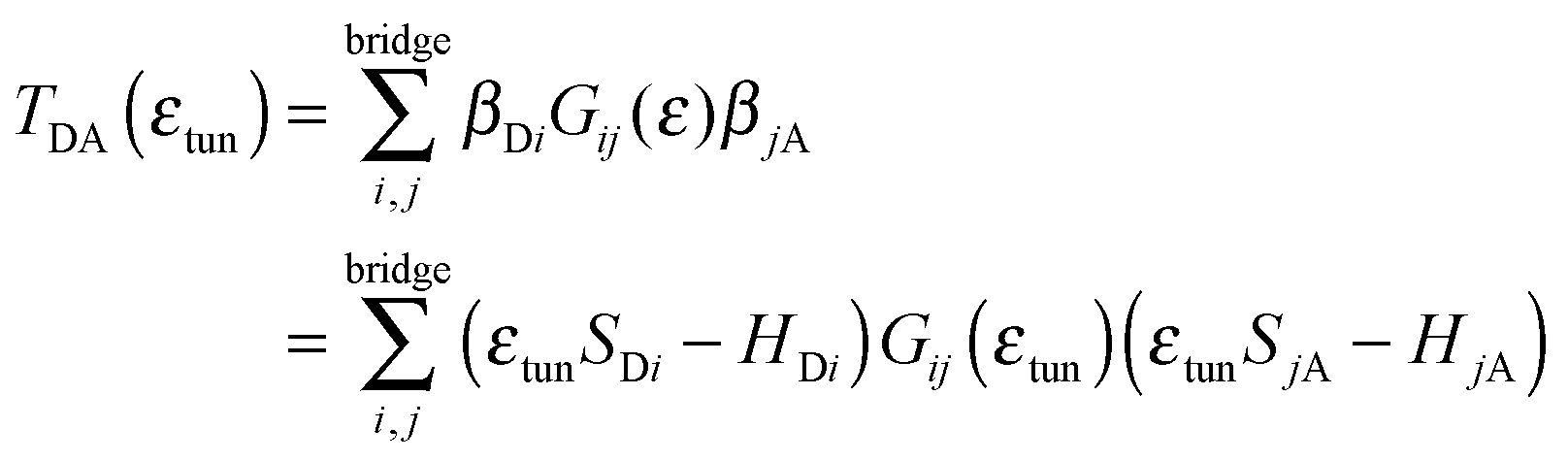 | (6) |
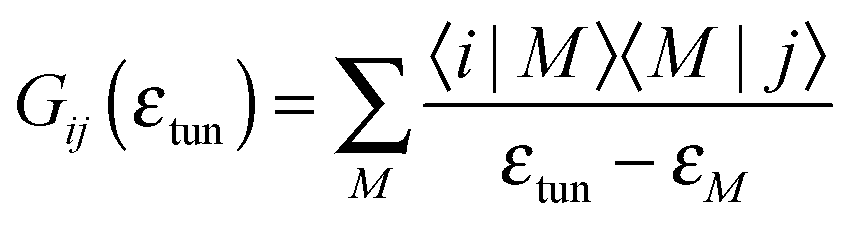 | (7) |
| G = S(εS − H)−1S | (8) |
This pathways approach for calculating the electronic couplings offers the ability to search through the various combinations of TB and/or TS pathways, to find those that maximize the D–A interaction through the bridge. In long hydrocarbon-like bridge constructions, the highest probability pathways are predominantly mediated by filled–filled orbital TB type interactions, with TS contributions playing a significant role at short bridge distances.32
Application of this model for prediction of TDAvia an effective Hamiltonian methodology relies on a localized representation of D, A, and B states. One finds, therefore, a dependency on the character and underlying quantum chemistry of the orbital localization routine. Related method developments from Skourtis et al. involving construction of an effective Hamiltonian have used non-orthogonal orbital localization schemes within the natural bonding orbital (NBO) analysis model.9,10,33,34 In the present work, several other localized molecular orbital (LMO) representations are evaluated, including the Edmiston–Ruedenberg (E–R),35,36 Boys,37 and Pipek–Mezey (P–M)38 localization routines, as implemented in GAMESS. While these routines generate orthogonal LMOs, the implemented methodology allows for either an orthogonal or a non-orthogonal LMO basis scheme.
In what follows, results of the effective Hamiltonian and pathways methodologies are characterized as a function of orbital localization method, basis set, and wavefunction type, providing a more detailed analysis of the features important for achieving reliable predictions of coupling elements with respect to experiment. Full details of all methodology can be found in available ESI.†
GFM pathway analysis techniques
In our previous work, electron transfer pathways were analyzed in terms of GFM decay elements, by association of the first atom in the pathway to each atom down the pathway,4,7,8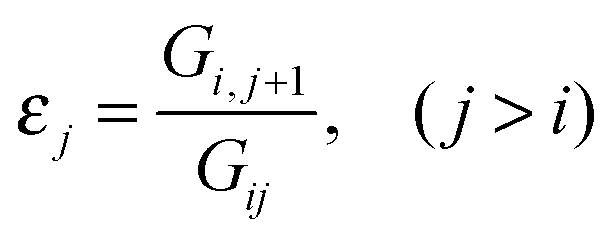 | (9) |
In previous studies, GFM elements were determined within an atomic orbital (AO) basis, with indices corresponding to the AO basis representation of the molecule.4,5 In the present work, formulation of the GFM within a localized molecular orbital (LMO) space is also developed. Transformation into a LMO basis yields a GFM showing virtual propagation through localized molecular orbitals along the bridge, offering an intuitive representation that appeals to the chemists' orbital-based description of TB and TS interactions.
Through-bond and through-space interactions
TB and TS interactions are responsible for the difference in energy between otherwise degenerate π orbitals separated by some distance within a molecule. Depending on the nature of the orbital interactions and their energy separation, the orbital levels are characteristically shifted, generating energy splittings.39 When two degenerate orbitals interact through space, two combination orbitals are formed: a higher energy antisymmetric form, π− = π1 − π2, and a lower energy symmetric form, π+ = π1 + π2. The magnitude of splitting of these near-degenerate orbitals is highly distance dependent; energy splittings are observed on the order of ∼4 eV at bonding distances, and exponentially diminish with increasing π–π orbital separation. When two such degenerate orbitals have an intervening σ bridge structure, there is the possibility for the nonbonding pair of orbitals to couple via TB interactions within the σ and σ* frameworks, as shown for the case of an even number of intervening bonds in Fig. 2. | ||
| Fig. 2 Orbital energy splitting resulting from TB and TS interactions in a simple hydrocarbon system with an even number of intervening σ bonds. | ||
In the case where coupling to a central σ bond plays a dominant role, the ordering of the energy levels can switch, such that the symmetric combination is higher in energy than the antisymmetric combination. This case is shown in Fig. 3 for an odd number of intervening bonds.
 | ||
| Fig. 3 Orbital energy splitting resulting from TB and TS interactions in a simple hydrocarbon system with an odd number of intervening σ bonds. | ||
As a result, the order of S/A orbitals can become a diagnostic of the type and relative magnitude of the electronic coupling. In terms of tunneling through a D–B–A system, the degree of TB and TS interactions provides important mechanistic information towards understanding electronic pathways.
Results and discussion
Extensive photoemission spectroscopy (PES) and electron transmission spectroscopy (ETS) studies40,41 on candidate D–B–A systems provide experimental ionization potential (IP) data comparisons for the present work. In particular, the set of molecules illustrated in Fig. 4 represent a test series of D–B–A systems characterized by ethylene D/A groups with intervening σ-bonded norbornyl-unit bridge units of varying length (Set A: 1–6) and construction (Set B: 4–11). All model test system geometries were optimized at the B97-D/Def2-TZVPP level of theory, and confirmed to be positive definite minima with subsequent Hessian analyses. | ||
| Fig. 4 Simple hydrocarbon ethylene model ET systems, representing Set A: 1–5 and Set B: 4–11. | ||
In Set A: 1–5, the intervening bridge length is systematically increased, providing the opportunity to investigate the correlation of electron transfer rate as a function of bridge length. Experimentally, ETS-measured vertical IP values reveal an exponential decay of the ET rate (and electronic coupling) with respect to the bridge length, as typically characterized by the value β. In such a series, long-range TB interactions (Fig. 5) are primarily responsible for mediating the extraordinarily rapid thermal ET rates between ethylene D/A groups across comparatively long hydrocarbon bridges (i.e., >6 Å).42
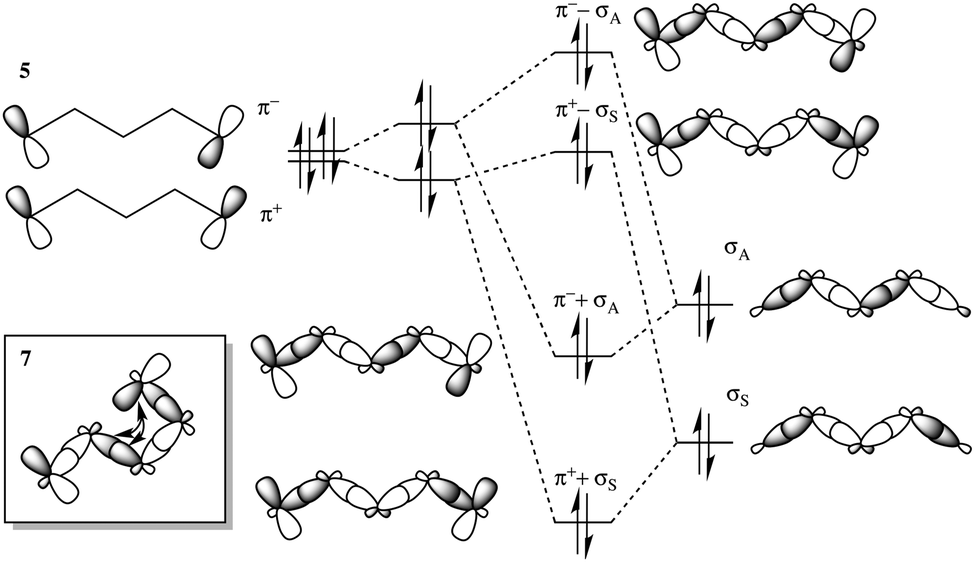 | ||
| Fig. 5 Long-range TB and TS interactions in representative systems 5 and 7. | ||
Investigations of Set B: 4–11, enable better understanding of how variation in bridge construction as a function of bridge length (and therefore contributions of TB and TS interactions), modulates electron transfer (e.g., see inset in Fig. 5). Reference experimental IP values enable investigation of how underlying QM methodology affects prediction with the current approach, through the relationship,
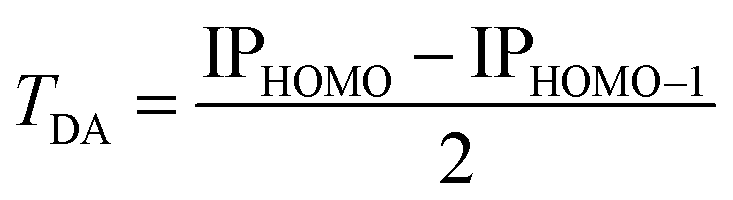 | (10) |
Coupled to the effective Hamiltonian approach, DFT methods are of particular interest for their capacity to produce reliable predictions with comparatively low computational costs. A few works have explored the use of BLYP or B3LYP for estimation of electronic coupling using KT,3 and for related chemical hardness predictions,44 and more recently, long-range corrected density functionals have been suggested as improved methods for prediction of IPs for small molecules.45
Effective Hamiltonian method performance
A detailed investigation of basis set and wavefunction type was carried out to determine the sensitivity of the effective Hamiltonian strategy on the underlying theoretical methodology. The basis set investigation was carried out for molecules of Set A, across a series of twenty-five basis sets, ranging from single to triple-ζ with varying diffuse and polarization functionality, with the dispersion-corrected density functional of Grimme, B97-D.46 Resulting electronic couplings for each level of theory in the form of the decay curves for lengthening bridges from 1 to 5 are summarized in Fig. 6 (all references and raw data provided as ESI†).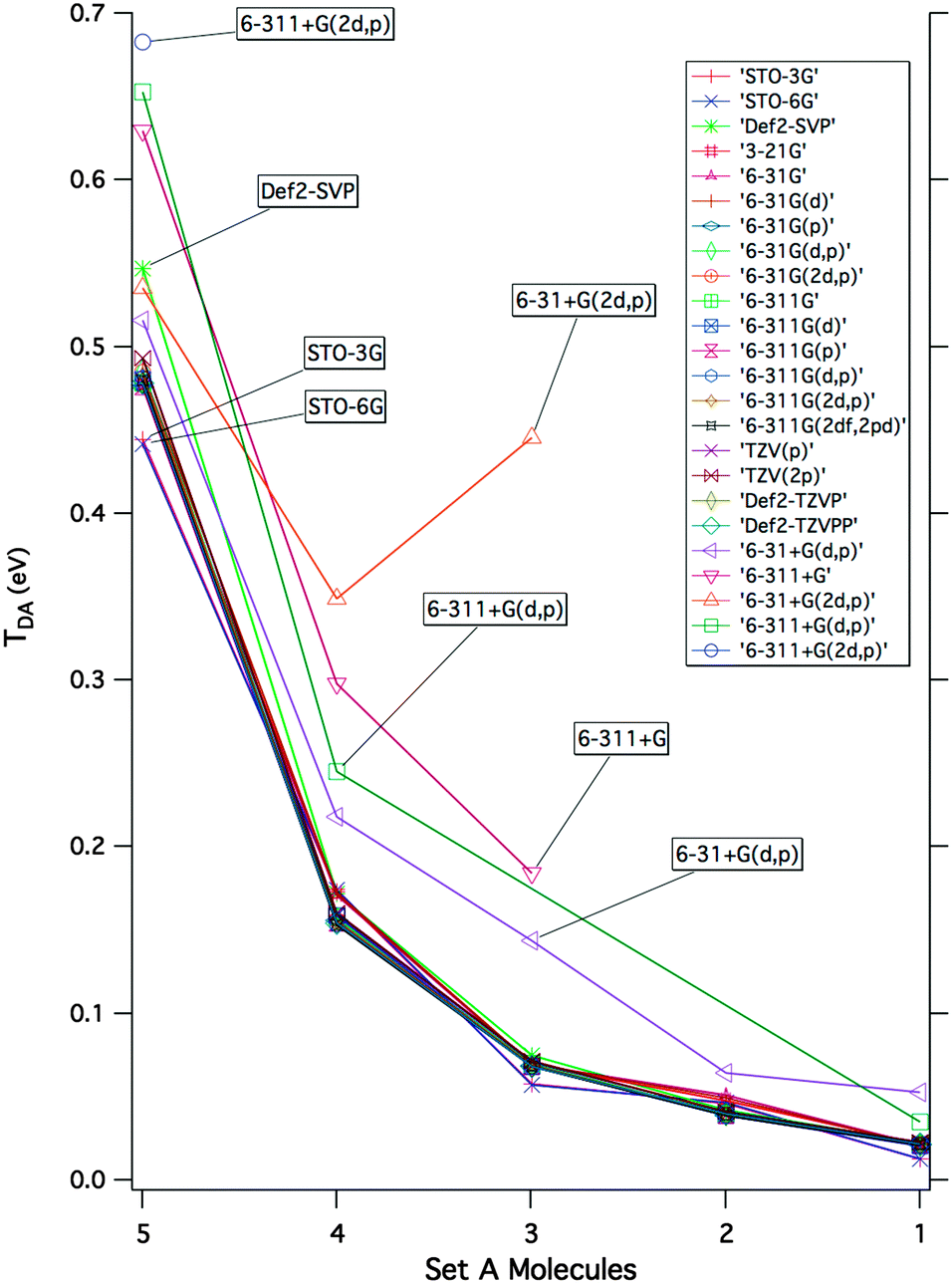 | ||
| Fig. 6 Decay curves generated from |TDA| predictions for the increasing D/A distance of Set A molecules 1–5, presented as a function of basis set, X, calculated at the theory level B97-D/X//B97-D/Def2-TZVPP. | ||
As a general observation, one finds a relative consistency in predictions across basis set, with the exception of when diffuse functionality is incorporated into the basis set. A minor sensitivity is observed across the various forms of single-ζ bases, and once at least a balanced double-ζ representation is attained (e.g., 6-31G(d,p)), predicted values are consistent through to the triple-ζ with large polarization extent. When diffuse functionality is incorporated, one finds chaotic behavior in the effective Hamiltonian convergence, and a significant overestimation of coupling energies. To further verify this effect, the basis set study was repeated within the Hartree–Fock ansatz, therefore eliminating the variability of DFT functionals and any DFT error cancellation issues. As presented in the ESI,† the erroneous overestimation of TDA with diffuse basis sets is still observed. This phenomenon, also noted in the literature,3,47–50 can be attributed to the local nature of the property, which is essentially lost when diffuse functionality expands the D/A areas for the electron to occupy into regions too delocalized onto the bridge.
With regard to the actual magnitude of the coupling, one finds that insufficiently small single-ζ basis sets without polarization functions, such as 3-21G and 6-31G, used in many early studies,51–53 underestimate TB coupling and therefore yield poor prediction of coupling elements. The present findings emphasize that reliable TDA prediction is supported by localized but sufficient representations of the electronic structure on the D/A, and B, with double-ζ or triple-ζ basis sets and adequate polarization to accommodate the orbital overlap along the bridge system. In the current findings, a triple-ζ polarized basis representation, such as Def2-TZVPP or 6-311G(2df,2pd), offers good choice for reliable prediction of TDA.
Following the basis set study, the impact of HF theory and density functional type, with molecular orbital localization scheme, were jointly analyzed for determination of TDA elements. Together with the 6-311G(2df,2pd) basis set, the RHF wave-function and DFT functional types, B3LYP, BLYP, PBE, PBE0, M06-L, M06, M06-2X, M06-HF, B97-D, and wB97x-D were evaluated. The orbital localization method choice is of particular importance, given both the range in theory and the need to have precise selection and representation of D/A orbitals. As electronic coupling elements are a function of orbital overlap, one expects to find variation as a function of orbital localization method. Each use different criteria together with a unitary transformation to transform standard delocalized canonical MOs into localized MOs, while still preserving the total electronic wavefunction. Resulting differences in orbital overlap along the σ-bonding network of the bridge will be manifested in variations of TDA, which is a product of the couplings for each step along the pathway. Here we investigate three orbital localization procedures as implemented in GAMESS: Pipek–Mezey (P–M) localization, Boys localization, and Edmiston–Ruedenberg (E–R) localization.
Fig. 7 summarizes mean absolute errors, MAEs, for prediction of electronic coupling with respect to experimental vertical ionization measurements,40 for molecules in Set B (for additional detail, see ESI†). Most notably, TDA is significantly overestimated when employing either the Boys or E–R localization methods. Although the Boys and E–R schemes are widely used orbital localization methods, both methods have localization criteria that are not necessarily optimal for representing the local σ-bond bridge frame-work, nor the weak coupling and orthogonality between the π-type D/A and σ-type bridge states. By maximizing the separation of the orbital centroids, the Boys method results in a high degree of mixing of the σ and π space, for example as illustrated in Fig. 8. The E–R method maximizes orbital self-repulsion energies, and also does not provide a rigorous σ–π separation.54 Accordingly, localization schemes that do not provide a highly localized description of D and A and/or a mixing of the σ and π space, tend to result in poor prediction of TDA regardless of wavefunction type.
 | ||
| Fig. 7 MAE (eV) of calculated TDA for systems 4–11 with reference to PES experiments, as a function of wavefunction/functional type and localization method, using the 6-311G(2df,2pd) basis set. | ||
 | ||
| Fig. 8 Localized highest occupied orbitals of molecule 6 across three orbital localization methods, depicting varying extents of σ- and π-orbital mixing. | ||
In contrast, the P–M orbital localization scheme provides well-defined, highly orthogonal σ and π spaces (Fig. 8). TDA predictions for Set B using P–M LMOs show MAE values reduced to 0.04 eV, a significant improvement over results with Boys or E–R LMOs (MAE errors of 0.340 and 0.347 eV, respectively). Very recent studies also suggest the choice of P–M orbital localization for other ET-relevant properties, and further show the relatively low scaling in computational cost compared to other localization schemes.55,56
Further analysis of TDA predictions across density functional type using the P–M orbital localization scheme together with the 6-311G(2df,2pd) basis set was next carried out. One finds reliable results with the BLYP and B97-D functionals, closely followed by PBE, and M06-L. The PBE exchange–correlation functional is a simplified non-empirical GGA functional, whereas PBE0 is a modified hybrid functional that includes 25% exact HF exchange. The Minnesota density functional series also enables a more systematic comparison of the impact of the percentage of HF exchange, from 0% (M06-L), to 27% (M06), to 54% (M06-2X), to full 100% (M06-HF). Within this series, the meta-GGA functional M06-L,57 and M06 yielded the lowest MAE for this test set, with proportionally increasing error moving to M06-2X and M06-HF. The same trend is observed in comparisons between PBE versus PBE0 hybrid functionals, and between BLYP with B3LYP functionals. Together, these results suggest that the effective Hamiltonian methodology achieves improved accuracy with DFT functionals that include a relatively low percentage (e.g., ∼25%) of HF exchange, similar to recent findings for prediction of TDA using different methodologies.58
As previously shown for other molecular properties,59 we suspect that the performance of several DFT functionals would further improve in this context with inclusion of the semi-empirical dispersion correction of Grimme.46 For example, the B97-D functional captures the weak TS and TB electronic interactions in the D–B–A model systems and provides overall good predictions with respect to experiment. Overall, the use of the well-performing meta and hybrid functionals together with P–M LMOs, the effective Hamiltonian approach achieves agreeable accuracy of TDA predictions, with relative low basis set sensitivity.
Effective Hamiltonian predictions for delocalized D/A groups
Systems of interest in D–B–A ET studies typically involve D/A groups of greater complexity than the ethylene D/A functionalities of Sets A and B. More complicated D–B–A systems, which typically involve delocalized π networks can introduce difficulties in determining D and A LMOs important to the ET process. To explore the effects of D/A choice on prediction of electronic coupling in more complex systems, we developed a method to scan through all possible D/A combinations of LMOs. This capability is exemplified for the Set C: 12–16 series of D–B–A systems (Fig. 9), which also have experimental comparisons available. This series is characterized by a dimethoxynaphthalene donor coupled to dicyanovinyl, with varying length norbornyl-type bridge. For a sequence of increasing bridge lengths, one expects to find an exponential decrease in D/A electronic coupling with increase in bridge length, associated with a sharp decay of TS interactions, and a progressive weakening in TB coupling. Experimental ET reaction rate measurements have been determined for this series enabling comparison to theoretical predictions viaeqn (1).32,42,53 | ||
| Fig. 9 Spectroscopically characterized series of D–B–A systems, Set C: 12–16, for variable bridge lengths. | ||
Analysis of all possible D/A LMO pairs reveals a particular LMO from each D and A, which dominates contributions to the electronic coupling. Selections among the various π-type LMOs within the 1,1-dicyanovinyl acceptor moiety reveal near-zero couplings with all selections except for the ethyne π-type LMO perpendicular to the plane of the cyano-groups. Pairing this acceptor LMO to all possible LMOs on the dimethoxynaphthalene, again reveal negligible couplings for all but the central ethylene LMO (‘k’ in Table 1).
|
|
|||||
|---|---|---|---|---|---|
| D–B–A system in Set C | |||||
| LMO | 12 | 13 | 14 | 15 | 16 |
| i | 0.0006 | 0.0053 | 0.0052 | 0.0001 | 0.0000 |
| j | 0.0163 | 0.0054 | 0.0005 | 0.0002 | 0.0069 |
| k | 0.3689 | 0.1126 | 0.0253 | 0.0080 | 0.0022 |

Calculated values of TDA with this LMO selection correlate well to the experimentally observed decay curve as a function of increasing bridge length. The experimental ET rate decay factor, β, a three-point correlation, was determined to be 0.85.32,42,60 This result can be compared to the effective Hamiltonian's predicted value for Set C of ∼0.64 (Fig. 10). This is in acceptable agreement with the experimentally observed decay, and is well within the proposed range of 0.4–0.7 for these types of D–B–A systems.5,6 These findings suggest that when using an appropriate combination of D and A LMOs, the effective Hamiltonian method can be useful in describing the electronic coupling properties for this form of covalently-bonded D–B–A system. Further investigation is warranted to evaluate the performance of this approach with more complex conjugated D and A groups, to support the use of such an approach for a broad spectrum of applications.
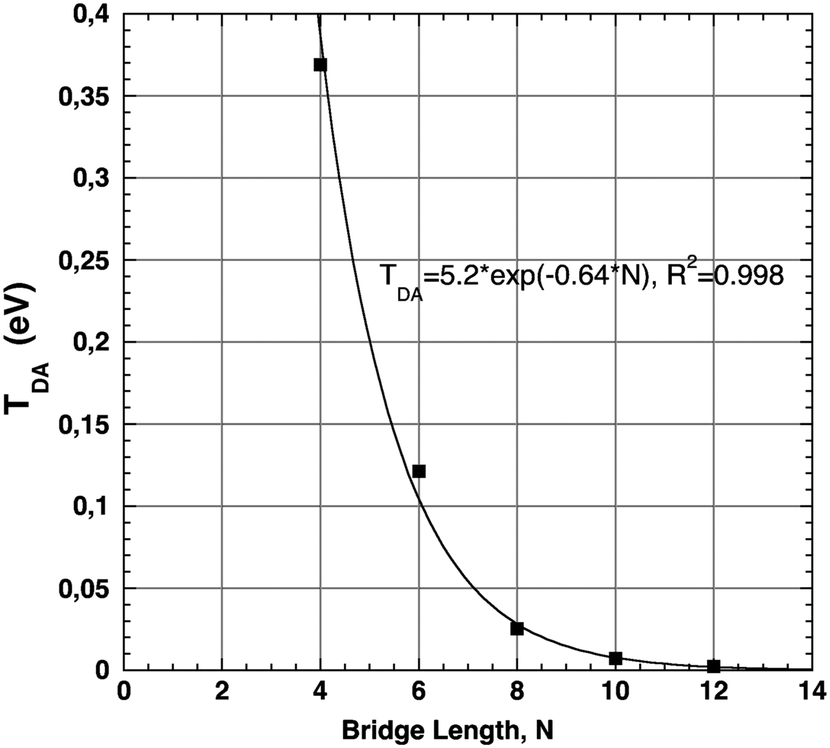 | ||
| Fig. 10 T DA of systems of Set C: 12–16 of increasing bridge length, N, calculated at B97-D/6-311G(2df,2pd) yielding exponential decay curve of β decay factor 0.64. | ||
Pathways model: AO-based approach
In typical D–B–A systems, highly interconnected bridge structures impart a large number of possible tunneling pathways, hampering the ability to predict the impact of variations in pathway electronic structure. Design of a simplified model D–B–A system with a limited number of tunneling pathways enables a more controlled exploration of the influence of bridge structure and substituents on the magnitude of electronic coupling. Set D: 17–26 shown in Fig. 11 was designed for such an investigation. | ||
| Fig. 11 One- and two-pathway model systems, Set D: 17–26. | ||
All structures in Set D are based on the two-pathway hydrocarbon system, 22, which has two identical pathways between the ethylene D to A groups. This system was fully optimized in C2v symmetry with B97-D/Def2-TZVPP and confirmed a positive definite minimum with Hessian analysis. Modified from this parent system, the additional two-pathway systems, 23–26, were created by freezing the identical backbone and optimizing the added electron withdrawing group (EWG) or electron donating group (EDG), to differentiate the two pathway options. Each substituent was optimized in two rotational conformations preserving Cs symmetry. For example, the EWG, NO2, is considered with the π orbital plane oriented both parallel and perpendicular to the tunneling pathway, and the EDG, NH2, is considered with the lone pair oriented up or down with respect to the curvature of the hydrocarbon chain. For comparison, the analogous one-pathway systems, 17–21, were also investigated with an identical framework to a single branch of the 2-path systems. With the bridge geometry preserved among all one- and two-pathway systems, the impact of the substituent on the calculated TDA is isolated. B97-D/Def2-TZVPP level TDA and pathways calculations on P–M LMOs were conducted for each system in the set.
Previous efforts using the pathways model have predominantly involved either Hückel theory, semi-empirical, or Hartree–Fock theory, together with minimal single-ζ basis set descriptions.4,7–10 In those studies, pathway decay coefficients were selected based on the single ‘2s’ orbital representations for each nucleus along the bridge backbone. However, as demonstrated in the previous sections, single-ζ levels of theory are not particularly reliable for capturing the weak TB and TS interactions, and are typically quantitatively inaccurate. When moving to a double- or triple-ζ basis sets, complications arise as to which groupings of AO representations appropriately characterize the GFM decay or propagation coefficients. For example, unlike a single-ζ basis representation, a triple-ζ basis has 3 valence ‘2s’ orbital coefficients, resulting in 9 GFM elements for each step along the bridge. Several approaches were considered in the present work for treating the multiple cross-terms in the GFM corresponding to correlations of each valence ‘s’ and ‘p’ type orbital. The most consistent measure of probability coefficients through the pathway was found to be an average of all 9 cross terms of the ‘2s’ valence AOs. The resulting TDA predictions are summarized in the insets of Fig. 12, as well as corresponding propagation pathway coefficients along each designed bridge of Set D.
 | ||
| Fig. 12 Pathway propagation elements for the 6 TB steps of each branch of one-branch 17–26 (left) and two-path systems 22–26 (right) calculated with B97-D/Def2-TZVPP. Inset tables compare the overall TDA prediction (eV). | ||
In all systems of Set D, the 6 TB pathway steps are palindromic due to the underlying symmetry (Cs) of the minimum energy structure. The propagation coefficients across the pathway of the simplest one-path system, 17, maintain nearly equal magnitudes, due to the near equivalence of these sp3-hybridized carbon-to-carbon steps. Similarly, both (equivalent) branches of the unsubstituted two-pathway system, 22, yield a series of propagation coefficients with consistently high magnitudes. The availability of a constructively interfering second pathway with high tunneling propagation magnitudes is reflected in the nearly two-fold increase in TDA over the one-pathway system.
Addition of a central substituent along a branch of a pathway results in interesting modulations in TDA and associated pathway coefficients. Incorporating the EDG into the one-pathway system in either orientation, 20 or 21, results in a slight increase in TDA. This increase in TDA is expected due to increased electron donation onto the bridge, with commensurate increase in orbital overlap, facilitating propagation of the tunneling electron. The same pattern is observed in the two-pathway EDG systems, 25 and 26. The perturbation of the pathway coefficients along the unsubstituted branches of these bridges is negligible with respect to the unsubstituted case.
Incorporation of the EWG, NO2, results in an orientation-dependent effect. With the oxygen atoms of the EWG oriented parallel to the axis of the pathway, 18, the coupling energy increases with a similar magnitude as with the NH2 substitution. However, rotating the NO2 π-orbital plane perpendicular to the pathway, 19, results in a significant drop in TDA, and a corresponding decrease in the propagation coefficients closest to the NO2 group (TB steps 3 and 4). Again in this case, the two-path systems show the same trend, with a marked decrease in pathway coefficients as well as TDA predictions for the NO2 group in a perpendicular orientation, as in 24. In this system, both the H- and EWG-substituted pathway coefficients decrease with respect to 22, whereas the net coupling across the bridge in 23 increases. This observation points to an intriguing capacity for substituents to have a switch-like effect, depending on their orientation, either pulling or pushing electrons in a potential tunneling pathway.
The insight from the GFM analysis of bridge substituent effects is further supported by analysis of the change in total electron density. Fig. 13 depicts electron density contour plots calculated for an identical 2D plane through each system. These maps reveal that NO2, when oriented parallel to the bridge as in systems 18 and 23, has little impact on the electron density on the bridge with respect to the unsubstituted species 17 and 22. In contrast, contour plots of the structure with NO2 rotated 90° into the perpendicular orientation as in 19 and 24, depict increasing density directly onto the central carbon, and significantly reduced electron density on the subsequent atom centers towards the ethylene D/A groups. The notable decrease in electron density on carbons 2 and 4 along the bridge corresponds to decreases observed in propagation elements and overall decrease in TDA.
 | ||
| Fig. 13 B97-D/Def2-TZVPP electron density contour maps computed for the identical molecular plane for each of systems 17–26. | ||
Overall, this approach for predicting TDA and propagation coefficients enables comparative investigation of bridge modifications. Through these analyses across Set D systems, one can clearly identify the importance of EWG vs. EDG substituents as well as their geometrical orientation on pathway properties.
Tunneling pathways in a Gly5 model peptide
A simple model peptide was constructed for application of the GFM approach for investigation of tunneling propagation elements as a function peptide conformation. A peptide composed of five glycine residues with neutral-charged termini was optimized at the B97-D/Def2-TZVPP level of theory, with constraints to hold specific internal dihedral angles to idealized average values.61 Three different idealized peptide geometries were thus optimized: an α-helix (Φ = −57°, Ψ = −47°), 3–10 helix (Φ = −49°, Ψ = −26°), and β-strand (Φ = −129°, Ψ = −124°). As employed in other electronic coupling investigations across alkane bridge constructions,10 beryllium (Be) atoms were selected as model D/A entities. For this Gly5 model, Be atoms were systematically oriented with 2.5 Å separation from the final C–C bond of each the C- and N-terminus, a distance found to yield small D–B and B–A couplings agreeable to calculation within the non-adiabatic weak coupling regime.10With each Gly5 bridge conformation as the tunneling medium, TDA was calculated between the valence orbitals of the Be atoms, and the associated GFMs analyzed using the converged εtun value within the Green's function. The propagation coefficients, again taken as the average of the ‘2s’ orbital elements of the triple-ζ basis GFM, were recorded for each C, N and O atom along the TB-pathway of the peptide backbone, as well as for the TS steps through three H-bonding routes (accessible to only specific peptide conformations), for a total of four possible pathways. The H-bond connectivity a, (Fig. 14) bridging the first 1GLY to the final 5GLY residue, is accessible only to the α-helix conformation, whereas the 3–10 helix has H-bonding contacts in positions b and c, joining 1GLY–4GLY and 2GLY–5GLY, respectively. The product of the propagation coefficients along each pathway option provides an estimate of the relative contributions of each TB or TS pathway to the overall electronic coupling.
 | ||
| Fig. 14 Models of the three Gly5 conformations, their D–A distances (rDA), total electronic coupling, TDA, and contributions of each potential pathway, tDA, given as the product of propagation coefficients through each path, a, b and c, which include a through-space (TS) H-bond step, and the entirely through bond (TB) pathway. | ||
Significant contributions to the electronic coupling through the bridge are observed as a result of the H-bond connectivity in the helices. The predicted TDA value through the α-helix is the largest not only due to the shortest D/A distance of 12.25 Å, but also given that the H-bond, a, provides a pathway of fewest TB/TS steps between N- and C-termini. Analysis of propagation coefficient products, tDA in Fig. 14, through path a for each geometry clearly associates optimal accessibility in this pathway via the α-helix conformation. Comparatively, pathways b and c, which are most optimally oriented within the 3–10 helical structure, also yield propagation coefficient products that indicate accessibility to a tunneling electron. Nevertheless, the greater number of steps and tunneling distance in paths b and c results in an overall lower TDA than through the α-helix.
The D/A distance increases dramatically moving into the β-strand conformation, and therefore, the overall coupling TDA is reduced by several orders of magnitude. In this extended Gly5 form, H-bonding pathways, a, b and c, show negligible contributions to tunneling. As reflected through analysis of propagation coefficient products, the TB pathway is dominant. Further, the near-planarity of the torsion angles along the β-strand backbone generates more optimal overlap for the TB pathway through the Gly5 backbone, relative to the helical forms.
Using semi-empirical QM approaches within an adiabatic dipole transition model to estimate electronic coupling, Shin et al. also predict an analogous decay of four orders of magnitude as one moves from the α-helix backbone torsions to those of a β-strand in analogous Glyn bridges.62 Our DFT pathways and effective Hamiltonian analyses further support and detail these findings, by demonstrating the impact of different non-covalent interactions on the tunneling propagation elements. As such, these methods lend themselves as useful theoretical tools to guide and complement efforts to engineer peptide or other organic D–B–A systems with specific charge transfer properties.
Formulation of a LMO-based pathways approach
As discussed in the previous sections, the AO-based methodology for determination of pathway coefficients raises several questions regarding the most appropriate treatment of AO contributions in complex basis sets. Although this and previous works applying an AO-basis for pathways analysis demonstrate the utility of this method, we also propose alternative representations of the pathway states, which circumvent the issues of AO choices. Despite the potential appeal of a canonical molecular orbital (CMO) basis, CMOs pair poorly to the pathways model analysis for a covalently-bound bridge. The large extent of delocalization through the bridge orbitals results in a lack of sequential TB and TS steps to trace the electronic interaction between D and A as a pathway. Alternatively, LMOs offer a means for the partitioning of covalently-bound D–B–A systems, which resolves the delocalization challenge from CMOs. Furthermore, the use of LMOs provides provides an intuitive view of the underlying TB and TS interactions responsible in ET coupling. Accordingly, we have transformed the original GF pathways algorithm to accommodate the LMO basis, transforming eqn (8) as follows:| GLMO = SLMO(εSLMO − HLMO)−1SLMO | (11) |
| SLMO = ϕ†SAOϕ | (12) |
| HLMO = ϕ†HAOϕ | (13) |
 | ||
| Fig. 15 Possible tunneling pathways in 5, from a top-down view perspective. | ||
| Propagation step | Pathway propagation coefficients (a.u.) | ||||
|---|---|---|---|---|---|
| A | B | C | D | E | |
| 1 | −3.14 | −2.49 | −3.14 | −3.14 | −3.14 |
| 2 | 0.93 | 1.68 | 0.93 | 1.86 | 1.86 |
| 3 | −2.96 | 0.24 | 2.14 | 1.68 | 2.26 |
| 4 | −0.93 | 1.68 | −2.14 | 0.24 | −0.02 |
| 5 | 3.14 | 2.49 | 0.93 | 1.68 | −2.96 |
| 6 | — | — | 3.14 | 2.49 | 0.93 |
| 7 | — | — | — | — | 3.14 |
Path A, through four bonds along either side of the bridge, has a symmetry induced palindromic series of coefficient magnitudes. In addition to the oscillating sign of the coefficients, previously shown to be indicative of a dominant pathway,4 this pathway option is expected to have the highest probability, due to significant TB interactions. This is seen in the large magnitude coefficients through these σ bonds, and the fewest steps in the pathway (i.e. shortest tunneling distance).
Path C, which switches from one side of the molecule to the other through the central cross over bridge, also maintains similarly high coefficients, although it has one additional TB step compared to path A. In contrast to the relatively high magnitudes of coefficients in paths A and C, the hyperconjugation-like path B, has a marked drop-off midway along the path (step 3 of Table 2), corresponding to the low-overlap TS jump across the central C–H and H–C bonds. While paths D and E maintain higher overlap TB steps, these longer path lengths consequently have lower overall tunneling probabilities.
These results support further development of the LMO basis for analysis of tunneling pathways, particularly for more complex covalently-bound bridge systems. Also, analogous to the utility of the LMO description of D and A states in the effective Hamiltonian approach, the LMO-based pathways model offers a solution for partitioning bridge states into intuitive fragments, which complement the underlying TB and TS theory.
Conclusions
In this work, GFM approaches for calculation of tunneling pathways and associated electronic coupling, TDA, are investigated as a function of the underlying electronic structure theory methodology. Through these evaluations, we demonstrate that, with careful consideration of underlying wavefunction and DFT methodology, the effective Hamiltonian and pathways models are able to achieve the set out error tolerance of less than 0.1 eV with respect to experiment. We find improved accuracy of TDA predictions with a variety of modern meta-GGA and hybrid DFT functionals, when coupled to a sufficient non-diffuse basis sets, and an orbital localization scheme that avoids extensive mixing of σ and π spaces. Previous complications associated with extension of the methodology to larger basis sets is addressed when utilizing AO representations, by taking an average of all relevant matrix elements in the AO representations.Propagation elements extracted from the GFM pathways analysis enables quantitative comparison of tunneling probabilities along pathways in varying organic and biomolecular systems. Finally, a new LMO-GFM pathways model adaptation is presented offering a more intuitive understanding of electron tunneling through D–B–A systems in terms of TB and TS interactions.
Further developments of the described methodology to include effects of solvent are ongoing, which is particularly important for experimental models where solvent becomes crucial to interpretation of electron transfer phenomenon. Additional work is focused on coupling the methodology with calculation of n-dimensional Franck–Condon factors for prediction of electron transfer rates.
Acknowledgements
We acknowledge the University of Zürich and associated CMSZH, and the Swiss National Science Foundation, for support of this research. We are grateful to Prof. Jose Onuchic and Prof. Jay S. Siegel for helpful discussions.Notes and references
- I. A. Balabin and J. N. Onuchic, J. Phys. Chem. B, 1998, 102, 7497–7505 CrossRef CAS.
- E. Cukier and R. J. Cave, Chem. Phys. Lett., 2005, 402, 186–191 CrossRef CAS PubMed.
- L. A. Curtiss and J. R. Miller, J. Phys. Chem. A, 1998, 102, 160–167 CrossRef CAS.
- C. Kobayashi, K. K. Baldridge and J. N. Onuchic, J. Chem. Phys., 2003, 119, 3550–3558 CrossRef CAS PubMed.
- J. N. Onuchic and D. N. Beratan, J. Chem. Phys., 1990, 92, 722–733 CrossRef CAS PubMed.
- J. N. Onuchic, D. N. Beratan, J. R. Winkler and H. B. Gray, Annu. Rev. Biophys. Biomol. Struct., 1992, 21, 349–377 CrossRef CAS PubMed.
- J. N. Onuchic, C. Kobayashi and K. K. Baldridge, J. Braz. Chem. Soc., 2008, 19, 206–210 CrossRef CAS PubMed.
- J. N. Onuchic, C. Kobayashi, O. Miyashita, P. Jennings and K. K. Baldridge, Philos. Trans. R. Soc., B, 2006, 361, 1439–1443 CrossRef CAS PubMed.
- S. Priyadarshy, S. S. Skourtis, S. M. Risser and D. N. Beratan, J. Chem. Phys., 1996, 104, 9473–9481 CrossRef CAS PubMed.
- A. Teklos and S. S. Skourtis, J. Chem. Phys., 2006, 125, 244103 CrossRef PubMed.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265–322 CrossRef CAS.
- K. V. Mikkelsen and M. A. Ratner, Chem. Rev., 1987, 87, 113–153 CrossRef CAS.
- M. D. Newton, Chem. Rev., 1991, 91, 767 CrossRef CAS.
- M. D. Newton and N. Sutin, Annu. Rev. Phys. Chem., 1984, 35, 437–480 CrossRef CAS.
- J. J. Hopfield, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3640–3644 CrossRef CAS.
- J. Jortner, J. Chem. Phys., 1976, 64, 4860–4867 CrossRef CAS PubMed.
- A. T. Balaban, Bull. Soc. Chim. Belg., 1996, 105, 383–389 CAS.
- A. Heckmann and C. Lambert, Angew. Chem., Int. Ed., 2012, 51, 326–392 CrossRef CAS PubMed.
- D. N. Beratan and S. S. Skourtis, Curr. Opin. Chem. Biol., 1998, 2, 235–243 CrossRef CAS.
- W. B. Curry, M. D. Grabe, I. V. Kurnikov, S. S. Skourtis, D. N. Beratan, J. J. Regan, A. J. A. Aquino, J. N. Berza and J. N. Onuchic, J. Bioenerg. Biomembr., 1995, 27, 285–293 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus and S. T. Elbert, J. Comput. Chem., Jpn., 1993, 14, 1347 CrossRef CAS PubMed.
- D. N. Beratan, J. N. Onuchic, J. R. Winkler and H. B. Gray, Science, 1992, 258, 1740–1741 CAS.
- I. V. Kurnikov and D. N. Beratan, J. Chem. Phys., 1996, 105, 9561–9573 CrossRef CAS PubMed.
- P.-O. Löwdin, J. Math. Phys., 1962, 3, 969–982 CrossRef PubMed.
- P.-O. Löwdin, J. Mol. Spectrosc., 1964, 14, 131–144 CrossRef.
- T. R. Prytkova, I. V. Kurnikov and D. N. Beratan, J. Phys. Chem., 2005, 109, 1618–1625 CrossRef CAS PubMed.
- A. de la Lande, N. S. Babcock, J. Řezáč and B. Lévy, Phys. Chem. Chem. Phys., 2012, 14, 5902–5918 RSC.
- M. D. Newton, Chem. Rev., 1991, 91, 767–792 CrossRef CAS.
- M. Rust, J. Lappe and R. J. Cave, J. Phys. Chem. A, 2002, 106, 3930–3940 CrossRef CAS.
- S. S. Skourtis and D. N. Beratan, Adv. Chem. Phys., 1999, 106, 377 CrossRef CAS PubMed.
- I. A. Balabin and J. N. Onuchic, Science, 2000, 290, 114–117 CrossRef CAS.
- K. D. Jordan and M. N. Paddon-Row, J. Phys. Chem., 1992, 96, 1188–1196 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, 2001 Search PubMed.
- F. Weinhold, J. Comput. Chem., 2012, 33, 2363–2379 CrossRef CAS PubMed.
- C. Edmiston and K. Ruedenberg, in Quantum Th. of Atoms, Mols. & Solid St, ed. P. O. Löwdin, Academic Press, New York, London, 1966, p. 263 Search PubMed.
- R. C. Raffenetti, K. Ruedenberg, C. L. Jonssen and H. F. Schaefer, Theor. Chim. Acta, 1993, 86, 149–165 CrossRef CAS.
- S. F. Boys, in Quantum Th. of Atoms, Mols. & Solid St, ed. P. O. Löwdin, Academic Press, New York, London, 1966, p. 253 Search PubMed.
- J. Pipek and P. Z. Mezey, J. Chem. Phys., 1989, 90, 4916 CrossRef CAS PubMed.
- K. K. Baldridge, T. R. Battersby, R. VernonClark and J. S. Siegel, J. Am. Chem. Soc., 1997, 119, 7048–7054 CrossRef CAS.
- V. Balaji, L. Ng, K. D. Jordan, M. N. Paddon-Row and H. K. Patney, J. Am. Chem. Soc., 1987, 109, 6957–6969 CrossRef CAS.
- K. Kim, K. D. Jordan and M. N. Paddon-Row, J. Phys. Chem., 1994, 98, 11053–11058 CrossRef CAS.
- H. Oevering, M. N. Paddon-Row, M. Heppener, A. M. Oliver, E. Cotsaris, J. W. Verhoeven and N. S. Hush, J. Am. Chem. Soc., 1987, 109, 3258–3269 CrossRef CAS.
- T. Koopmans, Physica, 1934, 1, 104 CrossRef.
- R. Shankar, K. Senthilkumar and P. Kolandaivel, Int. J. Quantum Chem., 2009, 109, 764–771 CrossRef CAS PubMed.
- T. Tsuneda, J.-W. Song, S. Suzuki and K. Hirao, J. Chem. Phys., 2010, 133, 174101 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- X. Li, Z. Cai and M. D. Sevilla, J. Chem. Phys. A, 2002, 106, 1596–1603 CrossRef CAS.
- A. A. Voityuk, J. Chem. Phys., 2005, 034903 CrossRef PubMed.
- C. Herrmann, G. C. Solomon, J. E. Subotnik, V. Mujica and M. A. Ratner, J. Chem. Phys., 2010, 132, 024103 CrossRef PubMed.
- S.-H. Ke, H. U. Baranger and W. Yang, J. Chem. Phys., 2007, 127, 144107 CrossRef PubMed.
- A. H. A. Clayton, G. D. Scholes, K. P. Ghiggino and M. N. Pasddon-Row, J. Phys. Chem., 1996, 100, 10912–10918 CrossRef CAS.
- L. A. Curtiss, C. A. Naleway and J. R. Miller, J. Phys. Chem., 1993, 97, 4050–4058 CrossRef.
- K. D. Jordan and M. N. Paddon-Row, Chem. Rev., 1992, 92, 395–410 CrossRef CAS.
- J. W. Boughton and P. Pulay, J. Comput. Chem., 1993, 14, 736–740 CrossRef CAS PubMed.
- I.-M. Hoyvik, B. Janisk and P. Jørgensen, J. Comput. Chem., 2013, 34, 1456–1462 CrossRef CAS PubMed.
- S. Lehtola and H. Jonsson, J. Chem. Theory Comput., 2014, 10, 5324–5337 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- A. Kubas, F. Hoffmann, A. Heck, H. Oberhofer, M. Elstner and J. Blumberger, J. Chem. Phys., 2015, 140, 104105 CrossRef PubMed.
- E. G. Hohenstein, S. T. Chill and C. D. Sherrill, J. Chem. Theory Comput., 2008, 4, 1996–2000 CrossRef CAS.
- G. L. Closs and J. R. Miller, Science, 1988, 240, 440–447 CAS.
- G. A. Petsko and D. Ringe, Protein Structure and Function, New Science Press, London, 2004 Search PubMed.
- Y.-G. K. Shin, M. D. Newton and S. S. Isied, J. Am. Chem. Soc., 2003, 125, 3722–3732 CrossRef CAS PubMed.
- B. M. Bode and M. S. Gordon, J. Mol. Graphics Modell., 1999, 16, 133–138 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of computational methodology and results not shown in main text are available free of charge. See DOI: 10.1039/c5cp01861g |
| ‡ Molecular graphics were prepared with MacMolPlt.63 |
| This journal is © the Owner Societies 2015 |