
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct measurement of DNA-mediated adhesion between lipid bilayers
S. F.
Shimobayashi
a,
B. M.
Mognetti
b,
L.
Parolini
c,
D.
Orsi
d,
P.
Cicuta
*c and
L.
Di Michele
*c
aDepartment of Physics, Graduate School of Science, Kyoto University, Kyoto 606-8502, Japan
bInterdisciplinary Center for Nonlinear Phenomena and Complex Systems & Service de Physique des Systémes Complexes et Mécanique Statistique, Universitéibre de Bruxelles, Campus Plaine, CP 231, Blvd du Triomphe, B-1050 Brussels, Belgium
cBiological and Soft Systems Sector, Cavendish Laboratory, University of Cambridge, CB3 0HE, Cambridge, UK. E-mail: pc245@cam.ac.uk; ld389@cam.ac.uk
dDepartment of Physics and Earth Sciences, University of Parma, I-43124, Parma, Italy
First published on 12th May 2015
Abstract
Multivalent interactions between deformable mesoscopic units are ubiquitous in biology, where membrane macromolecules mediate the interactions between neighbouring living cells and between cells and solid substrates. Lately, analogous artificial materials have been synthesised by functionalising the outer surface of compliant Brownian units, for example emulsion droplets and lipid vesicles, with selective linkers, in particular short DNA sequences. This development extended the range of applicability of DNA as a selective glue, originally applied to solid nano and colloidal particles. On very deformable lipid vesicles, the coupling between statistical effects of multivalent interactions and mechanical deformation of the membranes gives rise to complex emergent behaviours, as we recently contributed to demonstrate [Parolini et al., Nat. Commun., 2015, 6, 5948]. Several aspects of the complex phenomenology observed in these systems still lack a quantitative experimental characterisation and a fundamental understanding. Here we focus on the DNA-mediated multivalent interactions of a single liposome adhering to a flat supported bilayer. This simplified geometry enables the estimate of the membrane tension induced by the DNA-mediated adhesive forces acting on the liposome. Our experimental investigation is completed by morphological measurements and the characterisation of the DNA-melting transition, probed by in situ Förster Resonant Energy Transfer spectroscopy. Experimental results are compared with the predictions of an analytical theory that couples the deformation of the vesicle to a full description of the statistical mechanics of mobile linkers. With at most one fitting parameter, our theory is capable of semi-quantitatively matching experimental data, confirming the quality of the underlying assumptions.
Due to both fundamental and practical interest, the complex phenomenology of multivalent selective interactions has recently been a hot research topic in Soft Matter and Physical Chemistry. One of the main driving forces behind this effort has been the development of self-assembly strategies based on DNA-mediated multivalent interactions.1 Introduced by the seminal works of Mirkin2 and Alivisatos,3 who nearly two decades ago demonstrated the selective DNA-mediated self-assembly of gold nanoparticles, this approach has been optimised to the point of mastering the structure of multicomponent crystal lattices4–8 and amorphous materials.9–11 Applications of these self-assembly strategies span from photonics and plasmonics,12 to biosensing,13 and gene therapy.14–16 An abundance of experimental studies called for the development of models to understand the complex statistical mechanics of DNA-mediated multivalent interactions between solid particles.17–22 More recently, the same self-assembly strategy has been applied to compliant Brownian units, including emulsion droplets,23,24 and lipid vesicles.25–29 Especially for the case of liposomes, which are significantly more deformable than emulsion droplets, DNA-mediated adhesion plays at a similar magnitude as other forces, leading to rich coupling.28,30 Moreover, on these liquid surfaces, DNA linkers can freely diffuse, and the confinement induced by binding results in significant entropic contributions to the hybridisation free energy.
We recently demonstrated that the coupling between mechanical deformability of Giant-Unilamellar-Vesicles (GUVs) and DNA-mediated interactions results in unexpected emergent response to temperature changes, leading to negative thermal expansion and tuneable porosity of DNA–GUV assemblies.30 By means of an analytical theory, that jointly describes the geometrical features of the GUVs and the statistical mechanics of DNA tethers,30,31 we rationalised experimental evidence and highlighted the importance of translational entropy in coupling the DNA-binding free energy to the morphology of the substrates. Nonetheless, a further experimental investigation is necessary to assess all the aspects of the complex phenomenology of DNA–GUV conjugates, and test the accuracy of the theoretical framework applied to their description. In particular, a direct measurement of the DNA-mediated adhesive forces and the quantitative characterisation of the melting transition would represent new strong tests of the current understanding.
In this article, we present experiments aimed at measuring the temperature-dependence of the membrane tension induced on giant liposomes by DNA-mediated adhesion. Membrane tension is accessed by flickering measurements, in which we reconstruct the spectrum of thermal fluctuations of the liposomes at the equatorial cross-sections.32–34 This is a classic technique widely used to study vesicle tension and bending rigidity. To make these measurements possible, we adopt an experimental geometry in which liposomes do not interact with each other, instead they adhere to Supported Lipid Bilayers (SBLs) fabricated on rigid glass substrates, as demonstrated in Fig. 1. This allows imaging of the equator of the vesicle through confocal microscopy. Various morphological observables are also precisely assessed via 3-dimensional confocal imaging. Finally, the use of suitably labelled DNA tethers enables the direct assessment of the relative number of formed DNA bonds by means of in situ Förster Resonant Energy Transfer (FRET) spectroscopy. Experimental results are compared to predictions from our analytical theory, extended from ref. 30 to apply to this new geometry. Using at most one fitting parameter, we find semi-quantitative agreement for all the measured quantities, confirming the accuracy of the underlying assumptions of our model.
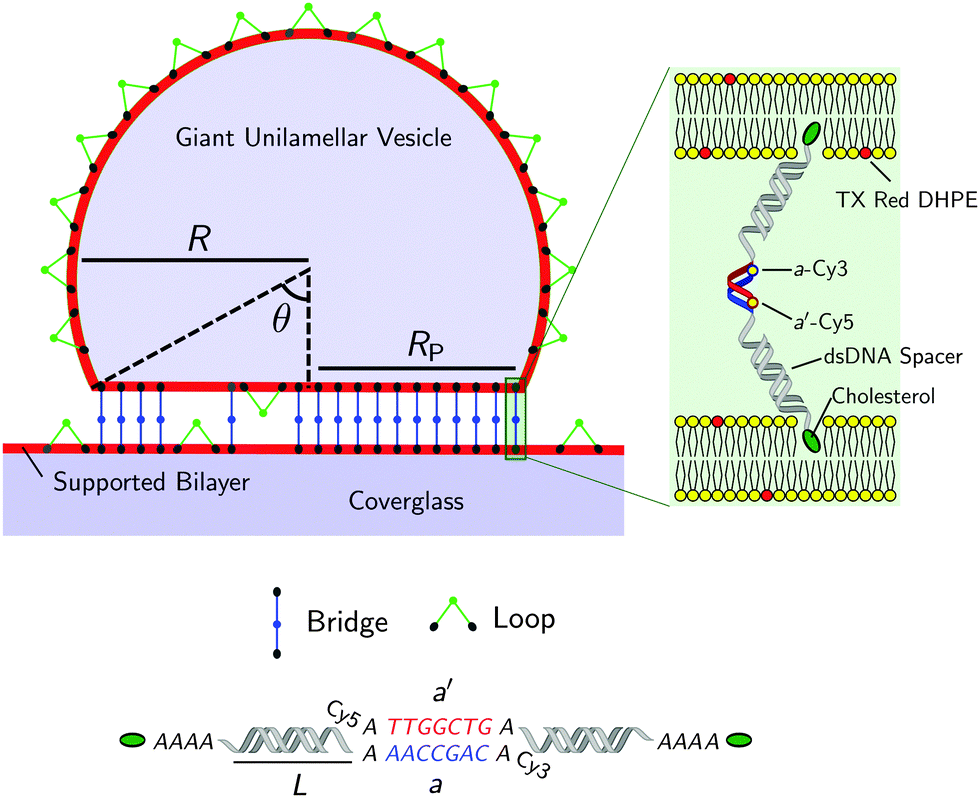 | ||
| Fig. 1 Not-to-scale schematics of a DNA-functionalised GUV adhering to a supported bilayer and details of the functionalisation. | ||
The remainder of this article is structured as follows. First we describe the experimental setup, including sample preparations protocols, materials, imaging and image analysis techniques. Then we outline our theoretical model focusing on the novel aspects introduced here for the GUV-plane geometry. Finally we discuss the experimental results and compare them with theoretical predictions.
1 Experimental
Fig. 1 shows a schematic of a DNA-functionalised GUV adhering to a DNA-functionalised SBL. GUVs and SBLs are prepared and separately functionalised with cholesterol labeled DNA constructs. The hydrophobic cholesterol inserts into the lipid bilayer allowing DNA tethers to freely diffuse. Connected to the cholesterol anchor there is a section of length L = 9.8 nm (29 base pairs35) of double-strands DNA (dsDNA), terminating in a single-stranded DNA (ssDNA) sticky end, which mediates the attractive interactions. To further facilitate the pivoting motion of the DNA tethers, 4 unpaired adenine bases are left between the cholesterol anchor and the dsDNA spacer. One unpaired adenine is left between the dsDNA spacer and the sticky end. Two mutually complementary sticky ends are used: a and a′, of 7 bases each (see Fig. 1). GUVs and SBLs are functionalised with equal molar fractions of both a and a′. Tethers can therefore form intra-membrane loops and inter-membrane bridges. The latter are responsible for the observed adhesion and are confined within the contact area between the GUVs and the SBLs. We use FRET spectroscopy to estimate the fraction of formed DNA bonds. To enable these measurements the termini of the sticky ends a and a′ are functionalised with a Cy3 and a Cy5 molecules respectively, spaced by an unpaired adenine base. Note that the sticky ends used here are shorter than those adopted in our previous study30 (7 base-pairs instead of 9), this choice was made to lower the melting temperature of the DNA to within an experimentally accessible temperature, and reduce the overall strength of the DNA-mediated adhesion, making it easier to measure by flickering spectroscopy.1.1 Materials and sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 ratio in iso-osmolar solution containing TE buffer (10 mM tris(hydroxymethyl) aminomethane, 1 mM ethylenediaminetetra acetic acid, Sigma Aldrich), 5 mM MgCl2, and 272 mM glucose (Sigma Aldrich).
:9 ratio in iso-osmolar solution containing TE buffer (10 mM tris(hydroxymethyl) aminomethane, 1 mM ethylenediaminetetra acetic acid, Sigma Aldrich), 5 mM MgCl2, and 272 mM glucose (Sigma Aldrich).
Sample chambers are obtained by applying adhesive silicone-rubber multi-well plates (6.5 mm × 6.5 mm × 3.2 mm, Flexwell, Grace Biolabs) on glass coverslips (26 mm × 60 mm, no.1, Menzel-Glsäer), cleaned following a previously reported protocol.9 To form SBLs, the glass bottom of the cells is hydrophilised by plasma cleaning on a plasmochemical reactor (Femto, Diener electronic, Germany), operated at frequency of 40 kHz, pressure of 30 Pa, and power input of 100 W for 5 minutes. Within 5 minutes from plasma cleaning, each cell is filled with 100 μl of diluted SUV solution and incubated for at least 30 minutes at room temperature to allow bilayer formation. To remove excess lipid and magnesium, the cells are repeatedly rinsed with the experimental solution (TE buffer, 100 mM NaCl, 87 mM glucose). Care is taken in keeping the bilayers covered in buffer to avoid exposure to air.
For experiments not involving FRET spectroscopy GUVs and SBLs are stained with 0.8–1.2% molar fraction of Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (Texas Red DHPE, Life Technologies).
(i) 5′-CGT GCG CTG GCG TCT GAA AGT CGA TTG CGAAAA [Cholesterol TEG]-3′
(ii) 5′-GC GAA TCG ACT TTC AGA CGC CAG CGC ACGA [Sticky End] A Cy3/Cy5-3′.
The bold letters indicate the segments forming the dsDNA spacer, the italic letters the inert flexible spacers. DNA is purchased lyophilized from Integrated DNA Technologies, reconstituted in TE buffer, aliquoted, and stored at −20 °C. For assembling the constructs, we dilute equal amounts of the two single strands, (i) and (ii), to 1.6 μM in TE buffer containing 100 mM NaCl. Hybridization is carried out by ramping down the temperature from 90 °C to 20 °C at a rate of −0.2 °C min−1 on a PCR machine (Eppendorf Mastercycler).30
We tested samples at different DNA concentrations, obtained by setting X to Xlow = 0.05, Xmed = 0.5 and Xhigh = 1.5 pmoles. We have previously quantified the surface coverage of GUVs functionalised with Xmed in ρmedDNA = 390 ± 90 μm−2.30 Here we proportionally assume ρlowDNA = 39 ± 9 μm−2, ρhighDNA = 1200 ± 300 μm−2. Having estimated that the overall surface of the SBL is approximately equal to the surface of the GUVs used for each sample, we assume equal DNA coverages for the SBL. This assumption is confirmed by fluorescence emission measurements: at sufficiently high temperature, when no DNA bridges are formed between GUVs and SBL, DNA is uniformly distributed on both interfaces. In this regime the fluorescence emission from DNA located within the contact area between a GUV and the SBL approximately equals twice the intensity measured on the free SBL (2.1 ± 0.1), confirming that GUVs and SBL have, within experimental errors, the same DNA coverage.
1.2 Imaging and image analysis
The temperature-dependent morphology of adhering GUVs is captured via confocal z-stacks and reconstructed using a custom script written in Matlab. Briefly: each z-stack contains an adhering GUV. The plane of the SBL/adhesion patch is identified as the one with maximum average intensity, then the adhesion patch is reconstructed by thresholding, following the application of a bandpass filter to flatten the background and remove pixel-level noise. From the area Ap of the adhesion patch we extract the patch-radius as 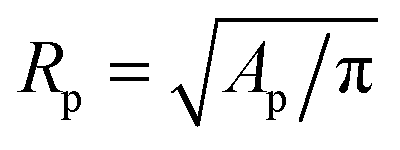 . The portion of the z-stack above the SBL is then scanned, and in each plane the contour of the vesicle, suitably highlighted by filtering and thresholding, is fitted with a circle. The slice featuring the largest circle is identified as the equatorial plane, determining the vesicle radius R. The contact angle is derived as θ = sin−1(Rp/R) (see Fig. 1).
. The portion of the z-stack above the SBL is then scanned, and in each plane the contour of the vesicle, suitably highlighted by filtering and thresholding, is fitted with a circle. The slice featuring the largest circle is identified as the equatorial plane, determining the vesicle radius R. The contact angle is derived as θ = sin−1(Rp/R) (see Fig. 1).
For flickering experiments, movies are recorded across the equatorial plane. Details of the flickering analysis are reported in the next section. To improve the quality of the signal, imaging for morphological characterisation and flickering analysis is carried out on samples stained with Texas Red DHPE.
FRET spectroscopy measurements are carried out by performing a spectral emission scan (λ-scan) of the contact area between an adhering GUV and the surrounding free SBL. While exciting the donor (Cy3), the emission of donor and acceptor is reconstructed by scanning the acquisition window from 530 to 785 nm, with intervals of 6.375 nm. Similarly to the case of z-stacks, the adhesion patch is identified in each image by filtering and thresholding. For FRET imaging, non-fluorescent lipids are used in order to prevent undesired energy transfer between the bilayer and the DNA tethers.
:9 ratio in iso-osmolar glucose solution to enable sedimentation.
(1) The position of the centre, and a rough-guess value of the radius R of the GUV are automatically detected on the first frame of the video by processing of the thresholded image (see Fig. 2a).
 | ||
| Fig. 2 Intermediate steps of the contour detection algorithm. (a) The position of the centre and the mean radius R of the vesicle define an annular region in the frame; (b) the annular region C(r,φ) extrapolated from the frame and fitted to find the membrane position with sub-pixel precision, as described in the text. The resulting contour is reported as a blue line; (c) colormap representation of the membrane fluctuations relative to the average radius. The colorbar reports the relative fluctuation as percentage in the range −5%, +5%. | ||
(2) The image of the radial profile C(r,φ), where r is the distance from the centre and φ the azimuthal angle, is selected within an annular region of user-defined width that contains the membrane. The annular region is then mapped onto a rectangular stripe using a cubic interpolation (see Fig. 2b).
(3) For each φ, the position of the membrane is roughly located as the maximum of C(r,φ). The precision of this estimate is limited by the pixel size and highly susceptible to noise.
(4) For each φ, the contour location is refined by evaluating the centroid of C(r,φ) within an interval of 6 pixel centred around the maximum found in step 3. This interval is chosen as about 3 times the spatial resolution of the microscope to include the entire radial section of the membrane. The centroid calculation is iterated 5 times, each time by re-centring the 6-pixel interval around the centroid found in the previous step. This procedure allows for sub-pixel resolution.
(5) The algorithm cycles three times from point 2 to 4, each time refining the estimate of the vesicle's centre and mean radius using the obtained contour. The resulting radial profile r(φ) is shown in Fig. 2b.
The procedure is repeated for every frame in a video of the fluctuating membrane, each time using the center and radius values of the previous frame as the starting point of the algorithm. An example of the temporal evolution of the membrane radial profile r(φ) is shown in Fig. 2c. The spectrum of the thermal fluctuations of the contour is calculated using Matlab Fast Fourier Transform algorithm, using the formula

2 Theoretical model
We present a quantitative model describing the DNA-mediated adhesion of GUVs on flat supported bilayers. The model is adapted from reference,30 where we treated the case of two identical adhering GUVs. Let us consider the interaction between an infinite SBL and a GUV adhering to it| U = Umembrane + UDNA + U0, | (1) |
2.1 Membrane deformation
In eqn (1), Umembrane summarises three contributions: stretching energy, bending energy, and the entropic cost of suppressing thermal fluctuations of the contact area between the GUV and the SBL.40 In the limit of strong adhesion the stretching energy dominates over the other two contributions, which we can neglect.40 As discussed later, for the case of DNA-mediated interactions, this condition is generally verified at low enough temperature. We can rewrite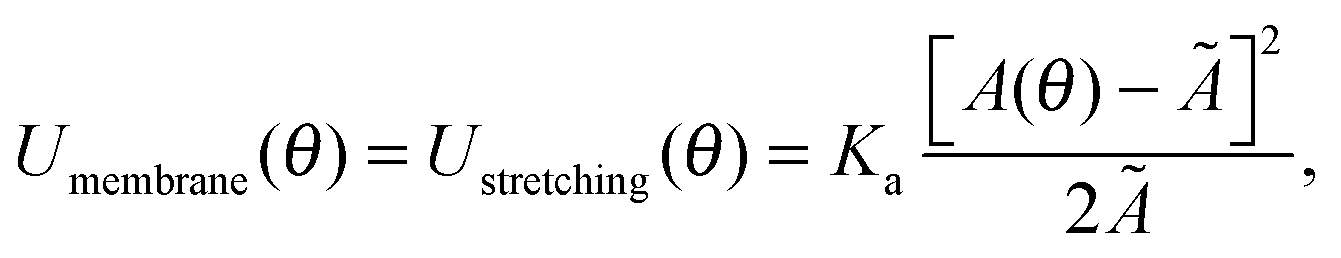 | (2) |
| Ã = A0[1 + α(T − T0)], | (3) |
2.2 DNA-mediated adhesion
We now focus on the DNA-mediated contribution to the interaction energy in eqn (1): UDNA.Given that the persistence length of dsDNA is ∼50 nm ≫ L = 9.8 nm,35 we can model the dsDNA spacers as rigid rods that, thanks to the fluidity of the membrane, can freely diffuse on the surface of the bilayers.30,31 Free pivoting motion is guaranteed by the flexibility of the joint between the cholesterol anchor and the dsDNA spacer (Fig. 1). As demonstrated in ref. 30, we can regard the sticky ends as point-like reactive sites, neglecting their physical dimensions. Moreover we can safely assume that the distance between the adhering membranes within the contact area is equal to L.30 Finally, we neglect excluded volume interactions between unbound DNA tethers.30,42 The last two assumptions guarantee a uniform distribution of unbound DNA tethers and loops over the GUV and SBL surfaces.
The free energy change associated to the formation of a single bridge (b) or a loop (![[small script l]](https://www.rsc.org/images/entities/char_e146.gif) ) within the GUV is
) within the GUV is
| ΔGb/ = ΔG0 − TΔSconfb/, | (4) |
accounts for the confinement entropic loss taking place when tethered sticky ends hybridise,18,20,45 and can be estimated as (see Appendix, Section A.2)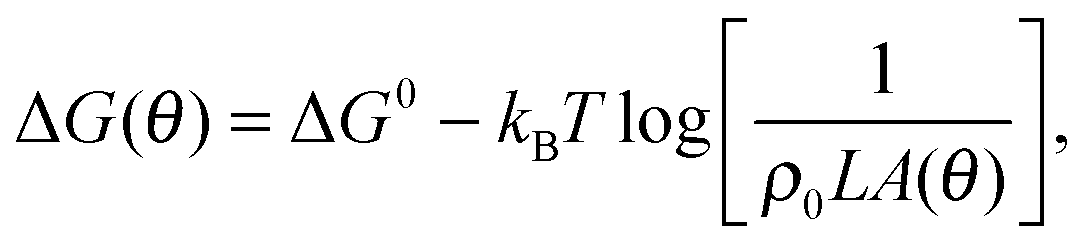 | (5) |
. The local roughness of the membranes could also influence ΔG (ref. 46) – this effect will be studied elsewhere.
We indicate with N the total number of a and a′ tethers on the GUVs, and model the SBL as an infinite reservoir of tethers. A combinatorial calculation detailed in the Appendix (Sections A.3 and A.4) allows the derivation of the overall hybridisation energy for the system of linkers
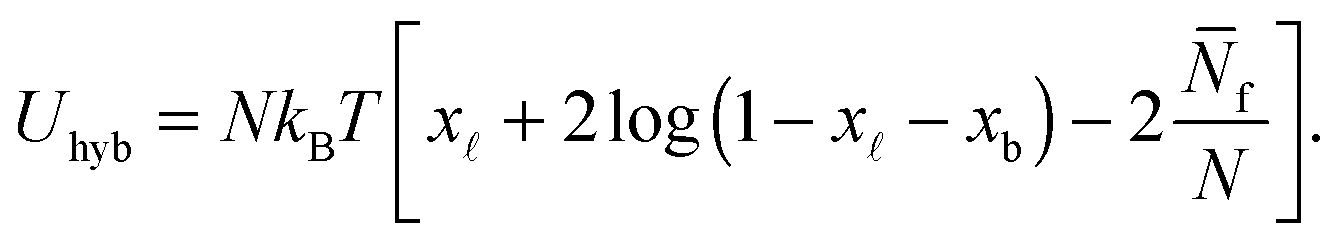 | (6) |
In eqn (6), xb and x are the fraction of tethers involved in bridges/loops, given by
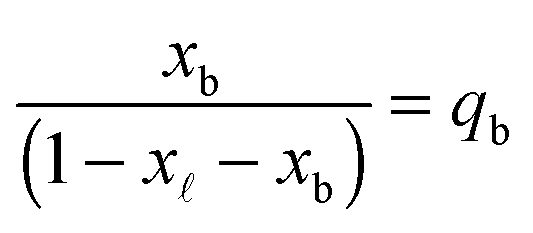 | (7) |
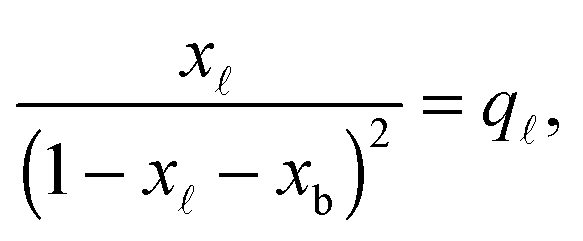 | (8) |
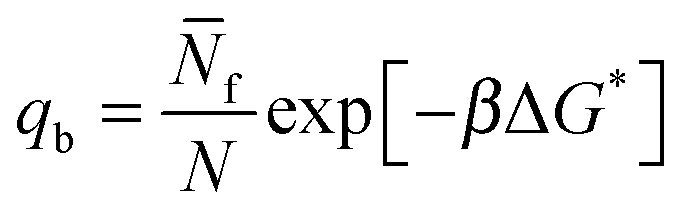 | (9) |
| q = exp[−βΔG*]. | (10) |
In eqn (9) and (10) the hybridisation free energy is re-defined as ΔG* = ΔG − kBTlogN. The combinatorial contribution −kBTlogN, typically estimated in ∼−10kBT,30 has a stabilising effect.
The quantity 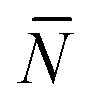 f appearing in eqn (6) indicates the average number of unbound DNA tethers anchored to the SBL available within the adhesion patch. The concentration cf of unbound tetheres of each type (a or a′) on the SBL, such that
f appearing in eqn (6) indicates the average number of unbound DNA tethers anchored to the SBL available within the adhesion patch. The concentration cf of unbound tetheres of each type (a or a′) on the SBL, such that 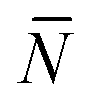 f(θ) = cfAp(θ), is given by
f(θ) = cfAp(θ), is given by
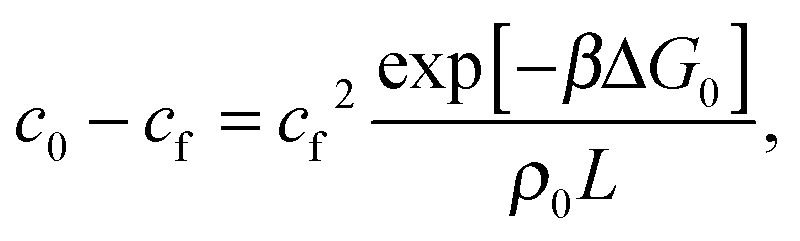 | (11) |
= c0 − cf, and the fraction of loops is xSBL = c/c0.
Eqn (6) generalises the expression found in ref. 30 to the case in which vesicles are in contact with an infinite reservoir of tethers. The overall DNA contribution to the interaction energy in eqn (1) is
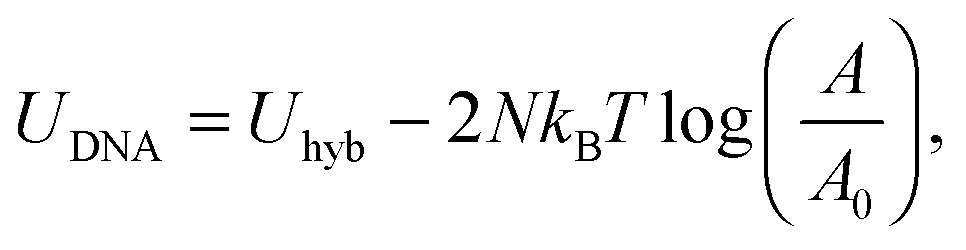 | (12) |
2.3 Overall interaction energy
By combining eqn (2), (6), and (12) into eqn (1), we obtain an analytical expression for the interaction energy of the GUV + SBL system as a function of the only independent variable θ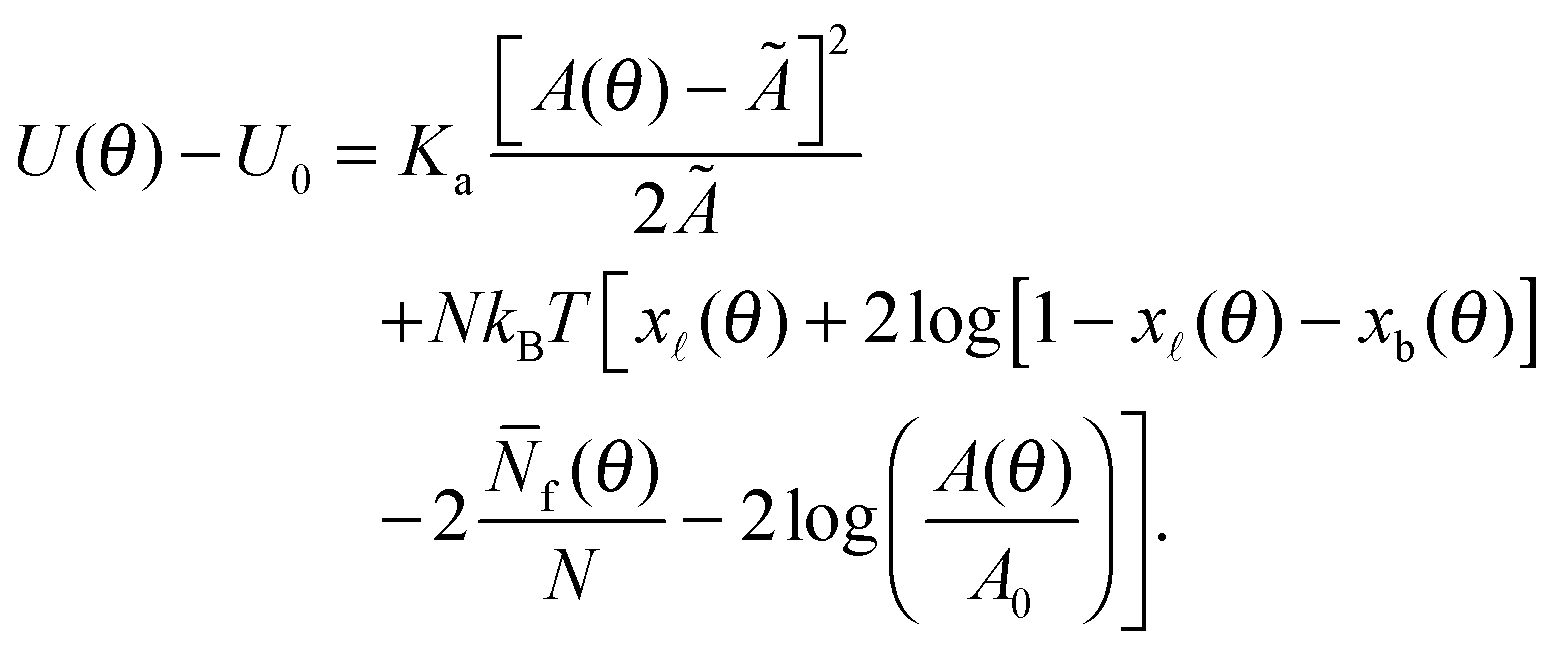 | (13) |
Note that U depends on θ though the adhesion area Ap (see eqn (A.22)) and the total area A of the GUV. On the other hand, the reference energy U0 is θ-independent (see derivation in the Appendix, Section A.5). The interaction energy can be minimised to calculate all the morphological observables (e.g. the contact angle θ, area of the adhesion patch Ap, area of the spherical section of the GUV A), as well as the fraction of formed bridges and loops (xb/).
2.4 Model parameters and error propagation
The model features seven input parameters: the thermal expansion coefficient α, the stretching modulus Ka, the length of the dsDNA tether L, the hybridisation enthalpy ΔH0 and entropy ΔS0 of the sticky ends, the DNA coating density ρDNA (used to calculate the overall number of strands per GUV: 2N), the neutral temperature T0 and radius R0.The stretching modulus is estimated from literature data as Ka = 240 ± 90 mN m−1, with the error bar covering the entire range of reported values.47–50 The thermal expansion coefficient has been experimentally estimated as α = 1.3 ± 0.7 K−1.30 The hybridisation enthalpy and entropy are estimated according to the nearest neighbours thermodynamic rules43 as ΔH0 = −54 ± 5 kcal mol−1 and ΔS0 = −154 ± 13 cal mol−1 K−1. It is not clear whether the stabilising effect of the non-hybridised dangling bases neighbouring the sticky ends, in the present case the adenine bases present at both sides of the sequences (see Fig. 1), should be taken into account. It is indeed possible that their attractive contribution is compensated or overwhelmed by the repulsive effect of the long inert DNA connected to the sticky ends.44 The errorbars in ΔH0 and ΔS0 are included to cover both these scenarios. The length L = 9.8 nm of the dsDNA spacers can be precisely estimated from the contour length of dsDNA (0.388 nm per base-pair35). The DNA coating density is estimated for different samples as explained in the experimental section. The neutral temperature T0 changes widely from vesicle to vesicle due to the polydispersity of electroformed samples. We generally use T0 as a fitting parameter. For a known T0, the “neutral radius” R0 is experimentally determined as R0 = R|T=T0. If T0 falls outside the experimentally accessible temperature range, we extrapolate
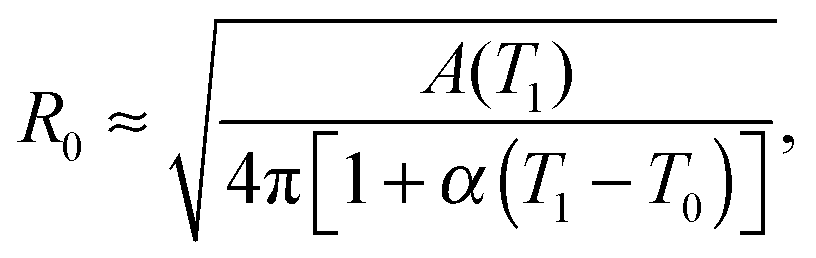 | (14) |
Errors in the input parameters are numerically propagated to the theoretical predictions.30 Briefly, we sample the results of the model using random values of the input parameters X ± ΔX extracted from a Gaussian distribution with mean equal to X and standard deviation equal to ΔX. From a sample of size of 10000, we estimate the theoretical prediction of each observable as the median of the sampled distribution. Errorbars cover the interval between the 16th and the 84th percentile.
3 Results and discussion
3.1 Qualitative observations
In Fig. 3 we can visually compare confocal images of a DNA-functionalised GUV adhering to a SBL (a) and a non-adhering GUV on a passivated glass surface (b). Both GUVs and SBL are stained with fluorescent lipids. | ||
| Fig. 3 From left to right: 3D reconstruction from confocal z-stack, and confocal cross section of an adhering GUV (a) and a non-adhering GUV on a glass substrate (b). For 3D rendering, confocal images have been acquired with a Leica HCX PL APO CS 63 × 1.4 NA oil immersion objective for better resolution, and deconvolved using the experimental point-spread function. Scale bars: 10 μm. | ||
It is clear from both the 3D reconstruction and the vertical cross section that the adhering GUV takes the shape of a truncated sphere, with a flat and circular contact region. This evidence confirms the assumption that, at low enough temperature, DNA-mediated adhesion is strong enough to guarantee the dominance of stretching over bending and other contributions to the deformation energy. The fluorescence intensity measured within the adhesion patches is almost exactly equal to twice the value measured on the SBL outside the adhesion region (1.95 times for the vesicle shown in Fig. 3a). This evidence confirms the presence of two lipid bilayers in close contact within the adhesion area and excludes the possibility of DNA-mediated fusion of the two membranes.51
The non-adhering GUV displayed in Fig. 3b does not exhibit a flat adhesion patch, as clear from the vertical cross-section. Note that for both the adhering and the non-adhering GUVs, the bottom part of the stacks appears brighter due to the z-dependent response of the instrument.
3.2 Temperature-dependence of the geometrical observables
In this section we discuss the temperature-dependence of the morphology of adhering GUVs. In Fig. 4a–c we show the experimentally determined contact angle θ, adhesion-patch radius Rp, and vesicle radius R for a typical adhering GUV in a sample with DNA concentration equal to ρmedDNA = 390 ± 90 μm−2. The contact angle displays a non-monotonic behaviour as a function of temperature, with a positive slope at low T, followed by a sudden decay for T higher than ∼30 °C. The adhesion radius follows the same trend, as does the vesicle radius, which however displays much smaller relative variations. The solid lines in Fig. 4a–c represent theoretical predictions calculated using the input parameters in Table 1, and using the neutral temperature T0 as a fitting parameter. Grey-shaded regions indicate propagated uncertainty in the theoretical predictions. The agreement between theory and experiments is quantitative at low temperatures. At high T, the theory fails to predict the drop in contact angle observed in experiments. This behaviour is expected since our theoretical description is valid in the limit of strong adhesion, where the attractive forces are sufficient to suppress bending contributions to the interaction energy. At high temperature the DNA, which in the present experiment features relatively short sticky ends, starts to melt, causing the loosening of the adhesive forces and a change in the GUV shape, detected as a shrinkage of the adhesion area. The temperature-dependence of the fraction of DNA bonds is quantified and discussed in the following sections.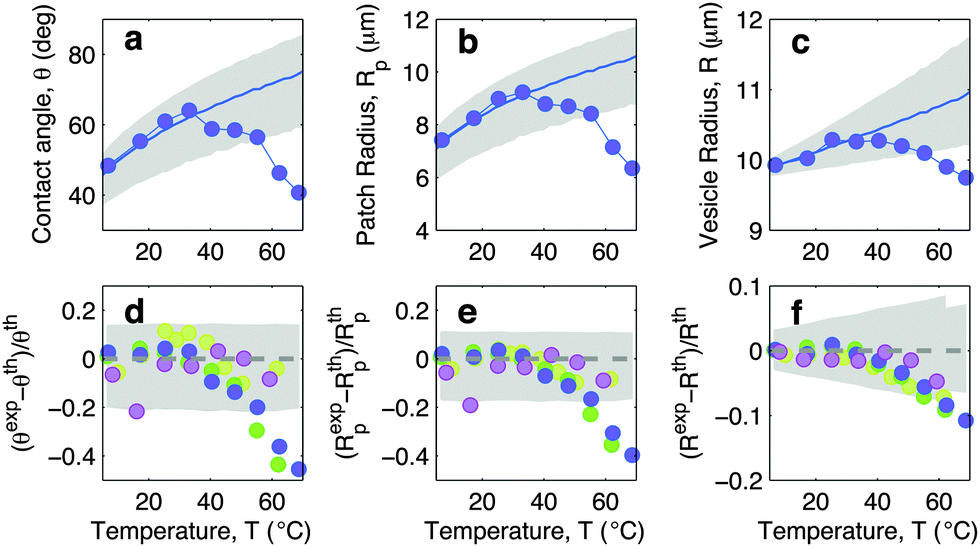 | ||
| Fig. 4 Experimental and theoretical temperature-dependent vesicle adhesion for samples with intermediate DNA coating density ρmedDNA = 390 ± 90 μm−2 (see Experimental section). In the top row we demonstrate the temperature-dependence of the contact angle θ (a), adhesion-patch radius Rp (b), and vesicle radius R (c) for a typical adhering GUV. Points indicate experimental data, solid lines mark theoretical predictions, with errorbars visualised as grey-shaded regions. Fitting parameter T0 = −10 °C. In the bottom row we summarise the results for 4 vesicles. Points represent the relative deviation of experimental data from theoretical predictions (Xexp − Xth)/Xth, with X = θ (d), Rp (e), and R (f). Grey-shaded regions mark the uncertainty interval of the theory. Model parameters are reported in Table 1. | ||
| α = 1.3 ± 0.7 K−1 |
| K a = 240 ± 90 mN m−1 |
| L = 9.8 nm |
| ΔH0 = −54 ± 5 kcal mol−1 |
| ΔS0 = −154 ± 13 cal mol−1 K−1 |
In Fig. 4d–f we show the relative deviations of the experimentally-determined morphological observables from the theoretical predictions, defined as (Xexp − Xth)/Xth, for X = θ, Rp, and R. The data, collected from various vesicles are consistent: the experimental data fall within theoretical errorbars at low T, deviating at higher temperature due to the failure of the strong adhesion assumption.
In Fig. 5 we show experimental, and theoretically predicted morphological observables for the case of low DNA concentration, ρlowDNA = 39 ± 9 μm−2. Similarly to the case of higher DNA concentration, experimental data are backed by theoretical predictions. In this case, however, the high-temperature deviation of the experiments from the theoretical predictions appears to be less evident, and shifted towards higher temperatures. This suggests the presence of an (however small) adhesive force hindering the partial detachment of the GUVs. We ascribe this behaviour to the effect of non-specific membrane–membrane adhesion, e.g. dispersion attraction, that for higher DNA coverage is suppressed by steric repulsion.
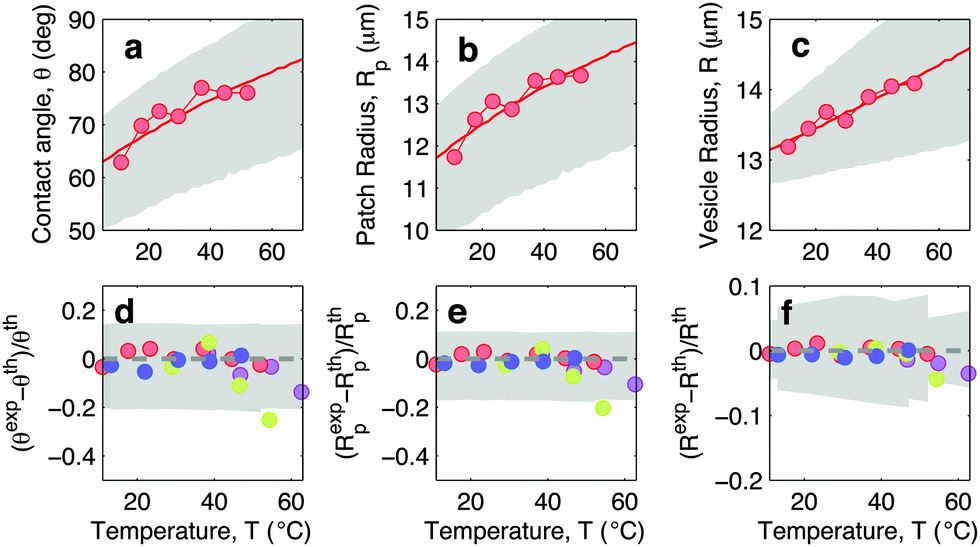 | ||
| Fig. 5 Experimental and theoretical temperature-dependent vesicle adhesion for samples with low DNA coating density ρlowDNA = 39 ± 9 μm−2 (see Experimental section). In the top row we demonstrate the temperature-dependence of the contact angle θ (a), adhesion-patch radius Rp (b), and vesicle radius R (c) for a typical adhering GUV. Points indicate experimental data, solid lines mark theoretical predictions, with errorbars visualised as grey-shaded regions. Fitting parameter T0 = −40 °C. In the bottom row we summarise the results for 4 vesicles. Points represent the relative deviation of experimental data from theoretical predictions (Xexp − Xth)/Xth for X = θ (d), Rp (e), and R (f). Grey-shaded regions mark the uncertainty interval of the theory. Model parameters are reported in Table 1. | ||
3.3 DNA-melting
We investigate the temperature-dependence of the fraction of formed DNA bonds via in situ FRET measurements. Cyanine fluorophores, Cy3 (donor) and Cy5 (acceptor), are connected to the 3′ termini of a and a′ sticky ends, as sketched in Fig. 1. FRET efficiency is described by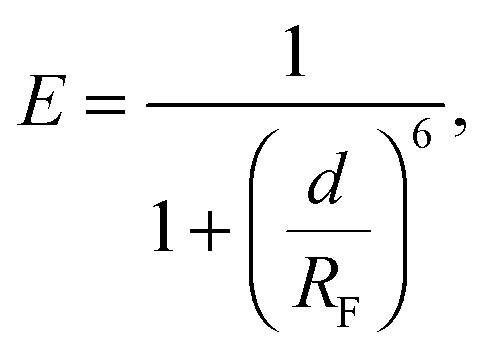 | (15) |
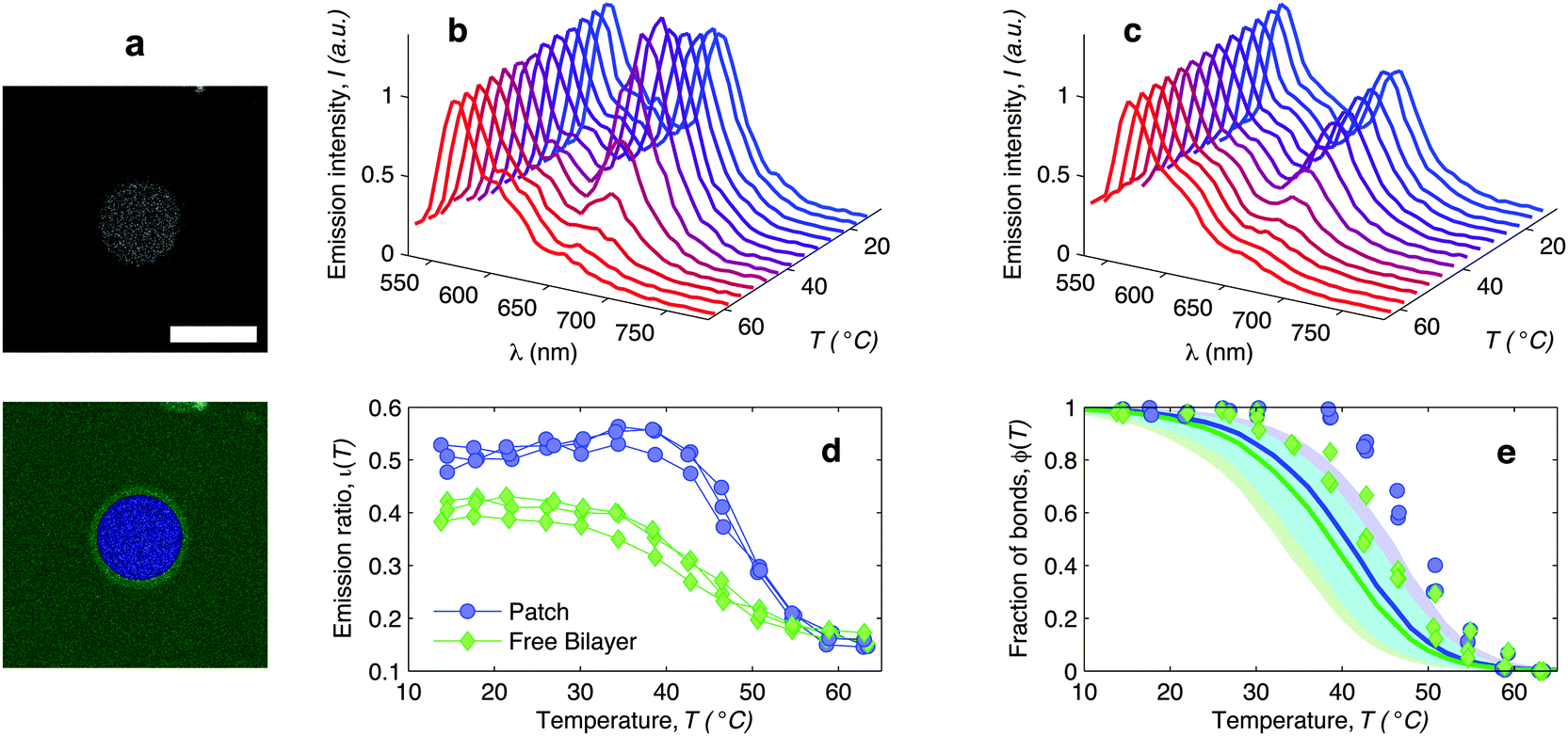 | ||
| Fig. 6 In situ FRET spectroscopy characterisation of the temperature-dependence of the fraction of DNA bonds. (a) Confocal image of the adhesion patch of a GUV. At the bottom we show the same image segmented with our software to separate the adhesion patch (blue) from the free supported bilayer (green). Scale bar: 15 μm. (b) Fluorescence emission spectra measured within the adhesion patch for a typical vesicle (blue area in panel (a)). Colours from blue to red mark low to high temperatures in the interval 14.5 ≤ T ≤ 62.9 °C. The curves are normalised to the emission peak of Cy3 (568 nm). In panel (c) we show the emission spectra measured on the free SBL (green area in panel (a)). (d) Relative intensity of the Cy5 emission peak (665 nm) measured within (blue circles) and outside (green lozenges) the adhesion patch. The curves are relative to 3 different vesicles. The amplitude of the Cy5 and Cy3 emission peaks is determined through a Gaussian fit of the 5 data points closest to the maximum. (e) Experimental (symbols) and theoretically predicted (solid lines) fraction of formed DNA bonds within (blue circles) and outside (green lozenges) the adhesion patch. Experimental data are extracted from the curves in panel (d) as described in the text. Theoretical curves are calculated using the parameters in Table 1, DNA-coating density ρhighDNA = 1200 ± 300 μm−2, T0 = −20, and R0 = 10 μm. Note that the value of T0 does not significantly affect these quantities. Blue and green shaded regions indicate propagated uncertainties of the blue and green solid lines. Cyan shaded region marks their overlap. | ||
The curves ι(T) can be used to semi-quantitatively estimate the temperature dependence of the overall fraction of DNA bonds. We fit the low temperature plateaus (T < 35 °C) in Fig. 6d with linear baselines B(T) and assume that ι(T) plateaus to a constant value for T > 57 °C. The fraction of formed DNA bonds is thereby estimated as
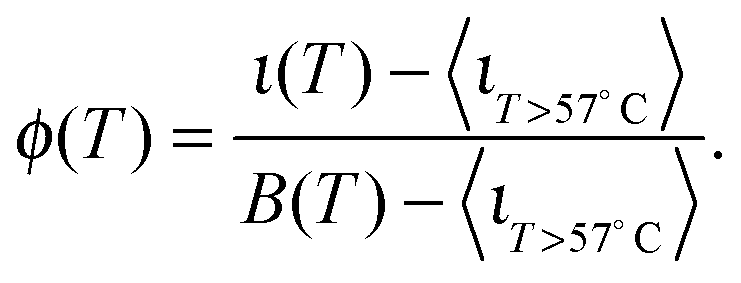 | (16) |
A better estimate of ϕ(T) could be obtained by measuring ι(T) up to higher temperatures, and fitting the high-temperature plateau with a second linear baseline. However, temperatures higher than 65 °C cannot be safely probed due to the risk of destabilisation of the dsDNA spacers. The experimental ϕ(T) data in Fig. 6e indicate that the DNA melting transition is relatively broad, spanning more than 30 °C.54 Moreover, the melting seems to occur at a higher temperature (by about 5 °C) within the adhesion patch.
The experimentally estimated fraction of DNA bonds can be compared with theoretical predictions. On the free SBL, only loops can form, and therefore ϕ(T) should be compared to the fraction of loops xSBL, calculated according to eqn (11), (A.17) and (A.18) (Appendix). Within the adhesion patch we count contributions from bridges, loops formed on the GUV, and loops formed on the SBL. By assuming evenly distributed loops, the overall number of DNA bonds found within the patch is
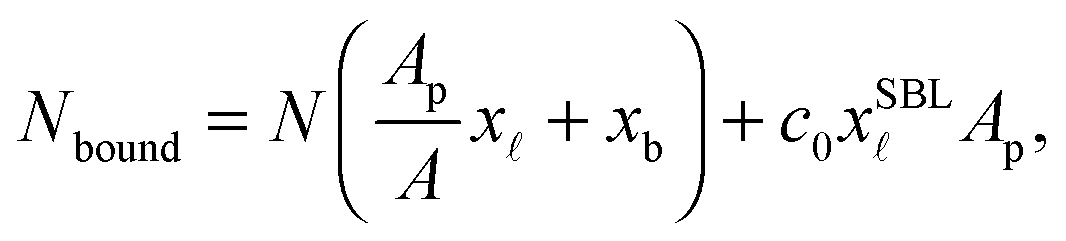 | (17) |
are the fractions of loops and bridges on the GUV, given by eqn (A.28) and (A.29) (Appendix). The overall number of DNA tethers of each species (a or a′), bound and unbound, found within the patch is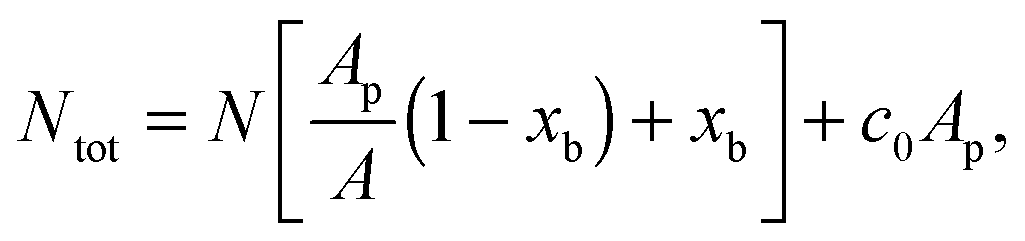 | (18) |
3.4 Membrane tension
The temperature dependence of the membrane tension is measured by flickering analysis of the equatorial cross sections of GUVs. The tension is extracted by fitting the power spectra of the thermal fluctuations, determined as explained in the experimental section, with the function | (19) |
![[script L]](https://www.rsc.org/images/entities/char_e144.gif) is the contour length of the equatorial cross-section of the GUV, σ is the membrane tension, κ the bending modulus, and qx the wave vector evaluated along the contour. Eqn (19) is derived from the original work of Helfrich,33 describing the fluctuations of an infinite 2D membrane, and corrected to account for the fact that, by imaging an equatorial cross-sections, only modes propagating along the horizontal direction should be considered.34 Of the discrete set of wave vectors qx(n) = 2πn/, modes with n < 6 are excluded from the analysis. Mode n = 0 and n = 1 correspond to size changes and translations of the GUV. Modes with n > 2 describe thermal fluctuations. However, eqn (19) is derived for a planar membrane, and should not be used to describe modes with n < 6, which are influenced by the spherical geometry of the GUV.34 For our analysis we fit the spectra for modes 6 < n < 16. At higher q we approach the resolution limits of the current method.
is the contour length of the equatorial cross-section of the GUV, σ is the membrane tension, κ the bending modulus, and qx the wave vector evaluated along the contour. Eqn (19) is derived from the original work of Helfrich,33 describing the fluctuations of an infinite 2D membrane, and corrected to account for the fact that, by imaging an equatorial cross-sections, only modes propagating along the horizontal direction should be considered.34 Of the discrete set of wave vectors qx(n) = 2πn/, modes with n < 6 are excluded from the analysis. Mode n = 0 and n = 1 correspond to size changes and translations of the GUV. Modes with n > 2 describe thermal fluctuations. However, eqn (19) is derived for a planar membrane, and should not be used to describe modes with n < 6, which are influenced by the spherical geometry of the GUV.34 For our analysis we fit the spectra for modes 6 < n < 16. At higher q we approach the resolution limits of the current method.
In Fig. 7a blue circles mark the tension measured as a function of temperature for adhering GUVs. In Fig. 7b we show examples of power spectra fitted by eqn (19). The tension typically lies in the interval 2 × 10−7–2 × 10−6 N m−1, with clear variations between different GUVs. In the tested range, the tension consistently displays a weak dependence on temperature changes.
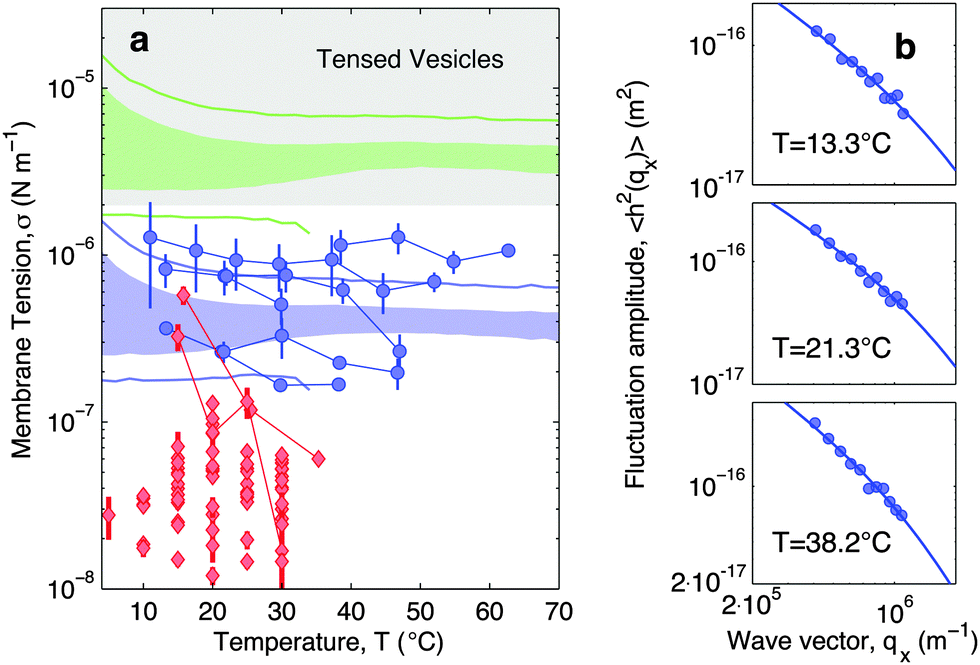 | ||
| Fig. 7 Experimental and theoretical temperature-dependent membrane tension. (a), Membrane tension σ measured from filckering experiments. Blue circles indicate adhering GUVs with low DNA coverage (ρlowDNA = 39 ± 9 μm−2). Red lozenges indicate non-adhering GUVs on a BSA-coated glass surface. Grey-shaded regions mark regimes in which membrane tension is currently inaccessible to our technique. The blue-shaded band indicates the theoretical prediction for σ calculated with the fitting parameter −60 ≤ T0 ≤ 0 °C and low DNA coverage. Blue lines mark the corresponding errorbars. The green-shaded band indicate the theoretical prediction for σ for higher DNA coverage (ρmedDNA = 390 ± 90 μm−2). Green lines mark the corresponding errorbars. Model parameters are reported in Table 1.30 (b) Fluctuation-amplitude spectra for adhering GUVs with low DNA coverage and various temperatures. Symbols indicate experimental data and solid lines indicate fits according to eqn (19). From top to bottom the fitted values of the membrane tension are σ = 3.6 ± 0.3, 2.6 ± 0.2, 1.7 ± 0.1 × 10−7 N m−1. | ||
For comparison, the membrane tension is measured on non-adhering GUVs supported by a passivated glass substrate. The values of σ measured for non-adhering GUVs (red lozenges in Fig. 7a) are significantly lower than those measured on adhering vesicles, falling within the range 10−8–5 × 10−7 N m−1, and being clustered around 2–3 × 10−8 N m−1. Furthermore, membrane tension of non-adhering GUVs typically displays a strong decrease upon increasing temperature. The large variability observed in the tension of adhering and, in particular, of free GUVs, is ascribed to the polydispersity of electroformed samples, which produces vesicle populations with very different excess areas (T0).
With the present technique we cannot access the tension of vesicles adhering to SBL for the case of higher DNA concentrations. Indeed, for values of σ in the grey-shaded region on Fig. 7a, the relevant portions of the fluctuation power spectra are masked by experimental noise deriving from the finite resolution of the contour-tracking procedure.
The membrane tension can be evaluated within the framework of our model. At equilibrium, the derivative of the interaction energy in eqn (1), taken with respect to the GUV area, is
 | (20) |
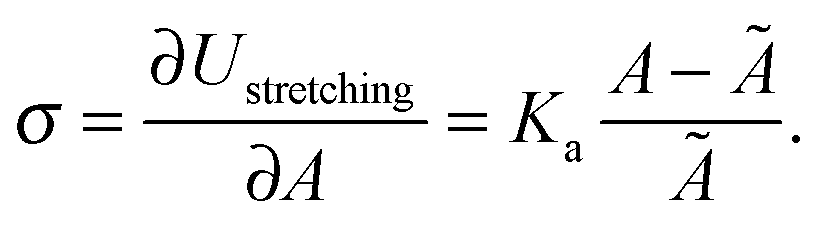 | (21) |
Eqn (20) suggests that by measuring σ we can directly probe the DNA-mediated forces. The blue-shaded region in Fig. 7a marks the model predictions for σ, calculated using the parameters in Table 1, ρlowDNA = 39 ± 9 μm−2, and values of the neutral temperature covering the experimentally observed range (−60 < T0 < 0 °C). The size of the vesicles does not impact the predictions of σ, therefore we fix R0 = 10 μm. Solid blue lines mark the uncertainty interval propagating from the errorbars of the model parameters (Table 1). With no fitting parameters, we observe a semi-quantitative agreement between theory and experiments. In particular, the theory predicts the weak temperature-dependence of σ observed in the experiments.
The green-shaded region in Fig. 7a indicates the theoretical prediction calculated using ρmedDNA = 390 ± 90 μm−2. The predicted tension falls within the non-accessible region.
4 Conclusion
In this work we experimentally investigated temperature-dependent adhesion of Giant-Unilamellar-Vesicles on supported lipid bilayers mediated by mobile DNA linkers. The simple geometry of the problem allows for an accurate characterisation of the morphology of adhering GUVs and the temperature dependent fraction of bound DNA tethers by means of confocal microscopy. For the first time to our knowledge, we quantify the temperature-dependent membrane tension induced by DNA bonds by analysing the thermal fluctuations of the GUVs imaged across their equatorial plane.The experimental results are compared to theoretical predictions from our recently developed model,30 which we here extend to the case of vesicle-plane adhesion. The model takes into account both the elastic deformation of the GUV and the statistical-mechanical details of the DNA-mediated interactions.
For sufficiently high DNA coverage, the adhesion contact angle exhibits a re-entrant temperature dependence. Upon heating from low temperature the contact angle increases, reaching a maximum at T ≃ 30–40 °C. Upon further temperature increase, the contact angle drops. The re-entrance is less pronounced or absent for lower DNA coverage. With a single fitting parameter, the model is capable of quantitatively predicting the low temperature regime and ascribes the increase in contact angle to the interplay between the temperature-dependent excess area of the GUV and the entropic coupling between the hybridisation free-energy of the mobile tethers and the adhesion area. The theory is developed in the limit of strong adhesion, therefore it fails to predict the re-entrant behaviour of the adhesion area, caused by the weakening of the DNA bonds. The less-pronounced re-entrance observed for low DNA concentrations is ascribable to non-specific adhesive interactions that kick-in at high temperature, and are suppressed by steric repulsion for samples with high DNA coverage.
The melting of DNA bonds is investigated in situ by FRET measurements. We observe a broad melting transition and find that bonds formed within the GUV-plane adhesion patch are more stable than in-plane bonds formed on free bilayers. With no fitting parameters our model can semi-quantitatively reproduce these features, although an underestimation of the melting temperature is observed.
Membrane tension measurements performed on adhering GUVs demonstrate a weak temperature dependence. In a similar range of temperatures, non-adhering GUVs exhibit significantly lower tension, rapidly decreasing upon heating. The differences in magnitude and trend demonstrates the role played by DNA in mediating membrane adhesion. Experimental results are in semi-quantitative agreement with theoretical prediction, which further demonstrates the accuracy of the model used to describe hybridisation free-energy of tethered mobile linkers, and in particular the translational-entropic contributions that couples it to the adhesion area.
Our experimental observations and the agreement with the theoretical predictions help to clarify the complex mechanisms controlling adhesion of soft units mediated by multiple linkers. Besides the fundamental interest for the still poorly understood physics of multivalent interactions, our findings can help the design of functional, responsive, tissue-like materials with promised applications in biosensing, encapsulation-release mechanisms, and filtration. Finally, the conclusions drawn for our model system can be adapted to the quantitative description of cell adhesion and spreading on solid substrates, with possible biomedical implications in prosthetics and scaffolds for tissue regeneration.56–62
A Details on model derivation
A.1 Geometrical expressions
In the limit of strong adhesion the GUV will take the shape of a truncated sphere with contact angle θ (Fig. 1), which we take as the independent variable of our model.40 In this simple geometry, the contact (patch) area, total area, and volume of the GUV are respectively| Ap = πR2sin2θ | (A.1) |
| A = πR2(1 + cosθ)(3 − cosθ) | (A.2) |
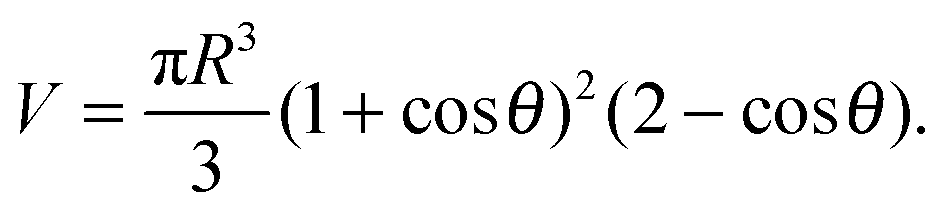 | (A.3) |
In the limit of water-impermeable membranes, the internal volume of the GUVs can be taken as a constant
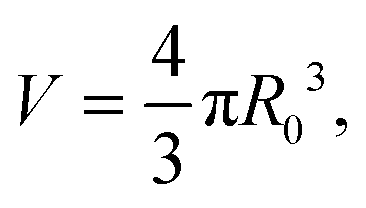 | (A.4) |
 | (A.5) |
Let us now recall the definition of reduced volume of a vesicle40,63
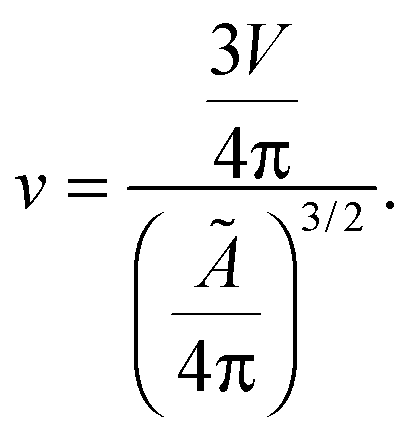 | (A.6) |
By combining eqn (A.6) with expression for the temperature-dependent unstretched area in eqn (3), we obtain
| v = [1 + α(T − T0)]−3/2. | (A.7) |
The reference temperature T0 is therefore defined as the temperature at which a GUV has reduced volume equal to 1. For T < T0, when v > 1, an isolated vesicle resembles a turgid sphere, with non-zero membrane tension whereas for T > T0, v < 1, it assumes the features of a “floppy” balloon, with excess area. At T = T0 an isolated vesicle is a perfect sphere with zero-membrane tension and radius equal to the reference radius R0.
By combining eqn (A.2), (A.3) and (A.5) we obtain an explicit, θ-dependent expression for the stretching energy in eqn (A.2). Note that eqn (A.5) has been derived under a constant-volume assumption (eqn (A.4)). Alternatively, an equivalent relation can be derived for water permeable – solute impermeable – GUVs, in which the volume is set by the balance between the osmotic pressure drop across the membrane and the Laplace pressure.40 In relevant experimental conditions the two assumptions lead to very similar results.30
A.2 Free energy for bridge and loop formation
For the case of mobile linkers, the configurational entropic contribution to the bridge/loop formation free energy ΔSconfb/ (eqn (A.4)) can be split into a rotational and translational contribution| ΔSconfb/ = ΔSrot + ΔStransb/. | (A.8) |
The rotational contribution takes the same expression for loop and bridge formation, and encodes for the reduction of configurational entropy following the hybridisation of two rigid tethers with fixed grafting sites22,30
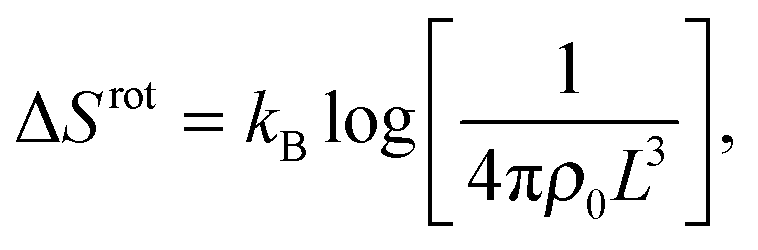 | (A.9) |
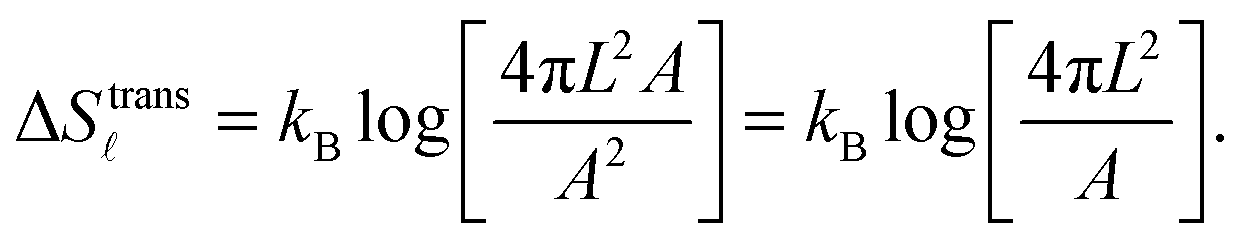 | (A.10) |
For the case of bridge formation, we consider a free linker on the SBL, initially located within the contact area Ap, and a second linker on the GUV, which is free to explore the entire surface area A. Upon binding, the area available to the pair is reduced to 4πL2Ap, resulting in
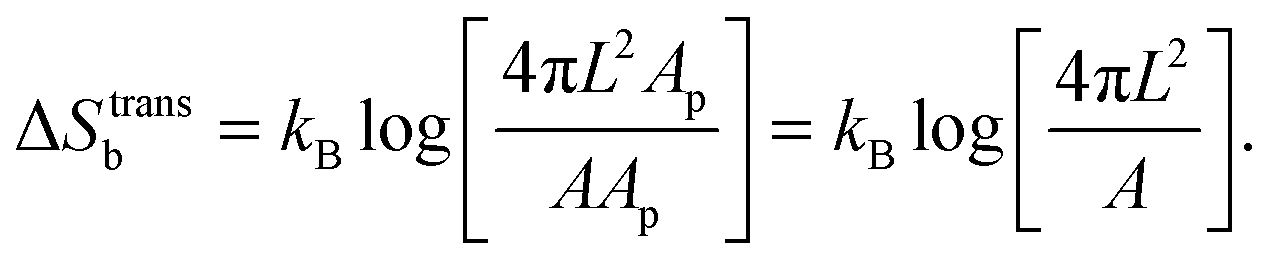 | (A.11) |
We notice that, contrary to the case of two adhering GUVs,30 here ΔStransb = ΔStrans. By combining eqn (A.8)–(A.11) with eqn (4), we obtain the hybridisation free energy of bridge formation on the loops formation on the GUV in eqn (5).
A.3 Fraction of loops and free tethers on the SBL
We now focus on the description of the tethers anchored to the SBL, and calculate the equilibrium fraction of formed loops in the absence of an adhering GUV. This information is needed for the calculation of the GUV–SBL adhesive interaction as well as for a direct comparison with experimental data.Let us consider a finite portion of the SBL of area Σ, containing two populations of N linkers with a and a′ sticky ends. Following eqn (5), the free energy for loop () formation on the SBL can be written as
 | (A.12) |
By indicating as N the number of loops within the SBL, and taking into account combinatorics, we can write the partition function of this systems as30
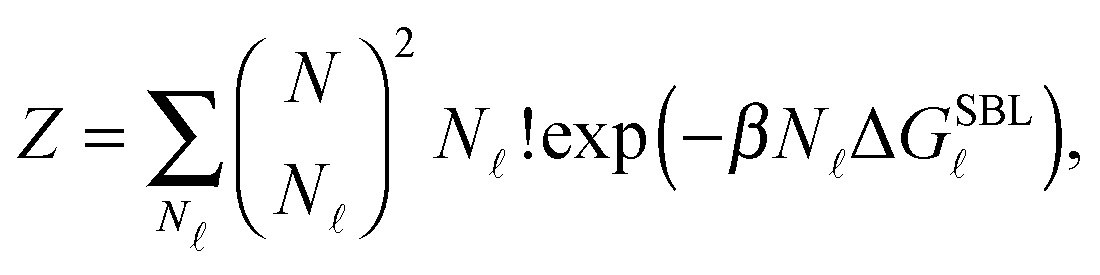 | (A.13) |
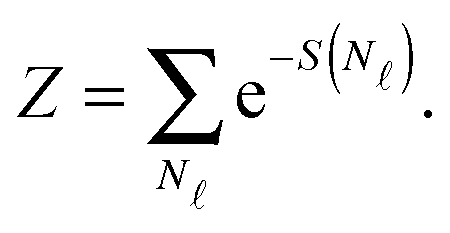 | (A.14) |
We now consider the limit of an infinite SBL with a constant DNA surface density c0, i.e. we take N,Σ → ∞, with c0 = N/Σ = ρDNA/2. By using the Stirling approximation we obtain
| S(c) = const·Σ[−clogc − 2(c0 − c)log(c0 − c) − cβΔG0 − clog(ρ0L) − c + const], | (A.15) |
=N/Σ. Within the saddle-point approximation,30 the sum in eqn (A.14) is dominated by the stationary point of S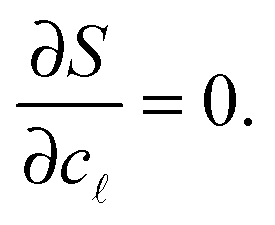 | (A.16) |
By solving eqn (A.16) we obtain the expression in eqn (11) in the text, where we introduce the concentration of free tethers on the SBL cf =c0 − c. Note that with eqn (11) we recover a simple mass-balance relation between loops and free tethers on the SBL, which ultimately results in
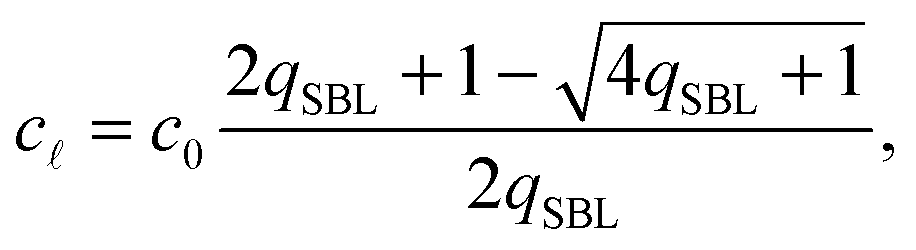 | (A.17) |
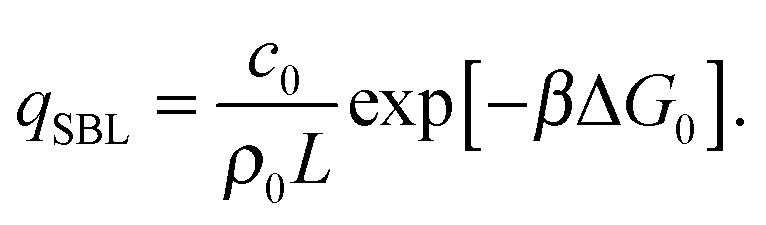 | (A.18) |
A.4 Combinatorial effects
Given the expressions for the hybridisation free-energy of a single bridge/loop (eqn (5)), a combinatorial approach is required to compute the overall DNA-mediated interaction energy.Following the derivation carried out to describe loop formation on the SBL, we indicate the total number of tethers with a (a′) sticky ends on the GUV as N, and define N as the number of those tethers linked in loops. We indicate as Nbi, with i = 1, 2, the number of tethers forming bridges with those on the SBL, with the index i referring to a and a′ sticky ends. We label as Nfi the number of free tethers on the SBL located within the adhesion patch, where the index i = 1, 2 now refers to a′ and a sticky ends. The partition function of the system of linkers can be written as
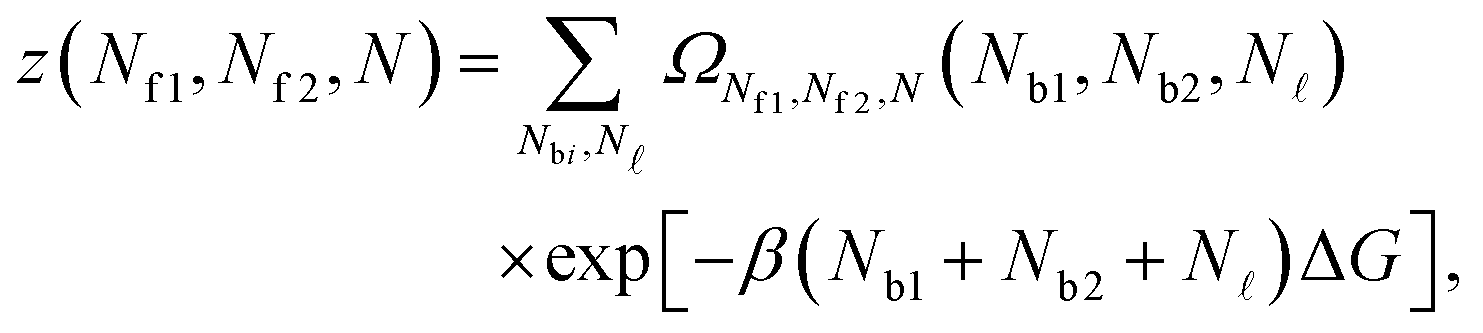 | (A.19) |
and Nbi is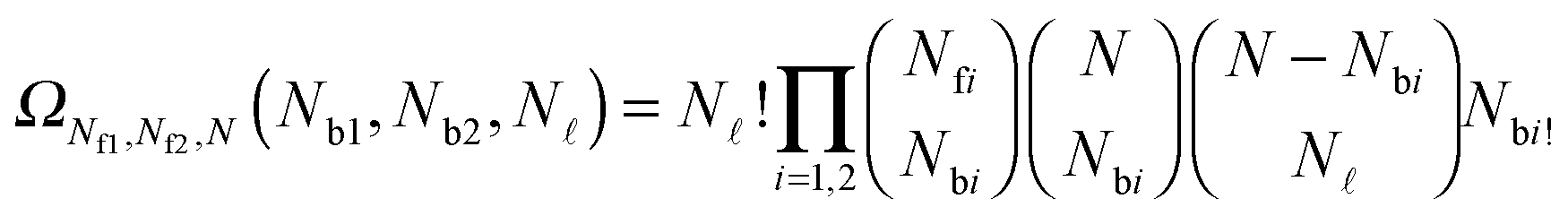 | (A.20) |
To account for strand-concentration fluctuations within the adhesion patch, we need to consider that Nfi is Poisson-distributed around its average value 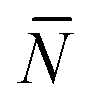 f
f
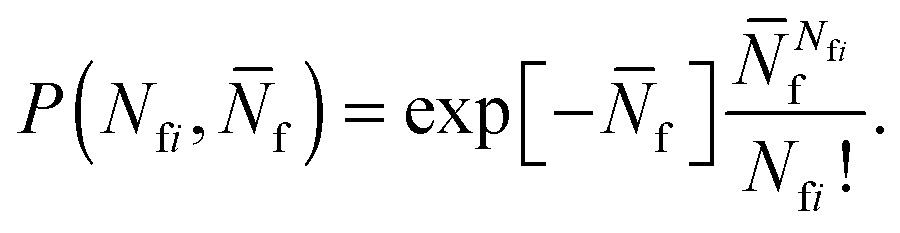 | (A.21) |
Using eqn (A.17), and recalling that cf = c0 − c is the concentration of free tethers within the SBL, we find
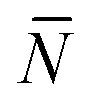 f(θ) = cfAp(θ), f(θ) = cfAp(θ), | (A.22) |
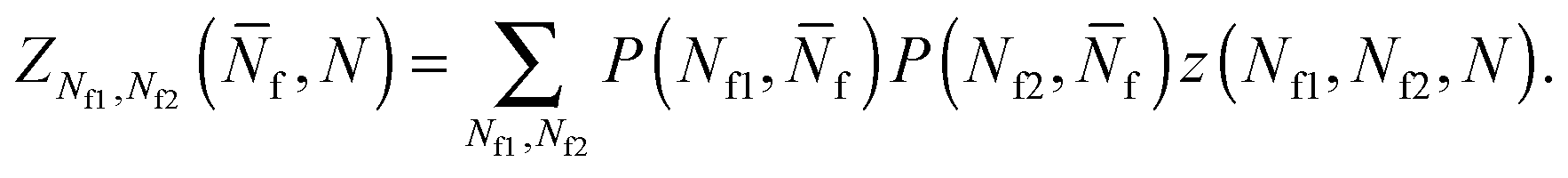 | (A.23) |
Eqn (A.23) can be rearranged as
 | (A.24) |
By defining the fractions xy = Ny/N (y = b1, b2, , f1, f2), and using the Stirling approximation, ![[scr A, script letter A]](https://www.rsc.org/images/entities/char_e520.gif) can be expressed as
can be expressed as
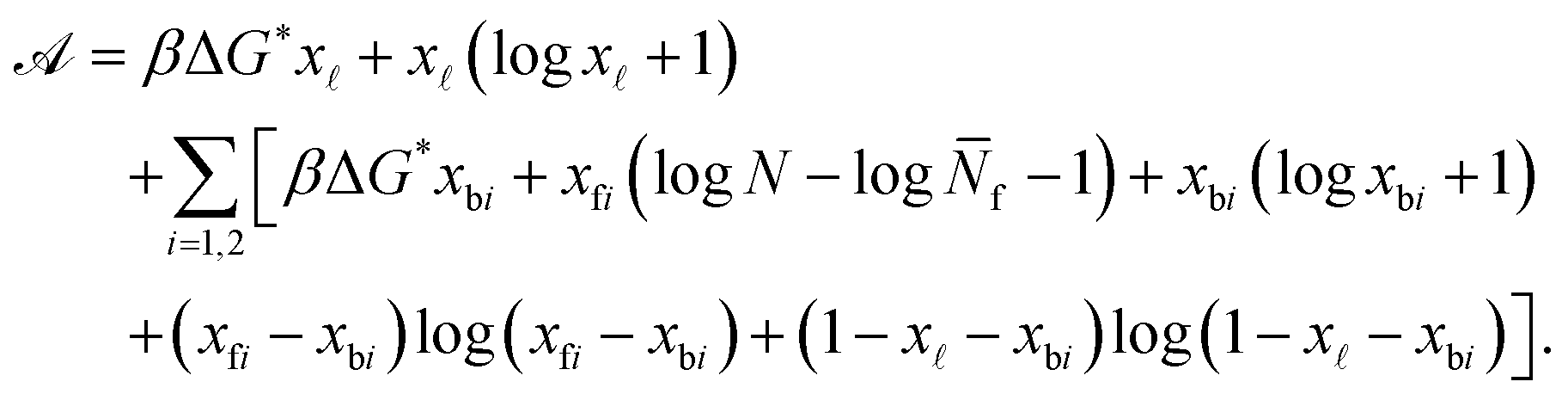 | (A.25) |
Note that in eqn (A.25) we re-defined the hybridisation free energy for bridge and loop formation as
| ΔG* = ΔG − kBTlogN. | (A.26) |
For typical experimental conditions, the attractive combinatorial term −kBTlogN in eqn (A.26) can be estimated in ∽−10kBT.30 Within the saddle-point approximation, the sum in eqn (A.24) is dominated by the stationary point of
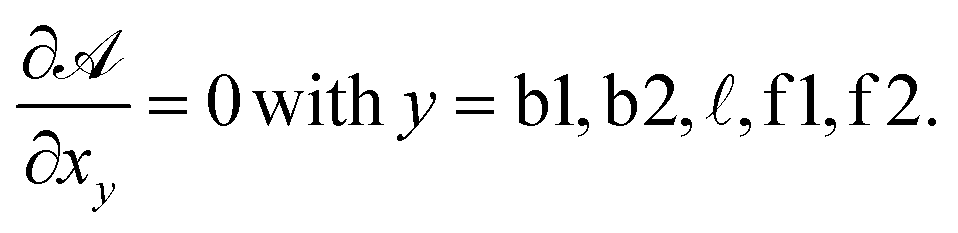 | (A.27) |
From the saddle-point equations eqn (A.27) we obtain eqn (7) and (8) in the text, where we find xb1 = xb2 = xb. By solving eqn (7) and (8) we obtain
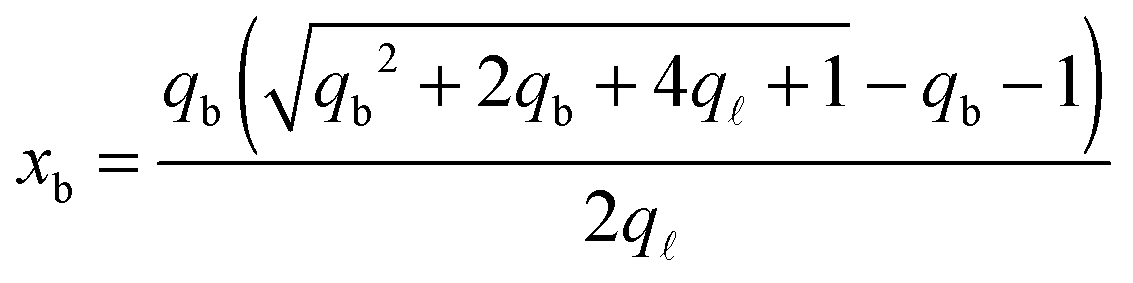 | (A.28) |
 | (A.29) |
Note that for simplicity the fraction of bridges and loops are indicated as xb/. The saddle point equations for xf (xf1 = xf2) read xf − xb = 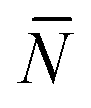 f/N, which confirms that the density of the free tethers in the patch region is equal to that of the reservoir, as expected.
f/N, which confirms that the density of the free tethers in the patch region is equal to that of the reservoir, as expected.
By inserting the saddle-point solutions for x, xb, and xf in eqn (A.24) and (A.25) we can calculate the free energy Uhyb (eqn (6)).19
A.5 Reference energy
The reference energy U0 in eqn (1) is calculated for isolated GUV and SBL and can be written as| U0 = Ustretching0 + UDNA0. | (A.30) |
The stretching term is30
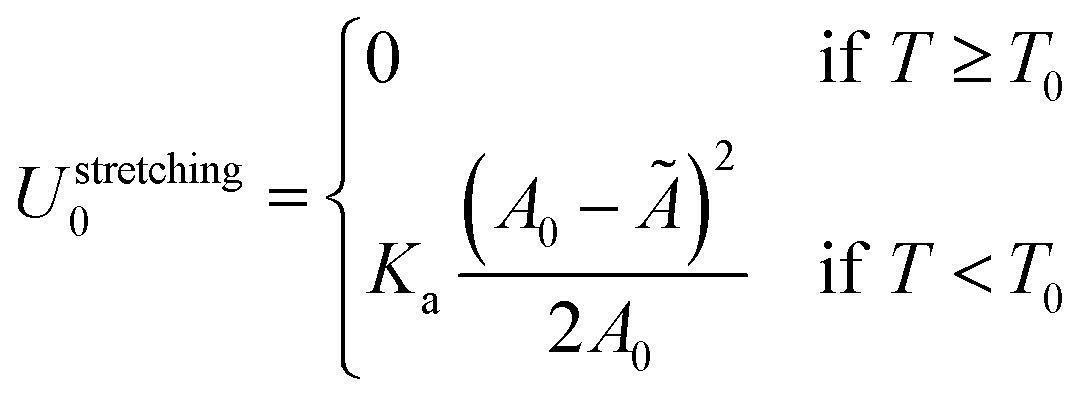 | (A.31) |
Note that the stretching contribution is only present for pre-stretched vesicles, i.e. if the reduced volume is v > 1 (i.e. T < T0).40 The DNA contribution is calculated for a GUV of area equal to the unstretched area Ã, in which only loops can form. By following the steps outlined in Section A.4 and in ref. 30 we calculate the fraction of tethers involved in loops
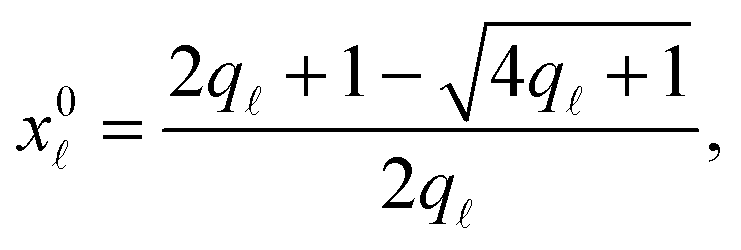 | (A.32) |
is given by eqn (10). The DNA part of the reference energy is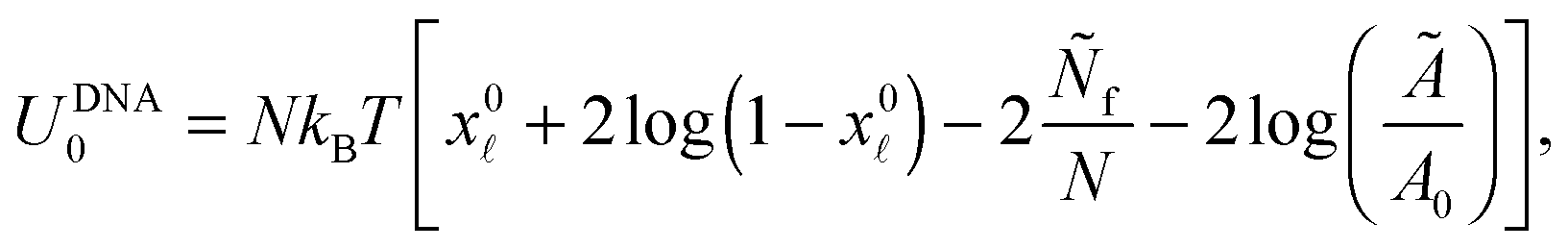 | (A.33) |
Note that U0 does not depend on the contact angle θ therefore its form does not influence the equilibrium features of the system.
Acknowledgements
The authors acknowledge financial support from the EPSRC Programme Grant CAPITALS number EP/J017566/1 (PC, LP, LDM), the Ernest Oppenheimer Fund and Emmanuel College Cambridge (LDM), Université libre de Bruxelles (BMM), and Grant-in-Aid for JSPS Fellows Grant (No. 25-1270) (SFS), Project SPINNER 2013, Regione Emilia-Romagna (It), European Social Fund (DO). All the data needed to support and reproduce these results are presented within the paper. Due to their size, sharing raw data (microscopy videos and images) online is not cost-effective.References
- L. Di Michele and E. Eiser, Phys. Chem. Chem. Phys., 2013, 15, 3115–3129 RSC.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS PubMed.
- A. P. Alivisatos, K. P. Johnsson, X. Peng, T. E. Wilson, C. J. Loweth, M. P. Bruchez and P. G. Schultz, Nature, 1996, 382, 609–611 CrossRef CAS PubMed.
- H. D. Hill, R. J. Macfarlane, A. J. Senesi, B. Lee, S. Y. Park and C. A. Mirkin, Nano Lett., 2008, 8, 2341–2344 CrossRef CAS PubMed.
- M. M. Maye, M. T. Kumara, D. Nykypanchuk, W. B. Sherman and O. Gang, Nat. Nanotechnol., 2010, 5, 116–120 CrossRef CAS PubMed.
- D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, Nature, 2008, 451, 549–552 CrossRef CAS PubMed.
- P. Cigler, A. K. R. Lytton-Jean, D. G. Anderson, M. G. Finn and S. Y. Park, Nat. Mater., 2010, 9, 918–922 CrossRef CAS PubMed.
- R. J. Macfarlane, B. Lee, M. R. Jones, N. Harris, G. C. Schatz and C. A. Mirkin, Science, 2011, 334, 204–208 CrossRef CAS PubMed.
- L. Di Michele, F. Varrato, J. Kotar, S. H. Nathan, G. Foffi and E. Eiser, Nat. Commun., 2013, 4, 2007 Search PubMed.
- F. Varrato, L. Di Michele, M. Belushkin, N. Dorsaz, S. H. Nathan, E. Eiser and G. Foffi, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19155–19160 CrossRef CAS PubMed.
- L. Di Michele, D. Fiocco, F. Varrato, S. Sastry, E. Eiser and G. Foffi, Soft Matter, 2014, 10, 3633–3648 RSC.
- K. L. Young, M. B. Ross, M. G. Blaber, M. Rycenga, M. R. Jones, C. Zhang, A. J. Senesi, B. Lee, G. C. Schatz and C. A. Mirkin, Adv. Mater., 2014, 26, 653–659 CrossRef CAS PubMed.
- C. S. Thaxton, R. Elghanian, A. D. Thomas, S. I. Stoeva, J.-S. Lee, N. D. Smith, A. J. Schaeffer, H. Klocker, W. Horninger, G. Bartsch and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18437–18442 CrossRef CAS PubMed.
- X. A. Wu, C. H. J. Choi, C. Zhang, L. Hao and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 7726–7733 CrossRef CAS PubMed.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027–1030 CrossRef CAS PubMed.
- J. I. Cutler, E. Auyeung and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 1376–1391 CrossRef CAS PubMed.
- R. Dreyfus, M. E. Leunissen, R. Sha, A. V. Tkachenko, N. C. Seeman, D. J. Pine and P. M. Chaikin, Phys. Rev. Lett., 2009, 102, 048301 CrossRef.
- P. Varilly, S. Angioletti-Uberti, B. M. Mognetti and D. Frenkel, J. Chem. Phys., 2012, 137, 094108 CrossRef PubMed.
- S. Angioletti-Uberti, P. Varilly, B. M. Mognetti, A. V. Tkachenko and D. Frenkel, J. Chem. Phys., 2013, 138, 021102 CrossRef PubMed.
- B. M. Mognetti, M. E. Leunissen and D. Frenkel, Soft Matter, 2012, 8, 2213–2221 RSC.
- B. M. Mognetti, P. Varilly, S. Angioletti-Uberti, F. J. Martinez-Veracoechea, J. Dobnikar, M. E. Leunissen and D. Frenkel, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E378–E379 CrossRef CAS PubMed.
- M. E. Leunissen and D. Frenkel, J. Chem. Phys., 2011, 134, 084702 CrossRef PubMed.
- M. Hadorn, E. Boenzli, K. T. Srensen, H. Fellermann, P. Eggenberger Hotz and M. M. Hanczyc, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20320–20325 CrossRef CAS PubMed.
- L. Feng, L.-L. Pontani, R. Dreyfus, P. Chaikin and J. Brujic, Soft Matter, 2013, 9, 9816–9823 RSC.
- M. Hadorn, E. Boenzli, K. T. Srensen, D. De Lucrezia, M. M. Hanczyc and T. Yomo, Langmuir, 2013, 29, 15309–15319 CrossRef CAS PubMed.
- M. Hadorn and P. Eggenberger Hotz, PLoS One, 2010, 5, e9886 Search PubMed.
- P. A. Beales and T. K. Vanderlick, J. Phys. Chem. A, 2007, 111, 12372–12380 CrossRef CAS PubMed.
- P. A. Beales, J. Nam and T. K. Vanderlick, Soft Matter, 2011, 7, 1747–1755 RSC.
- P. A. Beales and T. K. Vanderlick, Adv. Colloid Interface Sci., 2014, 207, 290–305 CrossRef CAS PubMed.
- L. Parolini, B. M. Mognetti, J. Kotar, E. Eiser, P. Cicuta and L. Di Michele, Nat. Commun., 2015, 6, 5948 CrossRef PubMed.
- S. Angioletti-Uberti, P. Varilly, B. M. Mognetti and D. Frenkel, Phys. Rev. Lett., 2014, 113, 128303 CrossRef.
- Y.-Z. Yoon, H. Hong, A. Brown, D. C. Kim, D. J. Kang, V. L. Lew and P. Cicuta, Biophys. J., 2009, 97, 1606–1615 CrossRef CAS PubMed.
- W. Helfrich and R. M. Servuss, Nuovo Cimento Soc. Ital. Fis., D, 1984, 3, 137–151 CrossRef.
- J. Pécréaux, H. G. Döbereiner, J. Prost, J. F. Joanny and P. Bassereau, Eur. Phys. J. E: Soft Matter Biol. Phys., 2004, 13, 277–290 CrossRef PubMed.
- S. B. Smith, Y. Cui and C. Bustamante, Science, 1996, 271, 795–799 CrossRef CAS.
- M. I. Angelova and D. Dimitrov, Faraday Discuss. Chem. Soc., 1989, 81, 303–311 RSC.
- M. I. Angelova, S. Soléau, P. Méléard, J.-F. Faucon and P. Bothorel, Prog. Colloid Polym. Sci., 1992, 89, 127–131 CAS.
- P. S. Cremer and S. G. Boxer, J. Phys. Chem. B, 1999, 103, 2554–2559 CrossRef CAS.
- A. A. Brian and H. M. McConnell, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 6159–6163 CrossRef CAS.
- A. Ramachandran, T. H. Anderson, L. G. Leal and J. N. Israelachvili, Langmuir, 2010, 27, 59–73 CrossRef PubMed.
- E. Evans and D. Needham, J. Phys. Chem., 1987, 91, 4219–4228 CrossRef CAS.
- D. Bracha, E. Karzbrun, G. Shemer, P. A. Pincus and R. H. Bar-Ziv, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4534–4538 CrossRef CAS PubMed.
- J. SantaLucia, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1460–1465 CrossRef CAS.
- L. Di Michele, B. M. Mognetti, T. Yanagishima, P. Varilly, Z. Ruff, D. Frenkel and E. Eiser, J. Am. Chem. Soc., 2014, 136, 6538–6541 CrossRef CAS PubMed.
- S. Angioletti-Uberti, B. M. Mognetti and D. Frenkel, Nat. Mater., 2012, 11, 518–522 CrossRef CAS PubMed.
- J. Hu, R. Lipowsky and T. R. Weikl, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15283–15288 CrossRef CAS PubMed.
- W. Rawicz, K. C. Olbrich, T. McIntosh, D. Needham and E. Evans, Biophys. J., 2000, 79, 328–339 CrossRef CAS.
- W. Rawicz, B. A. Smith, T. J. McIntosh, S. A. Simon and E. Evans, Biophys. J., 2008, 94, 4725–4736 CrossRef CAS PubMed.
- N. Fa, L. Lins, P. J. Courtoy, Y. Dufrêne, P. Van Der Smissen, R. Brasseur, D. Tyteca and M. P. Mingeot-Leclercq, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 1830–1838 CrossRef CAS PubMed.
- J. Pan, S. Tristram-Nagle, N. Kucerka and J. F. Nagle, Biophys. J., 2008, 94, 117–124 CrossRef CAS PubMed.
- G. Stengel, L. Simonsson, R. A. Campbell and F. Höök, J. Phys. Chem. B, 2008, 112, 8264–8274 CrossRef CAS PubMed.
- F. Yuan, L. Griffin, L. Phelps, V. Buschmann, K. Weston and N. L. Greenbaum, Nucleic Acids Res., 2007, 35, 2833–2845 CrossRef CAS PubMed.
- J. J. McCann, U. B. Choi, L. Zheng, K. Weninger and M. E. Bowen, Biophys. J., 2010, 99, 961–970 CrossRef CAS PubMed.
- S. A. J. van der Meulen and M. E. Leunissen, J. Am. Chem. Soc., 2013, 135, 15129–15134 CrossRef CAS PubMed.
- B. G. Moreira, Y. You, M. A. Behlke and R. Owczarzy, Biochem. Biophys. Res. Commun., 2005, 327, 473–484 CrossRef CAS PubMed.
- M. B. Dalva, A. C. McClelland and M. S. Kayser, Nat. Rev. Neurosci., 2007, 8, 206–220 CrossRef CAS PubMed.
- J. T. Parsons, A. R. Horwitz and M. A. Schwartz, Nat. Rev. Mol. Cell Biol., 2010, 11, 633–643 CrossRef CAS PubMed.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, New York, 5th edn, 2007 Search PubMed.
- G. Bao and S. Suresh, Nat. Mater., 2003, 2, 715–725 CrossRef CAS PubMed.
- D. E. Discher, P. Janmey and Y.-l. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS PubMed.
- E. Sackmann and A.-S. Smith, Soft Matter, 2014, 10, 1644–1659 RSC.
- S. F. Fenz and K. Sengupta, Integr. Biol., 2012, 4, 982–995 RSC.
- C. Tordeux, J. B. Fournier and P. Galatola, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2002, 65, 041912 CrossRef CAS.
| This journal is © the Owner Societies 2015 |