
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Peculiar morphological transitions induced by nanoparticles in polymeric blends: retarded relaxation or altered interfacial tension?
Shital Patangrao
Pawar
and
Suryasarathi
Bose
 *
*
Department of Materials Engineering, Indian Institute of Science, Bangalore-560012, India. E-mail: sbose@materials.iisc.ernet.in
First published on 12th May 2015
Abstract
Nanoparticles of different shapes can induce peculiar morphologies in binary polymer blends depending on their position. It is envisaged that the increased yield stress of the filled phase slows down the relaxation resulting in arresting the peculiar morphologies which otherwise is thermodynamically unfavourable due to the increased interfacial area. This essentially means that the highly irregular structures can be preserved even without altering the interfacial tension between the phases! On the other hand, in the case of interfacially adsorbed particles, the resulting solid-like interface can also preserve the irregular structures. These phenomenal transitions in filled blends are very different from the classical copolymer compatibilized polymer blends. Moreover, these irregular structures can further pave way in designing conducting polymer blends involving conducting nanoparticles and revisiting our understanding of the concept of double percolation!
1. Introduction
Blending existing polymers is a well-established strategy to design unique materials with myriad combination of properties. However, most polymer pairs are thermodynamically immiscible, owing to low entropy of mixing, resulting in phase separation in the macro scale. The various morphologies thus generated are governed primarily by the composition, viscosity ratio and the interfacial tension between the phases. During processing, the dispersed droplets deform significantly in flow fields leading to an increase in the interfacial area. In this process, the shape of the deformed droplets varies from ellipsoid to highly elongated fibril1 however, after the cessation of flow, the deformed droplets relax and return to their spherical shapes, driven by interfacial tension.2–4 In a typical processing time frame, the morphologies thus generated do not attain equilibrium and often coarsen upon annealing or post processing operations, again driven by the interfacial tension between the components. Though, the shape relaxation of deformed droplets has been studied mostly in Newtonian components, only few studies report it in viscoelastic medium.5,6 The matrix and the droplet viscoelasticity and elastic stresses at the interface seem to govern both the deformation dynamics and the relaxation processes. However, the shape relaxation of droplets in the presence of nanoparticles is scarcely studied. In polymer blends, the droplets undergo a more complex process when they restructure to a spherical shape in order to reduce the interfacial area, during which not only break-up and retraction but also coalescence of the droplets can occur. This phenomenon is even more complex in the case of blends with nanoparticles, especially if they are localized inside the droplet phase.7,8In order to stabilize the blend morphologies various strategies9–11 were adopted in the past where copolymers,12–20 mutually miscible polymers,21–25 reactive blending26–32 and various particles33–35 were introduced into the blend systems. In this perspective, we are mainly concentrating on nanoparticles, where introduction of particles not only stabilizes the phase morphology but also transforms the droplet–matrix morphology to peculiar structures. Thus nanoparticles act as compatibilizers similar to block co-polymers however the mechanism of stability is very different. The rationale behind this perspective is to highlight the enthalpic and the entropic driven localization of nanoparticles resulting in irregular phase structures. The latter phenomenon is more pronounced in rod like particles with large aspect ratio. In the case of classical compatabilization, improvement in mechanical properties is often realized through a tailored interface however, other functional properties like electrical conductivity; magnetic, optical etc. need incorporation of functional nanoparticles. Although, nanoparticles help in stabilizing bi-phasic structures the underlying mechanism greatly differs from the classical approach. When the nanoparticles with large aspect ratio localize in the minor phase, a peculiar phase morphology is often observed wherein the particles bridge the domains. In the case of blends with a conducting filler, this phenomenon can result in significant volume (bulk) electrical conductivity in the blends. This perspective highlights the peculiar morphologies triggered by the nanoparticles and their use in designing materials with tailor-made properties.
Given the brevity of this perspective we focused here mainly only on the effect of three different shaped nanoparticles in inducing peculiar morphology in binary blends. This will allow us to understand the key role of nanoparticles in inducing these phenomenal changes in the microstructure. In this context, three types of nanoparticles are considered such as carbon black (CB), clay and carbon nanotubes (CNT) representing spherical, two-dimensional platelets and one dimensional rod respectively. The mechanism and the critical concentration that induces these peculiar transitions are summarized from the literature in the last decade.
2. Mechanisms governing the selective localization of fillers in biphasic blends
Unlike fluid emulsions, in the case of highly viscous polymer blends, not only thermodynamics but various parameters like viscosities of individual phases and kinetics of mixing affect the localization of particles.36 Owing to their high viscosity, polymer phases and interfaces offer very high resistance to particle migration in a polymer matrix. This phenomenon further leads to a very high characteristic time required for the particles to attain thermodynamic equilibrium localization. Various mechanisms governing selective localization of particles are addressed below.2.1 Thermodynamics
Selective localization of particles in polymer blends can be predicted using the interfacial energy balance between particle and polymer phases. In this context, estimation of wetting coefficient, as proposed by Sumita et al.,37 can qualitatively predict the localization a priori.where γF1, γF2 and γ12 are the interfacial energies between filler/phase 1, filler/phase 2, and phase 1/phase 2 respectively. If ω12 > 1, fillers would selectively localize in phase 1, for ω12 < −1, fillers would preferentially localize in phase 2 and for −1 < ω12 < 1, fillers would localize at the interface. The interfacial tension between components was estimated with the help of theoretical models using well know Owens–Wendt38 and Wu's39 relations given below.
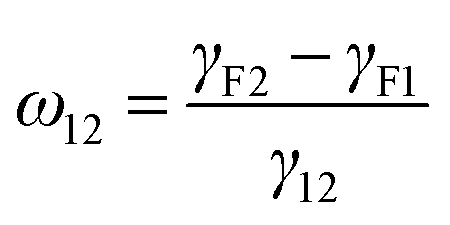

γ1, γ2 are surface free energies (SFE) of phase 1 and phase 2 respectively. γd1, γd2 are disperse and γp1, γp2 are the polar components of the surface free energies corresponding to phase 1 and phase 2 respectively.
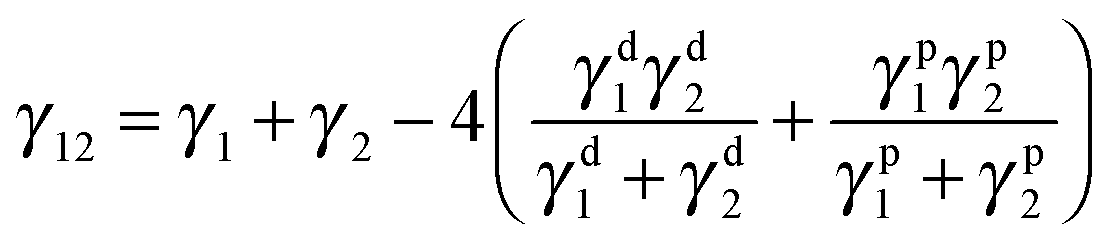
2.2 Kinetics
The sequence of polymer mixing has a prominent effect on the kinetics and state of mixing because of altered melt properties. Three different ways of particle loading and their effect on particle localization are addressed here. In the first case where all the blend components and the particles were charged simultaneously, the localization and migration of particles becomes a complex phenomenon because the melt extrusion consists of solids and viscous fluids as the morphology is still evolving. If one blend component melts at a considerably lower temperature than the other, particles can preferentially reside in the phase which melts first though this localization is thermodynamically not favored. With further mixing these fillers may migrate towards a thermodynamically preferential phase. In the second type, particles are generally incorporated after melting both the blend components. In this case, there is no solid polymer phase to exclude out and the particle may localize according to thermodynamic considerations. The third case is sequential mixing where particles can be first mixed with the one of blend component and then the second component is introduced later. In this approach, migration of particles can be realized if it was mixed with the non-preferred phase. The particles may migrate from one phase to the interface and finally in the favored phase. Various studies have been carried out to analyze the migration of particles.7,40,412.3 Viscosity of blend phases
In the case of highly viscous polymer fluids, the final localization of particles is greatly dependent on the shear viscosities of the blend components. Various studies were performed to evaluate this effect on the localization of particles however, the literature lacks in proper understanding due to the combined effect of thermodynamics and phase viscosity. In this context, Feng et al.42 systematically studied the effect of viscosity ratio on the localization of CB in PP/PMMA blends where PMMA with different viscosities was used. This study shows that when the viscosity ratio is one, CB resides in PMMA, the thermodynamically favored phase. With increasing viscosity of PMMA, CB migrated to the interface and at very high viscosity of PMMA, CB localized in the PP phase. The viscosity of the PP phase was not altered in this study. It can be concluded that particles prefer to localize in the low viscous phase.3. Mechanisms of filler induced deformation and stability of minor phases
The thermodynamic stability of elongated domains depends on the balance between polymer–polymer interfacial tension, which drives the minor phase to retract back to the spherical shape or to fragment into smaller droplets,43 and rheological stresses related to such morphological evolutions. This equilibrium is certainly altered upon addition of nanoparticles, which may affect both the interfacial tension and the flow behaviour depends on its position in the blends. However, the underlying mechanisms are still far from being understood. It has been reported that a major role is played by rheology.44–49 In the case of selective positioning within either of the polymers, the increased elasticity and viscosity of the host phase slowdown the shape relaxation and break up processes, stabilizing the irregularly shaped domains.5,6 The kinetic trapping of the filled phase may eventually result in stable bi-continuous morphologies. In addition, the self-networking capability of the nanoparticles is believed to be crucial, but many issues remain unresolved with respect to the shape of the particles. Among others, the relevance of the nanoparticles located at the polymer–polymer interface has been seldom systematically investigated. Theoretical and experimental studies suggest a compatibilizing action of the interfacially-adsorbed particles. A significant reduction of the average size of the minor phase of blends with drop-matrix morphology is usually reported as the evidence of compatibilization,50,51 but coalescence suppression by a shielding mechanism could generate the same effect without actually affecting the interfacial tension. Such an effect, however, would not be useful for the purpose of promoting co-continuity. A lowering of interfacial tension between the phases is actually needed to stabilize elongated domains of the minor phase.In a flow field, the total recovery time for the deformed droplet increases as the initial radius and the applied strain increase. This essentially means that a filled droplet will have an altered viscoelasticity which extends the time required to attain equilibrium both in deformation and relaxation. Highly deformed droplets can take an irregular shape when break-up occurs and the relaxation time of these droplets can increase significantly and more importantly this phenomenon depends largely on the concentration of nanoparticles. The time required to attain equilibrium for deformed droplets by coarsening is mainly dependent on the viscosity of the blend and interfacial tension. The kinetics of coarsening of unstable phases during steady state annealing can be related to the growth of droplets. As explained in Tomotika's theory,52 capillary pressure is responsible for phase coarsening and its kinetics can be estimated from the following equation,
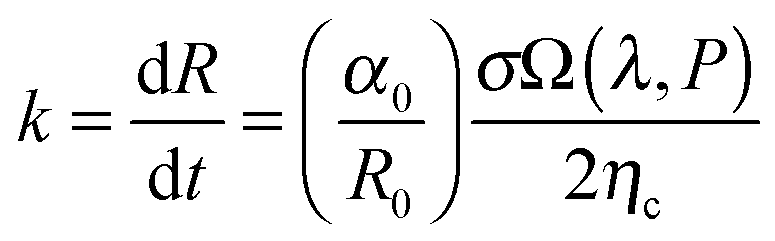
 is the rate of droplet growth.
is the rate of droplet growth. 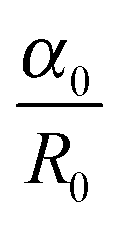 is the ratio of original amplitude to the phase radius and ηc is the viscosity of the blend. Veenstra et al.53 demonstrated a simplified relationship where the rate of droplet coarsening is inversely proportional to the viscosity of the blend and directly proportional to the interfacial tension of the components. The rate of droplet coarsening may be estimated from the following equation,
is the ratio of original amplitude to the phase radius and ηc is the viscosity of the blend. Veenstra et al.53 demonstrated a simplified relationship where the rate of droplet coarsening is inversely proportional to the viscosity of the blend and directly proportional to the interfacial tension of the components. The rate of droplet coarsening may be estimated from the following equation,where ηe and σ is blend's viscosity and interfacial tension respectively. c is dimensionless number and was found to be 0.07. It is expected that the elongated minor phase imbedded in another immiscible matrix can lead to break down into small droplets or coarsen to a sphere. Similarly, irregularly shaped unstable droplets can completely retract back into spheres and the characteristic time required for this phenomenal transformation can be estimated from the following equation as proposed by Carriere et al.,43

where ηm and ηd are the viscosity of matrix and droplet respectively. σ is the interfacial tension and D is the diameter of the elongated droplet. It is evident from these well established studies that minimization of interfacial tension and increase in blend viscosity are the key parameters to slow down or to completely suppress the relaxation of irregularly shaped phases. As mentioned earlier, elasticity also has a significant effect on relaxation kinetics of the elongated phase. In the case of blends with particles, elasticity may be induced by the interface and percolated network of the particles. It was observed that elasticity scales with particle fraction and is evident from the increased storage modulus at lower frequencies. The increased elasticity of the host polymer phase offers additional resistance to the relaxation process. In conclusion the increased elasticity and viscosity of host phase slows down the relaxation process. Fig. 1 shows retarded relaxation of PS droplets in the presence of nano-silica in PP/PS blends where relaxation is slowed down due to enhanced phase viscosity and reduced interfacial tension.6
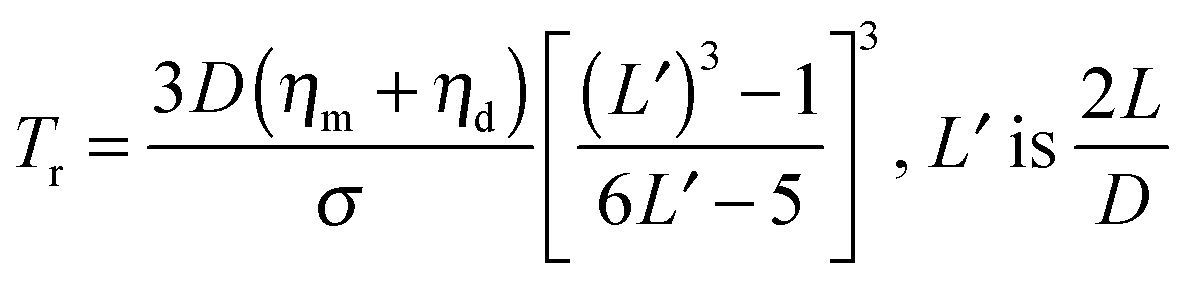
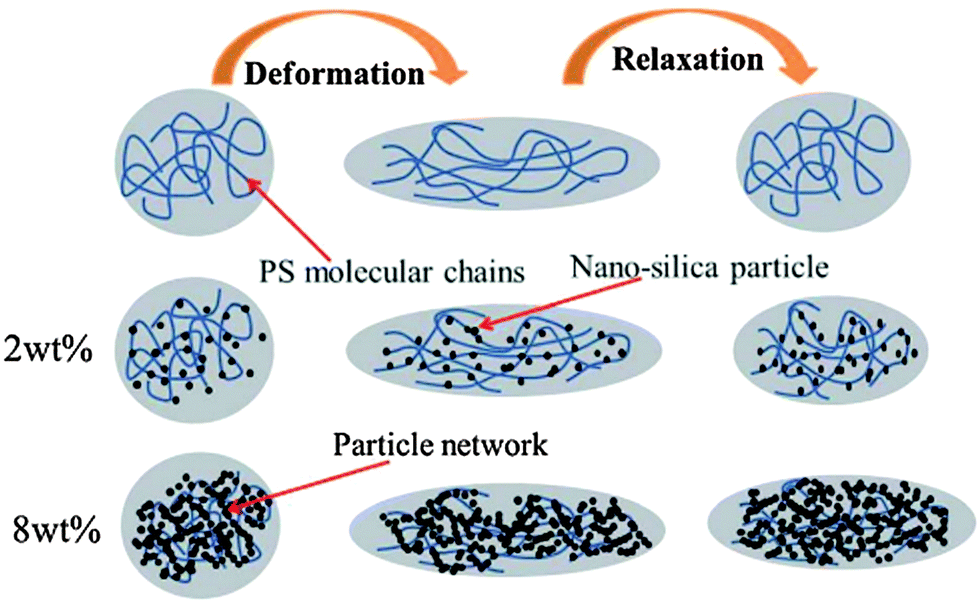 | ||
| Fig. 1 Cartoon illustrating retarded relaxation of PS droplets containing nano-silica particles in PP/PS blends (Liu et al.6 – reproduced by permission of Royal Society of Chemistry. All rights reserved). | ||
4. NPs induced peculiar phase structures
Among the different morphologies generated by blending, the bi-continuous microstructure has attracted a great deal of interest in the recent past. The interpenetration of the phases that characterizes this particular morphology often results in a synergistic combination of the properties. Table 1 summarizes the effect of filler induced phase transformation in various blends. These peculiar transformations of phases to a co-continuous network manifested a significant decrease in percolation threshold and enhanced electrical conductivity.54–58 Since, the concept of ‘double percolation’37 was proposed way back in 1991, improving the electrical conductivity in polymeric blends by distributing conducting particles either in one of the phases or at the blend's interface has been the subject of interest. This strategy reduces the percolation threshold of the conducting particle significantly in the blends and has gained tremendous popularity among the scientific community.59–64 The highly irregular microstructures induced by nanoparticles have also proven to reduce the percolation threshold especially with particles capable of self-networking.44,54,65,66 This strategy requires far less particles as the effective concentration increases with decreasing volume of the filled phase. These blends can be promising for various electrical,61,64,67–70 electromagnetic interference (EMI) shielding71–76 and electronics applications.| Blends | Filler | Filler fraction | Localization | Processing | Blend composition | Properties studied | Ref. |
|---|---|---|---|---|---|---|---|
| HDPE/PA6 | Organo clay | 4.8 wt% | PA6 | Melt extrusion | 75/25 | Rheology, mechanical | 45 |
| PPS/PBT | Organo clay | 4–6 phr | PBT | Melt extrusion | 60/40 | Rheology | 46 |
| PP/ABS | CB | 10 wt% | ABS | Melt extrusion | 70/30 | Rheology, conductivity | 44 |
| PP/PS | CB | 5 vol% | PS (butadiene of SBS) | Melt extrusion | 70/30 | Conductivity | 55 |
| PS/PMMA | Glass spheres | 30 vol% | PMMA | Melt extrusion | 50/50 to 70/30 | Rheology | 47 |
| HDPE/PS | CB | 1 wt% | HDPE | Melt extrusion | 5/95 | Rheology, conductivity | 65 |
| HDPE/PS | CB | 4 wt% | HDPE | Melt extrusion | 10/90 | Rheology, conductivity | 65 |
| ABS/PA6 | CB | 7.4 phr | PA6 | Melt extrusion | 60/40 | Rheology, conductivity and mechanical | 54 |
| PBT/LLDPE | Organo clay | Above 2 phr (8 phr) | PBT | Melt extrusion | 40/60 | Rheology | 48 |
| PPS/PA66 | MWNTs | 0.001–0.3 phr | PA66 | Melt extrusion | 60/40 | Morphology | 80 |
| PA6/ABS | Organo clay | 2 wt% (2–10 wt%) | PA6 | Melt extrusion | 40/60 | DMA | 81 |
| PLA/PA11 | OMMT, MS, CNT | 18, 6, 6 wt% respectively | PA11 | Melt extrusion | 70/30 | Rheology, DMA, conductivity | 66 |
| PPO/PA6 | Clay | >5 wt% | PA6 | Melt extrusion | 50/50 | Rheology | 49 |
| HDPE/PA6 | Clay | 5–20 wt% | PA6 | Melt extrusion |
75/25
25/75 |
Rheology, mechanical | 77 |
| HDPE/PA6 | MWNTs | 2 wt% | PA6 | Melt extrusion | 50/50 | Rheology, crystalinity | 82 |
| PEEK/TPI | MWNTs | 0.4 wt% | TPI | Melt extrusion | 50/50 | DMA, conductivity | 56 |
| PLLA/TPU | CNTs | 2 wt% | TPU | Melt extrusion | 70/30 | Tensile and impact test, DMA, rheology | 83 |
| PVDF/PA6 | CNTs | 1.2 wt% | PA6 | Melt extrusion | 50/50 | Electrical conductivity, mechanical | 57 |
| PA6/ABS | CNTs | 4 wt% | PA6 | Melt extrusion | 70/30 | Electrical conductivity | 58 |
4.1 Factors affecting the formation of peculiar morphologies
NP induced morphological evolutions are greatly dependent on the localization of particles and the composition. It is well known that localization of NPs in a particular phase not only affects the steady state shear viscosity but also alters the viscoelastic properties of the host polymer where elastic connotation is enhanced. Fig. 2a explains the effect of NPs localization in the minor and major phases of blends.77 In the case of blends where a particle resides in the minor phase, stabilized elongated structures are formed and beyond a critical concentration of NP results in a stable peculiar co-continuous structure. This phenomenon is observed mainly due to enhanced elastic connotation which hinders the relaxation of the elongated phase. Whereas, in blends where NP resides in the continuous phase the yield stress of the host phase is increased and restricts the coalescence process of the minor droplet phase. It is evident that packing of fillers in a continuous phase leads to smaller droplet size due to the higher force applied while processing through the highly elastic continuous phase and the suppressed or retarded coalescence phenomenon of droplets. Thus two main mechanisms which result in filler induced peculiar morphology can be envisioned, one is retarded relaxation of elongated droplets and second is reduction of interfacial energy, the effect of these mechanisms is completely dependent on the localization and nature of the NPs. In a study, the effect of stiff needle like sepiolite rods, and flexible CNTs was systematically investigated with respect to morphological transition in binary blends showed a different extent of co-continuity and mechanisms, where localization of both fillers was observed in the minor phase and at the interface.66 Interestingly, from contact angle experiments it was evident that sepiolite rods are effective in minimizing the interfacial energy and manifested in peculiar phases at low fractions. Whereas, since few flexible CNTs were found at the interface, it also manifested reduced interfacial energy and in addition viscoelasticity was enhanced dramatically over rods, which supports that enhanced continuity of phases is due to the synergetic effect of retarded relaxation with very high yield stress and minimization of interfacial energy. Since minimization of interfacial energy is a crucial parameter, tailoring the localization of fillers, especially with very high aspect ratio, at the interface is an important practice. | ||
| Fig. 2 (a) Schematic illustrating morphology evolution in the presence of particles in minor and major phase of blends (Filippone et al.77 – reproduced by permission of Elsevier, Copyright © 2010 Elsevier Ltd. All rights reserved), (b) morphology evolution in phase separating blends assisted through kinetics of mobile rod like particles (Li et al.79 – reproduced by permission of American Chemical Society, Copyright © 2011, American Chemical Society), (c and d) stitching of the interface using CNTs (Nuzzo et al.66 Reproduced by permission of Elsevier, Copyright © 2014 Elsevier Ltd. All rights reserved), (e) morphological transformation to peculiar phases in the presence of 4 wt% MWNTs in PA6/ABS blends, (f) co-continuity window without CNT in PA6/ABS blends and (g) co-continuity window and electrical conductivity in the presence of 4 wt% MWNTs in PA6/ABS blends ((e–g), Bose et al.58 – reproduced by permission of Springer,Copyright © 2015 Springer Ltd. All rights reserved). | ||
As we discussed about transformation of phase morphologies in the case of immiscible binary blends, a similar phenomenon is also applicable for miscible polymer blends which eventually phase separates. The kinetics of phase separation and morphological development was observed to be greatly influenced by the presence of fillers. Simulation studies of binary phase separating blends consisting of rod like fillers show network formation of minor phase through self-assembly of rod or needle like particles.78 The interconnected network formation is promoted by dynamics of phase separation of immiscible polymeric fluids where the minor phase selectively adsorbs on the mobile filler surface. Selective polymer–filler interaction and kinetics of rod motion determine the morphological development. When rod like particles specifically interact with one of the separating phases, kinetics of self-assembling rods results in generation of supramolecular phase structures. Such collaborative activity can provide ease to development of a connecting network of minor phase with an ultra low fraction of high aspect ratio fillers. In the case of temperature dependent phase separating polymer blends, phase structures are governed by various other parameters like phase separating temperature, quenching rate and annealing time. A similar type of phenomenon was observed in the case of PS/PVME blends with CdSe–TOPO nanorods where 2 vol% nanorods resulted in transformation of the droplet matrix to co-continuous morphology. Nanorod induced peculiar transformation of phases is presented in Fig. 2b.79 As the scope of this perspective is to understand the individual effect of filler on phase evolution, miscible polymer blends are not discussed.
4.2 Effect of particle anisotropy
The less understood effect is of course the shape of the nanoparticles. In the case of high aspect ratio particles like carbon nanotubes (CNT), the droplet relaxation time can further slowdown owing to the self-networking capability of CNTs. Moreover, droplet/domain bridging66,68 is frequently observed in the case of CNTs as their length is often larger than the domain size and hence, highly irregular structures are often developed mostly due to retarded relaxation. It is envisaged that in the case of CNTs, as the diameter is smaller than the radius of gyration of polymers in general, the reptation relaxation of the polymer chains is retarded due to polymer–CNT interactions.5,6 Moreover, anisotropic diffusion has been proposed owing to large specific interfacial areas in the case of CNTs such that any moderate perturbation in the vicinity of CNTs can have significant implication on the polymer reptation. In most cases CNTs are localized in a given phase and in few at the blend's interface. Unlike block-copolymers, even the surface functional groups present on the surface cannot reptate the parent CNT at the interface and hence, retarded relaxation possibly could be the main reason behind the peculiar morphology in polymer blends induced by CNTs or other high aspect ratio particles. The anisotropy of fillers localized at the interface plays a crucial role in the mechanism of phase continuity. Fig. 2c and d shows penetration of CNTs from one droplet to another and stitching of the interface.66 Owing to the very high aspect ratio, CNTs manifested ultra low critical concentration over isotropic fillers like carbon black and spherical particles to transform morphologies to peculiar structures.80 Along with alteration of viscoelastic properties, penetration of NPs from droplet interfaces results in minimization of interfacial surface energy and stitching of the interface leads to inhibition of the relaxation process. A similar effect was evident in PA6/ABS blends where addition of 4 wt% MWNTs manifested transformation of matrix droplets to peculiar phase structures (Fig. 2e) and the co-continuity window was observed to be increased over neat blends (Fig. 2f and g).58Fig. 3a summarizes the critical concentration of various particles that induced peculiar morphologies in the blends. Fig. 3b displays the fraction of particles required to induce such peculiar morphologies as a function of filled phase. It is evident from Fig. 3b that as the concentration of filled phase increases, the fraction of particles required for such phenomenal transition increases. From the literature, it is evident that the concentration required to achieve continuity in phases is very low for high aspect ratio particles like CNTs followed by two-dimensional platelets like clay and then spherical nanoparticles (see Fig. 3b). It is evident that the aspect ratio of particles is a dominating factor to enhance the connectivity of the minor phase where the fillers are selectively positioned in the minor phase of the blends.
 | ||
| Fig. 3 (a) Phase micrographs of various blends depicting phase structure transformation governed by filler loading (Steinmann et al.,47 Wu et al.,54 Li et al.49 and Zou et al.80 – reproduced by permission of Elsevier, Copyright © 2002, 2010, 2004, 2006, Elsevier Ltd. All rights reserved), and (b) critical concentration of various fillers required for generation of peculiar phase structures in blends. | ||
5. Conclusion and outlook
In summary, the shape of the nanoparticles seems to play a key role in preserving these peculiar morphologies. For instance, if the nanoparticles adsorb at the interface, the solid-like feature of the interface can stabilize the elongated or the irregular interface. In addition, if the nanoparticles pierce the interface, as in the case of rigid rod like particles due to high bending stress, a domain-bridging phenomenon can be observed again leading to highly irregular structures. CNTs often curl and reside inside the droplet, if it has specific affinity towards the minor phase. This may not reduce the interfacial tension but beyond a certain concentration can still induce a domain-bridging effect resulting in highly irregular structures. Hence, the increased yield stress of the filled phase and a slowdown of the relaxation seem to stabilize the peculiar morphologies in polymer blends which otherwise is thermodynamically unfavourable due to the increased interfacial area unless the nanoparticles reduce the interfacial tension.Abbreviations
| HDPE | High density polyethylene |
| PA6 | Polyamide 6 |
| PPS | Poly p-phenylene sulfide |
| PBT | Polybutylene terephthalate |
| PP | Poly propylene |
| ABS | Acrylonitrile butadiene styrene |
| PS | Poly styrene |
| PMMA | Polymethyl methacrylate |
| LLDPE | Linear low-density polyethylene |
| PA66 | Polyamide 66 |
| PA11 | Polyamide 11 |
| PPO | Poly phenylene oxide |
| PEEK | Polyether ether ketone |
| TPI | Thermoplastic polyimide |
| PLLA | Poly L-lactide |
| TPU | Thermoplastic polyurethane |
| PLA | Poly lactic acid |
| CNT | Carbon nanotubes |
References
- L. A. Utracki and Z. H. Shi, Polym. Eng. Sci., 1992, 32, 1824–1833 CAS.
- Z. Yuan and B. D. Favis, AIChE J., 2005, 51, 271–280 CrossRef CAS PubMed.
- J. R. Bell, K. Chang, C. R. López-Barrón, C. W. Macosko and D. C. Morse, Macromolecules, 2010, 43, 5024–5032 CrossRef CAS.
- S. P. Lyu, F. S. Bates and C. W. Macosko, AIChE J., 2000, 46, 229–238 CrossRef CAS PubMed.
- X.-Q. Liu, Q.-Y. Wang, R.-Y. Bao, W. Yang, B.-H. Xie and M.-B. Yang, RSC Adv., 2014, 4, 49429–49441 RSC.
- X.-Q. Liu, Z.-Y. Sun, R.-Y. Bao, W. Yang, B.-H. Xie and M.-B. Yang, RSC Adv., 2014, 4, 41059–41068 RSC.
- L. Elias, F. Fenouillot, J. C. Majesté and P. Cassagnau, Polymer, 2007, 48, 6029–6040 CrossRef CAS PubMed.
- X. Cai, B. Li, Y. Pan and G. Wu, Polymer, 2012, 53, 259–266 CrossRef CAS PubMed.
- C. Koning, M. Van Duin, C. Pagnoulle and R. Jerome, Prog. Polym. Sci., 1998, 23, 707–757 CrossRef CAS.
- M. Di Lorenzo and M. Frigione, J. Polym. Eng., 1997, 17, 429–460 CAS.
- A. Ajji and L. A. Utracki, Polym. Eng. Sci., 1996, 36, 1574–1585 CAS.
- C. W. Macosko, P. Guegan, A. K. Khandpur, A. Nakayama, P. Marechal and T. Inoue, Macromolecules, 1996, 29, 5590–5598 CrossRef CAS.
- D. Bikiaris and C. Panayiotou, J. Appl. Polym. Sci., 1998, 70, 1503–1521 CrossRef CAS.
- M. Heino, J. Kirjava and P. Hietaoja, J. Appl. Polym. Sci., 1997, 65, 241–249 CrossRef CAS.
- R. A. Kudva, H. Keskkula and D. R. Paul, Polymer, 1998, 39, 2447–2460 CrossRef CAS.
- S. H. Anastasiadis, I. Gancarz and J. T. Koberstein, Macromolecules, 1989, 22, 1449–1453 CrossRef CAS.
- B. Majumdar, H. Keskkula and D. R. Paul, Polymer, 1994, 35, 3164–3172 CrossRef CAS.
- W. Zhang, B. X. Fu, Y. Seo, E. Schrag, B. Hsiao, P. T. Mather, N.-L. Yang, D. Xu, H. Ade, M. Rafailovich and J. Sokolov, Macromolecules, 2002, 35, 8029–8038 CrossRef CAS.
- S. Thomas and R. E. Prud`homme, Polymer, 1992, 33, 4260–4268 CrossRef CAS.
- J. Kim, M. K. Gray, H. Zhou, S. T. Nguyen and J. M. Torkelson, Macromolecules, 2005, 38, 1037–1040 CrossRef CAS.
- N. Moussaif and R. Jérôme, Polymer, 1999, 40, 3919–3932 CrossRef CAS.
- V. Nelliappan, M. S. El-Aasser, A. Klein, E. S. Daniels and J. E. Roberts, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 3173–3181 CrossRef CAS.
- Y. Li and H. Shimizu, ACS Appl. Mater. Interfaces, 2009, 1, 1650–1655 CAS.
- D. F. Siqueira, F. Galembeck and S. P. Nunes, Polymer, 1991, 32, 990–998 CrossRef CAS.
- G. P. Kar, S. Biswas and S. Bose, Phys. Chem. Chem. Phys., 2015, 17, 1811–1821 RSC.
- M. Xanthos and S. S. Dagli, Polym. Eng. Sci., 1991, 31, 929–935 CAS.
- F. C. Chang and Y. C. Hwu, Polym. Eng. Sci., 1991, 31, 1509–1519 CAS.
- P.-C. Lee, W.-F. Kuo and F.-C. Chang, Polymer, 1994, 35, 5641–5650 CrossRef CAS.
- C. T. Maa and F. C. Chang, J. Appl. Polym. Sci., 1993, 49, 913–924 CrossRef CAS PubMed.
- B. Majumdar, H. Keskkula, D. R. Paul and N. G. Harvey, Polymer, 1994, 35, 4263–4279 CrossRef CAS.
- N. C. Liu, H. Q. Xie and W. E. Baker, Polymer, 1993, 34, 4680–4687 CrossRef CAS.
- N. C. Liu and W. E. Baker, Adv. Polym. Technol., 1992, 11, 249–262 CrossRef CAS PubMed.
- M. Si, T. Araki, H. Ade, A. L. D. Kilcoyne, R. Fisher, J. C. Sokolov and M. H. Rafailovich, Macromolecules, 2006, 39, 4793–4801 CrossRef CAS.
- A. E. Nesterov and Y. S. Lipatov, Polymer, 1999, 40, 1347–1349 CrossRef CAS.
- H. Essawy and D. El-Nashar, Polym. Test., 2004, 23, 803–807 CrossRef CAS PubMed.
- F. Fenouillot, P. Cassagnau and J. C. Majeste, Polymer, 2009, 50, 1333–1350 CrossRef CAS PubMed.
- M. Sumita, K. Sakata, S. Asai, K. Miyasaka and H. Nakagawa, Polym. Bull., 1991, 25, 265–271 CrossRef CAS.
- D. K. Owens and R. C. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS PubMed.
- S. Wu, J. Macromol. Sci., Rev. Macromol. Chem., 1974, 10, 1–73 CrossRef CAS PubMed.
- A. E. Zaikin, E. A. Zharinova and R. S. Bikmullin, Polym. Sci., Ser. A, 2007, 49, 328–336 CrossRef.
- A. E. Zaikin, R. R. Karimov and V. P. Arkhireev, Colloid J., 2001, 63, 53–59 CrossRef CAS.
- J. Feng, C. M. Chan and J. X. Li, Polym. Eng. Sci., 2003, 43, 1058–1063 CAS.
- C. J. Carriere, A. Cohen and C. B. Arends, J. Rheol., 1989, 33, 681–689 CrossRef CAS.
- S. Hom, A. R. Bhattacharyya, R. A. Khare, A. R. Kulkarni, M. Saroop and A. Biswas, J. Appl. Polym. Sci., 2009, 112, 998–1004 CrossRef CAS PubMed.
- G. Filippone, N. T. Dintcheva, D. Acierno and F. P. La Mantia, Polymer, 2008, 49, 1312–1322 CrossRef CAS PubMed.
- D. Wu, L. Wu, M. Zhang, W. Zhou and Y. Zhang, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 1265–1279 CrossRef CAS PubMed.
- S. Steinmann, W. Gronski and C. Friedrich, Polymer, 2002, 43, 4467–4477 CrossRef CAS.
- D. Wu, C. Zhou and M. Zhang, J. Appl. Polym. Sci., 2006, 102, 3628–3633 CrossRef CAS PubMed.
- Y. Li and H. Shimizu, Polymer, 2004, 45, 7381–7388 CrossRef CAS PubMed.
- P. Thareja, K. Moritz and S. S. Velankar, Rheol. Acta, 2010, 49, 285–298 CrossRef CAS PubMed.
- J. Huitric, M. Moan, P. J. Carreau and N. Dufaure, J. Non-Newtonian Fluid Mech., 2007, 145, 139–149 CrossRef CAS PubMed.
- S. Tomotika, Proc. Roy. Soc. Lond. Math. Phys. Sci., 1935, 150, 322–337 CrossRef.
- H. Veenstra, J. Van Dam and A. Posthuma de Boer, Polymer, 2000, 41, 3037–3045 CrossRef CAS.
- G. Wu, B. Li and J. Jiang, Polymer, 2010, 51, 2077–2083 CrossRef CAS PubMed.
- M. H. Al-Saleh and U. Sundararaj, Composites, Part A, 2008, 39, 284–293 CrossRef PubMed.
- C. Gao, S. Zhang, B. Han, H. Sun, G. Wang and Z. Jiang, RSC Adv., 2014, 4, 42175–42182 RSC.
- Y. Li and H. Shimizu, Macromolecules, 2008, 41, 5339–5344 CrossRef CAS.
- S. Bose, A. R. Bhattacharyya, R. A. Khare and A. R. Kulkarni, Handbook of Polymer Nanocomposites. Processing, Performance and Application, Springer, 2015, pp. 123-140 Search PubMed.
- S. P. Pawar, D. A. Marathe, K. Pattabhi and S. Bose, J. Mater. Chem. A, 2015, 3, 656–669 CAS.
- S. P. Pawar, K. Pattabhi and S. Bose, RSC Adv., 2014, 4, 18842–18852 RSC.
- M. Sumita, K. Sakata, Y. Hayakawa, S. Asai, K. Miyasaka and M. Tanemura, Colloid Polym. Sci., 1992, 270, 134–139 CAS.
- W. Thongruang, R. J. Spontak and C. M. Balik, Polymer, 2002, 43, 3717–3725 CrossRef CAS.
- C. Zhang, H. F. Han, X. S. Yi, S. Asai and M. Sumita, Compos. Interfaces, 1998, 6, 227–236 Search PubMed.
- C. Zhang, X. S. Yi, H. Yui, S. Asai and M. Sumita, Mater. Lett., 1998, 36, 186–190 CrossRef CAS.
- F. Gubbels, S. Blacher, E. Vanlathem, R. Jérôme, R. Deltour, F. Brouers and P. Teyssie, Macromolecules, 1995, 28, 1559–1566 CrossRef CAS.
- A. Nuzzo, E. Bilotti, T. Peijs, D. Acierno and G. Filippone, Polymer, 2014, 55, 4908–4919 CrossRef CAS PubMed.
- P. Pötschke, A. R. Bhattacharyya and A. Janke, Carbon, 2004, 42, 965–969 CrossRef PubMed.
- P. Pötschke, A. R. Bhattacharyya and A. Janke, Polymer, 2003, 44, 8061–8069 CrossRef PubMed.
- S. Bose, A. R. Bhattacharyya, A. R. Kulkarni and P. Pötschke, Compos. Sci. Technol., 2009, 69, 365–372 CrossRef CAS PubMed.
- R. A. Khare, A. R. Bhattacharyya, A. R. Kulkarni, M. Saroop and A. Biswas, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 2286–2295 CrossRef CAS PubMed.
- J. B. Bai and A. Allaoui, Composites, Part A, 2003, 34, 689–694 CrossRef.
- N. C. Das, T. K. Chaki, D. Khastgir and A. Chakraborty, Adv. Polym. Technol., 2001, 20, 226–236 CrossRef CAS PubMed.
- C. Y. Huang and C. C. Wu, Eur. Polym. J., 2000, 36, 2729–2737 CrossRef CAS.
- M. Rahaman, T. K. Chaki and D. Khastgir, J. Mater. Sci., 2011, 46, 3989–3999 CrossRef CAS.
- R. Rohini and S. Bose, ACS Appl. Mater. Interfaces, 2014, 6, 11302–11310 CAS.
- P. Xavier and S. Bose, RSC Adv., 2014, 4, 55341–55348 RSC.
- G. Filippone, N. Tz. Dintcheva, F. P. La Mantia and D. Acierno, Polymer, 2010, 51, 3956–3965 CrossRef CAS PubMed.
- G. Peng, F. Qiu, V. V. Ginzburg, D. Jasnow and A. C. Balazs, Science, 2000, 288, 1802–1804 CrossRef CAS.
- L. Li, C. Miesch, P. K. Sudeep, A. C. Balazs, T. Emrick, T. P. Russell and R. C. Hayward, Nano Lett., 2011, 11, 1997–2003 CrossRef CAS PubMed.
- H. Zou, K. Wang, Q. Zhang and Q. Fu, Polymer, 2006, 47, 7821–7826 CrossRef CAS PubMed.
- Y. Li and H. Shimizu, Macromol. Rapid Commun., 2005, 26, 710–715 CrossRef CAS PubMed.
- F. Xiang, Y. Shi, X. Li, T. Huang, C. Chen, Y. Peng and Y. Wang, Eur. Polym. J., 2012, 48, 350–361 CrossRef CAS PubMed.
- Y. Y. Shi, W. B. Zhang, J. H. Yang, T. Huang, N. Zhang, Y. Wang, G. P. Yuan and C. L. Zhang, RSC Adv., 2013, 3, 26271–26282 RSC.
| This journal is © the Owner Societies 2015 |