
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gas-phase structure and reactivity of the keto tautomer of the deoxyguanosine radical cation†
Linda
Feketeová
*abcd,
Bun
Chan
*be,
George N.
Khairallah
ab,
Vincent
Steinmetz
f,
Philippe
Maître
f,
Leo
Radom
be and
Richard A. J.
O'Hair
*ab
aSchool of Chemistry and Bio21 Institute of Molecular Science and Biotechnology, The University of Melbourne, 30 Flemington Road, Parkville, VIC 3010, Australia. E-mail: gkhai@unimelb.edu.au; rohair@unimelb.edu.au; Fax: +61 3 9347-5180; Tel: +61 3 8344-2452
bARC Centre of Excellence for Free Radical Chemistry and Biotechnology, Australia
cUniversité de Lyon, 69003 Lyon, France
dCNRS/IN2P3, UMR5822, Institut de Physique Nucléaire de Lyon, 69622 Villeurbanne, France. E-mail: l.feketeova@ipnl.in2p3.fr
eSchool of Chemistry, University of Sydney, NSW 2006, Australia. E-mail: bun.chan@sydney.edu. au; radom@chem.usyd.edu.au; Fax: +61 2 9351-3329; Tel: +61 2 9351-5361
fLaboratoire de Chimie Physique, Université Paris Sud, 15, Avenue Jean Perrin, Orsay Cedex, 91405, France. E-mail: philippe.maitre@u-psud.fr; Fax: +33 (0) 16915 6188; Tel: + 33 (0) 16915 3250
First published on 5th May 2015
Abstract
Guanine radical cations are formed upon oxidation of DNA. Deoxyguanosine (dG) is used as a model, and the gas-phase infrared (IR) spectroscopic signature and gas-phase unimolecular and bimolecular chemistry of its radical cation, dG˙+, A, which is formed via direct electrospray ionisation (ESI/MS) of a methanolic solution of Cu(NO3)2 and dG, are examined. Quantum chemistry calculations have been carried out on 28 isomers and comparisons between their calculated IR spectra and the experimentally-measured spectra suggest that A exists as the ground-state keto tautomer. Collision-induced dissociation (CID) of A proceeds via cleavage of the glycosidic bond, while its ion–molecule reactions with amine bases occur via a number of pathways including hydrogen-atom abstraction, proton transfer and adduct formation. A hidden channel, involving isomerisation of the radical cation via adduct formation, is revealed through the use of two stages of CID, with the final stage of CID showing the loss of CH2O as a major fragmentation pathway from the reformed radical cation, dG˙+. Quantum chemistry calculations on the unimolecular and bimolecular reactivity are also consistent with A being present as a ground-state keto tautomer.
Introduction
Reports on the carcinogenic effects of ionising radiation date back to as early as 1902, and Marie Curie and her daughter Irene are believed to have succumbed to complications from radiation-induced leukemia.1 Early research on the effects of ionising radiation on DNA2 predated Watson and Crick's revelation of its structure,3 which provided an important molecular foundation for all subsequent research.4 It is now widely recognised that oxidative damage to DNA, through nucleobase modifications and the formation of strand breaks, can cause mutagenesis, cancer and is involved in aging.5Extensive research focusing on a molecular understanding of the effects of ionising radiation on DNA and its consequences6 has revealed that the holes produced during the initial ionising event in DNA transfer mostly to the guanine (G) sites, which have the lowest ionisation energy (IE) of the four DNA bases.7 Thus, the electron-loss centre generated in duplex DNA ultimately ends up at guanine sites via hole migration through the DNA duplex. On loss of one electron, the resultant DNA base radical cation has a greatly increased acidity. For example, work on DNA models has demonstrated that the acidity of deoxyguanosine is enhanced in aqueous solution by around 5.6 orders of magnitude (5.6 pKa units) upon oxidation.7 Thus, proton-coupled electron and hole transfer becomes an important feature in the radiation damage process.6 One proposed pathway to strand breakage in DNA proceeds via species resulting from deprotonation of the guanine radical cation, which trigger specific hydrogen-atom abstraction reactions from the sugar moiety,8 thereby causing the heterolytic elimination of the phosphate–ester bond.9
Infrared (IR) spectroscopy integrated to tandem mass spectrometry has emerged as an important tool to characterise the structure of mass-selected and trapped molecular ions.10 The highly intense and tuneable IR beam delivered by Free Electron Lasers (FEL)11,12 is particularly well suited since it provides access to the so-called IR fingerprint region. As a result, isobaric ions bearing distinct functional groups can be clearly distinguished. Thus, the tautomers of protonated pyrimidic bases (thymine, cytosine, and uracil) have been characterised.13 It turns out that the CO stretching region around 1800 cm−1 is the most diagnostically useful part of the IR spectrum, since it is characteristic of the keto form. Although the spectral assignment in the 1500–1800 cm−1 region is more challenging, it also appears to be very informative for the structural assignment of tautomers.
We have recently shown that mass-spectrometry-based gas-phase studies that combine the use of IR spectroscopy, ion–molecule reactions and computational chemistry provide a powerful way of relating the structure of radical cations of relevance to biological damage to their fundamental chemistry.14,15 For example, we have used IR spectroscopy to show that the radical cation of 9-methylguanine (9MG) has the same ground-state keto tautomer found in DNA, and discovered via the use of ion–molecule reactions that its N–H acidity is enhanced by ∼470 kJ mol−1 in the gas phase.15 As noted in our previous paper,15 a key motivation for our studies is that Steenken has emphasised that the local environment experienced by nucleobases within DNA is different to model systems in bulk water, and suggested that gas-phase acidity data may be more appropriate to characterise the equilibrium position of the proton in oxidised GC base pairs.6c,7a
Given that sugar radicals have been implicated as products arising from ionisation at guanine sites, we examine here the gas-phase IR spectrum, and the bimolecular and unimolecular chemistry of the deoxyguanosine radical cation, dG˙+. In previous work, we showed that electrospray ionisation (ESI) of a solution of dG and Cu(NO3)2, coupled with multistage collision-induced dissociation (CID) in an ion-trap mass spectrometer, resulted in isomeric dG˙+ species A and B, which could be distinguished by their different fragmentation behaviour under CID (Scheme 1).16b Thus, the major fragment ion formed from A arises from cleavage of the glycosidic bond. In contrast, formaldehyde loss is the major fragmentation channel for B. We speculated that the difference in the unimolecular chemistry was due to the nucleobase moiety G, existing in different tautomeric forms in A and B.16b
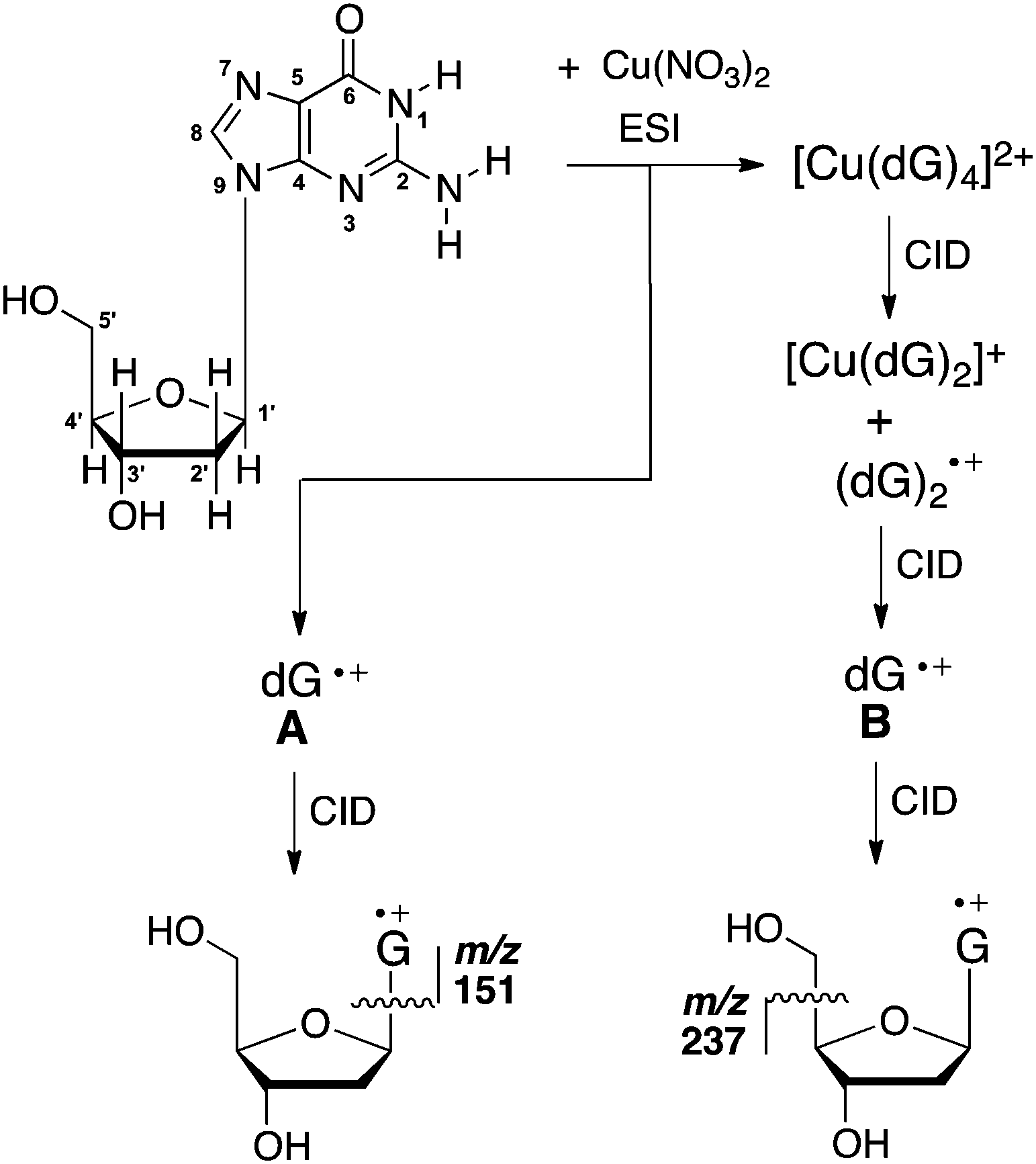 | ||
| Scheme 1 Structure and atom numbering of the anti form of the neutral keto isomer of deoxyguanosine (dG), together with the experimental procedure for the formation of isomeric dG˙+ species (A and B) via ESI/MSn, and their resultant fragmentation channels under conditions of collision-induced dissociation (CID). | ||
Here we use gas-phase IR spectroscopy, ion–molecule reactions and quantum chemistry calculations to interrogate the structure and reactivity of species A, which is formed via direct ESI/MS. Compared with the radical cation of 9MG,15 the presence of the sugar moiety greatly increases the number of isomers whose structures, energies and IR spectra need to be considered by quantum chemistry calculations. Apart from the keto and enol tautomers, there are the syn and anti conformational isomers associated with rotations around the C–N glycosidic bond.17 In addition, distonic ions can be formed via H-atom transfer from the sugar to the nucleobase radical cation.
Methods
Reagents were used as supplied: Cu(NO3)2 (Ajax chemicals, 99%) and deoxyguanosine (Sigma, 99%). Complexes were prepared by mixing 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mM solutions of the nucleoside: Cu(NO3)2, dissolved in 3:1 methanol:water, immediately before infusing the reaction mixture into the mass spectrometer.
:1 mM solutions of the nucleoside: Cu(NO3)2, dissolved in 3:1 methanol:water, immediately before infusing the reaction mixture into the mass spectrometer.
Infrared spectroscopy
IR spectroscopy in the 1000–2000 cm−1 spectral range was carried out using the FEL of CLIO12 coupled to a hybrid FT-ICR tandem mass spectrometer.18 Hybrid stands for the fact that prior to the transfer to the ICR cell, electrosprayed ions can be mass-selected in a quadrupole and accumulated in a linear hexapole pressurised with Ar (∼10−3 mbar) allowing for CID and/or thermalisation of the ions. As described previously, dG˙+ species A can be observed directly by ESI.16 Following mass-selection, the isolated dG˙+ (species A) was irradiated with the FEL IR beam for 250 or 500 ms. On the basis of previous results,18 it is important to emphasise that the mass-selected radical ions are subjected to multiple collisions with argon, thus ensuring an efficient thermalisation prior to the pulsed extraction towards the ICR cell.Upon resonant vibrational excitation, dissociation of the m/z 267 radical ions was monitored via the formation of the diagnostic m/z 151 fragment peak ions. The abundance of this photofragment and its corresponding precursor were recorded as a function of the IR wavelength in order to derive the IR action spectra, where the IR multiple-photon dissociation (IRMPD) efficiency is plotted against the photon energy, which was varied stepwise.
Mass spectrometry
All mass spectrometry experiments were carried out using a commercially available Finnigan-LTQ-FT (Thermo, Bremen, Germany) mass spectrometer equipped with ESI source19 described in detail elsewhere.20 The procedure used to generate dG˙+ species A was as described above. The sample was introduced into the mass spectrometer at 5.0 μL min−1via ESI. Typical ESI conditions used were: spray voltage, 3.3–5.0 kV; capillary temperature, 250 °C; nitrogen sheath pressure, 8–40 (arbitrary units). The capillary voltage and the tube lens offset were tuned to maximise the desired peak. The injection time was set using the automatic gain control function. The LTQ-FT mass spectrometer consists of (i) a linear ion trap (LTQ); (ii) ion-transfer optics; and (iii) an FT-ICR mass analyser. For the tandem mass spectrometry (MS/MS) experiments, dG˙+ species A produced via ESI was trapped in the LTQ and subjected to CID at an He bath gas pressure of ca. 2 × 10−3 Torr. CID was carried out by mass selecting the desired ions with a 1.5–6 m/z units window and subjecting them to the following typical conditions: normalised collision energy between 16% and 40%, which determines the translational kinetic energy of the ions; activation (Q), 0.25–0.35, which assigns the radio-frequency (RF) amplitude used to fragment ions, and activation time of 30 ms that is the time set to excite the ions via CID. The high resolution of the FT-ICR mass spectrometer was used to confirm the charge states of the mass-selected precursor ions. For high-resolution mass analysis, the ions were transferred via the ion optics transfer region (2 × 10−7 Torr) into an FT-ICR cell at a pressure below 1.5 × 10−9 Torr.Ion–molecule reactions
The mass spectrometry instrument described above has been modified to allow ion–molecule reactions (IMRs) to take place in the LTQ.21 dG˙+ species A was trapped in the LTQ and subjected to IMRs with the desired neutral reagents: diisopropylethylamine (iPr2NEt), triethylamine (Et3N), and diisopropylamine (iPr2NH). IMRs between the mass selected dG˙+ species A and the various bases were carried out with reaction times varying between 30 and 1000 ms. Branching ratios of the products were calculated by integrating the intensities under the appropriate peaks.Computational methods
Standard density functional theory calculations were carried out with Gaussian 09.22 Geometries were optimised at the B3-LYP/6-31+G(d,p) level. Improved single-point energies were obtained using the M06-2X/6-311+G(3df,2p) protocol.23 Zero-point vibrational energies and thermal corrections for enthalpies at 298 K were obtained using scaled B3-LYP harmonic vibrational frequencies.24 Unless otherwise noted, energies in the text refer to free energies at 298 K. The vibrational frequencies (B3-LYP) used for the simulated IR spectra were scaled according to literature recommendations.25 A scaling factor of 0.98 was applied to the calculated B3-LYP/6-31+G(d,p) harmonic frequencies in the 1000–2000 cm−1 region. Each calculated band was convoluted assuming a Gaussian shape with full width at half maximum of 20 cm−1.Results and discussion
IR spectroscopy of dG˙+ species A directly formed via ESI/MS on copper(II) solutions
The gas-phase deoxyguanosine radical cations, dG˙+A, formed via electrospray ionisation of a methanolic solution of a mixture of copper nitrate and dG,16 were mass selected and stored in the cell of a Fourier transform ion cyclotron resonance mass spectrometer, where they were subjected to infrared radiation. The resulting IR multiple-photon dissociation (IRMPD) spectrum shown in Fig. 1a was obtained. Table S1 (ESI†) lists the experimental band positions.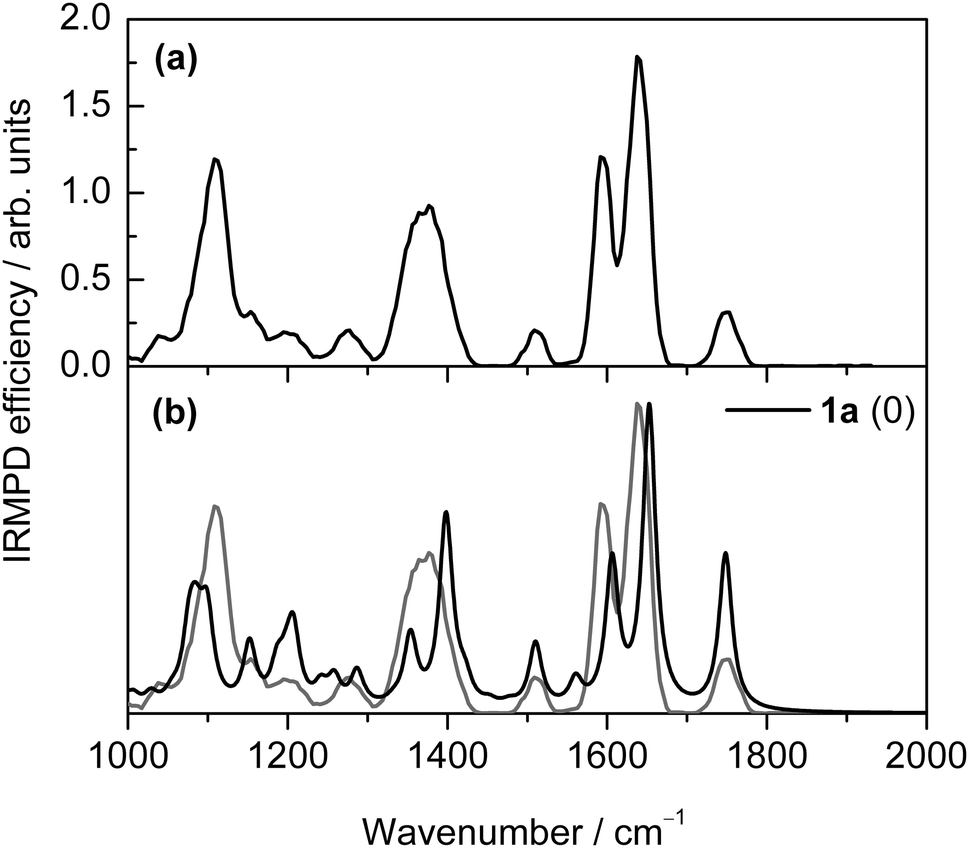 | ||
| Fig. 1 Experimental and (scaled) theoretical (B3-LYP/6-31+G(d,p)) IR spectra of dG˙+: (a) experimental spectrum of A with IRMPD efficiency, (b) theoretical spectrum for the lowest-energy keto isomer 1a. The experimental spectrum is included for comparison in gray in (b) and the scaling factor used is 0.98. | ||
The IR spectrum is dominated by an unresolved feature with two maxima at ∼1595 and ∼1650 cm−1. The band observed at ∼1750 cm−1 when dG˙+ ions are formed in the source region is characteristic of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode. Two other intense bands are observed at ∼1110 and ∼1372 cm−1. Finally, weaker features are observed at ∼1200, ∼1275, and ∼1511 cm−1.
O stretching mode. Two other intense bands are observed at ∼1110 and ∼1372 cm−1. Finally, weaker features are observed at ∼1200, ∼1275, and ∼1511 cm−1.
The experimental IR spectrum of A is compared with the spectra calculated (at the B3-LYP/6-31+G(d,p) level) for a number of different tautomers lying within 50 kJ mol−1 in free energy (at the M06-2X/6-311+G(3df,2p) level) of the lowest-energy isomer 1a, together with some higher-energy tautomers such as one proposed in our previous study.16b A total of 28 tautomers of dG˙+ were examined theoretically in this way, and their structures and IR spectra are given in Fig. S1–S3 (ESI†).
An examination of the calculated IR spectra (Fig. 1 and Fig. S3, ESI†) shows that the IR spectrum is very sensitive to the nature of the tautomers. The experimental spectrum (Fig. 1a) nicely matches the spectrum calculated for the lowest-energy keto tautomer 1a (Fig. 1b). On this basis, band assignments are proposed in Table S1 (ESI†). As mentioned above, the band observed at ∼1750 cm−1 is a signature of a keto isomer, and the corresponding CO stretching mode is predicted at 1748 cm−1 for the lowest energy isomer 1a. A very good agreement between theory and experiment can also be found for the lower energy bands, which are assigned to the ring deformation (N3–C4 stretching) (1a: 1653 cm−1, expt: 1640 cm−1), NH2 scissoring (1a: 1606 cm−1, expt: 1595 cm−1), guanine six-membered-ring breathing (1a: 1510 cm−1, expt: 1511 cm−1), multiply-coupled C–H/N–H bending (1a: 1353, 1355, and 1398 cm−1, expt: 1372 cm−1), and multiply-coupled guanine/sugar ring breathing (1a: 1076, 1084, 1089, 1099, and 1152 cm−1, expt: 1110 cm−1) modes. Thus we assign species A as the keto isomer 1a.
It is interesting to compare our results on the IR spectroscopic identification of the keto tautomer radical cation of dG with results for the parent neutral.26 Although the keto tautomer of dG is predicted to be more stable than the enol tautomer by 3.8 kJ mol−1, gas-phase IR spectroscopy of laser-desorbed dG only identified the enol tautomer,26a,b which has been ascribed to a short excited-state lifetime of the keto form in the two-photon ionization event required to generate the IR spectrum.26c In contrast, hydration of laser-desorbed dG by one or two water molecules, results in the keto form being observed in the gas-phase IR spectrum.26c Thus the way the dG radical cation or neutral is transferred to the gas phase appears to play a key role as to which tautomer is formed. Although the exact mechanism by which dG˙+A is formed during the ESI process, which transfers species from solution to the gas phase, remains unknown, our assignment of this species as the keto form in the gas phase is consistent with the following known solution-phase chemistry of dG: (i) NMR studies have demonstrated that it exists largely as the keto tautomer in its neutral form;27 and (ii) upon binding of Cu2+, the keto tautomer is maintained.‡28
Ion–molecule reactions of dG˙+ A
The acidity of dG˙+A was estimated via IMRs with neutral reference bases B with known proton affinities.31 Five types of reactions were observed: proton transfer (PT, eqn (1)), electron transfer (ET, eqn (2)), hydrogen-atom abstraction (HAA, eqn (3)), adduct formation (AddF, eqn (4)), and complexation-induced fragmentation (CplxFr, eqn (5)).| dG˙+ + B → (dG − H)˙ + (B + H)+ PT | (1) |
| → dG + B˙+ ET | (2) |
| → (dG + H)+ + (B − H)˙ HAA | (3) |
| → [dG + B]˙+ AddF | (4) |
| → [dG + (B − X)]+ + X˙ CplxFr | (5) |
Table 1 provides a summary of the branching ratios (BRs) for these reactions, and a sample spectrum of the IMR of dG˙+ with iPr2NEt at 100 ms reaction time is shown in Fig. 2. We found that during the IMRs with the bases, the reactions did not go to completion and that the dG˙+ at m/z 267 was still observed even at very long reaction times (e.g., 10000 ms). We attribute this observation to the complexation-induced isomerisation reaction of the dG˙+ radical cation, which produces a different isomer(s), which we call species C. We base this conclusion on the CID spectrum of C, which is formed in the following sequential MSn reactions: (i) the IMR between dG˙+A and a base produces the adduct [dG + B]˙+ (MS2); (ii) isolation of this adduct followed by CID results in loss of the base and generation of dG˙+C (MS3); (iii) isolation of dG˙+C allows its CID spectrum to be collected in an MS4 experiment.
| Base (B) | Proton affinitya (B) | BR PT (eqn (1))b (%) | BR ET (eqn (2))b (%) | BR HAA (eqn (3))b (%) | BR AddF (eqn (4))b (%) | BR CplxFr (eqn (5))b (%) |
|---|---|---|---|---|---|---|
| a Values in kJ mol−1 are taken from the NIST database (ref. 37). b Branching ratios (BR) were determined as average values from measurements at several 100–1000 ms reaction times. | ||||||
| iPr2NEt | 994.3 | 6 | 18 | 2 | 60 | 13 |
| Et3N | 981.8 | <1 | 6 | 50 | 1 | 42 |
| iPr2NH | 971.9 | <1 | 0 | 5 | 58 | 37 |
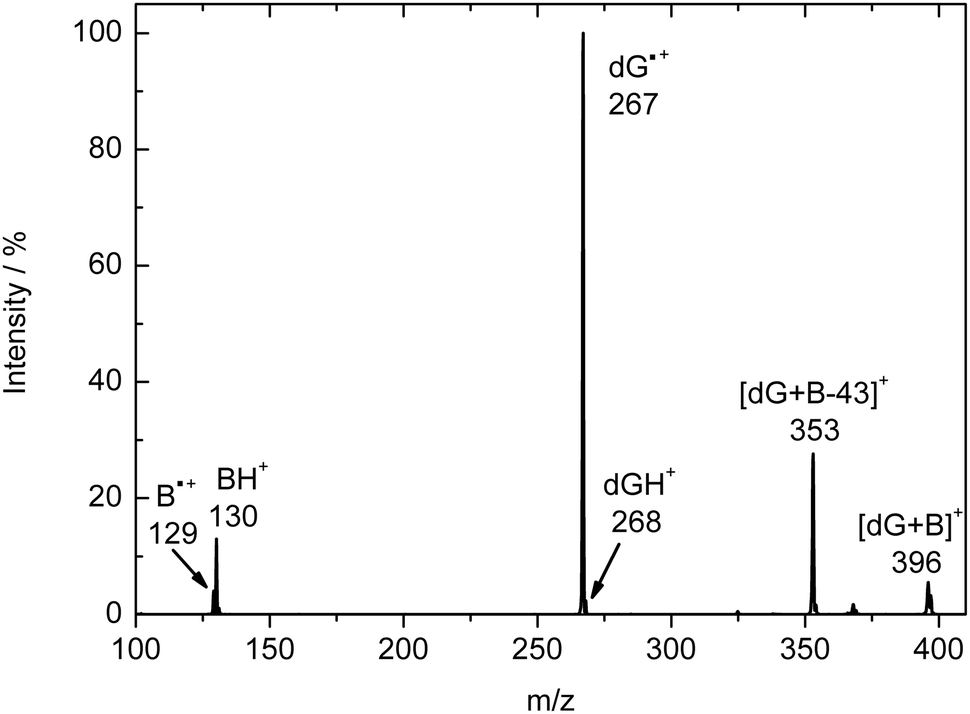 | ||
| Fig. 2 Ion–molecule reaction spectrum of dG˙+ formed in the ESI source reacting with diisopropylethylamine (iPr2NEt) at 100 ms reaction time. | ||
As an example, the MS4 spectrum of dG˙+C is shown in Fig. S4a (ESI†), where isomerisation has taken place through the reaction with iPr2NEt, and the resultant new species C fragments via the major loss of formaldehyde.§ In contrast, C is less reactive than A in IMRs with bases, and does not undergo reactions viaeqn (1)–(3). Thus it appears that in IMRs of dG˙+, A can not only react directly viaeqn (1)–(3), but it can also undergo isomerisation. Such reactions have been observed in the gas phase before and Bohme has classed them as proton-transport-catalysed reactions.32 Of particular relevance to the potential tautomers that might be formed in these reactions are previous observations of proton-transport-catalysed reactions that isomerise the keto form of amide radical cations to their enol forms.33
Theoretical modelling of the ion–molecule reactions of the keto and enol tautomers is summarised in Fig. S5 (ESI†). Note that the calculated M06-2X/6-311+G(3df,2p) proton affinities (PAs) of the bases are slightly underestimated by the theory (by 4–8 kJ mol−1) while the IEs are slightly overestimated. Nevertheless, the experimental IMR results are consistent with the proton affinities calculated for the keto tautomer.
The PA for the (9MG – HN1)˙ radical is 968.9 kJ mol−1 at the M06-2X/6-311+G(3df,2p) level. For the corresponding radical (1a – HN1)˙ it is 993.5 kJ mol−1. This is consistent with the increase in PA on going from the nucleobase G (951.4 kJ mol−1) to nucleoside dG (980.7 kJ mol−1).34 Hence iPr2NEt, which has the highest calculated PA of 990 kJ mol−1 (Fig. S5, ESI†), appears to possess the highest branching ratio for eqn (1) (6%, Table 1), whereas Et3N, with a calculated PA of 976 kJ mol−1 that is smaller than that of (1a – H)˙, shows a branching ratio for eqn (1) <1%. The most acidic proton of 1a is the C2N–H of the amino group, the proton that would be involved in a guanine–cytosine pairing, consistent with the results observed for 9MG˙+.15
The electron-transfer reaction (eqn (2)) depends on the IE of the bases versus the electron affinity (EA) of the dG˙+ radical cation. The calculated IEs (Fig. S5, ESI†) for iPr2NEt, Et3N and iPr2NH are 705, 744 and 756 kJ mol−1, respectively, whereas the EA of 1a is calculated to be 712 kJ mol−1. In this case also, theory is consistent with the experimental results, where the branching ratio for the iPr2NEt is the highest (18%, Table 1) and that of iPr2NH is the lowest (0%, Table 1). Our theoretical results are also consistent with the gas-phase IE of dG, which has been bracketed as 718 kJ mol−1 < IE dG < 751 kJ mol−1via the Cooks kinetic method,35 as well as with a literature value from DFT calculations of 730 kJ mol−1.36
The energetics of the HAA pathway (eqn (3)) were also determined theoretically by calculating the bond dissociation energies (BDE) for the bases and comparing them with H-atom-abstraction energies for the dG˙+ radical cation (Fig. S5, ESI†). The BDEs for iPr2NEt and iPr2NH are similar at 370 and 371 kJ mol−1, respectively, while the BDE for Et3N is 398 kJ mol−1. According to the calculations, the H atom of the αC can be readily abstracted only by the N7 of the dG˙+ radical cation in its keto form, i.e., 1a, where the energy for the abstraction is predicted to be 401 kJ mol−1 (Fig. S5, ESI†). The energy for the abstraction of the H atom for the enol form is predicted to be <397 kJ mol−1, which is also consistent with the assignment of 1a to A. Interestingly, the branching ratio for the HAA pathway (eqn (3)) is 50% for Et3N, while for iPr2NEt and iPr2NH it is only 2% and 5%, respectively. This can be attributed to the fact that formation of the adduct (eqn (4)) is in these cases more favourable, as shown by the high branching ratios for iPr2NEt and iPr2NH of 60% and 58%, as opposed to the branching ratio for Et3N of only 1%.
Overall, the results of our computational investigations show proton transfers to neutral reagents that range from mildly endothermic (+14 kJ mol−1 for iPr2NH) to mildly exothermic (−12 kJ mol−1 for iPr2NEt). The trends in these calculated reaction energies are consistent with the observed experimental yields for proton transfer. In comparison, hydrogen abstraction from these bases by 1a is generally exothermic, with reaction energies ranging from −3 kJ mol−1 for Et3N to −31 kJ mol−1 for iPr2NEt. The relatively small energies for these proton-transfer (eqn (1)) or hydrogen-atom-abstraction (eqn (3)) reactions suggest that either class of reaction might be involved as an intermediate step for the observed isomerisation of A to C. In any case, the calculated thermochemical quantities and the experimental branching ratios, as well as the very observation of C, indicates a complex set of processes induced by IMRs.
Finally, it is worth considering the results of these gas-phase reactions within the context of the proposed biological chemistry of one-electron-oxidised guanine residues within DNA. It has been proposed that oxidised guanine residues have a significantly enhanced acidity, which allows them to undergo proton transfer to the adjacent cytosine residues. Our experimental results suggest that the PA for the (dG − H)˙ radical 1a (978–996 kJ mol−1) is close to that of iPr2NEt (994.3 kJ mol−1). The gas-phase acidity of neutral dG has been recently determined to be 1409 kJ mol−1.35,36 Thus in the gas phase, ionisation of dG enhances its acidity by ∼415 kJ mol−1, somewhat less than what we found for 9MG (∼470 kJ mol−1), but still a significant enhancement of acidity. Indeed, the enhanced acidity of dG˙+ is close to the PA of dC (988.4 kJ mol−1)38 and this suggests that proton transfer within the ionised base pair may become viable. As noted in the introduction, a mechanism proposed for strand breaking in ionised DNA involves hydrogen-atom abstraction from the sugar moiety induced by deprotonation of the guanine radical cations.8 Our observation of a hydrogen-atom abstraction channel for dG˙+ suggests that ionised guanine sites within DNA may indeed be cable of inducing hydrogen-atom abstraction reactions.
A mechanism for the formation of m/z 151 in the unimolecular fragmentation of dG˙+ species A
As noted in the introduction, the way in which the various dG˙+ species fragment depends on the way that they are generated (Scheme 1). The main dissociation channel in the low-energy CID spectrum of A is the cleavage of the N-glycosidic bond to give the radical cation of the guanine base at m/z 151,18 (Fig. S4b, ESI†) which contrasts with the series of fragment ions observed in the EI mass spectrum39 and upon REMPI.40 The IR spectroscopy and IMR results suggest that A exists as the keto tautomer (1a). Thus we have used DFT calculations to examine the energetics for loss of C5H8O3 (observed in the CID spectrum of A) versus loss of CH2O (not observed in the CID spectrum of A) in the keto isomer 1a.C5H8O3 loss proceeds as follows: isomers 1a and 1b are connected by a conformational change of a single-bond rotation and can be presumed to have a small barrier (Fig. 3, pathway a, red). Fragmentation is triggered by N–C bond cleavage (1b–1c‡) to give a complex of the nucleobase–sugar fragments (1c). This is followed by hydrogen-atom abstraction from the dissociated sugar moiety with concerted ring closure (1c–1d‡) to yield the cyclised sugar (1d, 1e). In contrast, loss of CH2O is triggered by hydrogen-atom abstraction from the 5′OH to the N3 position (Fig. 3, pathway b, blue).¶ The free energy barrier for C5H8O3 loss is 108 kJ mol−1, which is slightly less than the barrier for loss of CH2O (110 kJ mol−1), consistent with C5H8O3 loss being the main fragmentation channel.||
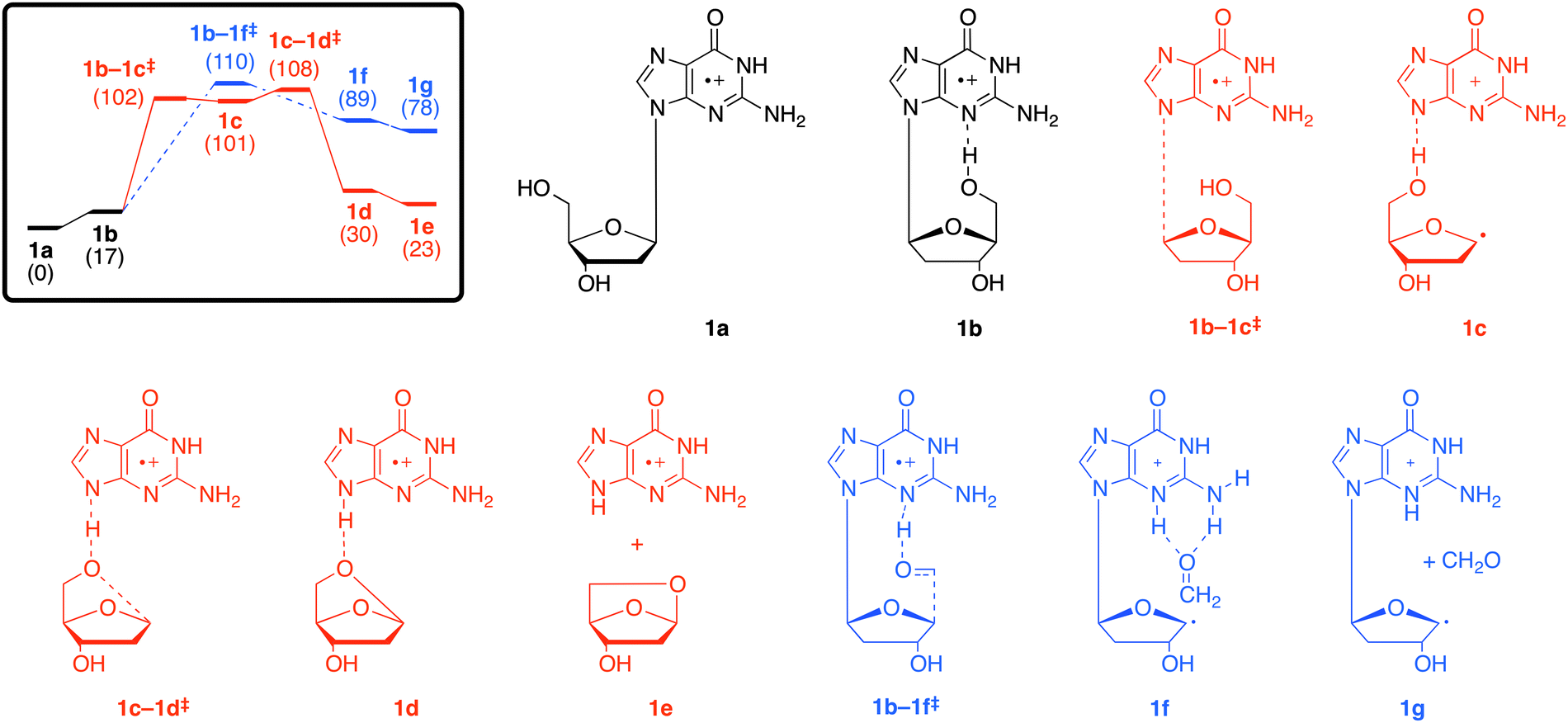 | ||
| Fig. 3 M06-2X/6-311+G(3df,2p) schematic free energy profile (kJ mol−1) associated with the fragmentation reactions of 1avia (a) loss of the cyclised sugar C5H8O3 (red), or (b) loss of CH2O (blue). | ||
Conclusions
The gas-phase structure and reactivity of dG˙+ species A has been investigated by a combination of IR spectroscopy, mass spectrometry experiments involving both ion–molecule reactions and CID, and computational quantum chemistry calculations. This powerful combination has allowed us to assign A as the ground-state keto form. It undergoes a number of reactions with amines, including hydrogen-atom abstraction, proton transfer, electron transfer and adduct formation. Thus, the PA of (dG − H)˙ radical has been experimentally bracketed as 994.3 kJ mol−1 > PA (dG − H)˙ > 981.8 kJ mol−1, consistent with our calculated value of 993.5 kJ mol−1 for removal of the N1 proton from the keto form of dG˙+. Similarly, the electron affinity of the dG˙+ radical cation (A) has been bracketed as 756 kJ mol−1 > electron affinity A >705 kJ mol−1, consistent with our calculated value of 712 kJ mol−1. Theoretical calculations on the unimolecular dissociation of the keto form (1a) of dG˙+ suggest that the favoured pathway involves cleavage of the glycosidic bond.On a final note, the dG˙+ species B and C remain to be fully characterised.§ A technical challenge is that they need to be formed in a series of multistage mass spectrometry experiments. Nonetheless, preliminary results suggest that B and C consist of isomer(s) that are different to A, and that they are less reactive than A in ion–molecule reactions.
Acknowledgements
Funding from the ARC (CoE program for RAJO and LR, APD for LF (CE0561607), ARF for GNK, and APF for LR), generous grants of computer time (to LR) from NCI NF and Intersect Australia Ltd, and (to RAJO) from VPAC, UPSud for a visiting Scholar Fellowship (to GNK). Financial support from the National FT-ICR network (FR 3624 CNRS) for conducting the research is gratefully acknowledged.Notes and references
- A. C. Upton, Historical perspectives on radiation carcinogenesis, in Radiation Carcinogenesis, ed. A. C. Upton, R. E. Albert, F. J. Burns and R. E. Shore, Elsevier, New York, 1986, pp. 1–10 Search PubMed.
- (a) A. H. Sparrow and F. M. Rosenfeld, Science, 1946, 104, 245 CAS; (b) G. Scholes, G. Stein and J. Weiss, Nature, 1949, 164, 709 CrossRef CAS; (c) G. Scholes, G. Stein and J. Weiss, Nature, 1950, 166, 640 CrossRef CAS; (d) B. E. Conway, L. Gilbert and J. A. V. Butler, J. Chem. Soc., 1950, 3421 RSC.
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737 CrossRef CAS.
- C. von Sonntag, The Chemical Basis of Radiation Biology, Taylor and Francis, London, 1987 Search PubMed.
- (a) C. Chatgilialoglu and P. O'Neil, Exp. Gerontol., 2001, 36, 1459 CrossRef CAS; (b) C. G. Fraga, M. K. Shigenaga, J.-W. Park, P. Degan and B. N. Ames, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 4533 CrossRef CAS.
- (a) A. O. Colson and M. D. Sevilla, Int. J. Radiat. Biol., 1995, 67, 627 CrossRef CAS PubMed; (b) A. Kumar and M. D. Sevilla, Chem. Rev., 2010, 110, 7002 CrossRef CAS PubMed; (c) S. Steenken, Chem. Rev., 1989, 89, 503 CrossRef CAS; (d) C. J. Burrows and J. G. Muller, Chem. Rev., 1999, 98, 1109 CrossRef PubMed.
- (a) L. P. Candeias and S. Steenken, J. Am. Chem. Soc., 1989, 111, 1094 CrossRef CAS; (b) S. Steenken and S. V. Jovanovic, J. Am. Chem. Soc., 1997, 119, 617 CrossRef CAS.
- D. Khanduri, S. Collins, A. Kumar, A. Adhikary and M. D. Sevilla, J. Phys. Chem. B, 2008, 112, 2168 CrossRef CAS PubMed.
- T. Melvin, S. W. Botchway, A. W. Parker and P. O. O'Neill, J. Am. Chem. Soc., 1996, 118, 10031 CrossRef CAS.
- L. Mac Aleese and P. Maitre, Mass Spectrom. Rev., 2007, 26, 583 CrossRef CAS PubMed.
- D. Oepts, A. F. G. Van der Meer and P. W. Van Amersfoort, Infrared Phys. Technol., 1995, 36, 297 CAS.
- R. Prazeres, F. Glotin, C. Insa, D. A. Jaroszynski and J. M. Ortega, Eur. Phys. J. D, 1998, 3, 87 CrossRef CAS.
- J. Y. Salpin, S. Guillaumont, J. Tortajada, L. MacAleese, J. Lemaire and P. Maître, ChemPhysChem, 2007, 8, 2235 CrossRef CAS PubMed.
- (a) S. Osburn, J. D. Steill, J. Oomens, R. A. J. O'Hair, M. V. Stipdonk and V. Ryzhov, Chem. – Eur. J., 2011, 17, 873 CrossRef CAS PubMed; (b) S. Osburn, G. Berden, J. Oomens, R. A. J. O'Hair and V. Ryzhov, J. Am. Soc. Mass Spectrom., 2011, 22, 1794 CrossRef CAS PubMed; (c) S. Osburn, G. Berden, J. Oomens, R. A. J. O'Hair and V. Ryzhov, J. Am. Soc. Mass Spectrom., 2012, 23, 1019 CrossRef CAS PubMed; (d) S. Osburn, T. Burgie, G. Berden, J. Oomens, R. A. J. O'Hair and V. Ryzhov, J. Phys. Chem. A, 2013, 117, 1144 CrossRef CAS PubMed; (e) S. Osburn, G. Berden, J. Oomens, K. Gulyuz, N. C. Polfer, R. A. J. O'Hair and V. Ryzhov, ChemPlusChem, 2013, 78, 970 CrossRef CAS PubMed.
- L. Feketeová, G. N. Khairallah, B. Chan, V. Steinmetz, P. Maître, L. Radom and R. A. J. O'Hair, Chem. Commun., 2013, 49, 7343 RSC.
- (a) P. Cheng and D. K. Bohme, J. Phys. Chem. B, 2007, 111, 11075 CrossRef CAS PubMed; (b) L. Feketeová, E. Yuriev, J. D. Orbell, G. N. Khairallah and R. A. J. O'Hair, Int. J. Mass Spectrom., 2010, 304, 74 CrossRef PubMed; (c) S. Wee, R. A. J. O'Hair and W. D. McFadyen, Rapid Commun. Mass Spectrom., 2005, 19, 1797 CrossRef CAS PubMed; (d) A. K. Y. Lam, B. F. Abrahams, M. J. Grannas, W. D. McFadyen and R. A. J. O'Hair, Dalton Trans., 2006, 5051 RSC.
- H. Asami, S.-H. Urashima, M. Tsukamoto, A. Motoda, Y. Hayakawa and H. Saigusa, J. Phys. Chem. Lett., 2012, 3, 571 CrossRef CAS.
- J. M. Bakker, T. Besson, J. Lemaire, D. Scuderi and P. Maître, J. Phys. Chem. A, 2007, 111, 13415 CrossRef CAS PubMed.
- R. Malek, W. Metelmann-Strupat, M. Zeller and H. Muenster, Am. Biotechnol. Lab., 2005, 23, 8 CAS; S. Horning, R. Malek, A. Wieghaus and M. W. Senko, J. E. P. Syka, Proc. 51st ASMS Conf. Mass Spectrom. Allied Top., Montreal, Canada, 2003.
- L. Feketeová, G. N. Khairallah and R. A. J. O'Hair, Eur. J. Mass Spectrom., 2008, 14, 107 CrossRef PubMed.
- (a) W. A. Donald, C. J. McKenzie and R. A. J. O'Hair, Angew. Chem., Int. Ed., 2011, 50, 8379 CrossRef CAS PubMed; (b) A. K. Y. Lam, C. Li, G. N. Khairallah, B. B. Kirk, S. J. Blanksby, A. J. Trevitt, U. Wille, R. A. J. O'Hair and G. da Silva, Phys. Chem. Chem. Phys., 2012, 14, 2417 RSC; (c) W. A. Donald, G. N. Khairallah and R. A. J. O'Hair, J. Am. Soc. Mass Spectrom., 2013, 24, 811 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- J. P. Merrick, D. Moran and L. Radom, J. Phys. Chem. A, 2007, 111, 11683 CrossRef CAS PubMed.
- Y. Bouteiller, J. C. Gillet, G. Grégoire and J. P. Schermann, J. Phys. Chem. A, 2008, 112, 11656 CrossRef CAS PubMed.
- (a) A. Abo-riziq, B. O. Crews, I. Compagnon, J. Oomens, G. Meijer, G. Von Helden, M. Kabelac, P. Hobza and M. S. de Vries, J. Phys. Chem. A, 2007, 111, 7529 CrossRef CAS PubMed; (b) E. Nir, I. Hünig, K. Kleinermanns and M. S. de Vries, ChemPhysChem, 2004, 5, 131 CrossRef CAS PubMed; (c) H. Asami, S.-H. Urashima and H. Saigusa, Phys. Chem. Chem. Phys., 2009, 11, 10466 RSC.
- (a) G. C. Y. Lee and S. I. Chan, J. Am. Chem. Soc., 1972, 94, 3218 CrossRef CAS; (b) C. Chang, D. J. Ashworth, L. Chern, J. DaSilva Gomes, L. Jose, C. Lee, P. W. Mou and R. Narayan, Org. Magn. Reson., 1984, 22, 671 CrossRef CAS PubMed.
- (a) H. Fritzsche, D. Tresselt and C. Zimmer, Experientia, 1971, 27, 1253 CrossRef CAS; (b) V. Andrushchenko and P. Bouř, J. Phys. Chem. B, 2009, 113, 283 CrossRef CAS PubMed.
- B. P. Pozniak and R. B. Cole, J. Am. Soc. Mass Spectrom., 2015, 27, 369 CrossRef PubMed and references cited therein.
- G. J. Van Berkel, S. A. McLuckey and G. L. Glish, Anal. Chem., 1992, 64, 1586 CrossRef CAS.
- E. P. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413 CrossRef CAS PubMed.
- D. K. Bohme, Int. J. Mass Spectrom., 1992, 115, 95–110 CrossRef CAS.
- (a) P. Mourgues, J. Chamot-Rooke, H. Nedev and H.-E. Audier, J. Mass Spectrom., 2001, 36, 102 CrossRef CAS PubMed; (b) M. A. Trikoupis, P. C. Burgers, P. J. A. Ruttink and J. K. Terlouw, Int. J. Mass Spectrom., 2001, 210/211, 489 CrossRef CAS; (c) D. Schroeder, J. Loos, R. Thissen, O. Dutuit, P. Mourgues, H.-E. Audier, C. Lifshitz and H. Schwarz, Angew. Chem., Int. Ed., 2002, 41, 2748 CrossRef; (d) M. A. Trikoupis, P. J. A. Ruttink, P. C. Burgers and J. K. Terlouw, Eur. J. Mass Spectrom., 2004, 10, 801 CrossRef CAS.
- F. Greco, A. Liguori, G. Sindona and N. Uccella, J. Am. Chem. Soc., 1990, 112, 9092 CrossRef CAS.
- A. Liguori, A. Napoli and G. Sindona, J. Am. Soc. Mass Spectrom., 2001, 12, 176 CrossRef CAS.
- C. E. Crespo-Hernandez, D. M. Close, L. Gorb and J. Leszczynski, J. Phys. Chem. B, 2007, 111, 5386 CrossRef CAS PubMed.
- NIST Chemistry WebBook, NIST Standard Reference Database Number 69, June 2005, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg, MD, 2005, http://webbook.nist.gov.au.
- S. Kumari, C. L. Devi, S. Prabhakar, K. Bhanuprakash and M. Vairaman, J. Am. Soc. Mass Spectrom., 2010, 21, 136 CrossRef CAS PubMed.
- (a) K. Biemann and J. A. McCloskey, J. Am. Chem. Soc., 1962, 84, 2005 CrossRef CAS; (b) NIST Standard Reference Database 69: NIST Chemistry WebBook.
- L. Li and D. M. Lubman, Int. J. Mass Spectrom. Ion Processes, 1989, 88, 197 CrossRef CAS.
- S. J. Shaw, D. M. Desiderio, K. Tsuboyama and J. A. McCloskey, J. Am. Chem. Soc., 1970, 92, 2510 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Calculated structures and relative energies of dG˙+ isomers (Fig. S1 and S2), calculated IR spectra of dG˙+ isomers (Fig. S3), assignment of the experimental IR spectrum, MS/MS CID spectra of dG˙+ (Table S1), calculated thermochemical properties of dG˙+ isomers (Fig. S4), and transition structure for keto–enol tautomerisation in dG˙+ (Fig. S5). See DOI: 10.1039/c5cp01573a |
| ‡ A reviewer has asked about the mechanism for the formation of the deoxyguanosine radical cation in the gas phase. They can be formed via gas-phase fragmentation of mass-selected Cu(II) clusters whose stoichiometry is known, as shown for species B in Scheme 1. Species A is directly formed via ESI/MS, but the precise mechanism for its formation remains unclear. Under gentle source conditions such as those used here, Chen and Bohme have suggested that the related guanosine radical cations16a are formed via intra-complex transfer from a coordinated ligand (in our case deoxyguanosine) to the Cu2+ centre within Cu(II) clusters whose exact stoichiometry is unknown, followed by dissociation within the ESI source to form the free radical cation. We cannot rule out the possibility of an outer-sphere electron-transfer process from deoxyguanosine to a Cu(II) species during the ESI process. Both mechanisms are consistent with the coordinated28 or free27 keto tautomer being present in solution. On a final note, the ESI source behaves as an electrolytic flow cell29 and while radical cations of other substrates have been observed previously,30 the presence of Cu(II) is required to observe the formation of Avia ESI/MS. |
| § Due to their similar CID spectra, it is tempting to speculate that species B and C are composed of the same isomers. Ideally, we would have liked to have measured the IR spectrum of C, but the instrument is not currently set up to allow gas-phase ion–molecule reactions. |
| ¶ The same mechanism has been proposed for the loss of CH2O from deoxyadenosine analogs.41 |
| || The barrier to keto–enol tautomerisation via an intramolecular 1,3-shift is calculated to be 162 kJ mol−1 (via the transition structure shown in Fig. S6, ESI†), and thus isomerisation of the keto to the enol form is unlikely to occur under the low-energy CID conditions used in our experiments. |
| This journal is © the Owner Societies 2015 |