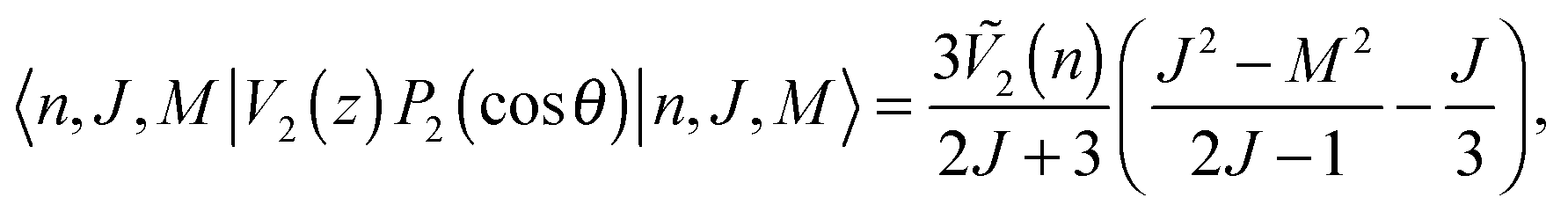Hindered rotational physisorption states of H2 on Ag(111) surfaces
Received
24th March 2015
, Accepted 23rd June 2015
First published on 23rd June 2015
Abstract
We have investigated the physisorption states of H2 on Ag(111) surfaces. To clarify the accurate adsorption properties of H2 on Ag(111), we performed first-principles calculations based on spin-polarized density functional theory (DFT) with the semiempirical DFT-D2 method and the newly-developed exchange functional with the non-local correlation functional vdW-DF2 (rev-vdW-DF2). We constructed exhaustive potential energy surfaces, and revealed that non-negligible out-of-plane potential anisotropy with a perpendicular orientation preference exists even for H2 physisorption on planar Ag(111), as predicted by previous results of resonance-enhanced multiphoton ionization spectroscopy and temperature-programmed desorption experiments. Therefore, the molecular rotational ground states of ortho-H2 split into two energy levels in the anisotropic potential. The obtained adsorption energy and the number of bound states, including the zero-point energies and the rotational energy shift, agree with diffractive and rotationally mediated selective adsorption scattering resonance measurements. The origin of the potential anisotropy on Ag(111) is a combination of the London dispersion interaction and the virtual transition of the metal electron to the unoccupied molecular state.
1. Introduction
One of the most important topics in surface science is to understand the behavior of hydrogen molecules on various surfaces. Hydrogen molecules appear in various catalytic reactions. Many experimental studies using high-resolution electron-energy-loss spectroscopy (HREELS),1,2 molecular beam scattering,3–8 and temperature-programmed desorption (TPD)9,10 have attempted to clarify the nature of physisorption states on various surfaces. However, there is little detailed information regarding hydrogen molecule behavior on various surfaces, including charge transfer, diffusion, hindered rotation, and physical bonding. This is because hydrogen molecules are difficult to directly observe because they are relatively inert and have weak molecule–surface interactions, which necessitate quite high energy resolution for unambiguous detection. Even with theoretical studies, it has been difficult to describe the physisorption states accurately. However, recent developments in theoretical techniques11–14 and computational power allow accurate description of these physisorption systems. In addition, resonant ionization experiments can achieve rotational-state-selective observation of hydrogen molecules on various surfaces.15,16 Thanks to these developments in theoretical and experimental techniques, we can achieve a full understanding of hydrogen molecule physisorption states using both theoretical and experimental methods.
Hydrogen also receives attention because of its minimal mass. Hydrogen systems have been investigated as they represent the most distinctive examples which show significant quantum effects. In fact, the quantum effects of hydrogen have been reported in various phenomena, such as tunneling effects in H2 dissociative adsorption and hydrogen recombination reactions.17 Furthermore, recent nuclear reaction analyses have directly observed zero-point vibration of hydrogen adsorbed onto Si and Pt surfaces.18 With its importance in catalysis and quantum mechanics, understanding the behavior of hydrogen on the surface is one of the most interesting topics in the condensed matter and physical chemistry fields.
In addition, understanding the rotational states of hydrogen molecules is important for the adsorption properties. Hydrogen molecules on surfaces have hindered rotational states in the anisotropic potential, while free rotational states appear in the gas phase. The adsorption energy in hindered rotational states changes from those in free rotational states because of the rotational energy difference between the gas phase and adsorption states. Despite its importance, the potential anisotropy and its molecular axis orientation preference are still controversial for physisorption systems. The conventional theory of the physisorption interaction, which consists of the attractive van der Waals interaction and the Pauli repulsion, leads to almost no potential anisotropy or a slight parallel preference toward the surface in the case of inert diatomic molecules on noble surfaces. In contrast, recent theoretical studies on multi-reference configuration interactions have reported that a non-negligible perpendicular preference exists in cases of H2 on Cu(100)19 and Ag(100)20 surfaces. Such a non-negligible perpendicular preference of H2 on Ag(111) has also been experimentally observed in resonant ionization experiments.21 Therefore, further studies are required to clarify the orientational preference and the origin of potential anisotropy of hydrogen molecules on noble surfaces.
In this study, we performed a first-principles investigation of H2 molecular adsorption onto Ag(111) surfaces by calculating the relevant potential energy surface (PES). We focused on H2/Ag(111) systems to compare our theoretical studies with previous experimental results.4,21 Although results have been reported for H2 on high symmetry adsorption sites,22,23 we performed exhaustive studies with a finer grid of adsorption sites. We also performed quantum mechanical computations of hydrogen nuclei with these PES results to take into account quantum effects and hindered rotation. From the computational results, we obtained accurate physisorption states of H2 on Ag(111) surfaces.
2. Computational methods
We performed total energy calculations based on spin-polarized DFT, using the generalized gradient approximation with the revised Perdew–Burke–Ernzerhof functional for the exchange–correlation energy,24 which is implemented in the plane-wave and projector-augmented wave method code, the Vienna Ab initio Simulation Package (VASP 5.3.3).25–30 To include the van der Waals interaction, we adopted the semiempirical DFT-D2 method reported by Grimme.11 In addition, we also adopted the second version of van der Waals density functional (vdW-DF2)31 as the nonlocal correlation functional. We used the newly developed exchange functional reported by Hamada14 with vdW-DF2 (rev-vdW-DF2). We applied a 600 eV cutoff to limit the plane-wave basis set without compromising the computational accuracy. A 7 × 7 × 1 Monkhorst–Pack special k-point grid32 for the first Brillouin zone sampling and a Gaussian smearing model of σ = 0.05 eV were used. The computed equilibrium gas phase H2 bond lengths using DFT-D2 and rev-vdW-DF2 are 0.747 and 0.750 Å, respectively. The Ag substrate was simulated as a slab of four fcc 2 × 2 Ag(111) layers. The supercell was constructed with a 15.5 Å vacuum separation between slabs to reduce the interaction between slabs. Our model with a single H2 molecule on a 2 × 2 Ag(111) surface corresponds to a surface coverage of 0.25 monolayers. The adsorption energy difference between results obtained from simulations using 2 × 2 and 3 × 3 Ag(111) surfaces is 1 meV. Lattice constants for this system were initially set based on the theoretically predicted equilibrium lattice constant of 4.24 Å with DFT-D2 and 4.10 Å with rev-vdW-DF2, obtained using a 15 × 15 × 15 k-mesh on the primitive one-atom fcc unit cell for bulk Ag. The corresponding lateral and interlayer Ag–Ag distances for DFT-D2 and rev-vdW-DF2 are 3.00 and 2.90 Å, respectively. For reference, the bulk Ag lattice constant obtained using powder diffraction experiments (JCPDS Card No. 04-783) is 4.0862 Å. To determine the optimized Ag(111) surface structure, we performed preliminary calculations in which the top two layers were relaxed until the forces on each atom were smaller than 0.02 eV Å−1. The supercell lattice vectors are held fixed during the relaxation. Because the H2 mass is much less than Ag, we expect a large difference in the time scales associated with surface relaxation and H2 adsorption dynamics. Thus, as a first step, we neglected surface relaxation.
We investigated the PES for H2 adsorption and diffusion by calculating the total energy of H2/Ag(111) systems as a function of the H2 center-of-mass positions. In Fig. 1, we show the H2 adsorption configurations on the 2 × 2 Ag(111) surface. To obtain an accurate PES, we adopted a 12 × 12 × 23 grid for H2 adsorption configurations. We considered H2 adsorbed from 2.6 to 7.0 Å from the surface. The step sizes for parallel and perpendicular directions to the surface are 0.5 and 0.2 Å, respectively. These step sizes are sufficiently small so that the accuracy of the calculated PES is ensured. All energies are potential energies with respect to the infinitely separated slab and H2.
 |
| | Fig. 1 Adsorption sites for potential energy calculations of H2 on Ag(111) surfaces. | |
In these calculations, we set the electric dipole correction layer in the vacuum area to compensate for the electric dipole interactions between repeated slabs.33 Electron transfer is obtained by taking the difference in the electron density distribution between two configurations – isolated and adsorbed onto the substrate – using Bader charge analysis.34
3. Results and discussion
First, we investigated the potential energy and potential anisotropy of H2 on four high-symmetry adsorption sites with the H–H axis oriented parallel and perpendicular to the surface. The computed DFT-D2 and rev-vdW-DF2 results are collected in Table 1. For reference, we also present the results obtained using the conventional PBE functional. The four high-symmetry adsorption sites considered are the top site, the bridge site, the fcc-hollow site, and the hcp-hollow site. At first, we point out that the most stable H2 center-of-mass height from the surface z over all adsorption configurations considered herein is 3.6 and 3.8 Å with DFT-D2 and rev-vdW-DF2, respectively. We found that the most stable H2 adsorption configuration with DFT-D2 is 3.8 Å above the surface at the fcc-hollow sites with the H2 bond oriented normal to the surface. Conversely, the most stable H2 adsorption configuration with rev-vdW-DF2 is 3.6 Å above the surface at the top site normal to the surface. From Table 1, we can also see that the corresponding potential energies for DFT-D2 and rev-vdW-DF2 are 38.1 and 39.8 meV, respectively. In comparison with the results obtained using the conventional PBE functional which does not include the van der Waals interaction accurately, both DFT-D2 and rev-vdW-DF2 can improve the potential energies of molecularly adsorbed H2 on Ag(111) surfaces. Though DFT-D2 corrected the potential energies by simple semiempirical dispersion terms, DFT-D2 gives similar results with rev-vdW-DF2 which includes the nonlocal correlation functional. The electron transfer from Ag(111) surfaces to H2 is only 0.01 e, which indicates that H2 physisorbs on Ag(111) surfaces. The H2 bond length r in the most stable H2 adsorption configuration becomes 0.747 and 0.752 Å with DFT-D2 and rev-vdW-DF2, respectively. In subsequent calculations used to construct the corresponding PES, we fixed the H2 bond length at these values. From the calculated exhaustive PES, we find that the potential energy difference between each adsorption site on Ag(111) surfaces is smaller than 1.3 meV with DFT-D2 and 0.1 meV with rev-vdW-DF2, which corresponds to the diffusion barrier height for in-plane diffusion on Ag(111). These values are negligibly small, so we conclude that H2 can diffuse without any barrier on Ag(111) surfaces under finite temperature conditions. In addition, as we mentioned, the adsorption height from the surface z over all adsorption configurations does not change. From these data, we can say that the adsorption properties do not change during diffusion processes.
Table 1 Calculated adiabatic potential energies and the most stable H2 center-of-mass height from the surface z for H2 at high-symmetry sites on Ag(111) surfaces. The results of DFT-D2, rev-vdW-DF2, and PBE computations are shown. ⊥ and ∥ indicate the molecular axis perpendicular and parallel to the surfaces, respectively
| |
|
Potential energy (meV) |
z (Å) |
| Top |
Bridge |
fcc-hollow |
hcp-hollow |
| DFT-D2 |
⊥ |
−36.8 |
−37.7 |
−38.1 |
−38.1 |
3.8 |
| ∥ |
−30.9 |
−30.1 |
−30.3 |
−30.3 |
3.8 |
| rev-vdW-DF2 |
⊥ |
−39.8 |
−39.7 |
−39.7 |
−39.7 |
3.6 |
| ∥ |
−35.3 |
−33.8 |
−33.4 |
−33.3 |
3.6 |
| PBE |
⊥ |
−10.1 |
−10.7 |
−10.8 |
−10.8 |
3.8 |
| ∥ |
−5.9 |
−4.9 |
−5.1 |
−5.0 |
3.8 |
We also focused on the hindered rotational states of H2 on Ag(111) surfaces. From Table 1, we can see that the corresponding potential anisotropy at the most stable adsorption heights vary from 6.7 to 9.2 meV with DFT-D2 and from 4.6 to 6.4 meV with rev-vdW-DF2.
Here, we calculated the molecular rotational wave function in the anisotropic potential. We defined the angle θ as the angle between the molecular axis and the surface normal. The φ rotation is not hindered in H2/Ag(111) systems, thus, we could define the φ angle as the angle between the molecular axis and an arbitrary reference axis parallel to the surface. In the case of H2 hindered rotation with out-of-plane potential anisotropy on Ag(111) surfaces, we calculated the wave functions of the hindered rotational states using the following Hamiltonian HR:35
| |  | (2) |
| | | Va(z) = V⊥(z) − V∥(z), | (3) |
where ℏ,
I, and
Va(
z) are the reduced Planck constant, the reduced mass of H
2, and the value of the out-of-plane potential anisotropy obtained from our DFT calculations, respectively. We adopted the potential anisotropy at the most stable adsorption configurations. The corresponding potential anisotropy values with DFT-D2 and rev-vdW-DF2 are −7.8 and −4.5 meV, respectively. As mentioned, such non-negligible orientation preference of H
2 normal to the surface has also been reported in other non-corrugated surface systems, for example, H
2/Cu(100),
19 H
2/Ag(100),
20 and H
2/Ag(110)
36 systems. We used a spherical harmonics basis to solve the Schrödinger equation with the Hamiltonian for the hindered rotational states
HR. We considered azimuthal quantum numbers of 0 to 20. From the obtained wave functions, we found that the hybridization of the spherical harmonics basis between deferent rotational states is less than 0.1%. The H
2 rotational constant, ℏ
2/2
I, which corresponds to 7.3 meV, is large owing to the small mass. Therefore, the rotational energy levels are well discretized, which leads to less hybridization of the rotational motion wave functions. In addition, according to the symmetry requirement for exchange of nuclei, the molecular rotational states of H
2 are strongly coupled with the nuclear spin states. Thus, the molecular rotational states can hybridize only in odd or even azimuthal rotational quantum numbers. From these points, we can assume that the molecular rotational motion wave function can be still treated as spherical harmonics even in the anisotropic adsorption potential on Ag(111) surfaces.
To compare our theoretical results with the previously reported experimental results, we expressed the potential energy with the isotropic and anisotropic terms using a Legendre polynomial. We define the potential energy as follows:
| | V(z, θ) = V0(z) + V2(z) P2(cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ), θ), | (4) |
| |  | (5) |
| |  | (6) |
where
V0(
z) and
V2(
z) are the isotropic and anisotropic potential terms, respectively.
P2(cos
θ) is the second-order Legendre polynomial. From
eqn (4), we can see that one-third of the anisotropic potential contributes to the isotropic potential energy. Thus, the isotropic potential energy, which corresponds to the potential energy of the molecular rotational ground states, includes the adsorption energy shift caused by the potential anisotropy. In addition, because H
2 is the lightest molecule, we take into account the zero-point energy. We assumed that H
2 is bound only in the direction perpendicular to the surfaces. Therefore, we calculated only the corresponding zero-point energy of the H
2–Ag(111) vibration along the surface-normal direction. We calculated the adsorption energy
Ṽ0(
n), which includes the zero-point energy and H
2–Ag(111) vibrational energy, from the Hamiltonian
Hv as follows:
| |  | (8) |
where |
n〉 and
m are the wave function of the
n-th H
2–Ag(111) vibrational state in the isotropic potential
V0(
z) and the total mass of H
2, respectively. In this paper, we adopted the Morse potential function for the potential energy curve fitting. Thus, we could obtain |
n〉 and
Ṽ0(
n) analytically. We show
Ṽ0(
n) for various vibrational states
n in
Table 2. We also show results obtained for D
2 to illustrate the isotope effect. The bound states of H
2 and D
2 exist up to
n = 5 and 7, respectively. The isotropic adsorption energy is equivalent to the adsorption energy of
para-H
2 and
ortho-D
2 in the rotational ground states. DFT-D2 and rev-vdW-DF2 predict zero-point energies of 4.6 and 5.8 meV, respectively. These values correspond to about 15% of the isotropic potential energy depth. Thus, we conclude that the zero-point energies cannot be neglected. Focusing on the isotope effect, the zero-point energy of D
2 is 3.3 and 4.2 meV with DFT-D2 and rev-vdW-DF2, respectively. The zero-point energy ratios between H
2 and D
2 for both methods are slightly smaller than

, which is generally considered as the conventional isotope effect. This originates from anharmonicity of the adsorption potential. The activation barriers for the in-plane diffusion with zero-point energy exist at the bridge site with both methods. The corresponding diffusion barrier heights are 0.2 meV with DFT-D2 and 1.1 meV with rev-vdW-DF2. These values are still negligibly small. Therefore, we posit that H
2 can diffuse freely on Ag(111) surfaces under finite temperature conditions. In
Table 2, we also show experimental results of resonance enhanced multiphoton ionization spectroscopy with temperature-programmed desorption (REMPI-TPD)
21 and diffractive selective adsorption and rotationally mediated selective adsorption (DSA-RMSA) scattering resonance measurements.
4 The value of
Ṽ0(
n) obtained from REMPI-TPD is −26 meV. In addition, we can also compare our DFT results of the H
2–Ag(111) vibrational excited states with the results of selective adsorption experiments. As we mentioned, the H
2–Ag(111) vibrational states of H
2 and D
2, which mean the bound states of H
2/Ag(111) systems, exist up to
n = 5 and 7 with both DFT methods, respectively. These values are consistent with the DSA-RMSA results. Each H
2–Ag(111) vibrational energy is also in good agreement with the corresponding experimental results. From these data, we assess that both DFT-D2 and rev-vdW-DF2 reproduce well the accurate physisorption states.
Table 2 Calculated adsorption energies of H2 and D2 on Ag(111) surfaces. Isotropic terms Ṽ0(n) and anisotropic terms Ṽ2(n) are shown. Experimental results of REMPI-TPD21 and DSA-RMSA4 are also included for comparison
| |
Vibrational state n |
Adsorption energy (meV) |
| DFT-D2 |
rev-vdw-DF2 |
REMPI-TPD21 |
DSA-RMSA4 |
|
Ṽ
0(n) |
Ṽ
2(n) |
Ṽ
0(n) |
Ṽ
2(n) |
Ṽ
0(n) |
Ṽ
2(n) |
Ṽ
0(n) |
| H2 |
0 |
−28.0 |
−5.2 |
−31.0 |
−3.1 |
−26 |
−5 |
−25.74 |
| 1 |
−19.7 |
−4.9 |
−20.9 |
−2.7 |
|
|
−16.87 |
| 2 |
−12.9 |
−4.4 |
−12.7 |
−2.1 |
|
|
−10.11 |
| 3 |
−7.6 |
−3.9 |
−6.6 |
−1.7 |
|
|
−5.47 |
| 4 |
−3.6 |
−3.4 |
−2.5 |
−1.3 |
|
|
−2.61 |
| 5 |
−1.1 |
−2.7 |
−0.3 |
−0.8 |
|
|
−1.21 |
|
|
| D2 |
0 |
−29.2 |
−5.0 |
−32.6 |
−3.1 |
|
|
|
| 1 |
−23.2 |
−4.8 |
−25.1 |
−2.8 |
|
|
−20.97 |
| 2 |
−17.8 |
−4.5 |
−18.5 |
−2.4 |
|
|
−15.25 |
| 3 |
−13.1 |
−4.2 |
−12.9 |
−2.1 |
|
|
−10.09 |
| 4 |
−9.1 |
−3.9 |
−8.3 |
−1.8 |
|
|
−6.57 |
| 5 |
−5.9 |
−3.5 |
−4.8 |
−1.5 |
|
|
−3.97 |
| 6 |
−3.3 |
−3.1 |
−2.2 |
−1.1 |
|
|
−2.39 |
| 7 |
−1.5 |
−2.6 |
−0.6 |
−0.7 |
|
|
−1.13 |
In anisotropic adsorption potential, the adsorption energy has rotational azimuthal and magnetic quantum number dependence. As we mentioned, the H2 rotational wave functions on Ag(111) surfaces still remain as spherical harmonics type functions. Thus, we treated the anisotropic term V2(z)P2(cosθ) as a perturbation to a free rotor. The contribution of the anisotropic term to the adsorption energy within first-order perturbation can be calculated as follows:37
| |  | (9) |
where
J and
M are the rotational azimuthal and magnetic quantum numbers, respectively. The calculated
Ṽ2(
n) values with DFT-D2 and rev-vdW-DF2 are −5.2 and −3.1 meV, respectively. These values correspond well with the experimental value of −5 meV obtained from REMPI-TPD measurements. For the molecular rotational ground states of
para-H
2 (
J = 0,
m = 0), there is no energy shift due to the anisotropic term. Conversely, the molecular rotational ground states of
ortho-H
2 (
J = 1,
m = 0, ±1), which originally degenerate in the isotropic potential, split into two energy levels. From
eqn (9), we can see that the molecular rotational energy shifts with
J = 1 and
m = 0 with DFT-D2 and rev-vdW-DF2 are −2.1 and −1.2 meV, and with
J = 1 and
m = ±1 are 1.0 and 0.6 meV, respectively. We present the adsorption energies of H
2 and D
2 on Ag(111) surfaces with vibrational and rotational ground states in
Table 3. We also show the results regarding
ortho-D
2 (
J = 0,
m = 0) and
para-D
2 (
J = 1,
m = 0, ±1), where the corresponding rotational constant ℏ
2/2
I is 3.7 meV. From
Table 3, we found that the molecular rotational states with
J = 1 and
m = 0, which are p
z orbital type wave functions, are the molecular rotational ground states of
ortho-H
2 and
para-D
2 on Ag(111) surfaces. This is because H
2 and D
2 prefer to have their molecular axis normal to the surface.
Table 3 Calculated adsorption energies of H2 and D2 on Ag(111) surfaces with vibrational and rotational ground states. J and m are the rotational azimuthal and magnetic quantum numbers, respectively
| |
J
|
M
|
Adsorption energy (meV) |
| DFT-D2 |
rev-vdw-DF2 |
| H2 |
0 |
0 |
−28.0 |
−31.0 |
| 1 |
0 |
−15.5 |
−17.6 |
| ±1 |
−12.4 |
−15.8 |
|
|
| D2 |
0 |
0 |
−29.2 |
−32.6 |
| 1 |
0 |
−23.9 |
−26.5 |
| ±1 |
−20.9 |
−24.7 |
We also discuss the origin of the potential anisotropy on Ag(111) surfaces. In the physisorption states, the van der Waals interaction is dominant. The main part of the attractive van der Waals interaction is the London dispersion interaction, which causes the nonlocal electron–electron correlation effect. In the case of H2/noble surface systems, the potential anisotropy caused by the London dispersion interaction, which is obtained from the polarizability along and transverse to the molecular axis, prefers the end-on molecular orientation and is 7% of the isotropic potential term V0(z).38 We can then assume that the values of the potential anisotropy from the contribution of the London dispersion interaction at the most stable adsorption configurations are −2.5 and −2.7 meV with DFT-D2 and rev-vdW-DF2, respectively. However, the obtained potential anisotropy Va(z) at the most stable adsorption configurations are −7.8 and −4.5 meV with DFT-D2 and rev-vdW-DF2, respectively. In addition, the repulsion caused by the electron overlap between H2 and the surface generally prefers the diatomic molecule to be oriented parallel to the surface. Thus, the origin of the potential anisotropy cannot be understood only through London dispersion interactions. The other important cause of the potential anisotropy is the interaction of metal surface electron states with H2 anti-bonding resonances.36 The energy shift from this resonance interaction δεum can be described in a second-order perturbation expression as follows:
| |  | (11) |
where |m〉 and |u〉 are wave functions of the metal surface electron states and H
2 anti-bonding states, respectively.
ε0m,
ε0u, and
νmol are one-electron of the metal surface electron states, H
2 antibonding states, and the attractive potential of H
2 nuclei, respectively. This term gives the energy gain from the virtual transition of the metal electron to the unoccupied molecular state. Here, we calculated
δεum using a simple two-level model. We described the wave functions of the Ag(111) surface electron using a nearly-free two-band model.
39 The wave functions of the H
2 anti-bonding orbitals consist of 1s orbitals of two hydrogen atoms that are orthogonal to each other.
40 From this calculation, we clarified that
δεum has the molecular axis orientation dependence. In the case of H
2/Ag(111) systems, this resonance interaction gives a perpendicular preference to the surface of about −10 meV with
z = 3.6 Å and
r = 0.75 Å. This is because the quite small product of the Fermi wave number of the surface electron and the bond length of H
2 in H
2/Ag(111) systems cancels the contribution of each H atom to the electron transfer in the parallel orientation.
41 In addition, DFT calculations using the conventional PBE functional, which does not include the London dispersion interaction, indicate a preferred orientation perpendicular to the surface. From these points, even in the physisorption states, not only the London dispersion interaction and the repulsion term, but also the resonance interaction is important for the potential anisotropy.
4. Conclusions
In this paper, we investigated the physisorption states of H2 on Ag(111) surfaces using first-principles calculations based on spin-polarized DFT. To include the van der Waals interaction, we adopted the semiempirical DFT-D2 and van der Waals density functional (rev-vdW-DF2) methods. First, we constructed exhaustive potential energy surfaces of H2 on Ag(111) surfaces. The calculated adsorption energy of H2 on Ag(111) surfaces, which includes the zero-point energy, is in good agreement with the results of DSA-RMSA experiments. The diffusion barrier of H2 on Ag(111) surfaces is negligible. We also clarified that the non-negligible out-of-plane potential anisotropy, which has a perpendicular preference to the surface, exists even on non-corrugated Ag(111) surfaces, as has been observed by REMPI-TPD experiments. We clarified that the molecular rotational ground states of ortho-H2 split into two energy levels in the anisotropic potential. From these points, we conclude that both semiempirical dispersion interaction terms and non-local correlation functionals can improve upon the description of the physisorption states of H2/Ag(111) systems. Finally, we revealed that the origin of the potential anisotropy on Ag(111) surfaces is a combination of the London dispersion interaction and the virtual transition of the metal electron to the unoccupied molecular state.
Acknowledgements
This work was supported by Japan Science and Technology Agency (JST) through the Strategic International Research Cooperative Program “Theoretical modeling and simulations of the structural, electronic and dynamical properties of surfaces and nanostructures in materials science research” and the Ministry of Education, Culture, Sports, Science and Technology (MEXT) through the Grant-in-Aid for Scientific Research on Innovative Areas Program (2507-26108502). Calculations were executed using computer facilities of the Information Initiative Center (Hokkaido University) and the ISSP Super Computer Center (the University of Tokyo). The authors are thankful to R. Brako of Ru![[d with combining overline]](https://www.rsc.org/images/entities/char_0064_0305.gif) er Bošković Institute for helpful discussions.
er Bošković Institute for helpful discussions.
References
- M. Gruyters and K. Jacobi, Chem. Phys. Lett., 1994, 225, 309 CrossRef CAS.
- K. Svensson and S. Andersson, Phys. Rev. Lett., 1997, 78, 2016 CrossRef CAS.
- H. Hoinkes, Rev. Mod. Phys., 1980, 52, 933 CrossRef CAS.
- C. f. Yu, K. B. Whaley, C. S. Hogg and S. J. Sibener, Phys. Rev. Lett., 1983, 51, 2210 CrossRef CAS.
- C. f. Yu, K. B. Whaley, C. S. Hogg and S. J. Sibener, J. Chem. Phys., 1985, 83, 4217 CrossRef CAS PubMed.
- S. Andersson, L. Wilzén and M. Persson, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 2967 CrossRef CAS.
- S. Andersson, L. Wilzén, M. Persson and J. Harris, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 8146 CrossRef CAS.
- S. Andersson and M. Persson, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 5685 CrossRef CAS.
- L. Amiaud, J. H. Fillion, S. Baouche, F. Dulieu, A. Momeni and J. L. Lemaire, J. Chem. Phys., 2006, 124, 094702 CrossRef CAS PubMed.
- S. Iwata, Y. Sato, K. Nakai, S. Ogura, T. Okano, M. Namura, A. Kasuya, K. Tohji and K. Fukutani, J. Phys. Chem. C, 2007, 111, 14937 CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- I. Hamada, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 121103(R) CrossRef.
- K. Fukutani, K. Yoshida, M. Wilde, W. A. Diño, M. Matsumoto and T. Okano, Phys. Rev. Lett., 2003, 90, 096103 CrossRef CAS.
- N. Watanabe, Y. Kimura, A. Kouchi, T. Chigai, T. Hama and V. Pirronello, Astrophys. J., Lett., 2010, 714, L233 CrossRef CAS.
- W. A. Diño, H. Kasai and A. Okiji, Prog. Surf. Sci., 2000, 63, 63 CrossRef.
- K. Fukutani, A. Itoh, M. Wilde and M. Matsumoto, Phys. Rev. Lett., 2002, 88, 116101 CrossRef CAS.
- G. Cilpa and G. Chambaud, Surf. Sci., 2007, 601, 320 CrossRef CAS PubMed.
- G. Cilpa, M. Guitou and G. Chambaud, Surf. Sci., 2008, 602, 2894 CrossRef CAS PubMed.
- T. Sugimoto and K. Fukutani, Phys. Rev. Lett., 2014, 112, 146101 CrossRef.
- Y. Kunisada, H. Nakanishi, W. A. Diño and H. Kasai, J. Vac. Soc. Jpn., 2012, 55, 115 CrossRef CAS.
- Y. Kunisada and H. Kasai, J. Phys. Soc. Jpn., 2013, 82, 023601 CrossRef.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater.
Sci., 1996, 6, 15 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- K. Lee, E. D. Murray, L. Kong, B. I. Lundqvist and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 081101 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- J. Neugebauer and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 16067 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jonsson, Comput. Mater. Sci., 2006, 36, 354 CrossRef PubMed.
- T. B. MacRury and J. R. Sams, Comput. Mol. Phys., 1970, 19, 337 CrossRef CAS PubMed.
- L. Wilzén, F. Althoff, S. Andersson and M. Persson, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 7003 CrossRef.
- S. Andersson and J. Harris, Phys. Rev. Lett., 1982, 48, 545 CrossRef CAS.
- J. Harris and P. J. Feibelman, Surf. Sci., 1982, 115, L133 CrossRef CAS.
- E. Ilisca, Phys. Rev. Lett., 1991, 66, 667 CrossRef CAS.
- Y. Takahashi, J. Phys. Soc. Jpn., 1977, 43, 1342 CrossRef CAS.
- A. Fukui, H. Kasai, H. Nakanishi and A. Okiji, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14136 CrossRef CAS.
|
| This journal is © the Owner Societies 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence



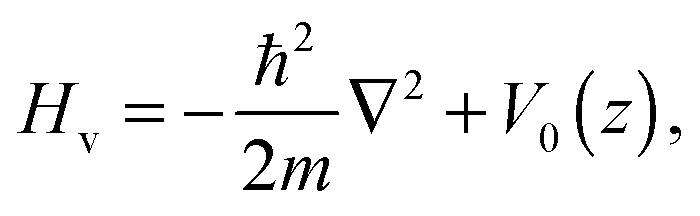
 , which is generally considered as the conventional isotope effect. This originates from anharmonicity of the adsorption potential. The activation barriers for the in-plane diffusion with zero-point energy exist at the bridge site with both methods. The corresponding diffusion barrier heights are 0.2 meV with DFT-D2 and 1.1 meV with rev-vdW-DF2. These values are still negligibly small. Therefore, we posit that H2 can diffuse freely on Ag(111) surfaces under finite temperature conditions. In Table 2, we also show experimental results of resonance enhanced multiphoton ionization spectroscopy with temperature-programmed desorption (REMPI-TPD)21 and diffractive selective adsorption and rotationally mediated selective adsorption (DSA-RMSA) scattering resonance measurements.4 The value of Ṽ0(n) obtained from REMPI-TPD is −26 meV. In addition, we can also compare our DFT results of the H2–Ag(111) vibrational excited states with the results of selective adsorption experiments. As we mentioned, the H2–Ag(111) vibrational states of H2 and D2, which mean the bound states of H2/Ag(111) systems, exist up to n = 5 and 7 with both DFT methods, respectively. These values are consistent with the DSA-RMSA results. Each H2–Ag(111) vibrational energy is also in good agreement with the corresponding experimental results. From these data, we assess that both DFT-D2 and rev-vdW-DF2 reproduce well the accurate physisorption states.
, which is generally considered as the conventional isotope effect. This originates from anharmonicity of the adsorption potential. The activation barriers for the in-plane diffusion with zero-point energy exist at the bridge site with both methods. The corresponding diffusion barrier heights are 0.2 meV with DFT-D2 and 1.1 meV with rev-vdW-DF2. These values are still negligibly small. Therefore, we posit that H2 can diffuse freely on Ag(111) surfaces under finite temperature conditions. In Table 2, we also show experimental results of resonance enhanced multiphoton ionization spectroscopy with temperature-programmed desorption (REMPI-TPD)21 and diffractive selective adsorption and rotationally mediated selective adsorption (DSA-RMSA) scattering resonance measurements.4 The value of Ṽ0(n) obtained from REMPI-TPD is −26 meV. In addition, we can also compare our DFT results of the H2–Ag(111) vibrational excited states with the results of selective adsorption experiments. As we mentioned, the H2–Ag(111) vibrational states of H2 and D2, which mean the bound states of H2/Ag(111) systems, exist up to n = 5 and 7 with both DFT methods, respectively. These values are consistent with the DSA-RMSA results. Each H2–Ag(111) vibrational energy is also in good agreement with the corresponding experimental results. From these data, we assess that both DFT-D2 and rev-vdW-DF2 reproduce well the accurate physisorption states.