
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optical writing and reading with a photoactivatable carbazole†
Jaume
Garcia-Amorós
*a,
Subramani
Swaminathan
a,
Yang
Zhang
a,
Santi
Nonell
 b and
Françisco M.
Raymo
*a
b and
Françisco M.
Raymo
*a
aLaboratory for Molecular Photonics, Department of Chemistry, University of Miami, 1301 Memorial Drive, Coral Gables, Florida 33146-0431, USA. E-mail: jgarciaamoros@ub.edu; fraymo@miami.edu
bGrup d'Enginyeria Molecular, Institut Químic de Sarrià, Universitat Ramón Llull, Via Augusta 390, E-08017, Barcelona, Spain
First published on 31st March 2015
Abstract
The fluorescence of a carbazole chromophore can be activated irreversibly under optical control with the photoinduced opening of an oxazine ring. In proximity to silver nanoparticles, the quantum efficiency of this photochemical transformation and that of the emissive process increase significantly. The plasmonic effects responsible for such enhancements, together with the photochemical and photophysical properties engineered into this particular photoactivatable fluorophore, permit the optical writing and reading of microscaled patterns at low illumination intensities.
The photochemical conversion of a nonemissive reactant into a fluorescent product permits the activation of fluorescence under optical control.1–5 Specifically, illumination of the reactant at an appropriate activation wavelength (λAc) induces the photochemical transformation and irradiation of the resulting product at a given excitation wavelength (λEx) generates fluorescence. Under these conditions, the interplay of two lasers operating at λAc and λEx can be exploited to switch fluorescence on within a defined region of space at a specific interval of time. In turn, such spatiotemporal control allows the monitoring of dynamic processes in real time with the sequential acquisition of fluorescence images after activation.6,7 Additionally, it also enables the reconstruction of fluorescence images with subdiffraction resolution after reiterative activation and excitation sequences.8,9 As a result, photoactivatable fluorophores are becoming invaluable probes for the investigation of the dynamic and structural properties of a diversity of biological specimens and synthetic materials.
The illumination of metallic nanostructures at appropriate wavelengths results in the coupling of incident radiations and surface plasmons to produce a local enhancement in electromagnetic field.10–12 This phenomenon facilitates electronic transitions in proximal chromophores, relative to distal counterparts, to promote photochemical transformations or enhance fluorescence within relatively short distances from the nanostructures. In this context, the concatenation of a photochemical reaction (activation) with a photophysical process (fluorescence), responsible for the operating principles of photoactivatable fluorophores, is particularly valuable to engineer plasmonic effects into photoresponsive materials. In fact, both the activation of the reactant and the emission of the product can be facilitated under the influence of metallic nanostructures. Indeed, we relied on these effects to implement autocatalytic loops with photoactivatable anthracenes,13 enable two-photon activation at low illumination intensities with a photoactivatable carbazole14 and enhance the production and radiative decay of singlet molecular oxygen.15 In the wake of these precedents, we realized that the irreversible activation of fluorescence under plasmonic assistance could be exploited to generate and detect fluorescent patterns efficiently with light. In this article, we report the synthesis of a carbazole–oxazine dyad specifically designed for this application and the experimental implementation of such a mechanism for optical writing and reading.
Compounds 1 and 2 (Fig. 1) were isolated in yields of 9 and 27% respectively, after the condensation of aldehyde 3 with oxazine 4 and indolenine 5 respectively, in the presence of trifluoroacetic acid (TFA). Electrospray ionization mass spectra (ESIMS) together with 1H nuclear magnetic resonance (NMR) spectra confirmed their structural identities.
 | ||
| Fig. 1 Synthesis of 1 and 2 and photochemical transformation of 1 into 2 and 6. | ||
The absorption spectrum (a in Fig. 2) of 1 shows an intense band at 285 nm for the carbazole chromophore. Illumination at a λEx positioned within this absorption does not cause any detectable fluorescence. Electron transfer from the excited carbazole to the adjacent 4-nitrophenoxy group is presumably responsible for preventing radiative deactivation. In fact, the oxidation potential (+0.92 V) of 9-methyl-9H-carbazole (7 in Fig. S1, ESI†), the reduction potential (−1.33 V) of 4 and the energy (3.32 eV) of the first singlet excited state of 7, suggest this process to be exergonic with a free-energy change of −1.1 eV.
 | ||
| Fig. 2 Absorption (a–c) and emission (d–f, λEx = 380 nm) spectra of MeCN solutions (10 μM, 20 °C) of 1, before (a and d) and after (b and e) irradiation (λAc = 350 nm, 4.88 mW cm−2, 10 min), and of 2 (c and f). | ||
Illumination of 1 at a λAc of 350 nm cleaves its 2-nitrobenzyl group to produce 2 and 6 (Fig. 1). Consistently, absorption and emission bands develop at 380 and 490 nm (b and e in Fig. 2) respectively, during the photolytic transformation. These bands resemble those observed in the spectra (c and f in Fig. 2) of 2 and confirm the photoinduced formation of this species. The fluorescence quantum yield (ϕ) of 2, however, is as low as 0.02 and only a relatively weak emission develops after photolysis of 1 (d and e in Fig. 2). Nonetheless, time-correlated single-photon counting (TCSPC) experiments confirm the assignment of the photogenerated band to 2. Specifically, the emission intensity, detected after photolysis of 1, decays biexponentially with lifetimes (τ1 and τ2 in Table S1, ESI†) of 0.65 and 3.6 ns (76 and 24%). Essentially the same temporal fluorescence evolution is also observed for 2 with τ1 and τ2 of 0.94 and 3.8 ns (66 and 34%). Presumably, the biexponential decay is a result of the coexistence of two emissive conformers in solution.
The square wave voltammogram (a in Fig. 3) of 1 shows peaks at −1.40 and −1.86 V for the two consecutive monoelectronic reductions of the nitro group.16 After illumination at λAc, both peaks disappear with the concomitant appearance of new ones at −1.18 and −2.08 V (b in Fig. 3). Literature data17,18 suggest that these additional peaks correspond to reductions of the nitroso and aldehyde groups of 6 respectively and confirm the photoinduced formation of this species.
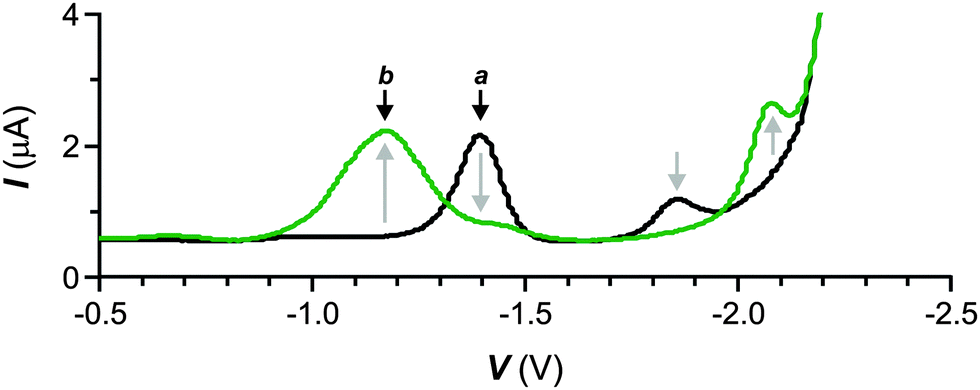 | ||
| Fig. 3 Square wave voltammograms [50 mV s−1, V vs. Ag/Ag+ (1 mM in AgNO3), Bu4NPF6 (50 mM)] of a MeCN solution of 1 (100 μM, 20 °C) before (a) and after (b) illumination (λAc = 350 nm, 4.88 mW cm−2, 10 min). | ||
The photoinduced conversion of 1 into 2 and 6, observed in acetonitrile solutions, occurs also within poly(butylmethacrylate) (PBMA) matrices with similar spectral changes (Fig. S2, ESI†). In PBMA, however, ϕ for 2 raises to 0.23, while τ1 and τ2 for the corresponding biexponential decay drop to 0.15 and 1.1 ns (87 and 13%). These trends suggest that the rigid polymer matrix accelerates the radiative deactivation of the excited state, while delaying its nonradiative decay. Presumably, the restricted rotation of the 3H-indole and carbazole fragments, around the adjacent [C–C] bonds, in the rigid environment is mostly responsible for this behavior. Essentially the same temporal fluorescence profile is detected also after illumination of a PBMA film, doped with 1, at λAc. Once again, the emission intensity decays biexponentially with τ1 and τ2 of 0.15 and 1.1 ns (87 and 13%), confirming that the fluorescent species photogenerated in the polymer matrix is indeed 2.
Fluorescence images of doped PBMA films further confirm the photoinduced formation of the fluorescent product. Specifically, illumination of an ellipsoidal region at a λAc of 405 nm for 90 s converts 1 into 2 exclusively in the irradiated area. Comparison of images (a and b in Fig. 4 and Fig. S3, ESI†), acquired by irradiating the entire field of view at the same wavelength (λEx = λAc) for only 432 ms before and after activation, shows significant emission only in the activated area. Furthermore, the acquisition of an additional image (c in Fig. 4 and Fig. S3, ESI†), 10 min after activation, does not reveal any noticeable change in the shape of the imprinted fluorescent pattern. Thus, the photogenerated fluorophores cannot diffuse within the rigid polymer matrix on this timescale. In turn, the lack of any diffusion offers the opportunity to induce and monitor the photochemical transformation within the very same area of the substrate. Specifically, the continuous illumination of a defined region of interest at λAc, with concomitant detection of the light emitted from the same area (a in Fig. 5), permits the acquisition of a temporal profile of the fluorogenic transformation. The resulting plot (c in Fig. 5) shows an increase in the emission of the fluorescent product with saturation reached after ca. 15 min. The time (t50) required to convert 50% of 1 into 2 is 294 s under these experimental conditions.
 | ||
| Fig. 4 Confocal laser-scanning fluorescence images (λEx = 405 nm, λEm = 430–650 nm, scale bar = 30 μm) of a PBMA film, doped with 1 (3% w/w) and deposited on a glass slide with silver nanoparticles on its surface, recorded before (a), immediately (b) and 10 min (c) after illumination (λAc = 405 nm, 0.1 mW, 90 s) of an ellipsoidal area within the imaging field. | ||
 | ||
| Fig. 5 Confocal laser-scanning fluorescence images (λEx = 405 nm, λEm = 430–650 nm, scale bar = 30 μm) of PBMA films, doped with 1 (3% w/w) and deposited on glass slides without (a) and with (b) silver nanoparticles on their surface, recorded after illumination (λAc = 405 nm, 0.1 mW, 15 min) of the entire imaging field and corresponding temporal evolutions (c and d, Δt = 432 ms) of the integrated emission intensity. | ||
The wavelength employed to activate 1 and excite 2 in PBMA happens to be positioned within the surface-plasmon band of silver nanoparticles (Fig. S4, ESI†).13a As a result, these particular metal nanostructures can have a pronounced influence on the photochemical transformation of 1 into 2 as well as on the photophysical behavior of the product. Indeed, a remarkable increase in fluorescence (b in Fig. 5) is actually observed upon activation of a polymer film deposited on a glass slide pre-coated with silver nanoparticles.‡ Under these conditions, the emission intensity, integrated within the activated area, is 6.0 times greater than that detected (a in Fig. 5) in the absence of metal nanostructures. This value is consistent with the ratio between the emission intensities recorded for 2 in PBMA with and without silver nanoparticles (Fig. S4, ESI†), which is 5.6. Thus, the interaction of the exciting radiations with the surface plasmons of the metal nanostructures clearly enhances the emission intensity of the fluorescence product. The local increase in electromagnetic field facilitates the excitation of proximal fluorophores. Additionally, the nanoparticles accelerate the radiative deactivation of the excited molecules. In fact, the fluorescence of 2 decays biexponentially also in the presence of the metal nanostructures with a τ1 of 0.15 ns (86%) once again, but τ2 decreases to only 0.79 ns (14%). Similarly, the very same plasmonic effects appear also to accelerate the photochemical reaction. In the presence of the nanoparticles, the temporal evolution of the emission intensity (d in Fig. 5) reaches saturation after only ca. 5 min and the corresponding t50 shortens to 66 s.
The ability of the silver nanoparticles to facilitate the photoinduced conversion of a nonemissive species into a fluorescent one and to enhance the emission of the latter can be exploited to write and read microstructures with visible light at relatively low illumination intensities. Arrays of dots (a in Fig. 6) with a diameter of ca. 20 μm or even letters (b in Fig. 6) with microscaled dimensions can be imprinted on a PBMA film, doped with 1, under the influence of an activating laser and then read by measuring the fluorescence of 2 photogenerated in the illuminated areas. Furthermore, the lack of any diffusion of the fluorescent species within the polymer matrix preserves the geometry of the written pattern and permits their optical reading, even after relatively long times. Specifically, the imprinted information can be read ca. 320 consecutive times before the signal-to-background ratio decreases from infinite to 1.25.
 | ||
| Fig. 6 Confocal laser-scanning fluorescence images (a and b, λEx = 405 nm, λEm = 430–650 nm, scale bar = 30 μm) of PBMA films, doped with 1 (3% w/w) and deposited on glass slides with silver nanoparticles on their surface, recorded after illumination (λAc = 405 nm, 0.1 mW, 300 s) of defined areas within the imaging field. Profile of the emission intensity (c) measured across some of the activated areas in a. | ||
In summary, our results demonstrate that the covalent connection of a carbazole fluorophore to a photoresponsive oxazine permits the activation of the emission of the former with the photoinduced cleavage of the latter. This photochemical transformation is facilitated if the carbazole–oxazine dyads are immobilized within a polymer matrix in proximity to silver nanoparticles. The plasmonic effects responsible for promoting the activation of the reactant cause also a pronounced enhancement in the emission of the product. As a result, microscaled fluorescent patterns can be generated and detected efficiently with far-field optics at relatively low illumination intensities. Thus, these operating principles can lead to the development of strategies for the optical writing and reading of information based on metallic nanostructures and photoactivatable fluorophores. In addition, the activated fluorescence of this system can be exploited to monitor conveniently the influence of plasmonic effects on photochemical reactions. Therefore, such structural design can evolve into valuable molecular probes to investigate the fundamental factors regulating the influence of metallic nanostructures on the excitation dynamics of organic chromophores.
JGA is grateful for a Beatriu de Pinós postdoctoral grant from the Generalitat de Catalunya (Spain, grant 2011 BP-A-00270). FMR acknowledges the NSF (CHE-1049860) for financial support.
Notes and references
- L. M. Wysocki and L. D. Lavis, Curr. Opin. Chem. Biol., 2011, 15, 752–759 CrossRef CAS PubMed.
- D. Puliti, D. Warther, C. Orange, A. Specht and M. Goeldner, Bioorg. Med. Chem., 2011, 19, 1023–1029 CrossRef CAS PubMed.
- W.-H. Li and W.-H. Zheng, Photochem. Photobiol. Sci., 2012, 11, 460–471 CAS.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- F. M. Raymo, Phys. Chem. Chem. Phys., 2013, 15, 14840–14850 RSC.
- Y. Xu, T. J. Melia and D. T. Toomre, Curr. Opin. Chem. Biol., 2011, 15, 822–830 CrossRef CAS PubMed.
- F. M. Raymo, ISRN Phys. Chem., 2012, 619251 Search PubMed.
- S. van de Linde, M. Heilemann and M. Sauer, Annu. Rev. Phys. Chem., 2012, 63, 519–540 CrossRef CAS PubMed.
- T. Ha and P. Tinnefeld, Annu. Rev. Phys. Chem., 2012, 63, 595–617 CrossRef CAS PubMed.
- R. A. Alvarez-Puebla, L. M. Liz-Marzán and F. J. García de Abajo, J. Phys. Chem. Lett., 2010, 1, 2428–2434 CrossRef CAS.
- H. Nabika, M. Takase, F. Nagasawa and K. Murakoshi, J. Phys. Chem. Lett., 2010, 1, 2470–2487 CrossRef CAS.
- J. C. Scaiano and K. Stamplecoskie, J. Phys. Chem. Lett., 2013, 4, 1177–1188 CrossRef CAS.
- (a) E. R. Thapaliya, S. Swaminathan, B. Captain and F. M. Raymo, J. Am. Chem. Soc., 2014, 136, 13798–13804 CrossRef CAS PubMed; (b) E. R. Thapaliya, B. Captain and F. M. Raymo, Asian J. Org. Chem., 2015, 4, 233–238 CrossRef CAS.
- J. Garcia-Amorós, S. Swaminathan, S. Sortino and F. M. Raymo, Chem. – Eur. J., 2014, 20, 10276–10284 CrossRef PubMed.
- X. Ragàs, A. Gallardo, Y. Zhang, W. Massad, C. D. Geddes and S. Nonell, J. Phys. Chem. C, 2011, 115, 16275–16281 Search PubMed.
- D. S. Silvester, A. J. Wain, L. Aldous, C. Hardacre and R. G. Compton, J. Electroanal. Chem., 2006, 596, 131–140 CrossRef CAS PubMed.
- N. R. Armstrong, R. K. Quinn and N. E. Vanderborgh, Anal. Chem., 1974, 46, 1759–1764 CrossRef CAS.
- L. J. Núñez-Vergara, J. A. Squella, C. Olea-Azar, S. Bollo, P. A. Navarrete-Encina and J. C. Sturm, Electrochim. Acta, 2000, 45, 3555–3561 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, voltammograms, absorption and emission spectra, fluorescence images, atomic force microscopy (AFM) images, fluorescence decays. See DOI: 10.1039/c5cp01336d |
| ‡ AFM images (Fig. S6, ESI†) show that the average diameter of the nanoparticles is ca. 20 nm. |
| This journal is © the Owner Societies 2015 |