
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning photochemistry: substituent effects on πσ* state mediated bond fission in thioanisoles†
Andreas M.
Wenge‡
,
Tolga N. V.
Karsili
,
Javier Diaz
Rodríguez§
,
Michael I.
Cotterell
,
Barbara
Marchetti
,
Richard N.
Dixon
and
Michael N. R.
Ashfold
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: mike.ashfold@bris.ac.uk
First published on 22nd May 2015
Abstract
We report a combination of experimental (velocity map imaging measurements of the methyl (Me) radical products) and ab initio electronic structure studies that explore the influence of substituents (Y) on the dynamics of S–Me bond fission following excitation to the first excited S1 states of thioanisole and three 4-substituted thioanisoles (4-YPhSMe, with Y = H, Me, MeO and CN). In all bar the case that Y = CN, the resulting 4-YPhS products are found to be formed predominantly in their excited (Ã) electronic state. In all cases, the relative yield of ![[X with combining tilde]](https://www.rsc.org/images/entities/i_char_0058_0303.gif) state products increases upon tuning to shorter excitation wavelengths and, in the specific case of bare thioanisole (as found previously by Lim and Kim, Nat. Chem., 2010, 2, 627), jumps when exciting on the parent resonance assigned to the S1(v7a = 1) level. Two conical intersections (CIs) in the RS–Me stretch coordinate are crucial to rationalising all of the observed dynamics. The first, (CI-1, between the diabatic 1ππ* and dissociative 1πσ* potential energy surfaces (PESs) at RS–Me ∼ 2 Å) lies above the S1(v = 0) level in energy, and the calculated minimum energy path through this barrier involves substantial deviations from planarity in all bar 4-CNPhSMe. Beyond this barrier, the potential is quite steeply repulsive, and Me + 4-YPhS(Ã) products are the inevitable products if the molecular framework is unable to re-planarise within the time it takes for the dissociating molecules to pass through the region of CI-2 (between the diabatic 1πσ* and ground (S0) states) where the product electronic branching is determined. The gradual increase in the yield of 4-YPhS() radicals upon tuning to shorter photolysis wavelengths, the much increased branching into PhS() products when exciting the PhSMe (S1, v7a = 1) level and the dominance of 4-CNPhS() products in the specific case that Y = CN can all be understood in terms of a (relative) lowering of the effective barrier associated with CI-1, thereby allowing access to the dissociative region of the PES at closer-to-planar geometries.
state products increases upon tuning to shorter excitation wavelengths and, in the specific case of bare thioanisole (as found previously by Lim and Kim, Nat. Chem., 2010, 2, 627), jumps when exciting on the parent resonance assigned to the S1(v7a = 1) level. Two conical intersections (CIs) in the RS–Me stretch coordinate are crucial to rationalising all of the observed dynamics. The first, (CI-1, between the diabatic 1ππ* and dissociative 1πσ* potential energy surfaces (PESs) at RS–Me ∼ 2 Å) lies above the S1(v = 0) level in energy, and the calculated minimum energy path through this barrier involves substantial deviations from planarity in all bar 4-CNPhSMe. Beyond this barrier, the potential is quite steeply repulsive, and Me + 4-YPhS(Ã) products are the inevitable products if the molecular framework is unable to re-planarise within the time it takes for the dissociating molecules to pass through the region of CI-2 (between the diabatic 1πσ* and ground (S0) states) where the product electronic branching is determined. The gradual increase in the yield of 4-YPhS() radicals upon tuning to shorter photolysis wavelengths, the much increased branching into PhS() products when exciting the PhSMe (S1, v7a = 1) level and the dominance of 4-CNPhS() products in the specific case that Y = CN can all be understood in terms of a (relative) lowering of the effective barrier associated with CI-1, thereby allowing access to the dissociative region of the PES at closer-to-planar geometries.
1. Introduction
An ability to ‘tailor’ photochemistry could impact on many fields of chemistry. Such ambitions have underpinned much of the recent interest in coherent control, i.e. the use of appropriately designed laser pulses to drive a photochemical process to some particular target outcome.1–3 Strategic substitution remote from the reaction centre offers another route to tuning the outcome of a chemical process, and much of the recent effort exploring and understanding the fragmentation dynamics of heteroaromatic and heteroatom containing aromatic molecules (azoles, phenols, thiophenols, etc.) following ultraviolet (UV) photoexcitation has focussed on such issues.4–11 Bond fission in these types of molecule often relies on the interaction between a bound 1ππ* excited state with high absorption cross-section and an optically ‘dark’ 1(n/π)σ* state, the potential energy surface (PES) for which is repulsive along the X–H (X = N, O, S) dissociation coordinate. Conical intersections (CIs) are central to the interpretation of such data. For example, long wavelength excitation of phenol populates vibrational levels of the 11ππ* state at energies below the CI between the 11ππ* and 11πσ* PESs, but O–H bond fission still occurs – on a nanosecond timescale – by tunnelling through the barrier under this CI.8,12,13 Broadly similar dynamics arise in the long wavelength photolysis of thiophenols, but the corresponding energy barrier is much smaller (comparable to the zero-point energy in the S–H stretch coordinate) and the rate of S–H bond fission is orders of magnitude faster. Recent studies have highlighted the way in which the rate of O–H bond fission (in phenols)10 and the electronic branching in the thiophenoxyl radical products (in the case of thiophenols)9 can be tuned by appropriate substitutions in the 4-(para-) position.An obvious question is whether these potentially important dynamical behaviours extend to molecular systems where the leaving species is not an H atom. Kim and coworkers have reported use of ion imaging methods to study the photodissociation of thioanisole14 (a derivative of thiophenol, in which the thiyl (also termed sulfenyl) hydrogen is replaced by a methyl group, henceforth denoted as PhSMe) and thioanisole-d315 following excitation to the 11ππ* state.16 The PhS fragments from S–Me bond fission are formed predominantly in their electronically excited Ã2B2(2A′) state but, in both cases, these workers also identified one or more parent vibronic resonances that yield strikingly different electronic branching between the à and ground 2B1(2A′′) states of the PhS product. These states of the radical are distinguished by having the odd electron in, respectively, the in-plane and out-of-plane singly occupied molecular orbitals which are largely localised on the sulphur atom. Thioanisole-h3 shows just one such resonance, 722 cm−1 above the S1 ← S0 origin, where the /à product branching ratio increases at least 3-fold,14 while the subsequent study of thioanisole-d3 photolysis found four resonances (at a similar excitation energy) that display similarly enhanced /à product branching ratios.15 Time-resolved imaging studies of the methyl radical photoproducts from photolysis of thioanisole-h3, allied with electronic structure calculations, sought to provide more insight into the possible role of S–Me torsional motion (τS–Me) in facilitating non-radiative transfer of population from the 11ππ* state to the dissociative 11πσ* state of PhSMe.17 One and two-dimensional (1- and 2-D) cuts through the respective singlet PESs in PhSMe (along RS–Me (the S–Me stretch coordinate) and/or ϕ (the dihedral angle between the S–Me bond and the ring plane)) have been reported previously, and will be revisited later in this study, but two regions of conical intersection (CI) are of particular note. The first, CI-1, between the 11ππ* and 11πσ* PESs is a true CI at both ϕ = 0° and at 90° and, since the two potentials are near-degenerate at intermediate ϕ, this is more correctly viewed as an effective ‘seam’ of intersection in this 2-D configuration space.17 The second, CI-2, between the 11ππ* and ground (1ππ) PESs at longer RS–Me is localised at ϕ = 0°; photo-excited molecules that pass through CI-2 would yield state PhS radical products, while those that approach this region of CI with non-planar geometries (ϕ ≠ 0°) would tend to follow the adiabatic path to à state products (plus a Me radical).
Here we report a combination of experimental studies (velocity map ion imaging (VMI)18,19 measurements of the Me radical fragments) and further high level, higher-dimensionality ab initio calculations that explore the influence of substituents (Y) on the dynamics of photoinduced S–Me bond fission in 4-substituted thioanisoles (4-YPhSMe). In addition to thioanisole itself, the systems chosen for detailed study involve Y = Me (which would be expected to be, at most, a mild perturber of the ring π system), MeO (a strong π electron donor) and CN (a strong π electron acceptor). Photoexcitation to the origin of the S1 state results in a marked population inversion between the à and states of the 4-YPhS radical products in all cases bar Y = CN – for which state products are dominant. In all cases, the relative yield of state products increases upon tuning to shorter excitation wavelength (i.e. upon increasing the vibrational energy within the predissociating S1 molecules). Such variation in the electronic branching in the products of a photochemical reaction (S–Me bond fission) with change of substituent, and/or by change of excitation wavelength, is considered in light of the detailed topographies of the relevant PESs and the way these influence the nuclear motions that lead to the eventual dissociation.
2. Methods
2.1 Velocity map imaging studies
2A′′ two-photon transition used to probe Me(v = 0) radicals by 2 + 1 resonance enhanced multiphoton ionization (REMPI).21 All experiments were performed in the leading edge of the molecular beam pulse with a 20 ns time delay between the photolysis and probe laser pulses in the interaction region. Unintended multiphoton excitation and dissociation of these precursors is a constant risk, given their relatively long S1 state lifetimes (2.2 ns in the case of the PhSMe(S1, v = 0) level22). The intensity of the pump laser was thus kept as low as possible in all the studies to minimize any background signal from such processes. Ions formed in the interaction region were accelerated with a velocity mapping ion-optics assembly, through a field free time-of-flight region, towards a position sensitive detector (double microchannel plates coupled to a phosphor screen and a CCD camera). The images were acquired and processed using event counting (LaVision, DaVis 6.2).
 | (1) |
 | (2) |
 | (3) |
 | (4) |
The experimental image is the projection of P(v,θ), which can be described mathematically by the inverse Abel transform. This transform is numerically stable, but the inversion is a known ill-posed problem. We avoid this difficulty by fitting a projection of eqn (3) directly to the measured data as outlined in ref. 23. These fits have been observed previously23–25 to be in very good agreement with the results of a direct Abel inversion by the matrix method26 given sufficient signal to noise ratio.
The velocity distributions were subsequently converted into total kinetic energy release (TKER) distributions using the conservation of momentum eqn (5)
 | (5) |
2.2 Ab initio calculations
Previous studies have increasingly recognised the multi-dimensional nature of the molecular evolution following S1 ← S0 excitation.14,15,17 Thus the present work has involved a hierarchy of electronic structure calculations.Molpro version 2010.127 was used to optimise the ground state 4-YPhSMe molecules, in Cs symmetry, using the Møller–Plesset level of theory at the second order (MP2), together with Dunning's augmented correlation consistent basis set of triple ζ quality: aug-cc-pVTZ (AVTZ), assigned to all atoms.28,29 For bare PhSMe only, 1-D potential energy cuts (PECs) along RS–Me for the S0 and first four singlet (and triplet) excited electronic states were calculated using complete active space self-consistent field (CASSCF) and with second order perturbation theory (CASPT2) and the above AVTZ basis set, with the phenyl ring frozen at its optimised ground state geometry. The resulting PECs were subsequently quasi-diabatised for ease of display and subsequent discussion.
1-D PECs along ϕ (the torsional coordinate) for the S0, S1 and S2 states of each (4-Y)PhSMe molecule were calculated also, using the equation-of-motion coupled cluster singles and doubles (EOM-CCSD) method and a smaller VTZ basis set. Again, the rest of the nuclear frame was maintained at the respective ground state equilibrium geometry and the resulting PECs were quasi-diabatised to aid discussion. These calculations also yielded transition dipole moments and oscillator strengths for the respective S1–S0 and S2–S0 excitations in each 4-YPhSMe molecule.
Further insights into the multidimensional nuclear distortions associated with the minimum energy path (MEP) to S–Me bond fission on the adiabatic S1 PES of both PhSMe and 4-CNPhSMe were obtained by calculating the fully ‘relaxed’ S1 PES using RS–Me as the driving coordinate using the CAM-B3LYP functional of time dependent density functional theory (TD-DFT) and the 3-21G basis set in Gaussian 09. The energies at various points along this MEP were then recalculated using Molpro 2010.1 at the CASPT2 level. Further details regarding all of these calculations are presented along with the relevant results in the later sections of the paper.
3. Results and discussion
This section is structured as follows. The experimental data illustrating the Y- and excitation wavelength (λex)-dependences of the electronic branching in the 4-YPhS products are presented first, and (substantially) rationalised in light of the available 1-D PECs along RS–Me (at ϕ = 0°) and in the S–Me torsional coordinate ϕ (with all other coordinates constrained to their values at the S0 minimum energy geometry). Remaining discrepancies are then highlighted, and recognised as manifestations of the multidimensional nature of the adiabatic S1 PES that governs the eventual product branching.3.1 Imaging results
The 1 + 1 REMPI spectrum of PhSMe shows resolved vibronic structure attributable to population of different Franck–Condon (FC) active vibrational levels of the S1 state.14,17 Similar spectra have been obtained for the various 4-substituted thioanisoles, as illustrated for Y = Me and CN in Fig. 1. Such spectra allow identification of the S1 ← S0 origin band (or bands in the case of 4-MeOPhSMe, given the presence of two different conformers), the wavenumbers of which are listed in Table 1. These spectra reveal numerous resonances in the first ∼1400 cm−1 above the S1 ← S0 origin (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) 0), including features close to (0 + 722) cm−1. The greater widths of these features (particularly in the case of 4-MePhSMe) compared with the case of PhSMe is attributed to the extra spectral density arising from internal rotation of the Me substituent. Photoexcitation of the (0 + 722) cm−1 feature in the spectrum of PhSMe (attributed to the v7a = 1 level, where ν7a is an in-plane mode involving ring shearing plus substantial S–Me stretching motion) has been shown previously to yield an anomalously high /Ã state product branching ratio, (ref. 14, and Table 1) – a point to which we return later.
0), including features close to (0 + 722) cm−1. The greater widths of these features (particularly in the case of 4-MePhSMe) compared with the case of PhSMe is attributed to the extra spectral density arising from internal rotation of the Me substituent. Photoexcitation of the (0 + 722) cm−1 feature in the spectrum of PhSMe (attributed to the v7a = 1 level, where ν7a is an in-plane mode involving ring shearing plus substantial S–Me stretching motion) has been shown previously to yield an anomalously high /Ã state product branching ratio, (ref. 14, and Table 1) – a point to which we return later.
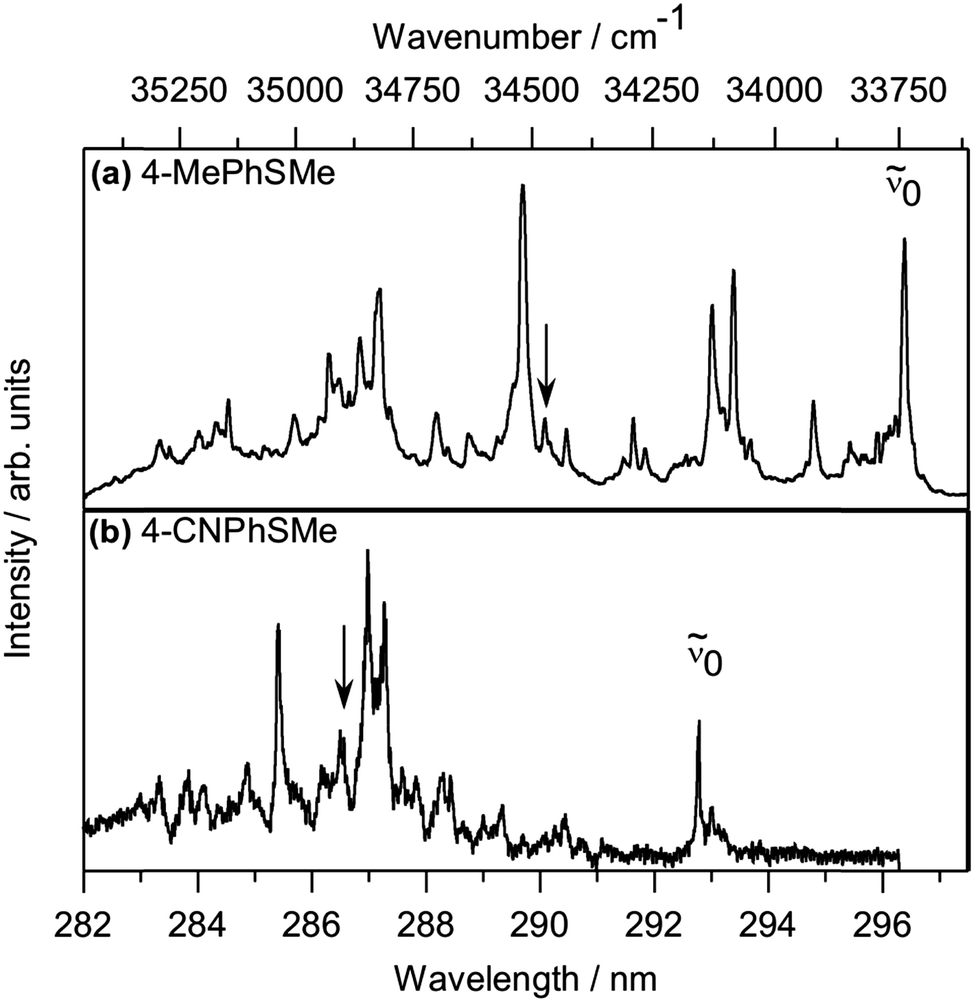 | ||
| Fig. 1 1 + 1 REMPI spectra of jet-cooled samples of (a) 4-MePhSMe and (b) 4-CNPhSMe, with the deduced origin band (0) indicated in each case. Peaks lying in the appropriate wavenumber range for the respective S1(v7a = 1) resonances on which images shown in Fig. 2 were recorded are marked by arrows. | ||
0) of PhSMe, 4-MePhSMe, 4-MeOPhSMe and 4-CNPhSMe determined by 1 + 1 REMPI spectroscopy, along with /à state product branching ratios following excitation at three different wavenumbers within the respective S1 ← S0 manifolds. Also shown are the S−Me bond dissociation energies (D0(4-YPhS–Me), in cm−1) and the energy separation of the and à states of the respective 4-YPhS radical fragments (in cm−1) derived from analysis of the measured TKER spectra (with the assumption that the vibrational energy disposals in both states of the radical are similar)
| Y | T 00(S1–S0)/cm−1 |
/Ã branching ratio |
D 0/cm−1 |
–Ã splitting/cm−1 |
||
|---|---|---|---|---|---|---|
| (v = 0) | ∼(0 + 722)/cm−1 (v7a = 1) |
∼(0 + 1400)/cm−1 |
||||
|
a Measurement made on resonance at (0 + 1000) cm−1.
b Equivalent branching ratios from Lim and Kim.14
|
||||||
| H | 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 504 504 |
0.08 | 0.52 | 0.15a | 24400 ± 200 |
2800 ± 200 |
| 0.05b | 0.43b | 0.15b | 3000 ± 7 (ref. 30) | |||
| Me | 33740 |
0 | 0.04 | 0.09 | 24000 ± 200 |
3200 ± 200 |
| 3320 ± 50 (ref. 9) | ||||||
| MeO | 32309 (anti) |
0 | 0 | 0.09 | 25100 ± 200 |
3400 ± 200 |
| 32133 (syn) |
4000 ± 50 (ref. 9) | |||||
| CN | 34155 |
1.33 | 2.23 | 4.56 | 25700 ± 200 |
1500 ± 200 |
Velocity map images were recorded for PhSMe and the three 4-YPhSMe molecules at many different excitation wavelengths. Fig. 2 shows raw (a) and fitted (b) images of the Me(v = 0) fragments from photolysis of PhSMe, 4-MePhSMe, 4-MeOPhSMe and 4-CNPhSMe at the respective S1 ← S0 origins (top row), on the v7a = 1 resonance (in the case of PhSMe) and on representative resonances at similar wavenumber in the other three cases (middle row), and on a resonance at higher wavenumber ∼(0 + 1400) cm−1 (for all except PhSMe, bottom row). Panel (c) in each case shows the corresponding TKER spectrum, and its decomposition into components attributable to formation of à and state radical products. Some data sets show a weak isotropic background signal (dashed line) which, after exhaustive optimisation of the beam conditions to minimise any cluster component, we attribute to multiphoton absorption and subsequent dissociation of the 4-YPhSMe parent. All of the images are slightly anisotropic; the recoil velocities of the Me fragments are preferentially parallel to ε (characterised by β ∼0.3–0.5), but none show any evidence of a component with opposite (negative) anisotropy on the leading edge of the TKER distribution as reported in an earlier study of PhSMe photolysis.14
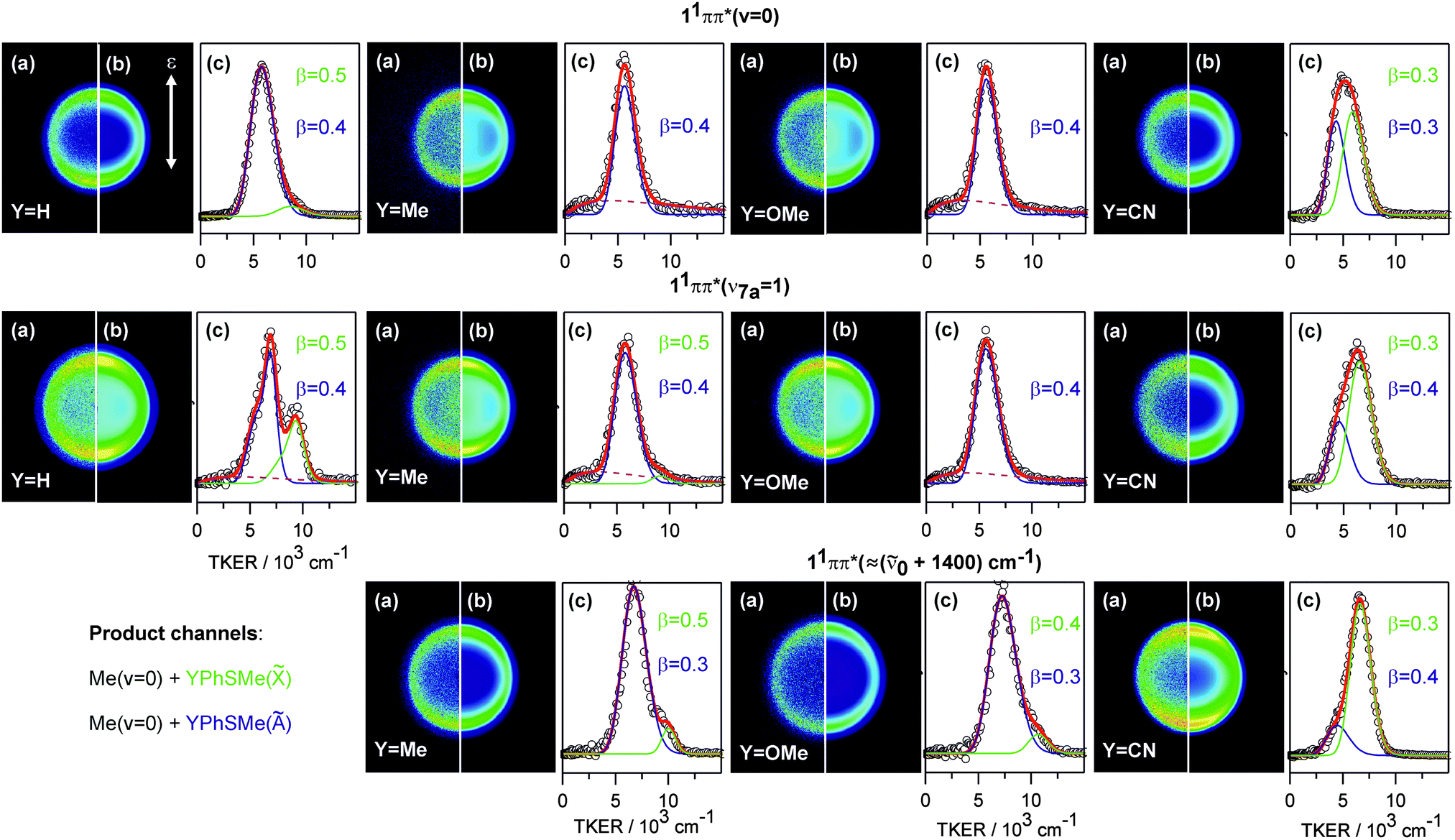 | ||
| Fig. 2 Velocity map images (a) and the fit results (b) of the Me(v = 0) fragments for PhSMe, 4-MePhSMe, 4-MeOPhSMe and 4-CNPhSMe following excitation at the respective S1 ← S0 origins, on the resonance associated with the S1(v7a = 1) level (in the case of PhSMe) or an illustrative resonance at similar wavenumber in the other three cases, and on a representative resonance at (0 + 1400) cm−1 (in all but PhSMe). The TKER spectra (c) are decomposed into features associated with formation of ground (, green) and excited (Ã, blue) state 4-YPhS co-fragments and, when necessary, to an isotropic background feature (dashed line) that we attribute to unintended but unavoidable multiphoton dissociation. | ||
The TKER spectrum obtained from photolysis of PhSMe at (0 + 722) cm−1 provides a convenient starting point for analysing and assessing these data. The faster and slower components are associated with formation of, respectively, PhS() and PhS(Ã) fragments (together with Me(v = 0) fragments). The separation between the respective maxima matches the documented energy splitting between these two states of the PhS radical (ΔE = 3000 cm−1, ref. 30). The previous VMI study concluded that most of the Me products are formed in their ground (v = 0) level.14 The breadth of the two TKER components implies that the PhS() and PhS(Ã) products are formed with a broad (∼2500 cm−1 full width at half maximum (FWHM)) spread of internal energies. This is understandable on FC grounds. S1 ← S0 excitation encourages expansion of the Ph ring, which would have to shrink again to attain the minimum energy geometry of the eventual PhS radicals; in-plane ring breathing vibrations can thus be expected to make a significant contribution to the observed internal excitation of these products. The fit of this particular TKER spectrum was noticeably improved by using a sum of two Gaussian functions (in velocity space) to describe each of the major product channels and a further Gaussian to describe the weak background signal. The best-fit decomposition in terms of these three components is shown in panel (c); the /Ã branching ratio so derived (0.52, Table 1) accords well with the earlier determination by Lim et al.14
The TKER spectrum obtained following excitation of PhSMe at the S1 ← S0 origin is dominated by a single peak which, on energetic grounds and in accord with previous analysis, we assign to formation of PhS(Ã) radicals.14 The weak pedestal at higher TKER is attributable to PhS() product formation. For simplicity, it proved sufficient to decompose this spectrum into just two Gaussian functions – one describing each product channel. The deduced /Ã branching ratio is small (∼0.08), as in the previous determination,14 implying a near-total population inversion between these two electronic states of the radical. Images taken on many other resonances in the range between 0 and (0 + 1400) cm−1 all appear very similar. Analysis of all such images returns a best-estimate of the S–Me bond dissociation energy, D0(PhS–Me), and shows the /Ã branching ratio increasing slowly as the excitation wavenumber is increased. Even at the highest wavenumber, however, this ratio is less than half that measured on the (0 + 722) cm−1 resonance.
Relative to PhSMe, 4-MePhSMe should be the least perturbed of the various substituted thioanisoles investigated in this work. However, the images of Me(v = 0) products from S–Me bond fission in this molecule taken on many different resonances within the S1 ← S0 absorption band all look very similar. The analogous images recorded when exciting on the S1 ← S0 origin, at (0 + 733) cm−1 (i.e. on one of the several resonances that might be expected to involve ν7a-like motion in the S1 state of 4-MePhSMe) and at ∼(0 + 1400) cm−1 are also displayed in Fig. 2. The best-fit value for the Ö energy splitting in the 4-MePhS radical is ∼400 cm−1 greater than in the unsubstituted thiophenoxyl radical, and the S–Me bond strength shows a similar decrease relative to that in bare thioanisole, both of which trends are consistent with some modest stabilisation of the ground state radical by the electron donating Me group.31 The associated TKER spectra are dominated by Me + 4-MePhS(Ã) products, the recoil anisotropy is again preferentially parallel, and the /à branching ratio is always small – increasing from 0 to ∼0.09 across this range (Table 1) without any obvious wavelength (or state) dependent discontinuity. We return to consider the very different electronic branching in the fragmentation of PhSMe and 4-MePhSMe in the region of the v7a = 1 resonance later.
The Me(v = 0) images from photolysis of 4-MeOPhSMe are also all dominated by the à state product channel; 4-MeOPhS() products are only recognisable in TKER spectra obtained at the shortest excitation wavelengths, by which point the /à branching ratio has increased to ∼0.09. Again, we find no evidence for any excitation wavelength (or state) dependent discontinuity in the branching ratio. The corresponding images from 4-CNPhSMe photolysis also look superficially similar, but more careful analysis of these data actually reveals very different behaviour. Inspection of Fig. 2 shows that the TKER spectrum obtained when exciting at the S1 ← S0 origin is broader than in the cases of PhSMe, 4-MePhSMe or 4-MeOPhSMe and, upon tuning to shorter wavelengths, the TKER profiles degrade in the opposite sense to those from the other photolyses. Decomposing these profiles leads to the conclusion that 4-CNPhS() fragments are the majority product at all wavelengths studied, with the /à branching ratio rising from ∼1.3 (at the S1 ← S0 origin) to ∼4.6 when exciting at ∼(0 + 1400) cm−1 – see Table 1 – but, again, we find no evidence for any excitation wavelength (or state) dependent discontinuity in this branching ratio. As Table 1 also shows, the Ö energy splitting in the 4-CNPhS radical is much reduced, and the S–Me bond correspondingly increased, relative to that found for the unsubstituted PhS radical and the PhSMe parent. Both differences can be traced to the electron withdrawing nature of the CN group, and the consequent destabilisation of the ground state radical.
3.2 Calculated 1-D PECs
The MP2 minimum energy geometries of PhSMe, 4-MePhSMe and 4-CNPhSMe are all planar, while that of 4-MeOPhSMe has the S–Me bond at a dihedral angle ϕ ∼ 70° to the ring plane. The latter geometry can be understood by recognising that the MeO substituent is a strong π donor, which delocalises electron density onto the ring. The SMe group is also a (milder) π donor. The presence of two counter-donating π–substituents on opposite sides of the ring overloads the π system in the HOMO, which can be relieved by breaking with the weaker π conjugator – in this case, the 3px (alternatively termed nπ in ref. 14) orbital centred on the sulfur atom. Similar behaviour was noted previously in the case of 4-MeOPhSH.9CASSCF and CASPT2 PECs along RS−Me were calculated for the ground and the first four excited singlet (and triplet) states of bare PhSMe. Fig. 3 shows the CASPT2 PECs, obtained by varying RS−Me while holding the rest of the nuclear framework at the ground state equilibrium geometry. These calculations used a state averaged (SA) CASSCF method, with the orbitals state-averaged over the above nine states and a (10/9) active space comprising the 3 ring centred π orbitals and their π* anti-bonding counterparts, σ and σ* orbitals localised on the S–Me bond and a diffuse px orbital supporting a lone pair on the S atom. A small imaginary level shift of 0.5 a.u. was required to aid convergence and circumvent the presence of intruder states.
 | ||
| Fig. 3 Cuts along RS−Me through the (quasi-diabatised) CASPT2 PECs of the ground and first few singlet and triplet excited states of PhSMe, with all other coordinates held fixed at the ground state equilibrium values. The diabatic states are labelled by their dominant electronic configuration in the vFC region (note, the first triplet state is best described as 3σσ* at long range (as discussed in the text)). CI-1 and CI-2 are also indicated. | ||
Both calculations reproduce the observed S1 ← S0 energy separation fairly well. The CASSCF calculations significantly underestimate the experimental D0(PhS–Me) value, while the CASPT2 calculations overestimate this quantity (by ∼0.3 eV). Nonetheless, the general shapes of the first few excited singlet and triplet PECs returned by both sets calculations are very reminiscent of those reported previously for PhSH.6 Inspecting the orbital coefficients confirms that these lower lying excited states are dominated by electron promotion from the highest occupied π orbital (at least in the vertical FC (vFC) region). The 11ππ*/11πσ* and 11πσ*/1ππ CIs (CI-1 and CI-2, respectively) are clearly evident in Fig. 3, as is the avoided crossing between the lowest lying 3A′(3ππ*) potential (henceforth T1) and the repulsive 3nσσ* PEC (at RS−Me ∼2.3 Å).
Fig. 4 shows EOM-CCSD PECs for the S0, 11ππ* and 11πσ* states of PhSMe, 4-MeOPhSMe and 4-CNPhSMe obtained by varying the S–Me torsion angle ϕ while holding all other degrees of freedom at their ground state equilibrium values. For the symmetrically substituted species (PhSMe and 4-CNPhSMe), ϕ = 0 and 180° represent equivalent planar geometries. The corresponding potentials for 4-MePhSMe are very similar to those for bare PhSMe. The diabatic 13ππ* and 13πσ* potentials show the same ϕ preference (i.e. planar and non-planar, respectively) as their singlet counterparts. The obvious asymmetry of the excited state PECs for 4-MeOPhSMe reflects the greater calculated stability of the anti-conformer (i.e. with the S–Me bond tilted away from the Me group in the MeO). These 1-D PECs highlight the different minimum energy geometries of the 11ππ* and 11πσ* states in these molecules at the equilibrium RS–Me value and, as noted in the previous studies of PhSMe,14,15,17 suggest a likely role for torsional motion in promoting coupling between these excited states. These EOM-CCSD calculations also confirm that the S1 ← S0 excitation dominates the long wavelength absorption in these thioanisoles and suggest that Y-substitution induces a (small) bathochromic shift of the S1 ← S0 origin (consistent with the measured origins listed in Table 1).
 | ||
| Fig. 4 Cuts along ϕ through the EOM-CCSD PESs of the S0 (black), 1ππ* (red) and 1πσ* (blue) states of (a) PhSMe, (b) 4-MeOPhSMe and (c) 4-CNPhSMe, with all other coordinates held fixed at their ground state equilibrium values. | ||
3.3 The dissociation mechanism
Key observations requiring explanation include the substituent dependence of the/Ã branching ratio, and two aspects of its photolysis wavelength dependence – the systematic increase in branching ratio with increasing excitation energy in all four molecules, and the atypical behaviour displayed by the PhSMe level resonant at (0 + 722) cm−1 (and by four levels of thioanisole-d3 at similar excitation energy.15)
As noted earlier, the recent literature contains several studies addressing non-radiative population transfer mechanisms from the optically ‘bright’ 11ππ* state to the dissociative 11(n/π)σ* PES in a range of heteroatom containing aromatic molecules. O–H bond fission by tunnelling through the barrier under the 11ππ*/11πσ* CI in the RO–H stretch coordinate in phenols has been studied extensively.8,10,12 The corresponding energy barrier in RS–H in the thiophenols is much smaller but, here too, tunnelling enables predissociation from the lowest S1 vibrational levels.9 Me is a much heavier leaving group; should S–Me bond cleavage in the thioanisoles be pictured in a similar way? Kim and Lim14 proposed a key role for torsional motion of the S–Me group in promoting dissociation from the S1 state of thioanisole – a view that has since been developed further17 – but the way in which the fragmentation dynamics might be tuned by strategic substitutions has not been explored hitherto.
In what follows, we first picture possible S–Me bond fission as a result of 4-YPhSMe(S1) molecules coupling to the dissociative 11πσ* PES, evolving towards CI-2 and eventually branching into and à state radical products. Such a mechanism provides a superficially satisfactory rationale for the observed behaviour but, as shown in the ESI,† wavepacket propagations on model 2-D(RS–Me,ϕ) PESs constructed by reference to the present ab initio data highlights inadequacies in such an approach. Thus we end this section by reporting the calculated MEP to S–Me bond fission on a fully relaxed, multi-dimensional adiabatic potential for the S1 state and showing that this can provide a plausible rationale for all of the observed product branching.
0 + 1400) cm−1.17 Such observations would be consistent with the expected poor overlap of the bound (S1(v = 0)) and continuum (11πσ*) wavefunctions, and would imply that intramolecular vibrational redistribution (IVR) within excited levels of the S1 state can aid passage through the seam of intersection associated with CI-1.
Me is a weak σ perturber and thus has little effect on the π-system; unsurprisingly, the 1-D PECs in ϕ space for 4-MePhSMe are similar to those of bare thioanisole. So, too, is the electronic branching in the 4-MePhS products and its variation with excitation wavelength (if we temporarily ignore the anomalous branching measured when exciting the v7a = 1 level of PhSMe). Introducing a strong π donor like MeO, in contrast, has an obvious effect on the torsional PECs. The minimum energy geometry of the S0 state of 4-MeOPhSMe is non-planar (ϕmin ∼ 70°). Vertical excitation will thus favour preparation of S1 molecules with non-planar geometries, where the calculated energy separation between the 11ππ* and 11πσ* PESs is reduced, though we note that the present EOM-CCSD calculations again place the 11πσ* PES above the 11ππ* PES at all ϕ (Fig. 4(b)). Thus, though the potentials are significantly modified by MeO substitution, we should again anticipate that 11ππ* ![[long arrow, wavy then straight]](https://www.rsc.org/images/entities/char_e0f6.gif) 11πσ* population transfer will be more efficient at non-planar geometries. As noted previously, the CN group withdraws π-density from the ring into its π* orbital, which is compensated by partial conjugation with the sulfur px lone-pair. The S0 state of 4-CNPhSMe thus has a planar equilibrium geometry, and vertical excitation will favour population of S1 molecules with ϕ = 0°. Nonetheless, the present analysis suggests that CN substitution should enhance the efficiency of 11ππ* 11πσ* population transfer – by virtue of the greater relative stabilisation of the 11πσ* PES (see Fig. 4(c)).
11πσ* population transfer will be more efficient at non-planar geometries. As noted previously, the CN group withdraws π-density from the ring into its π* orbital, which is compensated by partial conjugation with the sulfur px lone-pair. The S0 state of 4-CNPhSMe thus has a planar equilibrium geometry, and vertical excitation will favour population of S1 molecules with ϕ = 0°. Nonetheless, the present analysis suggests that CN substitution should enhance the efficiency of 11ππ* 11πσ* population transfer – by virtue of the greater relative stabilisation of the 11πσ* PES (see Fig. 4(c)).
![[X with combining tilde]](https://www.rsc.org/images/entities/b_i_char_0058_0303.gif) /Ã product branching following transfer to the 11πσ* PES.
Having reaffirmed previous suggestions that S–Me torsion might encourage radiationless transfer to the 11πσ* PES, we now consider factors that would influence the electronic branching in the resulting (4-Y)PhS + Me products. Here we distinguish global effects (which affect the branching over the whole range of substituents and photolysis energies investigated) and possible local effects, which only influence the branching in a particular molecule or specific energy region – notably the role of the ν7a mode in PhSMe.
/Ã product branching following transfer to the 11πσ* PES.
Having reaffirmed previous suggestions that S–Me torsion might encourage radiationless transfer to the 11πσ* PES, we now consider factors that would influence the electronic branching in the resulting (4-Y)PhS + Me products. Here we distinguish global effects (which affect the branching over the whole range of substituents and photolysis energies investigated) and possible local effects, which only influence the branching in a particular molecule or specific energy region – notably the role of the ν7a mode in PhSMe.
Global factors influencing the /Ã product branching following transfer to the 11πσ* PES can be envisaged by inspecting the calculated PECs shown in Fig. 3 and 4. To form 4-YPhS() radicals, the dissociating molecules must follow the diabatic path through CI-2 (which is localised at ϕ = 0°). 4-YPhS(Ã) product formation, in contrast, requires that the dissociating molecules follow the adiabatic path, avoiding CI-2 by virtue of having a non-planar geometry (ϕ ≠ 0°) at the relevant extended RS−Me bond lengths. Given these general guidelines, the influence of the various Y-substituents on the eventual product branching might appear relatively self-evident.
PhS(Ã) radicals are the dominant products from PhSMe photolysis at all wavelengths studied (see Table 1). This is consistent with the foregoing discussion, since CI-1 presents a larger barrier to dissociation at planar geometries and the coupling efficiency through this CI should increase at ϕ values away from 0°. state product formation would require that the anisotropy of the 11πσ* PES is sufficient to bring the molecular frame back close to planarity before the separating fragments reach RS−Me values associated with CI-2. The present observations could be accommodated by assuming that most dissociating molecules pass through the region near CI-2 with ϕ ≠ 0° and follow the adiabatic potential to PhS(Ã) products. The relative yield of state products increases upon tuning to shorter photolysis wavelengths, i.e. when populating higher vibrational levels of the S1 state. The S1 state lifetime is sufficient to allow some IVR prior to transfer to the 11πσ* PES. Any IVR that channels vibrational energy into modes that aid passage through CI-1 (e.g. S–Me stretching motion) would reduce the effective barrier at ϕ = 0° and thus encourage coupling through CI-1 at smaller ϕ; molecules evolving on the 11πσ* PES at smaller ϕ should have a higher probability of being drawn into the well associated with CI-2 and dissociating to state products. The observed electronic population inversion in the 4-MePhS products from 4-MePhSMe photolysis, and its excitation wavelength dependence, could be understood in the same way.
Rationalising the predominance of à state products in the presence of a strong π-donating substituent like MeO would be similarly straightforward. In this case, vertical excitation prepares S1 molecules with ϕ far from 0°, in regions of configuration space where 11ππ* 11πσ* coupling would be more favourable (recall Fig. 4(b)). Again, the topography of the 11πσ* PES seemingly offers little to encourage dissociating molecules to evolve towards near planar geometries, CI-2 is avoided, and à state products might thus be expected to dominate. Finally, we consider the influence of a strong π-withdrawing substituent (CN). As in PhSMe, the minimum energy geometries of the S0 and S1 states are both planar. The vertical energy separation of the 11πσ* and 11ππ* states is smaller, however, to the extent that the former is calculated to be the lower in energy at ϕ = 90° (Fig. 4(c)). Relative to PhSMe, we thus might anticipate enhanced 11ππ* 11πσ* coupling efficiencies at smaller ϕ and that, once on the 11πσ* PES, a larger fraction of the dissociating molecules would be drawn into the well associated with CI-2 and evolve to 4-CNPhS() radical products. Again, decreasing the excitation wavelength is seen to boost the relative yield of the diabatic dissociation products (Table 1) and, again, the same IVR based explanation could reasonably apply.
The finding that exciting the PhSMe S1(v7a = 1) level leads to an anomalously high PhS() product yield was first explained by assuming that this mode provided particularly direct access to the 11πσ* state via CI-1.14 This conclusion was supported by the deduced negative recoil anisotropy of Me products on the leading edge of the TKER distribution – consistent with products arising from direct excitation to the dissociative 11πσ* PES.14 As noted in Section 3.1, however, the present study finds no evidence of a velocity dependent Me product recoil anisotropy from PhSMe, or from any other 4-YPhSMe. Rather, we observe a small positive β value over the whole TKER range – consistent with dissociation following initial S1 ← S0 excitation. Nonetheless, the anomalously high /Ã branching ratio reported by Lim et al.,14 and reproduced in the present work, implies that the ν7a mode has a significant impact on the eventual dissociation dynamics. The special role of this resonance in PhSMe was explained by noting that this asymmetric in-plane vibration involves substantial S–Me stretching motion and matches closely with the derivative coupling (DC) component of the branching space associated with CI-1. As such, this mode would be particularly efficient at promoting vibronic coupling through the seam of intersection at geometries closer to ϕ = 0° – thus boosting the likelihood that dissociating molecules would subsequently pass through the localised region of configuration space associated with CI-2 and form state radical products.17 Broadly similar conclusions were reached regarding the nature of the ‘special’ resonances that yield anomalously large /Ã product branching ratios in the dissociation of thioanisole-d3, though the data analysis more clearly revealed the multi-dimensional facets of the radiationless transfer at the CI-1 seam.15
There remains a question as to why this particular resonance has such a dramatic effect on the /Ã branching ratio in PhSMe, but seemingly not in any of the 4-YPhSMe molecules investigated in the present work. MeO and CN substitutions both cause significant perturbation of the π system, so these are probably not ideal test-beds for exploring the nature of the special resonance. The Me group is not a direct π perturber, however. Me substitution in the 4-position has little influence on the ϕ-dependent PECs (cf. PhSMe) or on the deduced wavenumber of ν7a. Yet the relative yield of state products when exciting on any of the various features lying in the appropriate wavenumber range for v7a = 1 within the 1 + 1 REMPI spectrum of 4-MePhSMe (Fig. 1(a)) is small (see e.g.Fig. 2(a) and Table 1), and similar to that observed when exciting on other resonances at longer or shorter wavelength. A plausible explanation for this difference comes from analysing the nuclear motions involved in the respective ν7a modes. A Me group in the 4-position acts as a ‘mass anchor’, reducing the amplitude of the S−Me stretch component within the overall motion. As a result, and in contrast to PhSMe, the correlation between ν7a and the DC component of the branching space associated with CI-1 in 4-MePhSMe is weaker, and exciting the v7a = 1 level offers little benefit in terms of enhancing vibronic coupling at CI-1 at smaller ϕ – as would be required in this scenario if the dissociating flux was to sample CI-2 and evolve to state radical products. The current study suggests that each of the 4-substituents degrades the S–Me ‘local mode’ nature of ν7a (relative to the case in bare PhSMe), and thereby reduces the role of this mode in promoting coupling at CI-1.
Though persuasive, the foregoing interpretation is not without its difficulties. Like phenol,8,32 thioanisole and the symmetrically substituted species with Y = Me and CN should be viewed within the non-rigid G4 molecular symmetry group, which raises questions about the likely efficacy of torsion alone as a coupling mode at CI-1. The wavepacket calculations on model 2-D(RS–Me,ϕ) potentials summarised in the SI raise further questions. No prior assumptions were made regarding the magnitudes of the coupling terms at CI-1 or CI-2 (V12 and V02, respectively), and a wide range of possible values were explored. Given the assumed potentials, it was possible to find V12 values consistent with the S1 state having a ns lifetime but even assigning V02 its maximum possible value failed to generate anything approaching the extent of the observed population inversion observed in the PhS products. Given this inability to replicate the observed dominance of electronically excited PhS(Ã) products following excitation to the S1 state in this (model) 2-D framework, we now explore the problem in higher dimensionality by calculating minimum energy paths (MEPs) on the adiabatic S1 PESs for both PhSMe and 4-CNPhSMe.
 | ||
| Fig. 5 (a) MEP on the adiabatic S1 PES of PhSMe calculated by CASPT2 correcting the energies of the fully relaxed geometries (determined using TD-DFT) at a range of RS−Me bond extensions (solid red line). The evolving nuclear geometry is illustrated by the structures at four different RS−Me values superimposed above the MEP. The open red symbol shows the S1 energy calculated at the ground state equilibrium geometry, while the solid black line through the open symbols shows the S0 state energies calculated at the relaxed S1 geometries. The dashed curves extending from RS−Me ∼2 Å show the S1 and S0 energies calculated when extending RS−Me with all other coordinates fixed at their values at the local maximum on the fully relaxed S1 PES. (b) The corresponding MEP on the S1 PES of 4-CNPhSMe calculated by CASPT2 correcting the energies of the (CS constrained) geometries determined by TD-DFT at a range of RS−Me bond extensions (solid red line), and the S0 state energies calculated at these S1 geometries (solid black line). | ||
These calculations highlight the multi-dimensional nature of the dissociation. The TD-DFT calculations reaffirm the planar minimum energy geometry of the S1 state in the vFC region (i.e. near the minimum of the diabatic 1ππ* state) but also show substantial distortions by RS–Me ∼ 2.0 Å (i.e. in the region corresponding to (the lower half of) CI-1). The most obvious change is a torsion-like movement of the Me group around the C–S bond (as assumed previously14,17), but the calculations also show the S atom out of plane (on the opposite side of the ring-plane to the Me group) and some puckering of the benzene ring – all of which motions are pleasingly consistent with those identified by Han et al. in their recent state-selected photolysis studies of thioanisole-d3.15 Nuclear configuration 2 represents a local maximum on the calculated MEP, located ∼2500 cm−1 above the S1 minimum. Comparison with the rigid body CASPT2 PECs shown in Fig. 3 implies that this combination of orthogonal displacements enable a ∼30% reduction in the effective barrier height (measured relative to the S1 minimum). Extending RS–Me further, the calculated minimum energy geometry has reverted to planar by RS–Me = 2.2 Å but the anisotropy (around ϕ ∼ 0°) remains rather flat until approaching CI-2 (i.e. RS–Me > 3 Å). Calculated minimum energy structures at various RS–Me values are shown at the top of the figure. The solid black line through the open symbols shows the S0 state energies calculated at the relaxed S1 geometries. (The one filled black point shows the fully relaxed S0 state energy, and the single open red symbol shows the S1 state energy calculated at this ground state geometry). Two other PECs are shown by dashed lines extending from RS−Me ∼2 Å. These depict the S1 and S0 state energies calculated by extending RS−Me while holding all other degrees of freedom fixed at their values at the local maximum on the fully relaxed S1 PES. The red dashed curve captures the asymptotic limit associated with PhS(Ã) + Me products but, by virtue of the (fixed) non-planar geometry, shows no sign of the local minimum that would allow access to PhS() + Me products via coupling at CI-2.
Fig. 5(b) shows the results of similar calculations for 4-CNPhSMe. The corresponding S1 path along RS–Me proved hard to converge with C1 symmetry and was thus calculated with Cs symmetry constraints, using the same TD-DFT/CAM-B3LYP/3-21G level of theory. Wherever possible, the Cs constrained critical points (most notably the local maximum) along the relaxed path were then re-optimised without any symmetry constraint. These calculations returned negligible difference in the geometry and total energy when compared to the equivalent points computed with Cs symmetry constraints – implying that the minimum energy path in this case involves little (or no) distortion from planarity. As with PhSMe, the energies associated with the MEP were then recalculated at the CASPT2(12/10)/AVTZ level of theory in Molpro 2010.1; the results of these calculations are shown by the solid red curve in Fig. 5(b); as in Fig. 5(a), the solid black curve through the open black symbols shows the corresponding S0 energies calculated at the relaxed S1 geometries.
Thus we arrive at a refined interpretation for the observed dominance of electronically excited PhS(Ã) products following excitation to the S1(v = 0) levels of PhSMe (and, by analogy, 4-MePhSMe and 4-MeOPhSMe) and the very different product branching in the case of 4-CNPhSMe. Notwithstanding the more approximate nature of the TD-DFT calculations that underpin the MEP shown in Fig. 5(a), they serve to reinforce the view that S–Me bond fission occurs via tunnelling under CI-1, but the nuclear distortions required to minimise the effective barrier to dissociation are much more complicated than a simple torsion of the S–Me bond. Transit through the barrier is the rate limiting process, which is responsible for the ∼2.2 ns lifetime of PhSMe(S1).22 Post-barrier, the molecules emerge with non-planar geometries. The minimum energy path quickly reverts to (near)-planar geometries. However, the mean TKER of the asymptotic products (∼6000 cm−1) implies that it only takes ∼100 fs for RS–Me to increase from the exit side of the barrier to ∼3.5 Å and this is just too fast for the necessary heavy nuclear motions to replanarise the dissociating molecule before it passes through the RS–Me values associated with CI-2. Hence the observed propensity for remaining on the S1(adiab) potential and dissociating to PhS(Ã) + Me products. As argued previously, parent motions that facilitate tunnelling (e.g. motions in the RS–Me stretch coordinate) will allow barrier penetration at closer-to-planar geometries, thereby boosting the probability of non-adiabatic coupling at CI-2 and formation of PhS() products.
In the case of 4-CNPhSMe, the calculated MEPs shown in Fig. 5(b) broadly reinforce the conclusions reached previously from considering the 1-D PECs. Relative to PhSMe, the addition of an electron withdrawing CN group in the 4-position reduces the electron density on the ring and the driving force for non-planar distortions in the region of CI-1. After transiting the barrier, the dissociating molecules thus traverse the S1 PES with an overall geometry that is planar (or much closer to planar) and the enhanced non-adiabatic coupling at CI-2 and dissociation of 4-CNPhS() products inevitably follows.
4. Conclusions
Selective substitution of heteroaromatic molecules, even at positions remote from the reaction coordinate, can significantly affect their photochemical behaviour. In the case of phenols10 and thiophenols,9 the rates of O–H/S–H bond fission (by tunnelling) and/or the product branching can show marked sensitivity to the introduction of π–donor/acceptor groups on the ring. Here we have sought to generalise such concepts by investigating S–Me bond rupture following excitation to low vibrational levels within the S1 state in a range of 4-substituted thioanisoles – experimentally (by VMI studies of the Me(v = 0) products) and theoretically (by ab initio electronic structure calculations). The branching between the and à states of the 4-YPhS radical products provides a key measurable: à state products are found to dominate in the case that Y = H, Me or MeO (i.e. the products show an inverted electronic state population distribution), but not when Y = CN. Further, the relative yield of state products in all cases increases upon tuning to shorter excitation wavelengths and, in the specific case of bare PhSMe, jumps dramatically when tuning to the parent resonance assigned to the S1(v7a = 1) level.14
The companion theory builds a self-consistent picture of the S–Me bond fission process, which shows obvious parallels with the mechanism underpinning S–H bond rupture when exciting thiophenols at their S1−S0 origin.9 S–Me bond fission following excitation to the S1 state involves coupling via a conical intersection (CI-1) with the repulsive 1πσ* PES, in the RS–Me stretch coordinate, and subsequent branching into and à state radical products at CI-2, between the diabatic 11πσ* and the 1ππ PESs at longer S–Me bond lengths. As in the thiophenols, CI-1 lies at an energy above that of the S1(v = 0) level. Tunnelling through the barrier under CI-1 is facile in the thiophenols, but the corresponding barrier in thioanisole presents a real impediment to dissociation – as reflected by the orders of magnitude difference in the excited state lifetimes of PhSH (sub-ps)9 and PhSMe (∼2.2 ns).22 PhSMe(S1) molecules have a planar equilibrium geometry in the vFC region, but the calculated MEP through the barrier under CI-1 requires opposing out-of-plane motions of the S atom and the Me group, and some puckering of the benzene ring. Having emerged from this barrier region, the S–Me bond extends rapidly, to the extent that the dissociating molecules pass through RS–Me values associated with the CI-2 too quickly for the replanarisation of the nuclear framework required to enable coupling at CI-2 to take effect: Me + PhS(Ã) products are the inevitable result of such dissociations. The gradual increase in PhS() radical yield upon tuning to shorter photolysis wavelengths, and the marked increase in PhS() yield when exciting on a parent resonance (v7a = 1) involving substantial S–Me stretching motion, can both be understood in terms of an enhanced probability for barrier passage at closer-to-planar geometries. The dominance of 4-CNPhS() products when exciting low vibrational levels of the 4-CNPhSMe(S1) state can be traced to the reduced π-electron density on the ring, which is a driver for the out-of-plane distortions in the region of CI-1. Molecules dissociating on the adiabatic S1 PES thus propagate at much closer-to-planar geometries and thus have a greater opportunity for non-adiabatic coupling at CI-2.
The present study provides further illustration of the added benefits of investigating families of related molecules, at a number of different excitation wavelengths, and of combining careful experiments with appropriate high level theory when seeking to develop a more complete understanding of molecular photofragmentation dynamics.
Acknowledgements
The authors are grateful to the Marie Curie Initial Training Network ICONIC (contract agreement no. 238671) and the EPSRC (Programme Grants EP/G00224X and EP/L005913) for funding, and to Stephanie Harris and Drs Daniel Murdock and Dimitrios Zaouris for helpful discussions.Data accessibility: The underlying data for this paper has been placed in the University of Bristol's research data repository and can be accessed using the following DOI: 10.5523/bris.13z36fefmpje81i5jlryfrxuva.
References
- C. Brif, R. Chakrabarti and H. Rabitz, in Adv. Chem. Phys., ed. S. A. Rice and A. R. Dinner, Wiley-Blackwell, Malden, 2012, vol. 148, pp. 1–76 Search PubMed.
- M. Shapiro and P. Brumer, Phys. Rep., 2006, 425, 195–264 CrossRef CAS.
- T. Brixner and G. Gerber, ChemPhysChem, 2003, 4, 418–438 CrossRef CAS PubMed.
- A. L. Sobolewski, W. Domcke, C. Dedonder-Lardeux and C. Jouvet, Phys. Chem. Chem. Phys., 2002, 2, 1093–1100 RSC.
- M. G. D. Nix, A. L. Devine, B. Cronin, R. N. Dixon and M. N. R. Ashfold, J. Chem. Phys., 2006, 125, 133318 CrossRef PubMed.
- I. S. Lim, J. S. Lim, Y. S. Lee and S. K. Kim, J. Chem. Phys., 2007, 126, 034306 CrossRef PubMed.
- M. N. R. Ashfold, G. A. King, D. Murdock, M. G. D. Nix, T. A. A. Oliver and A. G. Sage, Phys. Chem. Chem. Phys., 2010, 12, 1218–1238 RSC and references therein.
- R. N. Dixon, T. A. A. Oliver and M. N. R. Ashfold, J. Chem. Phys., 2011, 134, 194303 CrossRef PubMed.
- T. A. A. Oliver, G. A. King, D. P. Tew, R. N. Dixon and M. N. R. Ashfold, J. Phys. Chem. A, 2012, 116, 12444–12459 CrossRef CAS PubMed.
- T. N. V. Karsili, A. M. Wenge, S. J. Harris, D. Murdock, J. N. Harvey, R. N. Dixon and M. N. R. Ashfold, Chem. Sci., 2013, 4, 2434–2446 RSC.
- G. M. Roberts and V. G. Stavros, Chem. Sci., 2014, 5, 1698–1722 RSC.
- G. A. Pino, A. N. Oldani, E. Marceca, M. Fujii, S. I. Ishiuchi, M. Miyazaki, M. Broquier, C. Dedonder and C. Jouvet, J. Chem. Phys., 2010, 133, 124313 CrossRef CAS PubMed.
- G. M. Roberts, A. S. Chatterley, J. D. Young and V. G. Stavros, J. Phys. Chem. Lett., 2012, 3, 348–352 CrossRef CAS PubMed.
- J. S. Lim and S. K. Kim, Nat. Chem., 2010, 2, 627–632 CrossRef CAS PubMed.
- S. Han, J. S. Lim, J. H. Yoon, J. Lee, S. Y. Kim and S. K. Kim, J. Chem. Phys., 2014, 140, 054307 CrossRef PubMed.
- For clarity, we use the labels 1ππ*, 1πσ*, etc., to represent diabatic states, S1, S2, etc. when discussing the adiabatic states, and use 11ππ* and S1 interchangeably when referring to vibronic levels in the potential well associated with the first excited singlet state. In contrast to the thiophenols, the first dissociative 1πσ* state in thioanisole is probably better pictured as 1nπσ*, as electron promotion is from a sulphur px dominated orbital rather than the more delocalised π orbital (in thiophenol, ref. 9). Nonetheless, to facilitate transferability across the various molecular systems, we persist with the ‘traditional’ 1πσ* descriptor in the present work.
- G. M. Roberts, D. J. Hadden, L. T. Bergendahl, A. M. Wenge, S. J. Harris, T. N. V. Karsili, M. N. R. Ashfold, M. J. Paterson and V. G. Stavros, Chem. Sci., 2013, 4, 993–1001 RSC.
- D. W. Chandler and P. L. Houston, J. Chem. Phys., 1987, 87, 1445–1447 CrossRef CAS.
- A. T. J. B. Eppink and D. H. Parker, Rev. Sci. Instrum., 1997, 68, 3477–3484 CrossRef CAS.
- E. Wrede, S. Laubach, S. Schulenburg, A. Brown, E. R. Wouters, A. J. Orr-Ewing and M. N. R. Ashfold, J. Chem. Phys., 2001, 114, 2629–2646 CrossRef CAS.
- J. W. Hudgens, T. G. DiGiuseppe and M. C. Lin, J. Chem. Phys., 1983, 79, 571–582 CrossRef CAS.
- M. Hoshino-Nagasaka, T. Suzuki, T. Ichimura, S. Kasahara, M. Baba and S. Kawauchi, Phys. Chem. Chem. Phys., 2010, 12, 13243–13247 RSC.
- A. M. Wenge, U. Kensy and B. Dick, Phys. Chem. Chem. Phys., 2010, 12, 4644–4655 RSC.
- D. K. Zaouris, A. M. Wenge, D. Murdock, T. A. A. Oliver, G. Richmond, G. A. D. Ritchie, R. N. Dixon and M. N. R. Ashfold, J. Chem. Phys., 2011, 135, 094312 CrossRef CAS PubMed.
- A. M. Wenge, A. Schmaunz, U. Kensy and B. Dick, Phys. Chem. Chem. Phys., 2012, 14, 7076–7089 RSC.
- G. Pretzler, H. Jäger, T. Neger, H. Philipp and J. Woisetschläger, Z. Naturforsch., 1992, 47a, 955–970 Search PubMed.
- H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov and G. Rauhut, et al. MOLPRO, version 2010.1, a package of ab initio programs, University of Cardiff, Cardiff, UK, 2010 Search PubMed.
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- D. E. Woon and T. H. Dunning, Jr., J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- J. B. Kim, T. I. Yacovitch, C. Hock and D. M. Neumark, Phys. Chem. Chem. Phys., 2011, 13, 17378–17383 RSC.
- J. H. Yoon, J. S. Lim, K. C. Woo, M. S. Kim and S. K. Kim, Bull. Korean Chem. Soc., 2013, 34, 415–420 CrossRef CAS.
- T. N. V. Karsili, A. M. Wenge, B. Marchetti and M. N. R. Ashfold, Phys. Chem. Chem. Phys., 2014, 16, 588–598 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp01660f |
| ‡ Carl Zeiss SMT GmbH, Rudolf-Eber-Straße 2, 73447 Oberkochen, Germany. |
| § Departamento de Química Física I, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain. |
| This journal is © the Owner Societies 2015 |